介孔硅酸镁/聚丁二酸丁二醇酯复合支架及其制备方法和应用与流程
本发明涉及一种介孔硅酸镁/聚丁二酸丁二醇酯复合支架及其制备方法和应用。
背景技术:
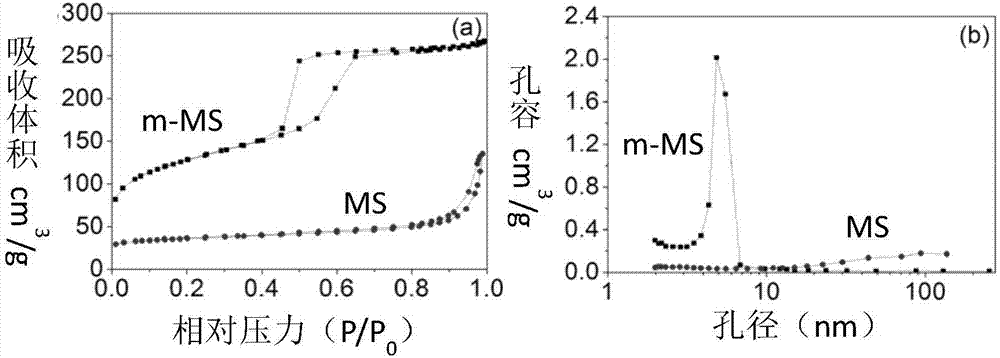
:由于自然灾害、交通事故或骨科疾病及人口老龄化等导致骨结构完整性遭到破坏,临床上骨缺损的病例日益增多。传统的治疗方式,包括自体骨移植和异体骨移植,具有材料来源等局限性,并有潜在风险。因此,越来越多的人开发并深入研究人工合成骨替代生物材料。目前临床常用的骨修复材料由于细胞响应慢、骨向分化能力差以及营养不足等呈现活性低、骨再生和骨整合速度慢等突出问题。例如,有机/有机复合材料(如壳聚糖/pbsu)的体外生物活性和降解性相对较差。用于骨修复领域的无机活性材料,包括β-磷酸三钙、羟基磷灰石等,其比表面积和孔容较小,体外生物活性较差。经研究,硅基材料具有较好的生物活性,可促进细胞增殖和分化,并有效激活骨的相关基因表达,在骨修复领域中一直备受瞩目。镁离子可以调节细胞与材料表面的亲和力,并促进成骨细胞的增殖。研究发现具有高表面积和孔容的生物活性材料在模拟体液中更有利于羟基磷灰石的形成。但介孔硅镁基生物活性材料存在脆性大,力学性能低,成型困难等缺点。近年来,生物降解高分子材料已广泛用于具有三维结构的骨组织工程支架材料。聚丁二酸丁二醇酯(poly(butylenesuccinate),pbsu)是一种可完全生物降解的脂肪族聚酯类材料。相比于可用于制备支架的有机材料,包括聚乳酸、聚己内酯等可生物降解材料等,聚丁二酸丁二醇酯(pbsu)价格较低,生物相容性良好、力学和机械性能优异、热稳定性好、易加工以及降解的产物无毒性等优点。然而,pbsu也有一些缺点,限制了其在生物医学领域的应用。例如,pbsu是疏水性材料,会影响pbsu表面的细胞粘附行为;pbsu是生物惰性材料,从而阻碍了其与宿主骨的结合,影响其成骨性能,缺乏对骨组织再生的明显促进作用。但目前尚未有关于介孔硅镁基生物活性材料与pbsu的复合骨修复材料的报道。技术实现要素:本发明所要解决的技术问题是克服现有技术中常用的骨修复材料细胞响应慢、骨向分化能力差、营养不足等呈现活性低、骨再生和骨整合速度慢等缺陷,提供了一种介孔硅酸镁/聚丁二酸丁二醇酯复合支架及其制备方法和应用。本发明的介孔硅酸镁/聚丁二酸丁二醇酯复合支架可以明显促进mc3t3-e1细胞的粘附、增殖和alp活性表达,并促进体内成骨,本发明的制备方法操作简便,制得的介孔硅酸镁/聚丁二酸丁二醇酯复合支架,孔连通性良好、孔径均一,具有优良的体外降解性和生物活性,还可释放出硅、镁离子,有利于细胞成骨分化。本发明通过下述技术方案解决上述技术问题。本发明提供了一种介孔硅酸镁/聚丁二酸丁二醇酯复合支架的制备方法,其包括下述步骤:(1)将聚丁二酸丁二醇酯和有机溶剂混合,得混合物ⅰ;(2)将介孔硅酸镁和致孔剂与步骤(1)中的混合物ⅰ混合,得混合物ⅱ;(3)将步骤(2)中的混合物ⅱ压制成型后除去有机溶剂,之后除去致孔剂,干燥即可。步骤(1)中,所述聚丁二酸丁二醇酯为本领域常规使用的所述聚丁二酸丁二醇酯,较佳地,所述聚丁二酸丁二醇酯的数均分子量为4×104~7×104,分子量分布(mw/mn)为1.2~2.4。步骤(1)中,所述有机溶剂为本领域常规使用的用于溶解聚丁二酸丁二醇酯的有机溶剂,例如二甲基甲酰胺、三氯甲烷、二氯甲烷、1,4-二氧己环或间甲基苯酚中的一种或多种,较佳地为二甲基甲酰胺。步骤(2)中,所述介孔硅酸镁为本领域常规使用的介孔硅酸镁,宏观以粉末态存在。较佳地,所述介孔硅酸镁符合下述条件:所述介孔硅酸钙镁呈非晶态,平均孔径为6~7nm,比表面积为445~455m2/g,孔容为0.3~0.5cm3/g。步骤(2)中,所述介孔硅酸镁可采用本领域常规用于制备介孔硅酸镁的方法制备而得,一般采用溶胶凝胶法。在本发明一较佳实施例中,所述介孔硅酸镁采用包括如下步骤的方法制备:s1、在45~55℃下,将模板剂与水和盐酸混合,搅拌,至溶液澄清;s2、在45~50℃下,将六水硝酸镁(mg(no3)2·6h2o)、正硅酸乙酯(teos)加入上述溶液中,搅拌4~6h后静置抽滤,之后干燥得白色粉末;s3、将所得白色粉末于500~700℃下煅烧除去模板剂即可。其中,步骤s1中,所述模板剂为本领域常规使用的模板剂,通常采用聚环氧乙烷-聚环氧丙烷-聚环氧乙烷三嵌段共聚物,例如sigmaaldrich提供的平均重均分子量为5800的p123三嵌段共聚物。所述水可为去离子水。在本发明一实施例中,步骤s1按照如下操作进行:将5.0~7.0gp123嵌段共聚物与20~40ml水和110~130ml浓度为1.7~2.5mol/l的盐酸混合,搅拌,至溶液澄清。其中,步骤s2中,向上述溶液中加六水硝酸镁和正硅酸乙酯可采用分步加入,先加入六水硝酸镁,再加入正硅酸乙酯。在本发明一实施例中,步骤s2按照如下操作进行:在45~50℃下,依次将4.6~5.0g六水硝酸镁和8.3~8.7g正硅酸乙酯加入上述溶液中,搅拌4~6h后静置抽滤。其中,步骤s2中,所述抽滤可进行多次,一般为2~5次,较佳地为3次。每次抽滤结束后一般采用去离子水进行清洗,之后再进行抽滤。其中,步骤s2中,所述干燥的操作和条件为本领域常规的干燥的操作和条件,一般在50~70℃的电热鼓风干燥箱中进行,较佳地在60℃的电热鼓风干燥箱中进行。其中,步骤s3中,所述煅烧的操作和条件为本领域常规的煅烧的操作和条件,较佳地在600℃的马弗炉中进行。步骤(2)中,所述致孔剂为本领域常规使用的致孔剂,一般采用氯化钠(nacl),所述氯化钠的颗粒粒径不做特殊限定,较佳地为300~500μm。所述氯化钠的用量例如可为相对于介孔硅酸镁和聚丁二酸丁二醇酯总质量的7~9倍,如8倍。步骤(2)中,将介孔硅酸镁和致孔剂与步骤(1)中的混合物ⅰ混合可采用分步进行,先将所述介孔硅酸镁与所述混合物ⅰ混合,再将致孔剂加入进行混合。本发明中,所述介孔硅酸镁与所述聚丁二酸丁二醇酯的质量比较佳地为(20~40):(80~60),更佳地为30:70。本发明中,所述有机溶剂的用量不做特殊限制,该用量的选用只要使得到的所述混合物ⅱ的物理状态能够进行压制成型的操作即可,例如所述聚丁二酸丁二醇酯和所述有机溶剂的用量比为1g:(2~4)ml,较佳地为1g:3ml。步骤(2)中,所述混合可采用搅拌混合,所述混合一般为混合至各组分均匀。步骤(3)中,所述压制成型本领域公知需要使用模具,一般为不锈钢模具,通常可根据所要制备的样品选择不同规格的模具。步骤(3)中,所述压制成型的操作和条件为本领域常规的压制成型的操作和条件,所述压制成型的压力较佳地为1~3mpa,如2mpa。所述压制成型的时间较佳地为1~3分钟,如2分钟。步骤(3)中,所述除去有机溶剂的操作和条件为本领域常规的除去溶剂的操作和条件,较佳地采用在通风橱中自然晾干10~14小时使所述有机溶剂挥发干净,较佳地为晾干12小时。步骤(3)中,所述去除致孔剂的操作和条件为本领域常规的除去致孔剂的操作和条件,较佳地在能够溶解致孔剂但不会使介孔硅酸镁和聚丁二酸丁二醇酯溶解的溶剂中浸泡后去除,所述溶剂一般采用水,例如去离子水;其中,所述浸泡的时间不作特殊限定,例如可为44~50h,较佳地为48h。浸泡期间,为使致孔剂更好地去除,一般可每隔一段时间更换一次溶剂,例如每6~10h更换一次溶剂,较佳地为每8h更换一次溶剂。步骤(3)中,所述干燥的操作和条件为本领域常规的干燥的操作和条件,一般为在35~39℃烘箱中干燥10~14小时,较佳地为在37℃烘箱中干燥12小时。本发明还提供了一种由上述方法制备的介孔硅酸镁/聚丁二酸丁二醇酯复合支架。本发明还提供了一种所述介孔硅酸镁/聚丁二酸丁二醇酯复合支架在制备骨修复材料中的应用。在符合本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。本发明所用试剂和原料均市售可得。本发明的积极进步效果在于:本发明制得的介孔硅酸镁/聚丁二酸丁二醇酯复合支架,孔连通性良好、孔径均一,具有优良的体外降解性和生物活性,还可释放出硅、镁离子,有利于细胞成骨分化,可以明显促进mc3t3-e1细胞的粘附、增殖和alp活性表达,并促进体内成骨,本发明的制备方法具有操作简便,孔径分布可调等优点。附图说明图1为本发明实施例1中的介孔硅酸镁(m-ms)和对比例1中的非介孔硅酸镁(ms)的氮吸附/脱附等温线(a)和孔径分布曲线(b)。图2为本发明实施例1中的m-ms的微观结构的透射电镜图。图3为本发明实施例1中的m-ms和对比例1中的ms的扫描电镜图(a,b)和eds谱图(c,d)。图4为本发明实施例1中的m-ms的x射线衍射图谱。图5为本发明实施例1中的m-ms和对比例1中的ms的吸水率(a),在tris-hcl中浸泡不同时间后的失重率(b)。图6为本发明实施例1中的m-ms和对比例1中的ms在sbf中浸泡7天前(a,c)后(b,d)的扫描电镜照片。图7为本发明实施例1中的m-ms在sbf中浸泡7天后的能谱分析。图8为本发明实施例1中的m-ms和对比例1中的ms在sbf中浸泡不同时间后的mg和si(a),ca和p(b)离子浓度的变化。图9为本发明实施例1中的m-ms和对比例1中的ms在sbf中浸泡不同时间后的ph的变化。图10(a)为mc3t3-e1细胞在本发明实施例1中的m-ms和对比例1中的ms及组织培养板(tcp)上分别培养1,4和7天后的mtt分析结果,(b)为mc3t3-e1细胞在本发明实施例1中的m-ms和对比例1中的ms及组织培养板(tcp)上分别培养4天和7天后的alp表达情况。图11为本发明实施例1和2及对比例2中的c20、c40、c0和介孔硅酸镁四种材料的ftir图谱。图12为本发明实施例1和2及对比例2中的c20、c40、c0和介孔硅酸镁四种材料的广角x-射线衍射图谱。图13为本发明实施例1和2及对比例2中的c20、c40和c0的表面微观形貌结构。图14为本发明实施例1和2及对比例2中的c20、c40和c0的eds图谱。图15为本发明实施例1和2及对比例2中的c20、c40和c0的srμct电子切片照片。图16为本发明实施例1和2及对比例2中的c20、c40和c0在去离子水中浸泡不同时间后的吸水率。图17为本发明实施例1和2及对比例2中的c20、c40和c0浸泡在tris-hcl溶液中不同时间的降解程度。图18为本发明实施例1和2及对比例2中的c20、c40和c0的tris-hcl溶液ph值变化。图19为本发明实施例1和2及对比例2中的c20、c40和c0在sbf中浸泡5天后的扫描电镜照片。图20为本发明实施例1和2及对比例2中的c20、c40和c0在sbf中浸泡5天后的eds图谱。图21为本发明实施例1和2及对比例2中的c20、c40和c0在sbf溶液浸泡过程中离子浓度的变化。图22为mc3t3-e1细胞接种到本发明实施例1和2及对比例2中的c20、c40和c0上3,6,12小时后cck-8测试结果。图23为本发明实施例1和2及对比例2中的c20、c40和c0在1,3和5天对mc3t3-e1细胞增殖的影响。图24为本发明实施例1和2及对比例2中的c20、c40和c0在7,10和14天对mc3t3-e1细胞alp活性表达的影响。图25为本发明实施例1和2及对比例2中的c20、c40和c0植入体内4、8和12周后股骨的ct三维重构照片。图26为本发明实施例1和2及对比例2中的c20、c40和c0植入动物体内4,8,12周后通过软件计算骨缺损区域新生骨面积。图27为本发明实施例1和2及对比例2中的c20、c40和c0植入动物体内4,8,12周后根据图26通过软件分析计算得出bmp-2阳性表达率的定量分析。具体实施方式下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。下述实施例1~4及对比例2中各制备两种不同规格的多孔支架材料,其中φ12×2mm的多孔支架用于材料表征及细胞实验,φ6×6mm的多孔支架用于抗压强度测试及动物实验。下述实施例1~4及对比例2中的聚丁二酸丁二醇酯(pbsu)购自安庆和兴化工有限责任公司,数均分子量为4×104~7×104,分子量分布(mw/mn)为1.2~2.4。实施例1(1)制备介孔硅酸镁(m-ms)首先,将30ml去离子水和120ml2.0mol/l稀盐酸的混合溶液,置于50℃水浴中。然后,准确称取4.0gp123(eo20po70eo20,5800,sigmaaldrich),将其加入上述混合溶液中,搅拌30分钟至澄清。随后,依次称取4.8g六水合硝酸镁(mg(no3)2·6h2o),准确称量8.5g正硅酸乙酯(teos)加入到上述澄清溶液中,搅拌5小时。静置后抽滤,去离子水清洗,再抽滤,重复3次后,置于60℃电热鼓风干燥箱(dhg-9070a,上海一恒科学仪器公司)中得到白色粉末。将粉末放入不锈钢模具中,采用压片机(yp-15t,天津金孚伦科技有限公司)在2mpa压力下制得圆片样品(φ12×2mm)。最后将上述粉末及圆片样品置于马弗炉(sgm2892a,上海西格马高温电炉公司)中,在600℃条件下进行烧结(升温速率:1℃/min),从而获得m-ms粉末及圆片样品(φ12×2mm)。(2)制备介孔硅酸镁/聚丁二酸丁二醇酯复合支架用称量瓶准确称取0.8g的pbsu,溶解在2.4ml二甲基甲酰胺溶液中。加入0.2g的介孔硅酸镁(m-ms)粉末并不断搅拌使其均匀分散,充分混合后加入8g的氯化钠颗粒(粒径为300-500μm),混合搅拌10分钟。然后将上述混合物加入到不锈钢模具中,2mp的压力下,2分钟压制成型。所得样品在通风橱中自然晾干12小时使溶剂挥发干净。然后将材料浸泡在去离子水中48小时,以去除致孔剂(氯化钠颗粒),每8小时更换一次去离子水。最后将样品置于37℃烘箱中12小时,获得骨修复支架材料c20。实施例2用称量瓶准确称取0.6g的pbsu,溶解在2ml二甲基甲酰胺溶液中。加入0.4g实施例1中制备的介孔硅酸镁(m-ms)粉末并不断搅拌使其均匀分散,充分混合后加入8g的氯化钠颗粒(粒径为300-500μm),混合搅拌10分钟。然后将上述混合物加入到不锈钢模具中,0.2mp的压力下,2分钟压制成型。所得样品在通风橱中自然晾干12小时使溶剂挥发干净。然后将材料浸泡在去离子水中48小时,以去除致孔剂(氯化钠颗粒),每8小时更换一次去离子水。最后将样品置于37℃烘箱中12小时,获得骨修复支架材料c40。实施例3(1)制备介孔硅酸镁(m-ms)首先,将40ml去离子水和130ml2.5mol/l稀盐酸的混合溶液,置于45℃水浴中。然后,准确称取5.0gp123(eo20po70eo20,5800,sigmaaldrich),将其加入上述混合溶液中,搅拌30分钟至澄清。随后,依次称取5.0g六水合硝酸镁(mg(no3)2·6h2o),准确称量8.7g正硅酸乙酯(teos)加入到上述澄清溶液中,搅拌6小时。静置后抽滤,去离子水清洗,再抽滤,重复3次后,置于60℃电热鼓风干燥箱(dhg-9070a,上海一恒科学仪器公司)中得到白色粉末。将粉末放入不锈钢模具中,采用压片机(yp-15t,天津金孚伦科技有限公司)在2mpa压力下制得圆片样品(φ12×2mm)。最后将上述粉末及圆片样品置于马弗炉(sgm2892a,上海西格马高温电炉公司)中,在600℃条件下进行烧结(升温速率:1℃/min),从而获得m-ms粉末及圆片样品(φ12×2mm)。(2)制备介孔硅酸镁/聚丁二酸丁二醇酯复合支架用称量瓶准确称取0.7g的pbsu,溶解在2.8ml二甲基甲酰胺溶液中。加入0.3g的介孔硅酸镁(m-ms)粉末并不断搅拌使其均匀分散,充分混合后加入9g的氯化钠颗粒(粒径为300-500μm),混合搅拌10分钟。然后将上述混合物加入到不锈钢模具中,1mp的压力下,3分钟压制成型。所得样品在通风橱中自然晾干12小时使溶剂挥发干净。然后将材料浸泡在去离子水中50小时,以去除致孔剂(氯化钠颗粒),每6小时更换一次去离子水。最后将样品置于39℃烘箱中10小时,获得骨修复支架材料。实施例4(1)制备介孔硅酸镁(m-ms)首先,将20ml去离子水和110ml1.7mol/l稀盐酸的混合溶液,置于55℃水浴中。然后,准确称取5.0gp123(eo20po70eo20,5800,sigmaaldrich),将其加入上述混合溶液中,搅拌30分钟至澄清。随后,依次称取4.6g六水合硝酸镁(mg(no3)2·6h2o),准确称量8.3g正硅酸乙酯(teos)加入到上述澄清溶液中,搅拌4小时。静置后抽滤,去离子水清洗,再抽滤,重复3次后,置于60℃电热鼓风干燥箱(dhg-9070a,上海一恒科学仪器公司)中得到白色粉末。将粉末放入不锈钢模具中,采用压片机(yp-15t,天津金孚伦科技有限公司)在2mpa压力下制得圆片样品(φ12×2mm)。最后将上述粉末及圆片样品置于马弗炉(sgm2892a,上海西格马高温电炉公司)中,在600℃条件下进行烧结(升温速率:1℃/min),从而获得m-ms粉末及圆片样品(φ12×2mm)。(2)制备介孔硅酸镁/聚丁二酸丁二醇酯复合支架用称量瓶准确称取0.7g的pbsu,溶解在1.4ml二甲基甲酰胺溶液中。加入0.3g的介孔硅酸镁(m-ms)粉末并不断搅拌使其均匀分散,充分混合后加入7g的氯化钠颗粒(粒径为300-500μm),混合搅拌10分钟。然后将上述混合物加入到不锈钢模具中,3mp的压力下,1分钟压制成型。所得样品在通风橱中自然晾干12小时使溶剂挥发干净。然后将材料浸泡在去离子水中44小时,以去除致孔剂(氯化钠颗粒),每10小时更换一次去离子水。最后将样品置于35℃烘箱中14小时,获得骨修复支架材料。对比例1首先,将30ml去离子水和120ml2.0mol/l稀盐酸的混合溶液,置于50℃水浴中。随后,依次称取4.8g六水合硝酸镁(mg(no3)2·6h2o),准确称量8.5g正硅酸乙酯(teos)加入到上述澄清溶液中,搅拌5小时。静置后抽滤,去离子水清洗,再抽滤,重复3次后,置于60℃电热鼓风干燥箱(dhg-9070a,上海一恒科学仪器公司)中得到白色粉末。将粉末放入不锈钢模具中,采用压片机(yp-15t,天津金孚伦科技有限公司)在2mpa压力下制得圆片样品(φ12×2mm)。最后将上述粉末及圆片样品置于马弗炉(sgm2892a,上海西格马高温电炉公司)中,在600℃条件下进行烧结(升温速率:1℃/min),从而获得非介孔硅酸镁(ms)粉末及圆片样品(φ12×2mm)。对比例2用称量瓶准确称取1g的pbsu,溶解在3ml二甲基甲酰胺溶液中,充分混合后加入8g的氯化钠颗粒(粒径为300-500μm),混合搅拌10分钟。然后将上述混合物加入到不锈钢模具中,2mp的压力下,2分钟压制成型。所得样品在通风橱中自然晾干12小时使溶剂挥发干净。然后将材料浸泡在去离子水中48小时,以去除致孔剂(氯化钠颗粒),每8小时更换一次去离子水。最后将样品置于37℃烘箱中12小时,获得骨修复支架材料c0。效果实施例1对实施例1中制备的介孔硅酸镁(m-ms)和对比例1中制备的非介孔硅酸镁(ms)进行性能表征:(1)氮气吸附脱附(bet)采用孔隙率仪(tristar3000,micro-metricsinstrumentcorp.,norcross,ga,usa)测定不同相对压力p/p0下n2等温吸附-脱附曲线,通过brunaueremmettteller(bet)计算材料的比表面积和孔容,并根据barrett-joyner-helen(bjh)公式计算平均孔径。(2)透射电镜(tem)将经研磨的m-ms粉末样品加入无水乙醇中超声分散15min,吸取一滴溶液滴在铜网上,干燥后采用透射电镜(tem;jem2010,日本)观察材料的微观结构。(3)扫描电镜(sem)将粉末样品研磨后黏附在导电胶上采用电子扫描电镜(s-3400n,hitachi,japan)观察样品表面形貌。(4)元素分析(eds)通过元素分析仪(eds,falcon,usa)上测得样品表面的元素组成。(5)x射线衍射分析(xrd)采用x射线衍射仪(xrd,rigakud/max2550,日本)在0.5~8°和10~80°分析了材料的微观结构及结晶状态。本实验通过溶胶凝胶法制得白色圆片m-ms样品,其表观实物形貌见图1,bet分析结果见图1。图1(a)是m-ms和ms粉末的n2吸附脱附等温线。根据iupac分类,m-ms呈现出iv型氮气吸附脱附等温线。m-ms的吸附等温线和脱附等温线分别在0.55–0.65和0.5–0.45p/p0区间呈陡峭的急剧变化,形成明显的h1型回滞环,是明显的吸附-解吸过程。同时,ms的吸附/脱附等温线相对较平缓。该结果表明m-ms具有有序的介孔结构。图1(b)显示的是采用bjh法按等温线计算的结果。从图中可以看出:m-ms具有很窄的孔径分布,其平均孔径、比表面积和孔容分别为5nm,451.0m2/g,0.41cm3/g。然而,ms的比表面积和孔容仅分别为75m2/g和0.21cm3/g。结果表明,相比于ms,m-ms具有很窄的孔径分布,其平均孔径、比表面积和孔容分别为5nm,451.0m2/g,0.41cm3/g,m-ms具有较大的表面积和较高的孔容。大的比表面积说明材料浸泡于模拟体液中或植入体内可以与周围液体有更大的接触面积,从而加快离子交换;同时,高孔容有利于该介孔材料负载药物或蛋白,赋予其更多的功能性。图2为m-ms微观结构的透射电镜图。由图2(a)中,可以清楚的观察到m-ms样品呈棒状,内部均匀排列圆柱型孔道,孔径均一且高度有序。图2(b)所示,m-ms样品中的孔结构呈现六边形排列。该tem结果,直观观察到材料内部的孔道结构,与bet结果相符。图3为m-ms(a,c)和ms(b,d)粉末样品的扫描电镜图(a,b)和eds谱图(c,d)。由扫描电镜图可知,粉末样品呈短棒状结构。eds分析结果表明,表明样品的主要元素均是o,mg和si。m-ms中镁元素的含量为15.2wt%,证明所合成的粉末为硅酸镁材料。图4是m-ms的x射线衍射图谱。其中,图4(a)是m-ms的广角x射线衍射谱图。图中可明显观察到在2θ=15-35°之间有一个宽的衍射峰,这是典型的无定形硅酸盐的衍射峰。结果表明m-ms呈无定性态,不含有结晶成分。图4(b)是m-ms的小角x射线衍射谱图。图中可见一个非常强的二维对称六角排列(p6mm)的(100)的衍射峰,和一个较弱的(110)的衍射峰。这表明m-ms有规整的介孔结构,与透射电镜的结果相符合。实施例3和4中的介孔硅酸镁(m-ms)的结构、形貌、平均孔径、比表面积、孔容与实施例1中的m-ms基本相当。效果实施例2实施例1中制备的介孔硅酸镁(m-ms)和对比例1中制备的非介孔硅酸镁(ms)的吸水性能和体外降解性能:材料的吸水性能是通过将材料浸泡在去离子水中一定时间后所吸收水的重量来测定。以m-ms圆片样品为例,具体步骤如下:首先准确称量m-ms圆片样品的重量,记为w0。之后将样品浸泡在去离子水中,分别在0.5,6,12,24和48小时后,擦去样品表面水份并对样品称重,记为wt。吸水率表示吸收水的重量占原重的百分比,按照公式计算:吸水率(%)=(wt-w0)/w0×100,通过将材料浸泡在tris-hcl缓冲液中,测量计算其重量损失率来表征体外降解性能。tris-hcl缓冲溶液(ph=7.4)的配制方法如下:42ml稀盐酸溶液(0.1mol/l)与50ml三羟甲基氨基甲烷(tris,0.1mol/l)溶液混合均匀后,用去离子水定容至100ml。首先将m-ms圆片样品超声清洗2次,100℃真空干燥至恒重,分析天平称其初始质量,记为w0。按照固体/液体为0.25g/50ml的比例,将样品浸泡在tris-hcl(ph=7.4)溶液中,置于恒温振荡箱(37℃,80r/min)中。分别在3,7,14,21,28,42,56,70和84天时取出样品,100℃真空干燥至恒重,称重并记为wi。则失重率(%)按照公式计算:失重率(%)=(w0-wi)/(w0)×100。图5显示了m-ms和ms浸泡在去离子水中不同时间后的吸水率(a)和在tris-hcl中浸泡不同时间后的失重率(b)。图5(a)所示,在开始的30分钟,m-ms和ms的吸水率分别达到69.5%和17.8%。随着时间增加,吸水率逐步提高。12小时后,m-ms和ms的吸水率分别增加到73.2%和25.3%。随后逐步平缓,最终在48小时,二者的吸水率分别停留在73.7%和26.7%。由此可见,m-ms和ms都具有较高的吸水率,而m-ms的吸水性能远远强于ms。图5(b)显示了m-ms浸泡在tris-hcl溶液中不同时间后的失重率,用于表征材料的体外降解性能。由图可见,在整个浸泡期间,随着时间的推移,m-ms和ms的失重率都不断提高;而在相同时间,m-ms的失重率明显高于ms。在浸泡84天后,m-ms和ms的失重率分别为41.1wt%和19.3wt%。结果表明,相比于ms,m-ms的降解速率更快;这是由于m-ms具有较高的比表面积,从而增大了m-ms和tris-hcl溶液的接触面积,最终加快了材料的降解。实施例3和4中的介孔硅酸镁(m-ms)的吸水性能和体外降解性能与实施例1中的m-ms基本相当。效果实施例3实施例1中制备的介孔硅酸镁(m-ms)和对比例1中制备的非介孔硅酸镁(ms)的体外生物活性和离子释放:模拟体液(simulatedbodyfluid,sbf)溶液中各离子浓度与人体体液的离子浓度相近。本实验通过测定样品浸泡在sbf溶液后表面生成磷灰石的量,对m-ms和ms的体外生物活性。按照固体/液体为0.25g/50ml的比例,将样品浸泡在sbf溶液中,置于恒温振荡箱(37℃,80r/min)中。7天后取出样品,用去离子水仔细漂洗,并在37℃的电热鼓风干燥箱中烘干。浸泡后,材料的表面形貌和组成分别采用sem和eds,xrd进行表征。将m-ms和ms圆片样品浸泡在sbf溶液中,置于转速80r/min的恒温振荡箱(37℃)中。分别在6,12,24,72和120小时后,用ph计(phs-2c,上海仪电科学仪器股份公司)测量溶液的ph值,用等离子体发射光谱仪(icp-aes;iris1000,thermoelemental,usa)测定sbf溶液中ca,p,mg和si离子的浓度。通过将m-ms和ms浸泡于sbf溶液中观察表明形貌变化来表征材料的体外生物活性。图6为m-ms(a,b)和ms(c,d)在sbf中浸泡7天前(a,c)后(b,d)的扫描电镜照片。由图6可以看出,浸泡前ms材料表面光滑;浸泡7天后,材料表面生成了少量的蠕虫状物质,这是典型的磷灰石形貌。同样情况下,浸泡前m-ms材料表面同样光滑;浸泡7天后,材料表面基本完全被蠕虫状物质所覆盖。结果表明,相比于ms,m-ms表面能够生成更多的磷灰石,具有更好的生物活性。这是由于,m-ms浸泡于sbf溶液后,溶液中的h+和m-ms的mg2+交换,使得材料表面形成硅烷醇基,从而诱导sbf溶液中的ca2+和po43-在材料表面沉积,最终生成磷灰石层。图7是m-ms在sbf中浸泡7天后的能谱分析。图8(a)eds结果显示,相比于浸泡前(图3(c)),浸泡7天后的材料表面除了o,mg,si元素外,出现了ca和p元素;表明有ca-p物质沉积在m-ms材料表面。图11(b)xrd结果显示,相比于浸泡前(图7(a)),浸泡7天后的材料表面出现了磷灰石的特征峰;表明m-ms材料表面有磷灰石相生成。结果表明,m-ms浸泡在sbf中具有表面生成磷灰石的能力,具有良好的体外生物活性,与扫描电镜结果一致。图8为m-ms和ms在sbf中浸泡不同时间后的mg和si(a),ca和p(b)离子浓度的变化。测试结果显示,溶液中mg和si离子浓度随着时间增长而不断增加。72小时后,浸泡m-ms和ms的溶液中mg离子浓度分别达到230mg/l和160mg/l,而si离子浓度分别达到54.7mg/l和46.5mg/l。然后,溶液中mg和si离子浓度维持稳定,基本没有发生变化。图8(b)结果显示,溶液中ca和p离子浓度随着时间增长而不断降低。72小时后,浸泡m-ms和ms的溶液中ca离子浓度分别达到52.6mg/l和27.9mg/l,而p离子浓度分别达到35.0mg/l和8.6mg/l。这说明,材料中的mg和si离子不断释放到溶液中,而溶液中的ca和p离子不断沉积在材料表面形成磷灰石。浸泡m-ms的溶液中离子浓度变化快,说明ca和p离子沉积在材料表面形成磷灰石的速度快。图9为m-ms和ms在sbf中浸泡不同时间后的ph的变化。ph的测试结果显示,浸泡m-ms的sbf溶液ph值在开始的24小时上升较快,然后缓慢上升到7.65,并稳定在该值。浸泡ms的sbf溶液ph值在开始的120小时逐步上升到7.58,然后基本稳定。结果表明,sbf溶液ph值变化范围为7.4-7.7,保持弱碱性环境。实施例3和4中的介孔硅酸镁(m-ms)的体外生物活性和离子释放性能与实施例1中的m-ms基本相当。效果实施例4实施例1中制备的介孔硅酸镁(m-ms)和对比例1中制备的非介孔硅酸镁(ms)的细胞实验:mc3t3-e1细胞(中国科学院,上海,中国)培养在dmem培基(dulbecco’smodifiedeaglemedium;thermofisherscientificinc.,usa)中。培基中含有10%(v/v)的胎牛血清(fbs;gibcobrl,grandisland,ny,usa),1%(v/v)的抗生素(100u/ml青霉素,100mg/ml链霉素)(gibcobrl,usa)。细胞生长环境为37.5℃,100%饱和湿度,5%co2。在使用细胞进行实验之前,使用0.25%胰酶对细胞进行消化,使其脱落下来呈悬浮状体,为后期实验计算细胞密度做准备。在进行细胞实验前,m-ms和ms圆片样品在高压灭菌器中120℃消毒30分钟后备用。将灭菌后的m-ms和ms圆片样品置于24孔板中进行细胞实验,细胞接种密度为l×104个细胞/孔,每隔两天更换一次培养液。(1)细胞增殖本实验采用mtt(3-(4,5-二甲基噻唑)-2,5-二苯基四氮唑溴盐)法检测材料表面细胞的增殖情况。细胞接种到m-m和ms圆片样品上,组织培养板(tcp)作为对照。培养1,4和7天后,用移液枪将培养基吸出,用pbs溶液轻轻冲洗材料三次。每个孔加入400μldmem培养液和40μl5mg/mlmtt后,将孔板置于37℃co2培养箱内培养4小时。小心弃去上清液,将100μl二甲基亚砜(dmso;国药集团,中国)加入到孔板中,置于培养箱中10分钟使紫色的结晶完全溶解后,将上清液转移到一个96孔板,立即使用elisa酶标仪(elx800,bio-tek)在490nm处测定吸光度(o.d.值)。图10(a)显示的是mc3t3-e1细胞在m-ms,ms和组织培养板(tcp)上分别培养1,4和7天后的mtt分析结果。由图可见,随着时间的延长,三种材料表面细胞的o.d.值逐渐增加,表明细胞数目逐渐增长。在7天时间内,m-ms表面细胞数目与ms和培养板(tcp)表面细胞数目没有明显差异。这表明,随着时间的延长m-ms可以促进细胞增殖,对细胞没有不良影响。碱性磷酸酶活性(alp)的表达常被用来当作成骨性能良好与否的标志[162]。图10(b)是mc3t3-e1细胞在m-ms,ms和tcp上分别培养4天和7天后的alp表达情况。由图可见,随着时间的延长,三种材料表面细胞的alp活性表达逐渐增强。在7天时,m-ms表面细胞的alp活性表达明显高于tcp表面细胞。结果表明,m-ms能够促进mc3t3-e1细胞向成骨细胞分化。测出的o.d.值显示,随着时间的延长,两种材料表面细胞的o.d.值逐渐增加,表明细胞数目逐渐增长。在7天时间内,m-ms表面细胞数目与培养板(tcp)表面细胞数目没有明显差异。这表明,随着时间的延长m-ms可以促进细胞增殖,对细胞没有不良影响。(2)碱性磷酸酶(alp)活性对培养在材料表面的mc3t3-e1细胞进行了碱性磷酸酶(alp)活性测定。细胞接种到m-ms圆片样品上,组织培养板(tcp)作为对照。培养4和7天后,弃去培养基,样品用pbs冲洗三次。每孔加入500μl的1%nonidetp-40(np-40)溶液,在室温下裂解细胞1小时,获得细胞裂解液。每孔加入50μl1mg/ml的硝基苯磷酸二钠盐(p-nitrophenylphosphate;songon,中国)溶液(含0.1mol/l甘氨酸和1mmol/l六水氯化镁,ph=9),置于37℃培养箱内孵育1小时。结束后,每孔加入100μl的0.1mol/lnaoh溶液终止显色反应。最后使用酶标仪(384spectramax,moleculardevices,美国)在405nm处,测定o.d.值。裂解液中总蛋白的含量采用牛血清白蛋白为标准蛋白,使用bca试剂盒(碧云天生物科技,中国)进行测定。alp活性表示为405nm处o.d.值/总蛋白含量。碱性磷酸酶活性(alp)的表达常被用来当作成骨性能良好与否的标志。测试结果显示,随着时间的延长,两种材料表面细胞的alp活性表达逐渐增强。在7天时,m-ms表面细胞的alp活性表达明显高于tcp表面细胞。结果表明,m-ms能够促进mc3t3-e1细胞向成骨细胞分化。实施例3和4中的介孔硅酸镁(m-ms)的细胞实验效果与实施例1中的m-ms基本相当。效果实施例5对实施例1和2及对比例2中的c20、c40和c0进行性能表征:(1)傅里叶红外分析(ftir)将约1mg多孔支架样品烘干后与100mg干燥的溴化钾粉末(kbr)混合研磨均匀,压成薄片,使用傅立叶变换红外光谱仪(nicolet380,thermo,usa)进行分析。图11为c0(a)、c20(b)、c40(c)和介孔硅酸镁(d)四种材料的ftir图谱。在c0的ftir谱线(a)中,在1730cm-1处出现的强吸收谱带,是pbsu中c=o键的对称伸缩振动吸收谱带。在m-ms的ftir谱线(d)中,1082cm-1,799cm-1和457cm-1处出现的强吸收谱带,分别是si-o-si反对称伸缩振动,对称伸缩振动和弯曲振动所致。此外,在1640cm-1处出现的强吸收谱带是h-oh弯曲振动所致,3450cm-1附近出现的较宽吸收峰是材料表面吸附水分子中游离的羟基吸收谱带。有趣的是,3450cm-1处的较宽吸收峰出现在c20和c40的谱线上,而1640cm-1处的吸收峰只出现在c40的谱线上。结果表明,c20和c40同时具有pbsu和m-ms的特征峰,从而确认m-ms已成功添加到pbsu中。(2)x射线衍射分析(xrd)采用x射线衍射仪(xrd,rigakud/max2550,日本)在10~80°分析了材料的微观结构及结晶状态。通过从x-射线衍射图谱的峰面积比来计算各组样品的结晶度。图12为c0(a)、c20(b)、c40(c)和介孔硅酸镁(d)四种材料的广角x-射线衍射图谱。在m-ms的xrd谱线(d)中,只能观察到在2θ=15-35°处有一个宽的衍射峰,表明典型的无定型硅酸盐。在c0的xrd谱图(a)中,可观察到在19.3°,22.2°,25.9°和29°处有明显的衍射峰,分别对应pbsu的(002),(110),(121)and(111)晶相。在c20和c40的xrd谱线(b,c)中,可以发现随着复合支架材料中m-ms含量的增加,衍射峰的位置并没有移动,但是衍射峰的强度却逐渐降低。通过计算可知,c0,c20和c40的结晶度分别为57.2,49.4和35.8%。结果表明,随着m-ms在复合支架材料中的含量增加,结晶度随之降低。(3)扫描电镜(sem)将多孔支架材料样品烘干后,黏附在导电胶上喷金后,采用电子扫描电镜(s-3400n,hitachi,japan)观察样品表面形貌。图13为c0(a,d)、c20(b,e)和c40(c,f)的表面微观形貌结构。由图可见,将上述的支架材料(c0,c20和c40)具有明显的大孔结构,孔径约为400μm,这与支架制备过程中所用的致孔剂(nacl)粒径大小一致。这说明,m-ms的加入,并没有对支架材料的大孔产生影响。在放大倍数的sem图(d,e,f)中,可以发现c0支架大孔孔壁表面相对光滑,而c20和c40支架大孔孔壁表面却是粗糙多孔的。并且,c40支架表面的微孔(1-10μm)数量远远多于c20支架。这些微孔是由于制备支架过程中溶剂挥发所导致的,使得前述的大孔相互贯通,从而形成了三维孔隙结构。结果表明,m-ms的加入,提升了孔的相互贯通性能。而m-ms本身具有约为5nm的小孔,总之c40支架具有了大孔-微孔-小孔的三级孔结构,这有利于支架植入体内后与周围组织液及血液的营养交换。(4)元素分析(eds)通过元素分析仪(eds,falcon,usa)上测得样品表面的元素组成。图14为c0(a)、c20(b)和c40(c)的eds图谱。由图中看出,co的eds谱图中,仅有碳和氧元素,代表了pbsu中的元素。c20和c40的eds谱图中,除了c和o元素,还有mg和si元素存在;且明显观察到,c40中mg和si元素的含量要比c20的高。结果表明复合多孔支架材料中含有pbsu和m-ms两种物质,与ftir分析结果一致。(5)同步辐射srμct成像采用上海同步辐射光源(shanghaisynchrotronradiationfacility,ssrf)的x-射线成像及生物医学应用线站(bl13w线站),对多孔支架材料样品进行x光扫描并成像观察,并通过一个高分辨率(9μm)的x射线电荷耦合相机接受信号。同步辐射电子束能量3.5gev,光斑尺寸45mm(h)×5mm(v)。选用x-射线能量为15kev,样品到接收器的距离为1.6m,角度180°。通过imageproplus6.0(mediacyberneticsco.)软件对所获得的图像进行去背景处理处理,并通过pitre(phase-sensitivex-rayimageprocessingandtomographyreconstruction)软件处理数据得到二维电子切片图片。图15为c0(a)、c20(b)和c40(c)的srμct电子切片照片。由图可见,三种多孔支架材料均具有400μm大小的大孔,呈网状分布,形成均匀的三维网络结构,且在宏观尺寸上没有明显的视觉差异。多孔支架材料表面与内部形貌没有明显的差别,均呈现三维网络结构,孔结构连通性良好。实验结果与扫描电镜结果相吻合。(6)孔隙率测定通过阿基米德原理测定多孔骨修复支架的孔隙率。首先将多孔支架材料(φ12×2mm)在烘箱中烘干并准确称量其重量记为m1;将多孔支架材料浸入水中以便水完全充满支架内部的孔隙,支架在水中的重量记为m3;从水中将多孔支架材料取出,擦干其表面的水分并准确称量其重量记为m2。孔隙率按下列公式计算:孔隙率(%)=(m2-m1)/(m2-m3)×100其中m1为多孔支架材料在空气的重量,m2为多孔支架材料与其内部所吸水的重量,m3为多孔支架材料悬浮在水中的重量。图16为c0(a)、c20(b)和c40(c)在去离子水中浸泡不同时间后的吸水率。由图可见,在开始的30分钟,c0的吸水率仅为190%,而c20和c40的吸水率分别高达345%和369%。多孔骨修复支架的吸水率随后持续快速增长,直到12小时后趋于稳定。最终在48小时,c0,c20和c40的吸水率分别为275%,381%和421%。结果表明,m-ms的加入大大提升了复合支架材料的吸水率。采用阿基米德原理,测得的c0,c20和c40多孔支架材料的孔隙率分别为66.5%,67.3%和69.7%。表明,m-ms的加入大大提升了复合支架材料的孔隙率,使得支架具有了更通透的孔结构。更高的吸水率,预示着将多孔支架材料植入体内后,可以再较短的时间内吸附大量的周围组织液,形成营养物质局部富集区,对骨缺损修复具有十分重要的影响和积极作用。(7)抗压强度测定将φ6×6mm的多孔支架在烘箱中烘干后,用万能试验机测量样品的抗压强度,施加载荷速率为1mm/min。实施例1和2及对比例2中的c20、c40和c0的抗压强度测定结果见表1。表1多孔支架的抗压强度实施例1(c20)实施例2(c40)对比例2(c0)抗压强度(mpa)3.0±1.24.1±1.92.2±0.8实施例3和4中的介孔硅酸镁/聚丁二酸丁二醇酯复合支架的结构、形貌、元素组成、同步辐射srμct成像、孔隙率抗压强度与实施例1和2中的介孔硅酸镁/聚丁二酸丁二醇酯复合支架基本相当。效果实施例6对实施例1和2及对比例2中的c20、c40和c0的吸水性能和体外降解性能的表征:材料的吸水性能是通过将三种多孔支架材料浸泡在去离子水中一定时间后的吸收水的重量来测定。具体步骤如下:首先准确称量m-ms和ms圆片样品的重量,记为w0。之后将样品浸泡在去离子水中,分别在0.5,6,12,24和48小时后,擦去样品表面水份并对样品称重,记为wt。吸水率表示吸收水的重量占原重的百分比,按照公式计算:吸水率(%)=(wt-w0)/w0×100%。通过将多孔支架材料(φ12×2mm)浸泡在tris-hcl溶液中,测量计算其重量损失率来表征体外降解性能。tris-hcl缓冲溶液(ph=7.4)的配制方法如下:42ml稀盐酸溶液(0.1mol/l)与50ml三羟甲基氨基甲烷(tris,0.1mol/l)溶液混合均匀后,用去离子水定容至100ml。首先将m-ms和ms圆片样品超声清洗2次,100℃真空干燥至恒重,分析天平称其初始质量,记为w0。按照固体/液体为0.25g/50ml的比例,将样品浸泡在tris-hcl(ph=7.4)溶液中,置于恒温振荡箱(37℃,80r/min)中。分别在3,7,14,21,28,42,56,70和84天时取出样品,100℃真空干燥至恒重,称重并记为wi。则失重率(%)按照公式计算:失重率(%)=(w0-wi)/(w0)×100%。图17为c0(a)、c20(b)和c40(c)浸泡在tris-hcl溶液中不同时间的降解程度,用于表征材料的体外降解性能。结果发现,随着时间的增加,三种多孔支架材料(c0,c20和c40)的失重率均逐渐提高;表明三种多孔支架材料都随时间增加而不断降解。相同时间点,复合多孔支架材料的失重率随m-ms含量的增加而增加。其中,c40的失重率曲线斜率最大,在浸泡在tris-hcl溶液中84天后失重率高达66.1%;而作为对照,c20和c0在浸泡84天后的失重率分别为38.1%和15.3%。结果表明,m-ms的加入提高了多孔支架材料的体外降解性能。这是因为m-ms的加入破坏了基体材料pbsu的结构,从而降低了复合多孔支架材料的结晶度。此外,m-ms具有良好的体外降解性能,随着时间的推移,m-ms逐步降解,提高复合多孔支架材料的失重率。另一方面,m-ms的降解进而导致复合多孔支架材料的大孔孔壁出现更多的微孔结构,使得tris-hcl溶液可以扩散到支架材料的内部,加速其降解过程。因此c40表现出最优异的体外降解性能。在选取的时间点,通过ph计(phs-2c,上海仪电科学仪器股份公司)测量tris-hcl溶液的ph值,记录其ph值变化。图18为c0(a)、c20(b)和c40(c)的tris-hcl溶液ph值变化。由图可见,浸泡c0的tris-hcl溶液ph值在前14天基本保持不变,随后逐渐降低,到84天时,ph值降低到6.83。而浸泡c20和c40的tris-hcl溶液ph值在前4天就大幅升高到7.62和7.70,随后显著降低到7.31和7.39(21天)。然后浸泡c20的tris-hcl溶液ph值逐渐降低到7.17(84天),而在此期间,浸泡c40的tris-hcl溶液ph值没有明显变化。这是由于,pbsu的主要降解产物(琥珀酸)随着降解时间的增加而逐渐增加,导致浸泡c0的tris-hcl溶液ph值不断降低。然而,随着m-ms的加入,复合多孔支架材料孔壁上的m-ms在前4天就迅速降解,释放出mg2+和sio44-,形成弱碱性环境。随着浸泡时间的延长,pbsu的主要降解产物(琥珀酸)逐渐增加,因此,ph值有所下降。而c40中m-ms含量较高,在浸泡21天后酸性和碱性降解产物达到动态平衡,从而ph值维持不变。总之,复合多孔支架材料中m-ms的降解,可以中和pbsu的酸性降解产物,从而维持弱碱性微环境,这将有利于细胞生长。实施例3和4中的介孔硅酸镁/聚丁二酸丁二醇酯复合支架的吸水性能和体外降解性能与实施例1和2中的介孔硅酸镁/聚丁二酸丁二醇酯复合支架基本相当。效果实施例7对实施例1和2及对比例2中的c20、c40和c0的体外生物活性的表征:将多孔骨修复支架材料(φ12×2mm)浸泡在sbf溶液中,5天后从溶液中把样品取出,用去离子水仔细润洗,并在37℃的电热鼓风干燥箱中烘干。材料的表面形貌和组成分别采用sem和eds进行表征。具体方法见2.2.3。用等离子体发射光谱仪(icp-aes;iris1000,thermoelemental,usa)测定sbf溶液中ca,p,mg和si离子的浓度。图19为c0(a,d)、c20(b,e)和c40(c,f)在sbf中浸泡5天后的扫描电镜照片。由图可见,c0表面仍旧光滑;c20的一部分表面生成蠕虫状物质;c40的支架表面则完全被蠕虫状物质所覆盖。可明显看出,c40表面生成的磷灰石层多于c20表面生成的磷灰石层,而c0表面没有生成磷灰石层。结果表明,m-ms的加入,改善了多孔支架材料的生物惰性;且复合支架材料随着m-ms含量的增加在sbf溶液中沉积磷灰石的能力随之增强。c40表现出良好的体外生物活性,适合作为骨修复材料使用。图20为c0(a)、c20(b)和c40(c)在sbf中浸泡5天后的eds图谱。相比于浸泡前(图5),c0在sbf溶液中浸泡5天后eds没有明显变化。c20和c40在sbf溶液中浸泡5天后,可以明显观察到ca和p峰的出现,表明多孔支架材料表面存在大量的ca元素和p元素,有ca-p物质生成。此外,ca/p比值为1.69,接近于羟基磷灰石的ca/p比值(1.67),证实了覆盖在多孔支架材料表面的蠕虫状物质为磷灰石。材料表面生成磷灰石层,有利于成骨细胞的粘附与增殖及植入体内后的成骨效果。图21为c0(a)、c20(b)和c40(c)在sbf溶液浸泡过程中离子浓度的变化。由图可见,sbf溶液中ca和p离子的浓度随着浸泡时间的延长逐渐下降,而mg和si离子浓度却逐渐增加。这是由于,c40中m-ms降解过程中释放出大量的mg和si离子,与溶液中的h3o+发生离子交换,在c40表面形成si-oh基团,进而诱导溶液中的ca2+和po43-在c40表面沉积生成磷灰石。这一结果与sem和eds结果一致。实施例3和4中的介孔硅酸镁/聚丁二酸丁二醇酯复合支架的体外生物活性与实施例1和2中的介孔硅酸镁/聚丁二酸丁二醇酯复合支架基本相当。效果实施例8实施例1和2及对比例2中的c20、c40和c0的细胞实验:本实验选用mc3t-e1细胞,mc3t3-e1细胞(中国科学院,上海,中国)培养在dmem培基(dulbecco’smodifiedeaglemedium;thermofisherscientificinc.,usa)中。培基中含有10%(v/v)的胎牛血清(fbs;gibcobrl,grandisland,ny,usa),1%(v/v)的抗生素(100u/ml青霉素,100mg/ml链霉素)(gibcobrl,usa)。细胞生长环境为37.5℃,100%饱和湿度,5%co2。在使用细胞进行实验之前,使用0.25%胰酶对细胞进行消化,使其脱落下来呈悬浮状体,为后期实验计算细胞密度做准备。在进行细胞实验前,c20、c40和c0在高压灭菌器中120℃消毒30分钟后备用。将灭菌后的c20、c40和c0置于24孔板中进行细胞实验,细胞接种密度为l×104个细胞/孔,每隔两天更换一次培养液。使用0.25%胰蛋白酶消化,然后加入含10%fbs的dmem培养液终止消化,离心后,使用含10%fbs,100u/ml青霉素,100μg/ml链霉素的dmem,配制mc3t3-e1悬液,充分吹打后计数,按需要的细胞密度进行接种。在进行细胞实验前,三组多孔支架样品采用环氧乙烷灭菌,储存在离心管中备用。将灭菌后的多孔支架样品(φ12×2mm)置于24孔板中进行细胞实验。培养基每三天更换一次。(1)细胞活性本实验采用cck-8法(cellcountingkit-8)测试多孔支架材料的细胞活性。细胞接种密度为3×104个细胞/孔,不加支架材料的空白孔作为对照。具体方法如下,将mc3t-e1细胞接种到多孔支架材料3、6和12小时后,使用ph=7.4无菌磷酸盐缓冲液(pbs)仔细冲洗支架三次,然后转移到一个全新的24孔板,以避免原24孔板内细胞的影响。以每500μldmem培养基中加入50μlcck-8检测液(dojindomoleculartechnologiesinc.,japan)的比例,向24孔板中加入检测液。在37.5℃,5%co2的环境下孵育3小时。孵育完成后,取上清液,转移到96孔板中,在酶标仪(synergyht,bio-tek,winooski,vermont,usa)上450nm处读取吸光度(od)值。(2)细胞形态观察本实验所用mc3t-e1细胞的接种密度为l×104个细胞/孔。细胞与多孔支架材料共培养3天后,弃去培基,样品经pbs溶液洗涤和戊二醛溶液固定后,使用激光共聚焦显微镜,考察mc3t-e1细胞在多孔支架材料表面及内部的粘附和铺展情况。图22为mc3t3-e1细胞接种到c0(a)、c20(b)和c40(c)3,6,12小时后cck-8测试结果。结果发现,c0的od值与空白对照没有明显区别,表明pbsu材料对细胞粘附没有影响。然而在3和6小时,c40和c20的od值明显高于c0;12小时,c20的od值仍旧明显高于c0,而c40的od值明显高于其他两组(c0和c20)。结果表明,粘附在c40上的细胞数目多于c0和c20。(3)细胞增殖细胞增殖实验所用mc3t-e1细胞的接种密度为5×103个细胞/孔。采用cck-8法测试多孔骨修复支架表面细胞细胞的增殖情况。将细胞在多孔支架材料上培养1,3和5天后,采用cck-8法在酶标仪上测定od值。本实验中,将各组多孔支架材料在1天的od值定义为1,从而得出3和5天的修订od值,最后细胞增殖情况以相对增值率表示。图23为在1,3和5天,c0(a)、c20(b)和c40(c)对mc3t3-e1细胞增殖的影响。由cck-8检测结果可以发现,三种多孔支架材料上细胞的相对增殖率都随时间而逐渐增加。整个培养期间,c0的细胞相对增殖率与空白对照无明显区别。3天,c0的细胞相对增殖率远远低于c40(p<0.05),而c20和c40间没有明显的差异。5天,c0的细胞相对增殖率远远低于c20(p<0.05)和c40(p<0.01)。结果表明,c40促进mc3t3-e1细胞增殖,具有最显著的作用。这是由于c40为细胞提供了的三维成长空间更为广阔,更好的孔隙结构利于营养物质和代谢产物的传输及细胞的长入;且其内部含有大量的m-ms粒子,在培养细胞过程中能够释放大量的镁和硅元素,能够促进mc3t3-e1细胞增殖。(4)碱性磷酸酶(alp)活性alp活性实验所用mc3t-e1细胞的接种密度为4×104个细胞/孔。细胞与多孔支架材料共培养24小时,待细胞完全贴壁后,将培基更换为成骨诱导培养基:dmem培基中含有10%(v/v)的胎牛血清(fbs;gibcobrl,grandisland,ny,usa),0.1μmol/l地塞米松(sigma),50μmol/l抗坏血酸(sigma)和10μmol/lβ-甘油磷酸钠(sigma)。培养7,10和14天后,进行alp活性测定。对培养在材料表面的mc3t3-e1细胞进行了碱性磷酸酶(alp)活性测定。细胞接种到m-ms和ms圆片样品上,组织培养板(tcp)作为对照。培养4和7天后,弃去培养基,样品用pbs冲洗三次。每孔加入500μl的1%nonidetp-40(np-40)溶液,在室温下裂解细胞1小时,获得细胞裂解液。每孔加入50μl1mg/ml的硝基苯磷酸二钠盐(p-nitrophenylphosphate;songon,中国)溶液(含0.1mol/l甘氨酸和1mmol/l六水氯化镁,ph=9),置于37℃培养箱内孵育1小时。结束后,每孔加入100μl的0.1mol/lnaoh溶液终止显色反应。最后使用酶标仪(384spectramax,moleculardevices,美国)在405nm处,测定o.d.值。裂解液中总蛋白的含量采用牛血清白蛋白为标准蛋白,使用bca试剂盒(碧云天生物科技,中国)进行测定。alp活性表示为405nm处o.d.值/总蛋白含量。图24为c0(a)、c20(b)和c40(c)在7,10和14天对mc3t3-e1细胞alp活性表达的影响。实验中材料诱导成骨性能高低的标志通常由alp活性表达作为参考。由图可见,三种多孔支架材料上细胞的alp活性表达都随时间的增加而增高。7和10天,c40(p<0.05)细胞的alp活性表达明显高于c0;14天,c20(p<0.05)和c40(p<0.01)细胞的alp活性表达明显高于c0。这是由于复合多孔支架材料中的m-ms能够持续释放镁和硅等元素,从而促进mc3t3-e1细胞alp活性表达。结果表明,c40对mc3t3-e1细胞向成骨细胞分化具有更明显的促进作用。实施例3和4中的介孔硅酸镁/聚丁二酸丁二醇酯复合支架的细胞实验效果与实施例1和2中的介孔硅酸镁/聚丁二酸丁二醇酯复合支架基本相当。效果实施例9实施例1和2及对比例2中的c20、c40和c0的动物实验:本实验采用兔右侧股骨末端缺损模型考察三种多孔支架材料(c0,c20和c40,φ6×6mm)的体内成骨效果。所有手术过程及动物饲养均在国家组织工程技术研究中心(上海,中国)进行,由当地伦理委员会批准。选取27只雌雄各半的实验用新西兰白兔(年龄5个月,平均体重为3kg)进行动物实验,随机平均分成三组,每组9只分别植入一种多孔支架材料。动物实验在无菌手术室进行。麻醉剂用量按动物体重计算(25mg/kg舒泰和0.15ml/kg2%rompon),经肌肉注射后进行全身麻醉。新西兰大白兔右后肢剃毛、消毒,按俯卧位放置在手术台上。右侧股骨髁部位取约长1cm的切口,逐层分离皮肤、肌肉、骨膜等组织,显露兔股骨外髁。选用骨科钻孔器,沿股骨纵轴和矢状轴垂直方向制造6mm×6mm圆柱腔骨洞缺损,上述三种多孔支架分别植入兔骨缺损模型,逐层严密缝合创口,在伤口表面涂抹碘伏进行伤口消毒。术后单笼饲养,正常喂食,连续3天肌肉注射青霉素以消炎。观察实验用新西兰白兔的术后恢复情况。分别于术后4,8和12周通过注射过量戊巴比妥钠处死动物,取出缺损修复部位,剥除肌肉组织后获得骨样标本。拍摄数码照片,对骨缺损情况进行大体观察。然后使用10%中性福尔马林缓冲液来固定标本,进行各种检测。大体观察:材料植入兔股骨缺损后三天,新西兰大白兔各种表现(进食、活动和排便情况)基本恢复正常,实验期间所有动物全部存活。按时取材,可以观察到新西兰大白兔无伤口感染、无骨折等并发症发生。三种多孔支架材料(c0,c20和c40)的植入对动物没有明显不良影响,生物安全性好。观察可知术后4周,三种多孔支架材料没有发生位移,周围组织没有明显的的炎症反应。c0和c20表面可以观察到明显的骨缺陷,而c40植入部位部分表面被组织覆盖,隐约可见多孔支架材料。术后8周,三种多孔支架材料植入部位表面均被组织覆盖。c0表面覆盖组织较薄,可明显看到凹陷;c20和c40表面覆盖组织较厚,骨缺损部位与周围骨组织的界面缩小。术后12周,c0植入部位与周围骨组织的界面明显缩小;c20植入部位只存有相对较小的疤痕;c40植入部位缺损基本愈合,新生骨组织与周围骨组织没有明显差别,修复完全。(1)同步辐射srμct成像为了更加直观地观察骨缺损部位内部植入材料降解及新骨组织生长的状况,采用上海同步辐射光源(ssrf)的bl13w线站,对骨样样品进行x光扫描并成像观察。具体步骤如下:首先将浸泡在戊二醛溶液中的骨头样品取出,并使用去自然水冲洗1小时,然后使用梯度乙醇溶液进行脱水处理,并置于通风橱中过夜至样品干燥。然后将骨头样品固定在bl13w线站的工作台上,选用x-射线能量为30kev,成像距离1.6m,旋转角度为180度。通过pitre软件得到二维电子切片图片,vgstudiomax2.0软件进行三维重构得到三维图像。图25为c0(a)、c20(b)和c40(c)植入动物体内4,8,12周后的ct三维重构照片。由图25a可知,术后4周,骨缺损部位清晰可见,周围骨组织向缺损部位空隙长入。c40植入部位有明显的新骨形成,骨缺损部位界面明显缩小,外部皮质骨基本闭合;然而,c0和c20植入部位只有很少的新生骨形成。由图25b可知,术后8周,各组缺损部位新生骨数量都有所增加,其中c0植入部位只有较少的矿化组织生成。而c20和c40植入部位有大量新生骨生成。且c40新生骨密度较c20新生骨密度更大,更接近于宿主骨。图25c可知,术后12周,c0植入部位新生骨数量最少,且有一个明显空洞存在,表明骨缺损仍未修复完全。与之相比,c20植入部位新生骨数量较多,未修复的骨缺损区域明显减少。c40植入部位新生骨组织与周围正常宿主骨组织之间没有明显区别,骨缺损修复完全。结果表明,c40植入体内12周后,支架完全降解,骨缺损修复效果良好。(2)组织学分析srμct检测完后,将骨样标本以0.9%nacl生理盐水冲洗后,用30%甲酸-福尔马林脱钙液脱钙,乙醇梯度脱水,石蜡包埋,使用薄片切片机制作5μm厚的切片。分别使用苏木精/伊红(he)处理组织切片,具体操作如下:he染色苏木素染液染色5-10分钟,0.5-1%盐酸酒精分色片刻,镜检控制直至细胞核及核内染色质清晰为止;流水冲洗半个小时,然后蒸馏水短洗;0.1-0.5%伊红染液染色1-5分钟;经梯度乙醇脱水,二甲苯透明两次,滴加适量中性树胶,加盖玻片封固。使用倒置显微镜(nikonte2000u,nikon,japan)观察,分析切片中材料残余、新生骨基质和炎症细胞等情况。将切片放置在倒置显微镜下,放大20倍,在每张切片上的支架材料植入部位选取4个不同的视野,视野的中心位置距宿主骨0.5mm。使用image-proplus6.0软件,分别测量新生骨面积和残余材料面积,取平均值作为每个标本的新生骨量和材料残余量。图26为通过软件计算c0(a)、c20(b)和c40(c)植入动物体内4,8,12周后的骨缺损区域新生骨面积。结果表明,新生骨组织随着植入时间的延长而逐渐增多;在相同植入时间,新生骨组织随多孔支架材料中m-ms含量的增加而增加(c40>c20>c0)。(3)免疫组织化学分析本实验进行骨形态发生蛋白-2(bonemorphogeneticprotein-2,bmp-2)免疫组织化学染色。首先将石蜡切片进行常规脱蜡处理,在3%h2o2(hydrogenperoxide)中孵育10分钟,以降低内源性过氧化物酶造成的非特异性背景染色。pbs缓冲液冲洗2次,用5%山羊血清封闭非特异性背景染色,室温下孵育5分钟后,pbs缓冲液冲洗2次,并加入一抗工作液,37℃孵育2小时。pbs缓冲液冲洗2次,加入二抗工作液,37℃孵育30分钟。用pbs缓冲液冲洗两次后,滴加适量的bmp-2标记的链霉卵白素工作液,37℃孵育30分钟。图27为根据图像通过软件分析计算得出bmp-2阳性表达率的定量分析。4周时,c20和c40的bmp-2阳性表达率均显著高于c0;8周时,三组多孔支架材料间没有明显区别;12周时,c40的bmp-2阳性表达率显著高于c20和c0。结果表明,随着时间的推移,三组多孔支架植入部位的bmp-2阳性表达率均增加,同时随着多孔支架材料中m-ms含量的增加而增加(c40>c20>c0)。实施例3和4中的介孔硅酸镁/聚丁二酸丁二醇酯复合支架的动物实验效果与实施例1和2中的介孔硅酸镁/聚丁二酸丁二醇酯复合支架基本相当。效果实施例10上述效果实施例1~10的数据统计分析所有数据均用平均值±标准差表示。数据组间差异统计采用origin8.0的onewayanova功能。p<0.05(*)和p<0.01(**)为具有显著性差异。实施例1中制备的介孔硅酸镁(m-ms)主要为无定形化合物;其平均孔径、比表面积和孔容分别为5nm,451.0m2/g和0.41cm3/g。m-ms具有优良的吸水性能,吸水率高达74%。同时,体外试验的结果证实,本实验中制备的介孔硅酸镁,和相同条件下未加入p123制备出的非介孔硅酸镁相比,具有更快的体外降解性能和更好的体外生物活性。体外降解实验结果显示,m-ms在tris-hcl缓冲液中浸泡84天后有40w%的失重率。将m-ms浸入sbf溶液中7天,材料表面有磷灰石层形成;且磷灰石生成量远远多于ms。更重要的是,由激光共聚焦显微镜观察,材料表面的细胞粘附生长情况良好,证明m-ms有很好的细胞相容性。alp活性实验结果表明,m-ms具有一定的成骨细胞诱导作用。在骨组织修复过程中,支架的作用是至关重要的,要求具有良好的孔结构,如高孔隙率和孔径(>300μm)。在本研究中,m-ms/pbsu复合支架(c40)的孔隙率约为70%。复合支架呈现出300-500μm的大孔结构,为盐浸出孔。支架表面上1μm至10μm的微孔可能是由于溶剂挥发的结果。另外,复合支架中加入的m-ms,具有5nm的均匀介孔通道。因此,c40具有大孔-微孔-介孔的多级孔结构,具有良好的孔连通性,有利于营养物和代谢产物的运输。孔隙率越大,从而导致复合支架的吸水率越高,利于细胞附着和扩散。骨修复支架材料需要具备适当的降解性。复合支架中,m-ms含量增加,失重率也有所增加。这是由于m-ms掺入支架后可能会破坏pbsu的结构,从而导致结晶度的降低。另一方面,大孔壁上的m-ms的降解可能在壁上产生更多的微孔,这可能有助于降解液扩散到支架内部,加速降解。pbsu的主要降解产物是琥珀酸,这导致含有c0的tris-hcl溶液的ph值持续降低。然而,通过加入m-ms,释放出mg2+和sio44-,导致弱碱性环境,中和来自pbsu的酸性降解产物。众所周知,诱导磷灰石形成的能力是植入物的体内生物活性的指示。磷灰石可以在m-ms/pbsu复合支架(c20和c40)的表面形成。这是由于,m-ms在sbf中开始降解,释放出si和mg离子,并且与来自sbf的h+之间发生快速离子交换,随后形成硅烷醇基团。这些基团进行缩聚以形成硅胶层,吸附溶液中的ca2+和po43-,导致在支架表面上形成无定形磷灰石层。细胞与生物材料表面的相互作用,被认为是生物材料评估中的重要因素。c40可以明显促进mc3t3-e1细胞在材料表面的扩散和迁移,增殖以及分化。因为据报道si和mg离子分别促进成骨细胞的有丝分裂和分化。一方面,在m-ms/pbsu复合支架的壁上存在微孔和介孔,有利于细胞的伪足锚定。另一方面,细胞优选复合支架的粗糙和更亲水的表面,而不是pbsu支架的光滑疏水表面。因此,改进的亲水性、孔隙率,si和mg离子的释放都有助于增强细胞生物反应。将支架植入兔股骨缺陷后,发现c40逐渐降解并被新骨组织替代,可观察到红色骨髓和更成熟的骨组织生成,且bmp-2阳性表达率最高。据报道,si离子有效地增强了骨基质合成和成骨细胞活性(形成矿化结节)相关的基因表达,刺激骨基质蛋白(col-i)合成;mg离子是最重要的矿物元素,调节活性钙的运输,稳定dna的结构,甚至影响骨骼代谢的所有阶段。因此,c40中m-ms的含量最高,其释放的mg和si离子有助于体内新骨再生。综上所述,上述效果实施例的结果表明,m-ms/pbsu复合多孔支架材料具有多级孔结构:大孔(300-500μm),微孔(1-10μm)和介孔(~5nm)。随着m-ms含量的增加,复合支架材料的结晶度降低,吸水率、体外降解性能和体外生物活性提高,可释放出mg和si等功能性离子。其中,c40支架可以明显促进mc3t3-e1细胞的粘附、增殖和alp活性表达,并促进体内成骨。这为其进一步将用于组织修复提供了基础,也为此类有机/无机硅镁基复合支架材料的制备和应用提供了新思路。虽然以上描述了本发明的具体实施方式,但是本领域的技术人员应当理解,这仅是举例说明,本发明的保护范围是由所附权利要求书限定的。本领域的技术人员在不背离本发明的原理和实质的前提下,可以对这些实施方式做出多种变更或修改,但这些变更和修改均落入本发明的保护范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1