一种香附四物颗粒的制备方法及其质量控制方法与流程
本发明属于中药配方颗粒技术领域,具体涉及一种香附四物颗粒的制备方法及其质量控制方法。
背景技术:
香附四物汤由香附、木香、延胡索、当归、川芎、熟地黄、白芍等七味药组成,为行气化瘀代表方之一,出自清代梁廉夫《不知医必要》卷四,具有养血调血,行气止痛之功效,主治气滞血瘀所致痛经、月经不调等症。
痛经的发生主要与经期子宫内膜合成和释放前列腺素增加有关,子宫肌反应性过高,继发子宫肌层缺血导致疼痛。中医认为,痛经的主要病机是由情志所伤、起居不慎或六淫为害等病因造成的气血运行不畅、气机阻滞,不通则痛。气血运行不畅是最主要的病理基础,临床上则以气滞血瘀型的痛经多见。
香附四物汤由四物汤加香附、木香、延胡索组成,具有养血调血行气止痛的功效,主治气滞血瘀所致痛经、月经不调等症。是行气化瘀代表方之一。香附味辛、甘、微苦,性平偏温,主要归肝经,辛散温通,入肝,能通行气血,善调理肝气之郁,又能调畅经血之滞,故有调肝理气,调经止痛之作用,李时珍称赞其为“气病之总司,女科之主帅”,说明对气滞血瘀型妇科病有特殊疗效。常与当归合用,一主气分,一主血分,气血并治,共奏理气活血之功。木香,味辛、苦,性温,主要归脾、胃、大肠、三焦经,辛香行散,苦降温通,能升可降,通理三焦,有行气止痛,健脾消食之功效,常与香附、延胡索等合用,以疏肝活血调经止痛。延胡索,味辛、苦性温,主要归心、肝、脾、肺经,辛散苦泄温通,即入心肝经走血分,又入脾肺经走气分,既能活血,又能行血中之气,而且又能止痛,故为活血行气止痛之要药,凡一身上下诸通之属与气滞血瘀者,均可用之。常配以当归等,以通络行滞祛痛。
生物效应研究表明香附四物汤对整体小鼠痛经模型具有显著的抑制作用,能显著抑制小鼠离体子宫的收缩频率及收缩活动力,并对COX-2酶的抑制作用显著增强;能够明显改善气滞血瘀模型大鼠血液流变学指标,干预的血虚小鼠模型改善外周血象作用不明显,在抑制痛经作用中以镇痛为主。临床研究表明,该方治疗气滞血瘀型原发性痛经的总有效率超过80%,较大品种月月舒痛经宝颗粒、布洛芬缓释胶囊更能够改善患者痛经症状。
中药汤剂作为传统的中医临床用药的主要剂型,具有随证加减、灵活组方、易于吸收、起效较快等优点,深受广大患者的信赖。却因调配、携带、临时煎煮、久置容易发生霉败变质、汤液味苦、量大等缺点不能适应现代人的生活要求。中药配方颗粒保持了单味药的多药效性,最大限度的保留了药物的有效成分,但目前中药配方颗粒都是单味中药提取物,使用时“单味提取,混合冲服”,存在“共煎”和“分煎”的差异性问题。复方配方颗粒传承了中药单味配方颗粒的优点,同时又考虑了饮片在煎煮过程中的相互作用,符合中医传统用药理论,具有较大的价值。
技术实现要素:
本发明的目的在于提供一种香附四物颗粒的制备方法及其质量控制方法。
本发明是通过以下技术方案实现的:
一种香附四物颗粒的制备方法,它是由重量比3:2:3:6:3:8:3的香附、木香、延胡索、当归、川芎、熟地和白芍制成,具体包括以下步骤:
a.按以上重量比称取当归、川芎、香附和木香饮片,加入药材总重量5-15倍重量的水,浸泡10-15小时后,然后水蒸气蒸馏提取8-12小时,提取挥发油;
b.取挥发油加入β-环糊精和水充分研磨,冷冻干燥得挥发油包合物;
c.取挥发油提取后所得残渣,加入上述重量比的熟地、白芍和延胡索,加水煎煮2~3次,每次加水量为总药材的6-14倍,煎煮1-3小时,合并药液,滤过,减压干燥至相对密度1.00-1.15的浓缩液,加入体积浓度95%乙醇至浓缩液中乙醇浓度为80~85%,静置,抽滤,取滤液,减压浓缩,干燥,得干膏粉;
d.取干膏粉按一定比例加入糊精、糖粉,混匀,制粒,干燥,整粒,最后加入步骤b制得的挥发油包合物,即得。
作为优选方案,以上所述的香附四物颗粒的制备方法,具体包括以下步骤:
a.按以上重量比称取当归、川芎、香附和木香饮片,加入药材总重量8倍重量的水,浸泡12小时后,水蒸气蒸馏提取10小时,提取挥发油;
b.取挥发油加入β-环糊精和水充分研磨,冷冻干燥得挥发油包合物;
c.挥发油提取后所得残渣,加入上述重量比的熟地、白芍和延胡索,加水煎煮2次,每次加水量为总药材的10倍,煎煮3小时,合并药液,滤过,减压干燥至相对密度1.05-1.08的浓缩液,加入体积浓度为95%乙醇至浓缩液中乙醇浓度为80%,静置24小时后,抽滤,滤液减压浓缩,干燥,得干膏粉;
d.取干膏粉加入糊精、糖粉,混匀,制粒,干燥,整粒,最后加入步骤b制备得到的挥发油包合物,即得。
本发明的当归、川芎、香附和木香含有挥发油,为最大限度的提取有效成分,本发明采用挥发油收集装置,获得挥发油,并对其进行包合处理。优选的步骤b中,所述β-环糊精的加入量为挥发油体积的5-20倍,特别优选8-15倍;水的加入量为挥发油体积的10-50倍,特别优选20-40倍。
优选的步骤d中,糊精的加入量为干膏粉重量的0-2倍,特别优选1~2倍,糖粉的加入量为干膏粉重量的0-2倍,特别优选1~2倍。
本发明还提供了一种上述制备方法制备的香附四物颗粒的质量控制方法,采用薄层色谱定性鉴别和高效液相色谱含量测定。
具体包括以下方法中的一种或几种:
所述的香附四物颗粒的质量控制方法,其特征在于,包括以下方法中的一种或几种:
(1)采用薄层色谱定性鉴别
取香附四物颗粒,研细,加碳酸氢钠溶液,超声处理,离心,取上清液用稀盐酸调节pH值至2~3,用乙醚振摇提取2~3次,合并乙醚液,挥干,残渣加甲醇使溶解,作为藁本内酯供试品溶液;
取香附四物颗粒,加乙醇超声溶解,置水浴上浓缩,用乙醚振摇提取2~3次,合并乙醚提取液,再向上述乙醚溶液中加碳酸钠溶液振摇提取,弃去乙醚液,提取液用稀盐酸调节pH值至2~3,再用乙醚振摇提取,合并乙醚提取液,挥干,残渣加甲醇溶解,作为阿魏酸供试品溶液;
取当归、川芎对照药材,加乙醇超声处理,置水浴上浓缩,用乙醚振摇提取2~3次,合并乙醚提取液,再向上述乙醚溶液中加碳酸钠溶液振摇提取,弃去乙醚液,提取液用稀盐酸调节pH值至2~3,再用乙醚振摇提取,合并乙醚提取液,挥干,残渣加甲醇溶解,制成对照药材溶液;
取香附、木香、延胡索、熟地和白芍,加乙醇超声处理,置水浴上浓缩,用乙醚振摇提取2~3次,合并乙醚提取液,再向上述乙醚溶液中加碳酸钠溶液振摇提取,弃去乙醚液,提取液用稀盐酸调节pH值至2~3,再用乙醚振摇提取,合并乙醚提取液,挥干,残渣加甲醇溶解,制成双阴性对照溶液;
取阿魏酸和藁本内酯对照品,加甲醇制成对照品溶液;
照薄层色谱法试验,吸取上述5种溶液,分别点于同一硅胶G薄层板上,以体积比为30:1:3的苯-冰醋酸-甲醇为展开剂,展开,取出,凉干,置紫外光灯下,365nm检视,结果,供试品与对照品的相应位置上,显相同颜色的斑点,阴性对照品无干扰;
(2)采用薄层色谱定性鉴别
取香附四物颗粒,研细,加乙醚加热回流,滤过,滤液挥干,残渣加乙酸乙酯溶解,作为供试品溶液;
取川芎药材粉末,加乙醚加热回流,滤过,滤液挥干,残渣加乙酸乙酯溶解,制成对照药材溶液;
取香附、木香、延胡索、熟地、当归和白芍,加乙醚加热回流,滤过,滤液挥干,残渣加乙酸乙酯溶解,制成阴性对照溶液;
取欧当归内酯A对照品,加乙酸乙酯制成对照品溶液;
照薄层色谱法试验,吸取上述4种溶液,分别点于同一硅胶GF254薄层板上,以体积比为3:1的正己烷-乙酸乙酯为展开剂,展开,取出,凉干,置紫外光灯下254nm检视,结果,供试品与对照品的相应位置上,显相同颜色的斑点,阴性对照品无干扰;
(3)采用薄层色谱定性鉴别
取香附四物颗粒,研细,加乙醚,放置1~2小时,时时振摇,滤过,滤液挥干,残渣加乙酸乙酯使溶解,作为供试品溶液;
取香附对照药材粉末,加乙醚,放置1~2小时,时时振摇,滤过,滤液挥干,残渣加乙酸乙酯使溶解制成对照药材溶液;
取木香、延胡索、当归、川芎、熟地和白芍药材,加乙醚,放置1~2小时,时时振摇,滤过,滤液挥干,残渣加乙酸乙酯使溶解制成阴性对照溶液;
取α-香附酮对照品,加乙酸乙酯制成对照品溶液;
照薄层色谱法试验,吸取上述4种溶液,分别点于同一硅胶GF254薄层板上,用体积比为80:1:1的二氯甲烷-乙酸乙酯-冰醋酸为展开剂,展开,取出,凉干,置紫外光灯254nm下检视,再喷以2,4-二硝基苯肼乙醇试液,可见光下检视;结果,供试品与对照品的相应位置上,显相同颜色的斑点,阴性对照品无干扰;
(4)采用薄层色谱定性鉴别
取香附四物颗粒,研细,加石油醚,冷浸30~60分钟,时时振摇,滤过,滤液挥干,残渣加醋酸乙酯使溶解,作为供试品溶液;
取木香对照药材的粉末,加石油醚,冷浸30~60分钟,时时振摇,滤过,滤液挥干,残渣加醋酸乙酯使溶解制成对照药材溶液;
取香附、延胡索、当归、川芎、熟地和白芍,加石油醚,冷浸30~60分钟,时时振摇,滤过,滤液挥干,残渣加醋酸乙酯使溶解制成阴性对照溶液;
取去氢木香内酯对照品和木香烃内酯对照品,加甲醇制成对照品溶液;
照薄层色谱法试验,吸取上述4种溶液,分别点于同一硅胶G薄层板上,用体积比15:5:1以环己烷-乙酸乙酯-甲酸为展开剂,展开,取出,凉干,喷以5%香草醛硫酸乙醇溶液,加热,至斑点显色清晰,日光检视,结果,供试品与对照品的相应位置上,显相同颜色的斑点,阴性对照品无干扰;
(5)采用薄层色谱定性鉴别
取颗粒15g,研细,加水100mL,温热使其充分溶散,放冷,用脱脂棉滤过,滤液用乙醚振摇提取2次,每次25mL,弃去乙醚液,再用乙酸乙酯振摇提取2次,每次25mL,合并乙酸乙酯液,蒸干,残渣加甲醇1mL使溶解,作为供试品溶液。取1g熟地药材的粉末,同法制成对照药材溶液;取缺熟地样品15g,同法制成阴性对照溶液。取5-羟甲基糠醛对照品,加甲醇制成每1mL含0.290mg的溶液,作为对照品溶液。照薄层色谱法(通则0502)试验,吸取上述4种溶液,分别点于同一硅胶G薄层板上,以二甲苯-乙酸乙酯(1:1)为展开剂,展开,取出,凉干,喷以2,4-二硝基苯肼乙醇溶液,日光检视。结果,供试品与对照品的相应位置上,显相同颜色的斑点。阴性对照品无干扰。
(6)采用薄层色谱定性鉴别
取香附四物颗粒,研细,加水,加热溶解,用水饱和正丁醇萃取2~3次,合并正丁醇液,以正丁醇饱和水洗涤,合并正丁醇液,水浴挥干,加甲醇溶解,采用硅胶柱,先以体积比为10:1的三氯甲烷和甲醇洗脱,再以体积比为9:1的三氯甲烷和甲醇洗脱,最好以体积比为8:1的三氯甲烷和甲醇洗脱,收集后续8:1洗脱液,蒸干,残渣加甲醇溶解,作为供试品溶液;
取白芍对照药材粉末,加水,超声处理,用水饱和正丁醇萃取2~3次,合并正丁醇液,以正丁醇饱和水洗涤,合并正丁醇液,水浴挥干,加甲醇溶解,采用硅胶柱,先以体积比为10:1的三氯甲烷和甲醇洗脱,再以体积比为9:1的三氯甲烷和甲醇洗脱,最好以体积比为8:1的三氯甲烷和甲醇洗脱,收集后续8:1洗脱液,蒸干,残渣加甲醇溶解,制成对照药材溶液;
取香附、木香、延胡索、当归、川芎和熟地药材,加水,超声处理,用水饱和正丁醇萃取2~3次,合并正丁醇液,以正丁醇饱和水洗涤,合并正丁醇液,水浴挥干,加甲醇溶解,采用硅胶柱,先以体积比为10:1的三氯甲烷和甲醇洗脱,再以体积比为9:1的三氯甲烷和甲醇洗脱,最好以体积比为8:1的三氯甲烷和甲醇洗脱,收集后续8:1洗脱液,蒸干,残渣加甲醇溶解,制成阴性对照溶液;
精密称取芍药苷对照品,加甲醇制成对照品溶液;
照薄层色谱法试验,吸取上述4种溶液,分别点于同一硅胶G薄层板上,以体积比为40:5:10:0.2三氯甲烷-乙酸乙酯-甲醇-甲酸为展开剂,展开,取出,凉干,喷以5%香草醛硫酸乙醇溶液,日光检视,结果,供试品与对照品的相应位置上,显相同颜色的斑点,阴性对照品无干扰。
(7)采用薄层色谱定性鉴别
取香附四物颗粒,研细,加甲醇,超声处理,滤过,滤液蒸干,残渣加水溶解,加浓氨试液调至碱性,用乙醚振摇提取,合并乙醚液,蒸干,残渣加甲醇溶解,作为供试品溶液;
取延胡索对照药材的粉末,加甲醇,超声处理,滤过,滤液蒸干,残渣加水溶解,加浓氨试液调至碱性,用乙醚振摇提取,合并乙醚液,蒸干,残渣加甲醇溶解制成对照药材溶液;
取香附、木香、当归、川芎、熟地和白芍,加甲醇,超声处理,滤过,滤液蒸干,残渣加水溶解,加浓氨试液调至碱性,用乙醚振摇提取,合并乙醚液,蒸干,残渣加甲醇溶解制成阴性对照溶液;
精密称取延胡索乙素对照品,加甲醇制成对照品溶液;
照薄层色谱法试验,吸取上述4种溶液,分别点于同一硅胶G薄层板上,以体积比为9:2甲苯-丙酮为展开剂,展开,取出,凉干,喷以改良碘化铋钾溶液,日光检视,结果,供试品与对照品的相应位置上,显相同颜色的斑点,阴性对照品无干扰。
本发明所述的香附四物颗粒的质量控制方法,包括以下方法中的一种或几种:
(1)香附四物颗粒的含量测定,采用高效液相色谱法,具体步骤如下:
色谱条件与系统适应性试验以十八烷基硅烷键合硅胶为填充剂;检测阿魏酸时以体积比20:80乙腈-1%冰乙酸溶液为流动相;检测香草酸时以体积比12:88的乙腈-1%冰乙酸溶液为流动相;检测波长分别为323nm和260nm;理论塔板数按阿魏酸和香草酸计算均不低于3000;
对照品溶液的制备取阿魏酸、香草酸对照品适量,精密称定,分别加甲醇制成每1mL含0.0316mg、0.0295mg的对照品溶液;
供试品溶液的制备取香附四物颗粒,精密称定,置具塞锥形瓶中,精密加甲醇,称重,放置过夜,超声,再称重,用甲醇补足损失的质量,摇匀,滤过,精密量取续滤液,水浴蒸干;残渣用氢氧化钠溶液溶解,转移入回流烧瓶中,水浴回流,放冷;移入分液漏斗中,用盐酸调pH=2,用乙醚萃取,合并乙醚液,回收乙醚,用甲醇溶解,摇匀,即得;
测定法分别精密吸取对照品溶液与供试品溶液各10μL,注入液相色谱仪,测定,即得;
本品每15g颗粒含当归和川芎以阿魏酸和香草酸计,不得少于0.714mg和0.439mg;
(2)香附四物颗粒的含量测定,采用高效液相色谱法,具体步骤如下:
色谱条件与系统适应性试验以十八烷基硅烷键合硅胶为填充剂;以体积比18:82乙腈-0.1%乙酸水为流动相;检测波长为230nm,理论塔板数按芍药苷和芍药内酯苷计算均不低于2000;
对照品溶液的制备取芍药苷、芍药内酯苷对照品适量,精密称定,分别加甲醇制成每1mL含0.300mg、0.200mg的对照品溶液。
供试品溶液的制备取香附四物颗粒,精密称定,置具塞锥形瓶中,精密加入甲醇,精密称定,超声处理,放冷,再称定,用甲醇补足减失质量,摇匀,滤过,取续滤液作为供试品溶液;
测定法分别精密吸取对照品溶液与供试品溶液各10μL,注入液相色谱仪,测定,即得。
本品每15g含白芍以芍药苷和芍药内酯苷计,不得少于52.5mg和17.9mg;
(3)香附四物颗粒的含量测定,采用高效液相色谱法,具体步骤如下:
色谱条件与系统适应性试验以十八烷基硅烷键合硅胶为填充剂;以体积比为55:45乙腈-0.1%乙酸水,并用三乙胺调pH=6.0作为为流动相;检测波长为280nm;理论塔板数按延胡索乙素和盐酸小檗碱计算均不低于3000;
对照品溶液的制备取延胡索乙素、盐酸小檗碱对照品适量,精密称定,分别加甲醇制成每1mL含0.246mg、0.297mg的对照品溶液;
供试品溶液的制备取香附四物颗粒15g,精密称定,置平底烧瓶中,精密加入体积比为1:20的浓氨试液-甲醇混合溶液,称定重量,冷浸,加热回流,放冷,再称定重量,用体积比为1:20浓氨试液-甲醇混合溶液补足减失的重量,摇匀,滤过;
精密量取续滤液75mL,蒸干,残渣加甲醇溶解,转移至10mL量瓶中,并稀释至刻度,摇匀,滤过,取续滤液,即得;
测定法分别精密吸取对照品溶液与供试品溶液各10μL,注入液相色谱仪,测定,即得;
本品每15g颗粒含延胡索以延胡索乙素和盐酸小檗碱计,不得少于2.67mg和1.22mg。
本发明与现有技术相比,具有如下有益效果:
(1)经典名方香附四物汤按照传统方法合煎制成复方配方颗粒,较目前单味配方颗粒服用时各药味简单相加,更能充分发挥中药配伍的优势,体现了中医药整体的观念,确保达到减毒增效的目的,为临床用药提供新的选择。
(2)本发明在煎煮提取过程中收集挥发油有效部位,挥发油用β-环糊精包合后再制成颗粒,挥发性降低,利用率增加,可促进非挥发性成分的吸收,提高药物生物利用度。
(3)本发明采用色谱法,即薄层色谱法和高效液相色谱法,对有效成分进行定性和定量分析,进行质量控制,可以很好的保证产品质量。
(4)本发明对香附四物颗粒建立了利用高效液相色谱法同时测定颗粒中阿魏酸、香草酸、芍药苷、芍药内酯苷、延胡索乙素和盐酸小檗碱六种主要指标性成分含量的质量控制方法,该质量标准实现了对处方中白芍、当归、川芎、延胡索四味饮片含量同时进行控制,具有较强的专属性和良好的重现性,真正体现药品安全有效、质量可控。
附图说明
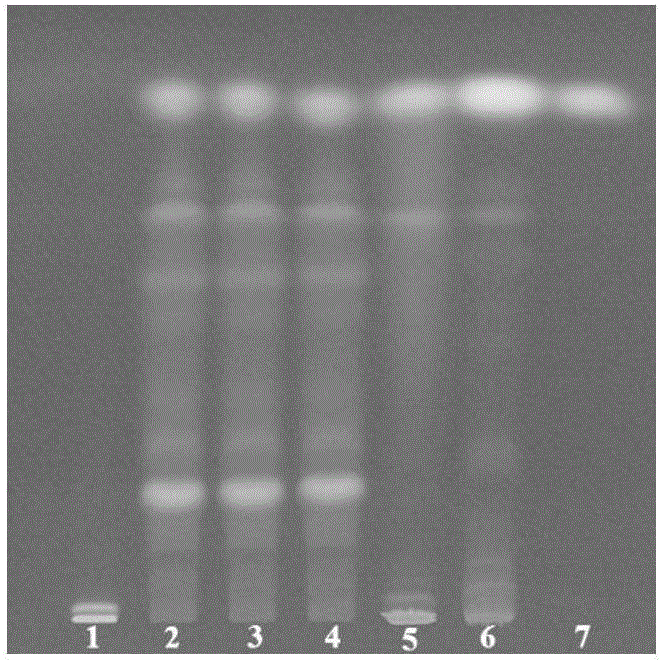
图1为实施例1的薄层色谱图,其中1为缺当归、川芎双阴性样品,2-4为3批次供试品,5为当归对照药材,6为川芎对照药材,7为阿魏酸对照品;
图2为实施例1的薄层色谱图,其中1为缺当归、川芎双阴性样品,2-4为3批次供试品,5为当归对照药材,6为川芎对照药材,7为藁本内酯对照品;
图3为实施例2的薄层色谱图,其中1为缺川芎阴性样品,2-4为3批次供试品,5为川芎对照药材,6为欧当归内酯A对照品;
图4为实施例3的薄层色谱图,其中1为缺香附阴性样品,2-4为3批次供试品,5为香附对照药材,6为α-香附酮对照品;
图5为实施例4的薄层色谱图,其中1为缺木香阴性样品,2-4为3批次供试品,5为木香对照药材,6为去氢木香内酯对照品,7为木香烃内酯对照品;
图6为实施例5的薄层色谱图,其中1为缺熟地阴性样品,2-4为3批次供试品,5为熟地对照药材,6为5-羟甲基糠醛对照品;
图7为实施例6的薄层色谱图,其中1为缺白芍阴性样品,2-4为3批次供试品,5为白芍对照药材,6为芍药苷对照品;
图8为实施例7的薄层色谱图,其中1为缺延胡索阴性样品,2-4为3批次供试品,5为延胡索对照药材,6为延胡索乙素对照品;
图9为实施例8的高效液相色谱图,其中A为阿魏酸对照品,B为供试品溶液,C为缺当归和川芎双阴性样品,1为阿魏酸;
图10为实施例8的高效液相色谱图,其中A为香草酸对照品,B为供试品溶液,C为缺当归和川芎双阴性样品,1为香草酸;
图11为实施例8的高效液相色谱图,其中A为芍药苷和芍药内酯苷混合对照品,B为供试品溶液,C缺白芍阴性样品,1为芍药内酯苷,2为芍药苷;
图12为实施例8的高效液相色谱图,其中A为延胡索乙素和盐酸小檗碱对照品,B为供试品,C为缺延胡索阴性样品,1为盐酸小檗碱,2为延胡索乙素。
具体实施方式
下面通过具体实施方式来进一步说明本发明,以下实施例为本发明具体的实施方式,但本发明的实施方式并不受下述实施例的限制。
实施例1:
一种香附四物颗粒的制备方法,包括如下步骤:
a.取香附300g、木香200g、当归600g、川芎300g,加入14kg的水,浸泡12小时后,水蒸气蒸馏提取10小时,提取挥发油。
b.挥发油加入β-环糊精(加入量为挥发油体积量的10倍)和水(加入量为挥发油体积量的30倍)充分研磨2小时,冷冻干燥得挥发油包合物。
c.挥发油提取后所得残渣,加相应比例的熟地800g、白芍300g和延胡索300g,加水煎煮2次,每次加水量30kg,煎煮2小时,抽出药液,合并药液,滤过,减压干燥至相对密度1.05的浓缩液,加入95%乙醇至体系中乙醇浓度为80%(v/v),静置24小时后,抽滤,滤液减压浓缩,干燥,得干膏粉。
d.干膏粉加入糊精(与干膏粉等量)和糖粉(与干膏粉等量),混匀,制粒,干燥,整粒,最后加入挥发油包合物,即得香附四物颗粒。
对香附四物颗粒采用薄层色谱定性鉴别,具体方法如下:
1.样品的制备
1.1对照品溶液的制备
取阿魏酸和藁本内酯对照品,加甲醇制成每1mL含2.05mg和2.00mg的溶液,作为对照品溶液。
1.2对照药材溶液的制备
取当归、川芎对照药材粉末各3g,加1%碳酸氢钠溶液150mL,超声处理10min,离心,取上清液用稀盐酸调节pH值至2~3,用乙醚振摇提取2次,每次40mL,合并乙醚液,挥干,残渣加甲醇1mL使溶解,作为对照药材溶液。
1.3供试品溶液的处理方法
供试品溶液1:取上述香附四物颗粒15g,加1%碳酸氢钠溶液150mL,超声处理10min,离心,取上清液用稀盐酸调节pH值至2~3,用乙醚振摇提取2次,每次40mL,合并乙醚液,挥干,残渣加甲醇1mL使溶解,作为供试品溶液1;
供试品溶液2:取上述香附四物颗粒15g至烧杯中,加80%乙醇150mL超声溶解,置水浴上蒸去乙醇至约30mL,用乙醚振摇提取2次,每次40mL,合并乙醚提取液,再向上述乙醚溶液中加1%碳酸钠溶液振摇提取2次,每次40mL,弃去乙醚液,提取液用稀盐酸调节pH值至2~3,再用乙醚振摇提取2次,每次60mL,合并乙醚提取液,挥干,残渣加甲醇1mL溶解,作为供试品溶液2。
1.4阴性对照溶液的制备
按上述处方比例及制法,制成缺当归和川芎药材的双阴性对照样品。按供试品溶液1和2的制备方法,制成双阴性对照溶液。
2.展开剂的条件优化
为优化展开剂,吸取上述溶液分别点于同一硅胶G薄层板上,选用二种展开体系,展开,取出,晾干,置紫外光灯(365nm)下检视,结果显示,阿魏酸和藁本内酯在展开系统A中均有明显斑点,在展开系统B中阿魏酸斑点不明显且分离效果不好,故选择苯-冰醋酸-甲醇(30:1:3)为阿魏酸和藁本内酯的展开剂。
3.点样量的确定
3.1对照品溶液点样量的确定
分别吸取阿魏酸对照品溶液各1、2、4、6μL依次点于同一硅胶G薄层板上,分别吸取藁本内酯对照品溶液1、2、3、4μL依次点于同一硅胶G薄层板上,以确定好的TLC分析展开条件进行分析,两种对照品不同点样量都有明显斑点,阿魏酸在4μL时斑点最为清晰,藁本内酯在2μL时即有较明亮的斑点。所以选择4μL和2μL分别为阿魏酸和藁本内酯的点样量。
3.2供试品溶液点样量的确定
分别吸取供试品溶液1各4、6、8、10μL,分别吸取供试品溶液2各2、4、6、8μL依次分别点于同一硅胶G薄层板,以确定的TLC条件进行分析,供试品溶液1在4~10μL内斑点皆清晰,点样量可为4μL;供试品溶液2在8μL时斑点最明显,点样量可选8μL。
4.样品检验
根据确定的TLC鉴别条件,对3批次香附四物颗粒进行阿魏酸和藁本内酯的TLC鉴别,结果如图1和2所示,供试品与对照品的相应位置上,显相同颜色的斑点。阴性对照品无干扰。
实施例2:
一种香附四物颗粒的制备方法,包括如下步骤:
a.取香附300g、木香200g、当归600g、川芎300g,加入16.8kg的水,浸泡15小时后,水蒸气蒸馏提取8小时,提取挥发油。
b.挥发油加入β-环糊精(加入量为挥发油体积量的8倍)和水(加入量为挥发油体积量的20倍)充分研磨2小时,冷冻干燥得挥发油包合物。
c.挥发油提取后所得残渣,加相应比例的熟地800g、白芍300g和延胡索300g,加水煎煮2次,每次加水量28kg,煎煮2小时,抽出药液,合并药液,滤过,减压干燥至相对密度1.13的浓缩液,加入95%乙醇至体系中乙醇浓度为80%(v/v),静置24小时后,抽滤,滤液减压浓缩,干燥,得干膏粉。
d.干膏粉加入糊精(与干膏粉等量)和糖粉(与干膏粉等量),混匀,制粒,干燥,整粒,最后加入挥发油包合物,即得香附四物颗粒。
对香附四物颗粒采用薄层色谱定性鉴别,具体方法如下:
1.样品的制备
1.1对照品溶液的制备
称取欧当归内酯A对照品适量,加乙酸乙酯制成每1mL含0.116mg的溶液(置棕色量瓶中),作为对照品溶液。
1.2对照药材溶液的制备
取川芎药材粉末1g,加乙醚100mL,加热回流l h,滤过,滤液挥干,残渣加乙酸乙酯2mL使溶解,作为对照药材溶液。
1.3供试品溶液的处理方法
取上述香附四物颗粒15g,加乙醚100mL,加热回流l h,滤过,滤液挥干,残渣加乙酸乙酯2mL使溶解,作为供试品溶液。
1.4阴性对照溶液的制备
按照上述处方的比例及制法,制备缺少川芎的阴性对照样品,按1.3项下的供试品溶液的制备方法,制成阴性对照溶液。
2.展开剂的条件优化
比较正己烷-乙酸乙酯(3:1)、正己烷-乙酸乙酯-甲酸(3:1:0.05)、正己烷-乙酸乙酯-甲酸(7:1:0.05)三种展开体系,选择正己烷:乙酸乙酯(3:1)为欧当归内酯A的展开剂。
3.点样量的确定
3.1对照品溶液点样量的确定
分别吸取欧当归内酯A对照品溶液各1、2、4、5μL依次点于同一硅胶GF254板。对照品不同点样量都有明显斑点,在4μL是斑点已经很清晰,选择4μL为欧当归内酯A的点样量。
3.2供试品溶液点样量的确定
分别吸取供试品溶液各2、4、8、10μL,以确定的TLC条件进行分析,供试品溶液在8μL时斑点较为清晰规整,所以点样量选择为8μL。
4.样品检验
根据确定的TLC条件,对3批次香附四物颗粒进行欧当归内酯A的薄层鉴别,结果如图3所示,供试品与对照品的相应位置上,显相同颜色的斑点。阴性对照品无干扰。
实施例3:
一种香附四物颗粒的制备方法,包括如下步骤:
a.取香附300g、木香200g、当归600g、川芎300g,加入16.8kg的水,浸泡15小时后,水蒸气蒸馏提取8小时,提取挥发油。
b.挥发油加入β-环糊精(加入量为挥发油体积量的8倍)和水(加入量为挥发油体积量的20倍)充分研磨2小时,冷冻干燥得挥发油包合物。
c.挥发油提取后所得残渣,加相应比例的熟地800g、白芍300g和延胡索300g,加水煎煮2次,每次加水量28kg,煎煮2小时,抽出药液,合并药液,滤过,减压干燥至相对密度1.13的浓缩液,加入95%乙醇至体系中乙醇浓度为80%(v/v),静置24小时后,抽滤,滤液减压浓缩,干燥,得干膏粉。
d.干膏粉加入糊精(与干膏粉等量)和糖粉(与干膏粉等量),混匀,制粒,干燥,整粒,最后加入挥发油包合物,即得香附四物颗粒。
对香附四物颗粒采用薄层色谱定性鉴别,具体方法如下:
1.样品的制备
1.1对照品溶液的制备
取α-香附酮对照品,加乙酸乙酯制成每1mL含2.2mg的溶液,作为对照品溶液。
1.2对照药材溶液的制备
取香附对照药材粉末1g,加乙醚75mL,放置1h,时时振摇,滤过,滤液挥干,残渣加乙酸乙酯1mL使溶解,作为对照药材溶液。
1.3供试品溶液的处理方法
取上述香附四物颗粒15g,加乙醚75mL,放置1h,时时振摇,滤过,滤液挥干,残渣加乙酸乙酯1mL使溶解,作为供试品溶液。
1.4阴性对照溶液的制备
按照上述处方的比例及制法,制备缺少香附的阴性对照样品,按供试品溶液的制备方法,制成阴性对照溶液。
2.展开剂和显色条件的优化
二氯甲烷-乙酸乙酯-冰醋酸(80:1:1)为α-香附酮的最适展开剂。硅胶GF254板和硅胶G板所得斑点效果相当,为求精准,选择硅胶GF254展开,紫外灯(254nm)检视,再喷以2,4-二硝基苯肼乙醇试液观看斑点显色。
3.点样量的确定
3.1对照品溶液点样量的确定
分别吸取α-香附酮对照品溶液各1、2、4、5μL依次点于同一硅胶GF254薄层板,以确定好的TLC条件进行分析,2μL时斑点最规整,选择2μL为点样量。
3.2供试品溶液点样量的确定
分别吸取供试品溶液各4、6、8、10μL,以确定的TLC条件进行分析,供试品溶液在10μL时斑点较为清晰规整,所以点样量选择为10μL。
4.样品检验
根据确定的TLC条件,对3批次香附四物颗粒进行α-香附酮的薄层鉴别,结果如图4所示,供试品与对照品的相应位置上,显相同颜色的斑点。阴性对照品无干扰。
实施例4:
一种香附四物颗粒的制备方法,包括如下步骤:
a.取香附300g、木香200g、当归600g、川芎300g,加入14kg的水,浸泡12小时后,水蒸气蒸馏提取10小时,提取挥发油。
b.挥发油加入β-环糊精(加入量为挥发油体积量的10倍)和水(加入量为挥发油体积量的30倍)充分研磨2小时,冷冻干燥得挥发油包合物。
c.挥发油提取后所得残渣,加相应比例的熟地800g、白芍300g和延胡索300g,加水煎煮2次,每次加水量30kg,煎煮2小时,抽出药液,合并药液,滤过,减压干燥至相对密度1.05的浓缩液,加入95%乙醇至体系中乙醇浓度为80%(v/v),静置24小时后,抽滤,滤液减压浓缩,干燥,得干膏粉。
d.干膏粉加入糊精(与干膏粉等量)和糖粉(与干膏粉等量),混匀,制粒,干燥,整粒,最后加入挥发油包合物,即得香附四物颗粒。
对香附四物颗粒采用薄层色谱定性鉴别,具体方法如下:
1.样品的制备
1.1对照品溶液的制备
取去氢木香内酯对照品、木香烃内酯对照品适量,加甲醇分别制成每1mL含2.28mg、1.85mg的溶液,作为对照品溶液。
1.2对照药材溶液的制备
取0.5g木香对照药材的粉末,加石油醚(30~60℃)75mL,冷浸30min,时时振摇,滤过,滤液挥干,残渣加醋酸乙酯1mL使溶解,取滤液作对照药材溶液。
1.3供试品溶液的处理方法
取上述香附四物颗粒15g,加石油醚(30~60℃)75mL,冷浸30min,时时振摇,滤过,滤液挥干,残渣加醋酸乙酯1mL使溶解,作为供试品溶液。
1.4阴性对照溶液的制备
按上述处方比例及制法,制成缺木香药材的阴性对照样品。按供试品溶液的制备方法,制成双阴性对照溶液。
2.展开剂的条件优化
为优化展开剂,吸取上述溶液分别点于同一硅胶G薄层板上,选用三种展开体系,展开,取出,晾干,展开剂B和C所得斑点较模糊,展距接近前沿,展开剂A所得斑点清晰规整,展距较为适宜,故选择环己烷-乙酸乙酯-甲酸(15:5:1)为木香烃内酯和去氢木香内酯的展开剂。
3.点样量的确定
3.1对照品溶液点样量的确定
分别吸取去氢木香内酯对照品溶液各1、2、3、4μL,木香烃内酯对照品溶液各2、4、6、8μL,依次分别点于同一硅胶G薄层板,以确定好的TLC条件进行分析,去氢木香内酯对照品不同点样量都有明显斑点,在2μL时斑点最规整,所以选择2μL为其点样量。木香烃内酯对照品在8μL是斑点最明显,故选择8μL为其点样量。
3.2供试品溶液点样量的确定
吸取供试品溶液各4、6、8、10μL,依次分别点于同一硅胶G薄层板,以确定的TLC条件进行分析,结果供试品溶液在4~10μL时皆显示斑点,其点样量可为10μL。
4.样品检验
根据确定的TLC鉴别条件,对3批次香附四物颗粒进行去氢木香内酯和木香烃内酯的薄层鉴别,结果如图5所示,供试品与对照品的相应位置上,显相同颜色的斑点。阴性对照品无干扰。
实施例5:
一种香附四物颗粒的制备方法,包括如下步骤:
a.取香附300g、木香200g、当归600g、川芎300g,加入14kg的水,浸泡12小时后,水蒸气蒸馏提取10小时,提取挥发油。
b.挥发油加入β-环糊精(加入量为挥发油体积量的10倍)和水(加入量为挥发油体积量的30倍)充分研磨2小时,冷冻干燥得挥发油包合物。
c.挥发油提取后所得残渣,加相应比例的熟地800g、白芍300g和延胡索300g,加水煎煮2次,每次加水量30kg,煎煮2小时,抽出药液,合并药液,滤过,减压干燥至相对密度1.05的浓缩液,加入95%乙醇至体系中乙醇浓度为80%(v/v),静置24小时后,抽滤,滤液减压浓缩,干燥,得干膏粉。
d.干膏粉加入糊精(与干膏粉等量)和糖粉(与干膏粉等量),混匀,制粒,干燥,整粒,最后加入挥发油包合物,即得香附四物颗粒。
对香附四物颗粒采用薄层色谱定性鉴别,具体方法如下:
1.样品的制备
1.1对照品溶液的制备
取5-羟甲基糠醛对照品,加甲醇制成每1mL含0.29mg的溶液,作为对照品溶液。
1.2对照药材溶液的制备
取1g熟地药材的粉末,加水100mL,温热使其充分溶散,放冷,用脱脂棉滤过,滤液用乙醚振摇提取2次,每次25mL,弃去乙醚液,再用乙酸乙酯振摇提取2次,每次25mL,合并乙酸乙酯液,蒸干,残渣加甲醇1mL使溶解,作为对照药材溶液。
1.3供试品溶液的处理方法
取颗粒15g,加水100mL,温热使其充分溶散,放冷,用脱脂棉滤过,滤液用乙醚振摇提取2次,每次25mL,弃去乙醚液,再用乙酸乙酯振摇提取2次,每次25mL,合并乙酸乙酯液,蒸干,残渣加甲醇1mL使溶解,作为供试品溶液。
1.4阴性对照溶液的制备
按处方比例及制法,制成缺熟地黄药材的阴性对照样品。按供试品溶液的制备方法,制成双阴性对照溶液。
2.展开剂的条件优化
不同优化条件:GF254板与展开剂A,紫外(254nm)检视;G板与展开剂B,喷显色剂,日光观察;G板与展开剂C,喷显色剂,日光检视。显色剂:2,4-二硝基苯肼乙醇溶液。将这三种展开和显色体系进行比较。结果显示,展开剂A的Rf较小,展开剂B的展距接近前沿,所以选择展开剂C:二甲苯-乙酸乙酯(1:1)为5-羟甲基糠醛的展开剂,2,4-二硝基苯肼乙醇溶液为显色剂。
3.点样量的确定
3.1对照品溶液点样量的确定
分别吸取5-羟甲基糠醛对照品溶液各6、8、10、12μL,依次点于同一硅胶G薄层板,以确定好的TLC条件分析,对照品不同点样量都有明显斑点,8μL时斑点较为规整,所以选择8μL为点样量。
3.2供试品溶液点样量的确定
分别吸取供试品溶液2各1、2、3、4μL,依次点于同一硅胶G薄层板,以确定的TLC条件进行分析,供试品溶液在1~4μL时皆显示明显斑点,点样量可选2μL。
4.样品检验
根据确定的TLC鉴别条件,对3批次香附四物颗粒进行5-羟甲基糠醛的薄层鉴别,结果如图6所示,供试品与对照品的相应位置上,显相同颜色的斑点。阴性对照品无干扰。
实施例6:
一种香附四物颗粒的制备方法,包括如下步骤:
a.取香附300g、木香200g、当归600g、川芎300g,加入16.8kg的水,浸泡15小时后,水蒸气蒸馏提取8小时,提取挥发油。
b.挥发油加入β-环糊精(加入量为挥发油体积量的8倍)和水(加入量为挥发油体积量的20倍)充分研磨2小时,冷冻干燥得挥发油包合物。
c.挥发油提取后所得残渣,加相应比例的熟地800g、白芍300g和延胡索300g,加水煎煮2次,每次加水量28kg,煎煮2小时,抽出药液,合并药液,滤过,减压干燥至相对密度1.13的浓缩液,加入95%乙醇至体系中乙醇浓度为80%(v/v),静置24小时后,抽滤,滤液减压浓缩,干燥,得干膏粉。
d.干膏粉加入糊精(与干膏粉等量)和糖粉(与干膏粉等量),混匀,制粒,干燥,整粒,最后加入挥发油包合物,即得香附四物颗粒。
对香附四物颗粒采用薄层色谱定性鉴别,具体方法如下:
1.样品的制备
1.1对照品溶液的制备
称取芍药苷对照品适量,加甲醇制成每1mL含1.03mg的溶液,作为对照品溶液。
1.2对照药材溶液的制备
取0.5g白芍对照药材粉末,用1mL甲醇溶解,采用硅胶柱(300-400目,约20g,内径约2-2.5cm),以三氯甲烷:甲醇(10:1,44mL;9:1,50mL;8:1,45mL)洗脱,弃去前15mL洗脱液,收集后续8:1洗脱液。蒸干,加1mL甲醇溶解残渣,为对照药材溶液。
1.3供试品溶液的处理方法
取香附四物颗粒15g,加50mL水,加热溶解,水饱和正丁醇萃取2次,每次25mL,合并正丁醇液,以正丁醇饱和水洗涤2次,每次25mL,合并正丁醇液,水浴挥干,得残渣。残渣用1mL甲醇溶解,采用硅胶柱(300-400目,约20g,内径约2-2.5cm),以三氯甲烷:甲醇(10:1,44mL;9:1,50mL;8:1,45mL)洗脱,弃去前15mL洗脱液,收集后续8:1洗脱液,蒸干,残渣加1mL甲醇使溶解,作为供试品溶液。
1.4阴性对照溶液的制备
按照处方的比例及制法,制备缺少白芍的阴性对照样品,按供试品溶液的制备方法,制成阴性对照溶液。
2.点样量的确定
2.1对照品溶液点样量的确定
分别吸取芍药苷对照品溶液各4、6、8、10μL,依次点于同一硅胶G板,以确定的TLC条件进行分析,芍药苷对照品不同点样量都有明显斑点,在10μL时斑点较为规整,颜色较为清晰,所以选择10μL为芍药苷的点样量。
2.2供试品溶液点样量的确定
分别吸取供试品溶液各2、4、8、10μL,以确定的TLC条件进行分析,供试品溶液在8μL时斑点较为清晰规整,所以点样量选择为8μL。
3.样品检验
根据确定的TLC条件,对3批次香附四物颗粒进行芍药苷的薄层鉴别,结果如图7所示,供试品与对照品的相应位置上,显相同颜色的斑点。阴性对照品无干扰。
实施例7:
一种香附四物颗粒的制备方法,包括如下步骤:
a.取香附300g、木香200g、当归600g、川芎300g,加入16.8kg的水,浸泡15小时后,水蒸气蒸馏提取8小时,提取挥发油。
b.挥发油加入β-环糊精(加入量为挥发油体积量的8倍)和水(加入量为挥发油体积量的20倍)充分研磨2小时,冷冻干燥得挥发油包合物。
c.挥发油提取后所得残渣,加相应比例的熟地800g、白芍300g和延胡索300g,加水煎煮2次,每次加水量28kg,煎煮2小时,抽出药液,合并药液,滤过,减压干燥至相对密度1.13的浓缩液,加入95%乙醇至体系中乙醇浓度为80%(v/v),静置24小时后,抽滤,滤液减压浓缩,干燥,得干膏粉。
d.干膏粉加入糊精(与干膏粉等量)和糖粉(与干膏粉等量),混匀,制粒,干燥,整粒,最后加入挥发油包合物,即得香附四物颗粒。
对香附四物颗粒采用薄层色谱定性鉴别,具体方法如下:
1.样品的制备
1.1对照品溶液的制备
称取延胡索乙素对照品适量,加甲醇制成每1mL含1.25mg的溶液,作为对照品溶液。
1.2对照药材溶液的制备
取1g延胡索对照药材的粉末,加甲醇100mL,超声处理30min,滤过,滤液蒸干,残渣加水20mL使溶解,加浓氨试液调至碱性,用乙醚振摇提取3次,每次20mL,合并乙醚液,蒸干,残渣加甲醇1mL使溶解,作为对照药材溶液。
1.3供试品溶液的处理方法
取颗粒15g,加甲醇100mL,超声处理30min,滤过,滤液蒸干,残渣加水20mL使溶解,加浓氨试液调至碱性,用乙醚振摇提取3次,每次20mL,合并乙醚液,蒸干,残渣加甲醇1mL使溶解,作为供试品溶液。
1.4阴性对照溶液的制备
按照处方比例及制法,制成缺少延胡索药材的阴性对照样品。然后按供试品溶液的制备方法,得到阴性对照溶液。
2.显色方法的优化
为优化显色方法,吸取上述溶液各10μL,分别点于同一硅胶G薄层板上,方法一:以甲苯-丙酮(9:2)为展开剂,喷以改良碘化铋钾溶液,日光检视;方法二:以甲苯-丙酮(9:2)为展开剂,取出,挥干,置碘缸中约1min后取出,挥尽板上吸附的碘后,置紫外光灯(365nm)下检视,喷以改良碘化铋钾溶液能得到明显的斑点,而紫外灯检视下,供试品溶液中延胡索乙素的斑点不易分辨,被别的斑点所覆盖,所以选择显色剂显色的方法进行分析。
3.点样量的确定
3.1对照品溶液点样量的确定
分别吸取延胡索乙素对照品溶液各4、6、8、10μL,依次点于同一硅胶G板,以确定好的条件进行分析,延胡索乙素对照品不同点样量都有斑点,在8μL时斑点已经较为规整,所以选择8μL为延胡索乙素的点样量。
3.2供试品溶液点样量的确定
分别吸取供试品溶液各4、6、8、10μL,依次点于同一硅胶G板,以确定的条TLC件进行分析。供试品溶液在4~10μL时皆有斑点,点样量可选6μL。
4.样品检验
根据确定的TLC鉴别条件,对3批次香附四物颗粒进行延胡索乙素的薄层鉴别,结果如图8所示,供试品与对照品的相应位置上,显相同颜色的斑点。阴性对照品无干扰。
实施例8:
一种香附四物颗粒的制备方法,包括如下步骤:
a.取香附300g、木香200g、当归600g、川芎300g,加入16.8kg的水,浸泡15小时后,水蒸气蒸馏提取8小时,提取挥发油。
b.挥发油加入β-环糊精(加入量为挥发油体积量的8倍)和水(加入量为挥发油体积量的20倍)充分研磨2小时,冷冻干燥得挥发油包合物。
c.挥发油提取后所得残渣,加相应比例的熟地800g、白芍300g和延胡索300g,加水煎煮2次,每次加水量28kg,煎煮2小时,抽出药液,合并药液,滤过,减压干燥至相对密度1.13的浓缩液,加入95%乙醇至体系中乙醇浓度为80%(v/v),静置24小时后,抽滤,滤液减压浓缩,干燥,得干膏粉。
d.干膏粉加入糊精(与干膏粉等量)和糖粉(与干膏粉等量),混匀,制粒,干燥,整粒,最后加入挥发油包合物,即得香附四物颗粒。
对香附四物颗粒进行含量测定,采用高效液相色谱法,具体方法如下:
1.阿魏酸和香草酸的含量测定
1.1仪器与试药
WatersAlliance 2695高效液相色谱系统(四元梯度泵、自动进样器、柱温系统、2998光电二极管阵列检测器及Empower 2色谱工作站,美国Waters公司);
KH-500型超声波清洗机(昆山禾创超声仪器有限公司);
HH-S型数显恒温水浴锅(金坛市医疗仪器厂);
MS105分析天平(Mettler-Toledo仪器有限公司);
EPED超纯水系统(南京易普易达科技发展有限公司)。
香附四物颗粒样品(按本实施例上述方法自制样品,批号:20151101、20151102、20151103);
对照品阿魏酸(批号:110773-201313)、香草酸对照品(批号:110776-200402),均购自中国食品药品检定研究院。其余试剂均为分析纯。
1.2方法与结果
1.2.1色谱条件
AlltimaTM C18色谱柱(250mm×4.6mm,5μm,GRACE);
流动相:,阿魏酸(乙腈-1%冰乙酸溶液体积比为20:80),香草酸(乙腈-1%冰乙酸溶液体积比为12:88);
体积流量1.0mL/min;柱温为30℃;进样量10μL;
检测波长:阿魏酸为323nm,香草酸为260nm;
出峰时间:阿魏酸为16min,香草酸为18min。
1.2.2溶液的制备
对照品溶液的制备取阿魏酸、香草酸对照品适量,精密称定,分别加甲醇制成每1mL含0.0316mg、0.0295mg的对照品溶液,逐级稀释,配制成6个不同浓度的对照品溶液。
供试品溶液的制备取香附四物颗粒15g,精密称定,置具塞锥形瓶中,精密加甲醇150mL,称重,放置过夜,超声1h,再称重,用甲醇补足损失的质量,摇匀,滤过,精密量取续滤液50mL,水浴蒸干。残渣用0.5mol/L氢氧化钠溶液50mL分3次溶解,转移入回流烧瓶中,水浴回流2h,放冷。移入分液漏斗中,用盐酸调pH=2,用乙醚萃取4次,每次25mL,合并乙醚液,回收乙醚,用甲醇溶解至10mL,摇匀,即得。
阴性对照品溶液的制备按香附四物颗粒的处方和制备工艺,制备缺当归和川芎阴性对照样品。再按供试品溶液的处理方法制得双阴性溶液。
1.3测定法
分别吸取对照品溶液、供试品溶液和阴性对照品溶液各10μL,按1.2.1项下的色谱条件进行分析,结果见图9和图10。从图可知,样品色谱图中,在与阿魏酸对照品、香草酸对照品色谱相应的位置上,有一相同保留时间的色谱峰,而阴性对照品溶液在此保留时间处无干扰。
1.4方法学考察
1.4.1标准曲线与线性范围
分别精密吸取6个不同浓度的对照品各10μL,注入液相色谱仪,按1.2.1项下色谱条件测定。以对照品峰面积值Y(μV·s)为纵坐标,以质量浓度X(μg/mL)为横坐标,进行线性回归,得出回归方程。
阿魏酸:Y=5.04×107X-5185.4,r=0.9999,在6.32~47.4μg/mL质量浓度范围内与峰面积线性关系良好;
香草酸:Y=3.83×104X+12993,r=0.9999,在5.90~29.5μg/mL质量浓度范围内与峰面积线性关系良好。
1.4.2精密度试验
取同一对照品溶液,按1.2.1项下色谱条件下连续进样6次,每次10μL,观察其重复性。结果表明,阿魏酸和香草酸对照品峰面积的RSD分别为0.620%和0.560%,显示仪器精密度好。结果见表1。
表1阿魏酸和香草酸的精密度试验结果
1.4.3重现性试验
取香附四物颗粒(批号20151101)6份,分别精密称定,按供试品溶液的制备方法制成供试品溶液,精密吸取供试品溶液10μL注入液相色谱仪,按1.2.1项下色谱条件测定,计算供试品溶液中阿魏酸和香草酸的RSD分别为0.363%和0.844%,方法重现性良好。结果见表2。
表2阿魏酸和香草酸的重现性试验结果
1.4.4稳定性试验
分别于0、2、4、6、8、10、12、24h精密吸取阿魏酸、香草酸对照品溶液各10μL注入液相色谱仪测定,按1.2.1项下的条件测定。结果显示,阿魏酸对照品溶液峰面积的RSD为1.27%,香草酸对照品溶液峰面积的RSD为2.66%,表明两个对照品溶液在24h之内稳定。结果见表3。
表3阿魏酸和香草酸的稳定性试验结果
1.4.5加样回收率试验
精密称定已测定的香附四物颗粒样品(批号:20151101)6份,分别加入阿魏酸、香草酸对照品适量,按照供试品溶液的处理方法,制得供试品溶液,按1.2.1项下所述的条件测定,计算加样回收率。结果表明,阿魏酸和香草酸的平均回收率分别为99.6%和101%,RSD分别为0.822%和1.07%,二者均有良好的回收率。结果见表4和表5。
表4阿魏酸的加样回收率试验结果
表5 香草酸的加样回收率试验结果
1.5样品测定
精密称取3批香附四物颗粒样品(批号:20151101、20151102、20151103),按供试品溶液的制备方法制成供试品溶液,按1.2.1项下色谱条件测定,并计算含量,结果见表6。
表6阿魏酸和香草酸的含量测定结果
2.芍药苷和芍药内酯苷的含量测定
2.1仪器与试药
2.1.1仪器
同1.1.1项所述。
2.1.2试药
香附四物颗粒样品(按本实施例上述方法自制样品,批号:20151101、20151102、20151103);
对照品芍药苷(批号:110736-201438),购自中国食品药品检定研究院;
对照品芍药内酯苷(批号:39011-90-0),购自南京春秋生物工程有限公司。其余试剂均为分析纯。
2.2方法与结果
2.2.1色谱条件
AlltimaTM C18色谱柱(250mm×4.6mm,5μm,GRACE);
流动相:乙腈-0.1%乙酸水(18:82);
体积流量1.0mL/min;
柱温为30℃;
检测波长为230nm;
进样量10μL;
芍药苷和芍药内酯苷的出峰时间约为12min和10min。
2.2.2溶液的制备
对照品溶液的制备取芍药苷、芍药内酯苷对照品适量,精密称定,分别加甲醇制成每1mL含0.300mg、0.200mg的对照品溶液,逐级稀释,配制成6个不同浓度的对照品溶液。
供试品溶液的制备取香附四物颗粒15g,精密称定,置具塞锥形瓶中,精密加入甲醇75mL,精密称定。超声处理25min,放冷,再称定,用甲醇补足减失质量,摇匀,滤过,取续滤液作为供试品溶液。
阴性样品溶液的制备按香附四物颗粒的处方和制备工艺,制备缺白芍的阴性制剂。再按供试品溶液的制备方法制得阴性溶液。
2.3测定法
分别吸取对照品溶液,供试品溶液和阴性对照品溶液各10μL,按2.2.1项下的色谱条件进行分析,结果见图11。样品色谱图中,在与芍药苷、芍药内酯苷对照品色谱相应的位置上,有一相同保留时间的色谱峰。而阴性对照品溶液在此保留时间处无峰。
2.4方法学考察
2.4.1标准曲线与线性范围
分别精密吸取6个不同浓度的对照品各10μL,注入液相色谱仪,按2.2.1项下色谱条件测定。以对照品峰面积值Y(μV·s)为纵坐标,以质量浓度X(μg/mL)为横坐标,进行线性回归,得出回归方程。
芍药苷:Y=1.39×104X+98826,r=0.9992,在150~750μg/mL质量浓度范围内与峰面积线性关系良好。
芍药内酯苷:Y=1.24×104X+48408,r=0.9996,在100~500μg/mL质量浓度范围内与峰面积线性关系良好。
2.4.2精密度试验
取同一对照品溶液,按2.2.1项下色谱条件下连续进样6次,每次10μL,观察其重复性。结果表明,芍药苷对照品和芍药内酯苷对照品峰面积的RSD分别为1.19%和0.820%,表明仪器精密度好。结果见表7。
表7芍药苷和芍药内酯苷的精密度试验结果
2.4.3重现性试验
取香附四物颗粒(批号:20151101)6份,分别精密称定,按照供试品溶液的制备方法制成供试品溶液。精密吸取10μL供试品溶液注入液相色谱仪,按2.2.1项下的条件测定,得供试品溶液中芍药苷和芍药内酯苷的RSD分别为1.43%和1.24%,表明,方法重现性良好。结果见表8。
表8芍药苷和芍药内酯苷的重现性试验结果
2.4.4稳定性试验
分别于0、2、4、6、8、10、12、24h精密吸取芍药苷、芍药内酯苷对照品溶液各10μL注入液相色谱仪测定,按2.2.1项下的条件测定。结果显示,芍药苷和芍药内酯苷对照品溶液峰面积的RSD分为1.05%和1.48%,表明,两种对照品溶液在24h之内稳定。结果见表9。
表9芍药苷和芍药内酯苷的稳定性试验结果
2.4.5加样回收率试验
取已测定的香附四物颗粒样品(批号:20151101)6份,精密称定,分别加入芍药苷和芍药内酯苷对照品适量,按照供试品溶液的制备方法,制得供试品溶液。按2.2.1项下的条件进行测定,计算回收率。结果表明,芍药苷和芍药内酯苷的回收率为101%和102%,RSD为1.74%和1.52%,二者均有良好的回收率。结果见表10和表11。
表10芍药苷的加样回收率实验结果
表11芍药内酯苷的加样回收率实验结果
2.5样品测定
精密称取3批香附四物颗粒样品(批号:20151101、20151102、20151103),按供试品溶液的制备方法制成供试品溶液,按2.2.1项下的色谱条件测定,并计算含量,结果见表12。
表12芍药苷和芍药内酯苷的含量测定结果
3.延胡索乙素和盐酸小檗碱的含量测定
3.1仪器与试药
3.1.1仪器
同1.1.1项所述。
3.1.2试药
香附四物颗粒样品(按本实施例上述方法自制样品,批号:20151101、20151102、20151103);
对照品延胡索乙素(批号:110726-201414)和对照品盐酸小檗碱(批号:110713-201212)均购自中国食品药品检定研究院。其余试剂均为分析纯。
3.2方法与结果
3.2.1色谱条件
AlltimaTM C18色谱柱(250mm×4.6mm,5μm,GRACE);
流动相为体积比为55:45乙腈-0.1%乙酸水,三乙胺调pH=6.0;
体积流量1.0mL/min;
柱温为30℃;检测波长为280nm;进样量10μL;
延胡索乙素和盐酸小檗碱的出峰时间约为11min和7min。
3.2.2溶液的制备
对照品溶液的制备:取延胡索乙素、盐酸小檗碱对照品适量,精密称定,分别加甲醇制成每1mL含0.246mg、0.297mg的对照品溶液,逐级稀释,配制成6个不同浓度的对照品溶液。
供试品溶液的制备取香附四物颗粒15g,精密称定,置平底烧瓶中,精密加入浓氨试液-甲醇(1:20)混合溶液150mL,称定重量,冷浸1h后加热回流1h,放冷,再称定重量,用浓氨试液-甲醇(1:20)混合溶液补足减失的重量,摇匀,滤过。精密量取续滤液75mL,蒸干,残渣加甲醇溶解,转移至10mL量瓶中,并稀释至刻度,摇匀,滤过,取续滤液,即得。
阴性样品溶液的制备根据香附四物颗粒处方和制备工艺,制成缺延胡索药材的阴性制剂。再按供试品溶液的制备方法制得阴性溶液。
3.3测定法
分别吸取对照品溶液、供试品溶液和阴性对照品溶液各10μL,按3.2.1项下的条件进行分析,结果见图12。在样品色谱图中,与延胡索乙素和盐酸小檗碱对照品色谱相应的位置上,有一相同保留时间的色谱峰。而阴性对照品溶液在此保留时间处无峰。
3.4方法学考察
3.4.1标准曲线与线性范围
分别精密吸取6个不同浓度的对照品10μL,注入液相色谱仪,按3.2.1项下的条件测定。以对照品峰面积值Y(μV·s)为纵坐标,以质量浓度X(μg/mL)为横坐标,进行线性回归,得出回归方程。
延胡索乙素:Y=8.42×103X-12481,r=0.9999,在98.4~492μg/mL质量浓度范围内与峰面积线性关系良好。
盐酸小檗碱:Y=3.08×104X-134654,r=0.9999,在59.4~446μg/mL质量浓度范围内与峰面积线性关系良好。
3.4.2精密度试验
取同一对照品溶液,按3.2.1项下色谱条件下连续进样6次,每次10μL,观察其重复性。结果表明,延胡索乙素对照品和盐酸小檗碱对照品峰面积的RSD分别为1.52%和1.00%,表明仪器精密度好。结果见表13。
表13延胡索乙素和盐酸小檗碱的精密度试验结果
3.4.3重现性试验
取香附四物颗粒(批号20151101)6份,分别精密称定,按照供试品溶液的制备方法,制成供试品溶液。精密吸取10μL,按3.2.1项下的条件测定,计算出供试品溶液中延胡索乙素和盐酸小檗碱的RSD分别为0.943%和1.14%,表明,方法重现性良好。结果见表14。
表14延胡索乙素和盐酸小檗碱的重现性试验结果
3.4.4稳定性试验
分别于0、2、4、6、8、10、12、24h精密吸取延胡索乙素、盐酸小檗碱对照品溶液各10μL按3.2.1项下条件测定。结果显示,延胡索乙素和盐酸小檗碱对照品溶液峰面积的RSD分别为0.830%和1.09%,表明,两种对照品溶液在24h内稳定。结果见表15。
表15延胡索乙素和盐酸小檗碱的稳定性试验结果
3.4.5加样回收率试验
取已测定的香附四物颗粒样品(批号:20151101)6份,精密称定,分别加入延胡索乙素和盐酸小檗碱对照品适量,按供试品溶液的制备方法,制成供试品溶液,按3.2.1项下的条件测定,计算回收率。结果表明,延胡索乙素和盐酸小檗碱的平均回收率为99.8%和100%,RSD为0.454%和0.835%,二者均有良好的回收率。结果见表16和表17。
表16 延胡索乙素的加样回收率试验结果
表17 盐酸小檗碱的加样回收率试验结果
3.5样品测定
精密称取3批香附四物颗粒样品(批号:20151101、20151102、20151103),按供试品溶液的制备方法,制成供试品溶液,按3.2.1项下的色谱条件测定,并计算含量,结果见表18。
表18延胡索乙素和盐酸小檗碱的含量测定结果
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!