一种酸响应性抗癌纳米药物的制备及应用的制作方法
本发明属于药物领域,更具体涉及一种酸响应性抗癌纳米药物的制备及应用。
背景技术:
在癌症治疗过程中,已经尝试了多种不同的治疗方式,化疗是肿瘤治疗的常用手段,由于化疗药物不具有肿瘤治疗的靶向性,导致产生很多毒副作用如恶心、呕吐、脱发、免疫力下降等,从而导致的病人生活质量下降,最终不得不放弃治疗的情况很常见。
纳米技术近几十年来应用于医药研究中,取得了初步的成果,纳米药物的粒径使它具有特殊的表面效应和小尺寸效应等,与常规药物相比,纳米药物的小粒径能够降低药物的使用量,提高药物的作用效率,降低药物的副作用,因此它具有许多常规药物不具备的优点。
目前研究发现,肿瘤组织的微环境中具有相对于正常细胞环境下较小的ph值(5.0-6.5),这是因为肿瘤细胞比正常细胞生长快,而在肿瘤组织中血管的供应往往跟不上肿瘤细胞快速扩增的脚步,供应的氧气和养料不足,肿瘤细胞总是处于缺氧和缺养料的微环境中生长,新陈代谢过程也与正常细胞不同,生成了更多的乳酸等酸性代谢产物,使得肿瘤组织周边的组织液ph值降低。
因此,本领域亟需开发一种能够降低抗肿瘤药物的毒性,提高抗肿瘤治疗效果和患者顺应性的药物。
技术实现要素:
本发明的目的是提供一种能够降低抗肿瘤药物的毒性,提高抗肿瘤治疗效果和患者顺应性的药物。
本发明的第一方面提供一种具有ph响应性神经肽y多肽复合物,所述的复合物包含:
(1)神经肽y多肽;和(2)纳米载药胶束,所述的纳米载药胶束具有羧基部分;
其中,所述的神经肽y多肽上的氨基部分通过与所述的纳米载药胶束的羧基部分形成酰胺键偶联于所述的纳米载药胶束上。
在另一优选例中,所述的复合物中,所述的神经肽y多肽位于所述的复合物表面,并暴露在复合物外侧。
在另一优选例中,所述的复合物在生理ph值(7.2~7.4)中释放5~20%的药物,在微酸环境(5.0~6.5)中释放40~90%的药物。
在另一优选例中,所述的神经肽y多肽是神经肽y的亚型,且所述神经肽y多肽的肽链长度为9~36个氨基酸。
在另一优选例中,所述的神经肽y多肽与纳米载药胶束的质量比为1:5-150,较佳地1:10-100,更佳地1:20-80。
在另一优选例中,所述的神经肽y多肽的肽链选自下组:npy、npy(28-36)、[arg6,pro34]pnpy、[phe6,pro34]pnpy、[asn6,pro34]pnpy、[cys6,pro34]pnpy、[phe6,pro34]pnpy、[d-his26,pro34]npy、[phe7,pro34]pnpy、[pro30,nle31,bpa32,leu34]npy(28-36)、[pro30,nal32,leu34]npy(28-36)、[pro30,nle31,nal32,leu34]npy(28-36)、[d-arg25]-npy、[d-his26]-npy、[d-arg25,d-his26]-npy、[arg7,pro34]pnpy、[leu31,pro34]pnpy、pyy(3-36)、(ahx5-24)npy、[ala31,aib32]pnpy、[d-trp34]-npy、[cpp1-7,pnpy19-23,ala31,aib32,gln34]hpp、npy(3-36)、npy(22-36),或其组合。
在另一优选例中,所述的复合物具有ph响应性部位,且所述的部位在ph改变时发生变化,所述的变化包括亲水性、疏水性、电性、结构中的一种或多种的改变。
在另一优选例中,所述的ph响应性部位在肿瘤细胞或组织的ph微环境中发生变化,所述的变化包括发生亲水性、疏水性、电性、结构中的一种或多种的改变。
在另一优选例中,所述复合物的ph响应部位含有酰胺键。
在另一优选例中,所述复合物的ph响应部位与神经肽y多肽通过酰胺键相连。
在另一优选例中,所述复合物的ph响应部位为酰胺键。
在另一优选例中,所述的纳米载药胶束选自下组:聚乙二醇-聚乳酸-羟基乙酸共聚物(peg-plga)、聚乙二醇-二硬脂酰基磷脂酰乙醇胺(peg-dspe)、二硬脂酰基磷脂酰乙醇胺-聚乳酸(dspe-pla)、聚乙二醇-聚己内酯(peg-pcl)、聚乙二醇-聚乙烯亚胺(peg-pei)、聚乳酸(pla)、聚乳酸-羟基乙酸共聚物(plga)、二硬脂酰基磷脂酰乙醇胺(dspe)、聚乙二醇(peg)、聚环氧乙烷-聚环氧丙烷-聚环氧乙烷(p123),或其组合。
本发明的第二方面提供一种载药纳米粒子,包含(a)本发明第一方面所述的具有ph响应性神经肽y多肽复合物;(b)有效治疗量的抗肿瘤活性成分。
在另一优选例中,所述的神经肽y多肽质量占载药纳米粒子总质量的百分比为0.01-60wt%。
在另一优选例中,所述的载药纳米粒子的粒径为5-200nm。
在另一优选例中,所述的抗肿瘤活性成分为小分子抗肿瘤药物,或大分子活性成分。
在另一优选例中,所述的抗肿瘤活性成分选自下组:阿霉素、顺铂、长春新碱、去甲长春碱、紫杉醇、丝裂霉素、长春地辛、曲妥珠单抗、替伊莫单抗、多西他赛,或其组合。
本发明的第三方面,提供一种本发明第二方面所述的载药纳米粒子的制备方法,所述的方法包含步骤:
(i)提供(a)本发明第一方面所述的具有ph响应性神经肽y多肽复合物;(b)有效治疗量的抗肿瘤活性成分;
(ii)将所述(a)、(b)进行复合,从而形成载药纳米粒子。
在另一优选例中,所述的载药纳米粒子中所含的抗肿瘤活性分子的含量为0.05-20mg/ml,较佳地0.1-10mg/ml,更佳地0.1-5mg/ml。
在另一优选例中,所述的载药纳米粒子的抗肿瘤活性成分包载率为40~95%,较佳地60-95%,更佳的80-95%。
在另一优选例中,所述的载药纳米粒子的载药量为0.1-40%,较佳的1-30%,更佳的4-25%。
在另一优选例中,所述的抗肿瘤活性成分与具有ph响应性神经肽y多肽复合物的质量比为1:1-40,较佳地1:5-25。
在另一优选例中,所述的制备方法选自下组:化学结合法、空白胶束载药法、透析法、乳化法、溶剂挥发法。
在另一优选例中,所述的溶剂挥发法中,所述的溶剂选自下组:蒸馏水、甲醇、乙醇、二氯甲烷、三氯甲烷、丙酮、二氯亚砜、n,n-二甲基甲酰胺,或其组合。
本发明的第四方面提供一种药物组合物,所述的组合物包含:
(i)本发明第二方面所述的载药纳米粒子;和
(ii)药学上可接受的载体。
本发明第五方面提供了一种本发明第二方面所述的载药纳米粒子,或本发明第四方面所述的药物组合物的用途,用于制备预防和/或治疗癌症的药物。
在另一优选例中,所述的药物的给药方式选自下组:口服、瘤内、直肠、肠胃外(静脉内、肌肉内或皮下)、和局部给药。
在另一优选例中,所述的癌症为细胞表面表达神经肽y多肽受体的癌症。
在另一优选例中,所述的癌症选自下组:乳腺癌、卵巢癌、肾癌、胃癌、脑癌。
本发明第六方面提供一种体外非治疗性的抑制肿瘤细胞的方法,包括步骤,在如本发明第二方面所述的载药纳米粒子的存在下,体外培养肿瘤细胞,从而抑制所述肿瘤细胞的生长。
本发明第七方面提供了一种抑制或治疗肿瘤的方法,给需要的对象施用如本发明第二方面所述的载药纳米粒子或本发明第四方面所述的药物组合物。
在另一优选例中,所述的对象为人或非人哺乳动物。
应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。
附图说明
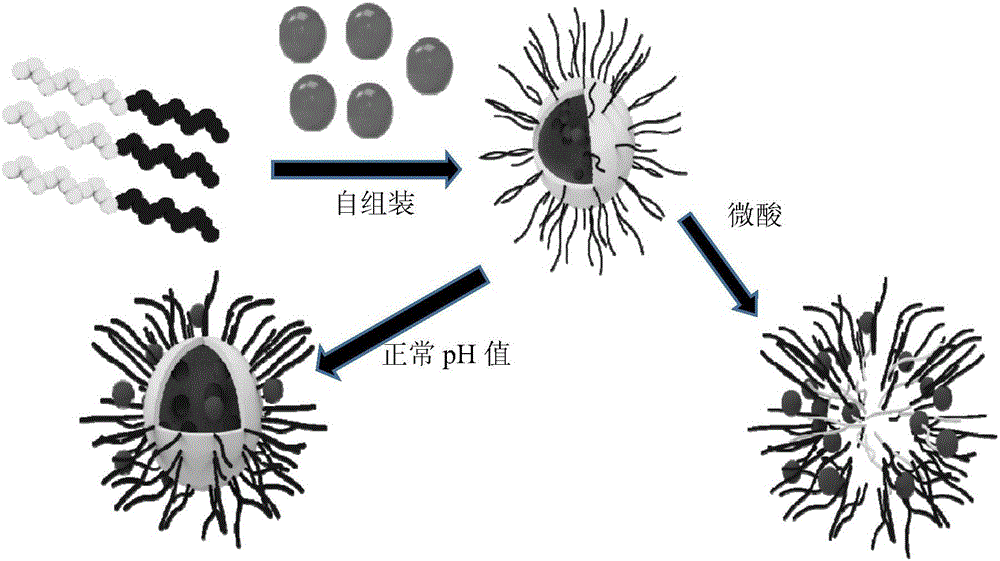
图1显示载药纳米粒子的制备及ph响应释放过程示意图。
图2显示本发明实施例1胶束的粒径分布图。
图3显示本发明实施例1胶束的药物释放曲线图。
图4显示本发明实施例2胶束的粒径分布图。
图5显示本发明实施例2胶束的药物释放曲线图。
图6显示本发明实施例3胶束的粒径分布图。
图7显示本发明实施例3胶束的药物释放曲线图。
具体实施方式
本发明人经过广泛而深入的研究,首次开发一种具有ph响应性神经肽y多肽复合物,所述的复合物包含:(1)神经肽y多肽;和(2)纳米载药胶束,所述的纳米载药胶束具有羧基部分;其中,所述的神经肽y多肽上的氨基部分通过与所述的纳米载药胶束的羧基部分形成酰胺键偶联于所述的纳米载药胶束上。所述的ph响应性神经肽y多肽复合物与抗肿瘤活性成分构建的载药纳米粒子具有良好肿瘤抑制作用。基于上述发现,发明人完成了本发明。
术语
除非另有定义,否则本文中所用的全部技术术语和科学术语均具有如本发明所属领域普通技术人员通常理解的相同含义。
如本文所用,术语“载药纳米粒子”是指粒度在1-100nm之间的粒子(纳米粒子又称超细微粒),属于胶体粒子大小的范畴。它们处于原子簇和宏观物体之间的过度区,处于微观体系和宏观体系之间,是由数目不多的原子或分子组成的集团,因此它们既非典型的微观系统亦非典型的宏观系统。
如本文所用,术语“有效治疗量”或“有效剂量”可互换使用,是指对人和/或动物产生功能或活性的且可被人和/或动物所接受的量。本领域的普通技术人员应该理解,所述的“有效量”或“有效剂量”可随着药物组合物的形式、给药途径、所用药物的辅料、疾病的严重程度以及与其他药物联合用药等情况的不同而有所不同。
如本文所用,术语“包含”、“含有”可互换使用,不仅包括封闭式定义,还包括半封闭和开放式的定义。换言之,所述术语包括了“由……构成”、“基本上由……构成”。
具有ph响应性神经肽y多肽复合物
本发明中,首次开发了一种具有ph响应性神经肽y多肽复合物,所述的复合物包含:
(1)神经肽y多肽;和(2)纳米载药胶束,所述的纳米载药胶束具有羧基部分;
其中,所述的神经肽y多肽上的氨基部分通过与所述的纳米载药胶束的羧基部分形成酰胺键偶联于所述的纳米载药胶束上。
在另一优选例中,所述的复合物中,所述的神经肽y多肽位于所述的复合物表面,并暴露在复合物外侧。
在另一优选例中,所述的复合物在生理ph值(7.2~7.4)中释放5~20%的药物,在微酸环境(5.0~6.5)中释放40~90%的药物。
神经肽y多肽
本发明的一个优选例中,神经肽y多肽是神经肽y的亚型,且所述神经肽y多肽的肽链长度为9~36个氨基酸。
本发明的另一个优选例中,发明人对所述的神经肽y多肽与纳米载药胶束的质量比行大量的实验筛选和优化,所述的神经肽y多肽与纳米载药胶束的质量比为1:5-150,较佳地1:10-100,更佳地1:20-80。
典型的,所述神经肽y多肽的肽链选自下组:npy、npy(28-36)、[arg6,pro34]pnpy、[phe6,pro34]pnpy、[asn6,pro34]pnpy、[cys6,pro34]pnpy、[phe6,pro34]pnpy、[d-his26,pro34]npy、[phe7,pro34]pnpy、[pro30,nle31,bpa32,leu34]npy(28-36)、[pro30,nal32,leu34]npy(28-36)、[pro30,nle31,nal32,leu34]npy(28-36)、[d-arg25]-np、[d-his26]-npy、[d-arg25,d-his26]-npy、[arg7,pro34]pnpy、[leu31,pro34]pnpy、pyy(3-36)、(ahx5-24)npy、[ala31,aib32]pnpy、[d-trp34]-npy、[cpp1-7,pnpy19-23,ala31,aib32,gln34]hpp、npy(3-36)、npy(22-36),或其组合。
纳米载药胶束
本发明中,所述纳米载药胶束是能够将药物包载的空白纳米胶束。代表性的,所述的纳米载药胶束选自下组:聚乙二醇-聚乳酸-羟基乙酸共聚物(peg-plga)、聚乙二醇-二硬脂酰基磷脂酰乙醇胺(peg-dspe)、二硬脂酰基磷脂酰乙醇胺-聚乳酸(dspe-pla)、聚乙二醇-聚己内酯(peg-pcl)、聚乙二醇-聚乙烯亚胺(peg-pei)、聚乳酸(pla)、聚乳酸-羟基乙酸共聚物(plga)、二硬脂酰基磷脂酰乙醇胺(dspe)、聚乙二醇(peg)、聚环氧乙烷-聚环氧丙烷-聚环氧乙烷(p123),或其组合。
在本发明的另一优选例中,所述的复合物具有ph响应性部位,且所述的部位在ph改变时发生变化,所述的变化包括亲水性、疏水性、电性、结构中的一种或多种的改变。
在另一优选例中,所述的ph响应性部位在肿瘤细胞或组织的ph微环境中发生变化,所述的变化包括发生亲水性、疏水性、电性、结构中的一种或多种的改变。
在另一优选例中,所述复合物的ph响应部位含有酰胺键。
在另一优选例中,所述复合物的ph响应部位与神经肽y多肽通过酰胺键相连。
在另一优选例中,所述复合物的ph响应部位为酰胺键。
载药纳米粒子
本发明提供一种载药纳米粒子,所述载药纳米粒子包含:
(a)具有ph响应性神经肽y多肽复合物;
(b)有效治疗量的抗肿瘤活性成分。
在另一优选例中,所述的神经肽y多肽质量占载药纳米粒子总质量的百分比为0.01-60wt%。
粒径作为评价载药纳米粒子的一项重要指标,粒径不仅影响载药纳米粒子的载药量、包封率等体外特性,还影响载药纳米粒子在体内的作用时间、稳定性等特性,最终影响抗肿瘤效果。载药纳米粒子直径可保持在1nm-10μm。因为正常组织周围的血管没有缝隙,而肿瘤组织周围的血管具有缝隙,所以小粒径的纳米粒子就会从这些缝隙中渗透出来,并利用增强的渗透保留效应聚集于肿瘤部位,然后攻击肿瘤细胞,但不会损害正常细胞,从而达到抑瘤效果。本发明中,发明人对载药纳米粒子的的粒径进行大量的筛选和优化,选择合适的粒径范围。在本发明的一个优选实施例中,所述载药纳米粒子的平均粒径为5-200nm。
在另一优选例中,所述的抗肿瘤活性成分为小分子抗肿瘤药物,或大分子活性成分。典型的,所述的抗肿瘤活性成分选自下组:阿霉素、顺铂、长春新碱、去甲长春碱、紫杉醇、丝裂霉素、长春地辛、曲妥珠单抗、替伊莫单抗、多西他赛,或其组合。
载药纳米粒子的制备方法
本发明提供了一种载药纳米粒子制备方法,所述的方法包含步骤:
(i)提供(a)具有ph响应性神经肽y多肽复合物;(b)有效治疗量的抗肿瘤活性成分;
(ii)将所述(a)、(b)进行复合,从而形成载药纳米粒子。
在另一优选例中,所述的载药纳米粒子中所含的抗肿瘤活性分子的含量为0.05-20mg/ml,较佳地0.1-10mg/ml,更佳地0.1-5mg/ml。所述含量为制备的液态载药纳米粒子中抗肿瘤活性分子的重量与液体体积的比值。
在另一优选例中,所述的载药纳米粒子的抗肿瘤活性成分的包载率为40-95%,较佳地60-95%,更佳的80-95%。
所述包载率指的是载药纳米粒子包载的抗肿瘤活性成分重量占总的抗肿瘤活性成分重量的百分含量,所述的总的抗肿瘤活性成分的重量为纳米粒子包载的抗肿瘤活性成分的量和未包载的(游离的)抗肿瘤活性成分的量的加和。
在另一优选例中,所述的载药纳米粒子的载药量为0.1-40%,较佳的1-30%,更佳的4-25%。所述载药量指的是载药纳米粒子包载的抗肿瘤活性成分的重量占载药纳米粒子的总重量的百分含量,
在另一优选例中,所述的抗肿瘤活性成分与具有ph响应性神经肽y多肽复合物的质量比为1:1-40,更佳地1:5-25。
在另一优选例中,所述的制备方法选自下组:化学结合法、空白胶束载药法、透析法、乳化法、溶剂挥发法。
在另一优选例中,所述的溶剂挥发法中,所述的溶剂选自下组:蒸馏水、甲醇、乙醇、二氯甲烷、三氯甲烷、丙酮、二氯亚砜、n,n-二甲基甲酰胺,或其组合。
一种优选的载药纳米粒子的制备方法,所述的方法包括步骤:
(i)将具有ph响应性神经肽y多肽复合物按照一定的质量比溶于有机溶剂
-8-中,加入(a)抗肿瘤活性成分溶液,混合均匀;
(ii)加入去离子水,搅拌,去除有机溶剂,得到载药纳米粒子。
在另一优选例中,在所述步骤(ii)中,去除有机溶剂后,经滤膜过滤,得到载药纳米粒子。
图1显示了载药纳米粒子的制备及其ph响应释放。
药物组合物、施用方法和应用
本发明提供了一种抑制或治疗肿瘤的药物组合物,所述的组合物包含(i)载药纳米粒子和(ii)药学上可接受的载体。
“药学上可接受的载体”指的是:一种或多种相容性固体、半固体、液体或凝胶填料,它们适合于人体或动物使用,而且必须有足够的纯度和足够低的毒性。“相容性”是指药物组合物中的各组分和药物的活性成分以及它们之间相互掺和,而不明显降低药效。
应当理解,在本发明中,所用的赋形剂没有特别的限制,可选用本领域常用的材料,或用常规方法制得,或从市场购买得到。
药学上可接受的载体的部分例子有纤维素及其衍生物(如羧甲基纤维素钠、甲基纤维、乙基纤维素等)、明胶、滑石粉、润滑剂(硬脂酸镁、硬脂酸钙)硫酸钙、多元醇(如甘露醇、山梨醇等)、糖类(如葡萄糖、蔗糖、乳糖、果糖等)、助悬剂、着色剂、调味剂、稳定剂、抗氧化剂、ph调节剂、防腐剂、注射用水等。
本发明中一个优选例中,药物组合物的制剂包括液体制剂、固体制剂或半固体制剂等。
代表性的,液体制剂可包含本领域常规采用的惰性稀释剂,如水或其他溶剂,增溶剂和乳化剂,例如乙醇、异丙醇、丙二醇、丁二醇、乙酸乙酯、以及油,特别是玉米油、棉籽油、花生油、蓖麻油、橄榄油以及这些物质的混合物等。除了这些惰性稀释剂外,药物组合物也可包含着色剂、调味剂、稳定剂、抗氧化剂、ph调节剂、防腐剂,如葡糖糖、蔗糖、甘露醇、维生素c、碳酸氢钠、苯甲醇等。
本发明中,优选的制剂为注射剂和粉针剂。对于胃肠外注射的注射剂可包含生理上可接受的无菌用水或无水溶液、分散液、悬浮液或乳液,和用于重新溶解成无菌的可注射溶液或分散液的无菌粉末。对于粉针剂,例如可用生理盐水或含有葡萄糖、甘露醇和其他赋形剂的水溶液通过常规方法进行制备,如冷冻干燥技术。
本发明的药物组合物的施用方法没有特地限制,代表性的施用方式包括(但不限于):静脉内、口服胃肠道内、瘤内、瘤旁、腹腔内、局部给药等。
一种优选的方式是将药物组合物通过注射给药,例如静脉注射、肌肉注射、皮内注射、皮下注射、腹腔注射、瘤内注射、瘤旁注射等。
本领域的技术人员应等理解的是,药物剂型应与给药方式相匹配。在一个优选例中,所述的药物的剂型选自下组:口服、瘤内、直肠、肠胃外(静脉内、肌肉内或皮下)、和局部给药。
在本发明的药物组合物中,活性成分的给药量是治疗有效量,例如每天约1微克/千克体重-20毫克/千克体重。在使用药物组合物时,将安全有效量的药物适用于人或哺乳动物,其中安全有效量至少为10微克微克/千克体重,而且在大多数情况下不超过10毫克/千克体重,较佳的为10微克/千克体重-5毫克/千克体重。当然具体剂量还要考虑给药途径、病人健康状况等因素,这些都是熟练医师技能范围之内的。此外本发明药物还可以与其他治疗剂一起使用(包括之前、之中或之后)。
在本发明中,所述载药纳米粒子或所述药物组合物适用于制备预防和/或治疗癌症的药物,优选适用于制备预防和/或治疗细胞表面表达神经肽y多肽受体的癌症的药物。代表性的,所述的癌症选自下组:乳腺癌、卵巢癌、肾癌、胃癌、脑癌。在另一优选例中,所述的药物的给药方式选自下组:口服、瘤内、直肠、肠胃外(静脉内、肌肉内或皮下)、和局部给药。
体外非治疗性的抑制肿瘤的方法
本发明提供一种体外非治疗性的抑制肿瘤的方法,包括步骤:在载药纳米粒子的存在下,体外培养肿瘤细胞,从而抑制所述肿瘤细胞的生长。
抑制或治疗肿瘤的方法
本发明中提供了一种抑制或治疗肿瘤的方法,通过给所需的对象施用所述载药纳米粒子或所述药物组合物。在另一优选例中,所述的对象为人或非人哺乳动物。
本发明的主要优点包括:
(1)本发明的载药纳米粒子具有良好的靶向性;
(2)本发明的载药纳米粒子具有高癌细胞抑制活性;
(3)通过偶联的方法可以很好的控制多肽占载药纳米粒子总质量的比例;
(4)载药纳米粒子采用的的高分子材料均通过fda验证,可以用于人体治疗。
(5)本发明的载药纳米粒子,具有ph响应型药物控制释放的特性,可以提高药物在肿瘤部位的富集。
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数是重量百分比和重量份数。
实施例1
[arg6,pro34]pnpy-peg-dspe材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-peg-dspe、nhs、edc混合活化2小时后加入1摩尔比的[arg6,pro34]pnpy,在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
[arg6,pro34]pnpy-peg-dspe与mpeg-dspe复合的负载阿霉素胶束的制备
(1)取[arg6,pro34]pnpy-peg-dspe与mpeg-dspe质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
对本发明实施例1所得胶束粒径和不同ph值条件下的药物释放度进行测定,并对有无靶分子[arg6,pro34]pnpy的载药胶束进行肿瘤细胞抑制率的测定。
结果如下:
粒径结果:
图2为本实施例1的胶束粒径分布图,胶束的平均粒径在50nm以下,多分散系数为0.214。
不同ph值条件下的药物释放度结果:
图3所示本实施例1的胶束在不同ph值下药物释放结果,从图中可以看出,胶束中的药物在ph5.0条件下的释放速率明显比ph7.4的条件下快,表明,实施例1的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
肿瘤细胞抑制率结果:
本实施例1中[arg6,pro34]pnpy-peg-dspe与mpeg-dspe复合的负载阿霉素胶束为靶向胶束,mpeg-dspe负载阿霉素胶束为非靶向胶束,靶向胶束和非靶向胶束以阿霉素剂量时与u87-mg细胞共孵育24小时后测定细胞抑制率。
肿瘤细胞抑制率结果如表1所示:
表1胶束的细胞抑制率
表1所示,与非靶向胶束相比,靶向胶束对脑胶质瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为8.793微克/毫升,非靶向胶束组的半数致死量为11.825微克/毫升,结果表明,靶分子[arg6,pro34]pnpy通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例2
[pro30,nle31,bpa32,leu34]npy(28-36)-peg-plga材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的mpeg-plga、nhs、edc混合活化2小时后加入1摩尔比的[pro30,nle31,bpa32,leu34]npy(28-36),在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
[pro30,nle31,bpa32,leu34]npy(28-36)-peg-plga与mpeg-plga复合的负载阿霉素胶束的制备
(1)取[pro30,nle31,bpa32,leu34]npy(28-36)-peg-plga与mpeg-plga质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌五分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到产物。
对本发明实施例2所得胶束粒径和不同ph值条件下的药物释放度进行测定,并对有无靶分子[pro30,nle31,bpa32,leu34]npy(28-36)的载药胶束进行肿瘤细胞抑制率的测定。
结果如下:
粒径结果:
图4为本实施例2的胶束粒径分布图,胶束的平均粒径在50nm以下,多分散系数为0.247。
不同ph值条件下的药物释放度结果:
图5所示本实施例2的胶束在不同ph值下药物释放结果,从图中可以看出,胶束中的药物在ph5.0条件下的释放速率明显比ph7.4的条件下快,表明,实施例2的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
肿瘤细胞抑制率结果:
本实施例2中[pro30,nle31,bpa32,leu34]npy(28-36)-peg-plga与mpeg-plga复合的负载阿霉素胶束为靶向胶束,mpeg-plga负载阿霉素胶束为非靶向胶束,靶向胶束和非靶向胶束以阿霉素剂量时与u87-mg细胞共孵育24小时后测定细胞抑制率。
肿瘤细胞抑制率结果如表2所示:
表2胶束的细胞抑制率
表2所示,与非靶向胶束相比,靶向胶束对脑胶质瘤细胞具有较好的抑制效果,半数致死量为9.248微克/毫升,非靶向胶束组的半数致死量为12.498微克/毫升,结果表明,靶分子[arg6,pro34]pnpy通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例3
[phe6,pro34]pnpy-mpeg-dspe材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-peg-dspe、nhs、edc混合活化2小时后加入1摩尔比的[phe6,pro34]pnpy,在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
[phe6,pro34]pnpy-mpeg-dspe与mpeg-dspe复合的负载多西他赛胶束的制备
(1)取[phe6,pro34]pnpy-peg-dspe与mpeg-dspe质量比为1:20的混合物共25毫克溶于2毫升乙醇,加入到1毫升多西他赛(5毫克每毫升)的乙醇溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到10毫升去离子水中,搅拌五分钟,在37摄氏度下搅拌挥发去除乙醇;
(3)将步骤(2)中得到的溶液在10000g条件下用透析分子量为10kda的超滤离心管离心40分钟取上清液。
对本发明实施例2所得胶束粒径和不同ph值条件下的药物释放度进行测定,并对有无靶分子[phe6,pro34]pnpy的载药胶束进行肿瘤细胞抑制率的测定。
结果如下:
粒径结果:
图6为本实施例2的胶束粒径分布图,胶束的平均粒径在50nm以下,多分散系数为0.254。
不同ph值条件下的药物释放度结果:
图7所示本实施例2的胶束在不同ph值下药物释放结果,从图中可以看出,胶束中的药物在ph5.0条件下的释放速率明显比ph7.4的条件下快,表明,实施例2的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
肿瘤细胞抑制率结果:
本实施例3中[phe6,pro34]pnpy-mpeg-dspe与mpeg-dspe复合的负载多西他赛胶束为靶向胶束,mpeg-dspe负载阿霉素胶束为非靶向胶束,靶向胶束和非靶向胶束以多西他赛剂量时与mcf-7细胞共孵育24小时后测定细胞抑制率。
肿瘤细胞抑制率结果如表3所示:
表3胶束的细胞抑制率
表3所示,与非靶向胶束相比,靶向胶束对乳腺癌细胞具有较好的抑制效果,半数致死量为7.253微克/毫升,非靶向胶束组的半数致死量为13.4987微克/毫升,结果表明,靶分子[phe6,pro34]通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例4
[phe6,pro34]pnpy-peg-dspe材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-peg-dspe、nhs、edc混合活化2小时后加入1摩尔比的[phe6,pro34]pnpy,在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
[phe6,pro34]pnpy-peg-dspe与mpeg-dspe复合的负载阿霉素胶束的制备
(1)取[phe6,pro34]pnpy-peg-dspe与mpeg-dspe质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为50.43%,释放速率明显比ph7.4的条件下为20.14%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为7.793微克/毫升,非靶向胶束组的半数致死量为11.825微克/毫升,结果表明,靶分子[arg6,pro34]pnpy通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例5
[asn6,pro34]pnpy-peg-dspe材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-peg-dspe、nhs、edc混合活化2小时后加入1摩尔比的[asn6,pro34]pnpy,在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
[asn6,pro34]pnpy-peg-dspe与mpeg-dspe复合的负载阿霉素胶束的制备
(1)取[asn6,pro34]pnpy-peg-dspe与mpeg-dspe质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为54.43%,释放速率明显比ph7.4的条件下为19.14%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为7.993微克/毫升,非靶向胶束组的半数致死量为11.825微克/毫升,结果表明,靶分子[arg6,pro34]pnpy通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例6
[cys6,pro34]pnpy-peg-dspe材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-peg-dspe、nhs、edc混合活化2小时后加入1摩尔比的[cys6,pro34]pnpy,在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
[cys6,pro34]pnpy-peg-dspe与mpeg-dspe复合的负载阿霉素胶束的制备
(1)取[cys6,pro34]pnpy-peg-dspe与mpeg-dspe质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为53.43%,释放速率明显比ph7.4的条件下为20.14%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为8.293微克/毫升,非靶向胶束组的半数致死量为11.825微克/毫升,结果表明,靶分子[cys6,pro34]pnpy通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例7
[phe6,pro34]pnpy-peg-dspe材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-peg-dspe、nhs、edc混合活化2小时后加入1摩尔比的[phe6,pro34]pnpy,在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
[phe6,pro34]pnpy-peg-dspe与mpeg-dspe复合的负载阿霉素胶束的制备
(1)取[phe6,pro34]pnpy-peg-dspe与mpeg-dspe质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为52.43%,释放速率明显比ph7.4的条件下为21.14%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为8.793微克/毫升,非靶向胶束组的半数致死量为11.825微克/毫升,结果表明,靶分子[phe6,pro34]pnpy通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例8
[d-his26,pro34]npy-peg-dspe材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-peg-dspe、nhs、edc混合活化2小时后加入1摩尔比的[d-his26,pro34]pnpy,在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
[d-his26,pro34]npy-peg-dspe与mpeg-dspe复合的负载阿霉素胶束的制备
(1)取[d-his26,pro34]pnpy-peg-dspe与mpeg-dspe质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为51.43%,释放速率明显比ph7.4的条件下为21.44%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为8.293微克/毫升,非靶向胶束组的半数致死量为11.825微克/毫升,结果表明,靶分子[d-his26,pro34]npy通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例9
[phe7,pro34]pnpy-peg-dspe材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-peg-dspe、nhs、edc混合活化2小时后加入1摩尔比的[phe7,pro34]pnpy,在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
[phe7,pro34]pnpy-peg-dspe与mpeg-dspe复合的负载阿霉素胶束的制备
(1)取[phe7,pro34]pnpy-peg-dspe与mpeg-dspe质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为53.43%,释放速率明显比ph7.4的条件下为20.44%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为7.893微克/毫升,非靶向胶束组的半数致死量为11.825微克/毫升,结果表明,靶分子[phe7,pro34]pnpy通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例10
[pro30,nle31,bpa32,leu34]npy(28-36)-peg-dspe材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-peg-dspe、nhs、edc混合活化2小时后加入1摩尔比的[pro30,nle31,bpa32,leu34]npy(28-36),在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
[pro30,nle31,bpa32,leu34]npy(28-36)-peg-dspe与mpeg-dspe复合的负载阿霉素胶束的制备
(1)取[pro30,nle31,bpa32,leu34]npy(28-36)-peg-dspe与mpeg-dspe质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为53.83%,释放速率明显比ph7.4的条件下为20.74%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为7.493微克/毫升,非靶向胶束组的半数致死量为11.825微克/毫升,结果表明,靶分子[pro30,nle31,bpa32,leu34]npy(28-36)通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例11
[pro30,nal32,leu34]npy(28-36)-peg-dspe材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-peg-dspe、nhs、edc混合活化2小时后加入1摩尔比的[pro30,nal32,leu34]npy(28-36),在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
[pro30,nal32,leu34]npy(28-36)-peg-dspe与mpeg-dspe复合的负载阿霉素胶束的制备
(1)取[pro30,nal32,leu34]npy(28-36)-peg-dspe与mpeg-dspe质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为51.83%,释放速率明显比ph7.4的条件下为22.74%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为7.893微克/毫升,非靶向胶束组的半数致死量为11.825微克/毫升,结果表明,靶分子[pro30,nle31,bpa32,leu34]npy(28-36)通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例12
[pro30,nle31,nal32,leu34]npy(28-36)-peg-dspe材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-peg-dspe、nhs、edc混合活化2小时后加入1摩尔比的[pro30,nle31,nal32,leu34]npy(28-36),在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
[pro30,nle31,nal32,leu34]npy(28-36)-peg-dspe与mpeg-dspe复合的负载阿霉素胶束的制备
(1)取[pro30,nle31,nal32,leu34]npy(28-36)-peg-dspe与mpeg-dspe质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为54.83%,释放速率明显比ph7.4的条件下为19.74%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为7.493微克/毫升,非靶向胶束组的半数致死量为11.825微克/毫升,结果表明,靶分子[pro30,nle31,nal32,leu34]npy(28-36)通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例13
[d-arg25]-npy-peg-dspe材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-peg-dspe、nhs、edc混合活化2小时后加入1摩尔比的[d-arg25]-npy,在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
[d-arg25]-npy-peg-dspe与mpeg-dspe复合的负载阿霉素胶束的制备
(1)取[d-arg25]-npy-peg-dspe与mpeg-dspe质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为52.83%,释放速率明显比ph7.4的条件下为21.84%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为8.193微克/毫升,非靶向胶束组的半数致死量为11.825微克/毫升,结果表明,靶分子[d-arg25]-npy通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例14
[d-his26]-npy-peg-dspe材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-peg-dspe、nhs、edc混合活化2小时后加入1摩尔比的[d-his26]-npy,在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
[d-his26]-npy-peg-dspe与mpeg-dspe复合的负载阿霉素胶束的制备
(1)取[d-his26]-npy-peg-dspe与mpeg-dspe质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为51.83%,释放速率明显比ph7.4的条件下为22.86%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为8.493微克/毫升,非靶向胶束组的半数致死量为11.825微克/毫升,结果表明,靶分子[d-his26]-npy通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例15
[d-arg25,d-his26]-npy-peg-dspe材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-peg-dspe、nhs、edc混合活化2小时后加入1摩尔比的[d-arg25,d-his26]-npy,在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
[d-arg25,d-his26]-npy-peg-dspe与mpeg-dspe复合的负载阿霉素胶束的制备
(1)取[d-arg25,d-his26]-npy-peg-dspe与mpeg-dspe质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为54.83%,释放速率明显比ph7.4的条件下为18.86%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为7.826微克/毫升,非靶向胶束组的半数致死量为11.825微克/毫升,结果表明,靶分子[d-arg25,d-his26]-npy通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例16
[arg7,pro34]pnpy-peg-dspe材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-peg-dspe、nhs、edc混合活化2小时后加入1摩尔比的[arg7,pro34]pnpy,在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
[arg7,pro34]pnpy-peg-dspe与mpeg-dspe复合的负载阿霉素胶束的制备
(1)取[arg7,pro34]pnpy-peg-dspe与mpeg-dspe质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为52.67%,释放速率明显比ph7.4的条件下为19.24%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为8.124微克/毫升,非靶向胶束组的半数致死量为11.825微克/毫升,结果表明,靶分子[arg7,pro34]pnpy通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例17
[asn6,pro34]pnpy-peg-plga材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-peg-plga、nhs、edc混合活化2小时后加入1摩尔比的[asn6,pro34]pnpy,在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
[asn6,pro34]pnpy-peg-plga与mpeg-plga复合的负载阿霉素胶束的制备
(1)取[asn6,pro34]pnpy-peg-plga与mpeg-plga质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为91.87%,释放速率明显比ph7.4的条件下为19.24%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为4.152微克/毫升,非靶向胶束组的半数致死量为9.647微克/毫升,结果表明,靶分子[asn6,pro34]pnpy通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例18
npy-peg-dspe材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-peg-dspe、nhs、edc混合活化2小时后加入1摩尔比的npy,在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
npy-peg-dspe与mpeg-dspe复合的负载阿霉素胶束的制备
(1)取npy-peg-dspe与mpeg-dspe质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为54.67%,释放速率明显比ph7.4的条件下为17.26%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为7.156微克/毫升,非靶向胶束组的半数致死量为11.825微克/毫升,结果表明,靶分子npy通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例19
npy(28-36)-peg-dspe材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-peg-dspe、nhs、edc混合活化2小时后加入1摩尔比的npy(28-36),在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
npy(28-36)-peg-dspe与mpeg-dspe复合的负载阿霉素胶束的制备
(1)取npy(28-36)-peg-dspe与mpeg-dspe质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为53.16%,释放速率明显比ph7.4的条件下为18.29%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为7.325微克/毫升,非靶向胶束组的半数致死量为11.825微克/毫升,结果表明,靶分子npy(28-36)通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例20
[leu31,pro34]pnpy-peg-dspe材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-peg-dspe、nhs、edc混合活化2小时后加入1摩尔比的[leu31,pro34]pnpy,在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
[leu31,pro34]pnpy-peg-dspe与mpeg-dspe复合的负载阿霉素胶束的制备
(1)取[leu31,pro34]pnpy-peg-dspe与mpeg-dspe质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为55.16%,释放速率明显比ph7.4的条件下为16.29%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为8.192微克/毫升,非靶向胶束组的半数致死量为11.825微克/毫升,结果表明,靶分子[leu31,pro34]pnpy通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例21
pyy(3-36)-peg-dspe材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-peg-dspe、nhs、edc混合活化2小时后加入1摩尔比的pyy(3-36),在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
pyy(3-36)-peg-dspe与mpeg-dspe复合的负载阿霉素胶束的制备
(1)取pyy(3-36)-peg-dspe与mpeg-dspe质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为53.46%,释放速率明显比ph7.4的条件下为18.19%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为8.596微克/毫升,非靶向胶束组的半数致死量为11.825微克/毫升,结果表明,靶分子pyy(3-36)通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例22
(ahx5-24)npy-peg-dspe材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-peg-dspe、nhs、edc混合活化2小时后加入1摩尔比的(ahx5-24)npy,在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
(ahx5-24)npy-peg-dspe与mpeg-dspe复合的负载阿霉素胶束的制备
(1)取(ahx5-24)npy-peg-dspe与mpeg-dspe质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为54.21%,释放速率明显比ph7.4的条件下为19.27%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为8.135微克/毫升,非靶向胶束组的半数致死量为11.825微克/毫升,结果表明,靶分子(ahx5-24)npy通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例23
[ala31,aib32]pnpy-peg-dspe材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-peg-dspe、nhs、edc混合活化2小时后加入1摩尔比的[ala31,aib32]pnpy,在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
[ala31,aib32]pnpy-peg-dspe与mpeg-dspe复合的负载阿霉素胶束的制备
(1)取[ala31,aib32]pnpy-peg-dspe与mpeg-dspe质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为51.29%,释放速率明显比ph7.4的条件下为20.71%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为8.426微克/毫升,非靶向胶束组的半数致死量为11.825微克/毫升,结果表明,靶分子[ala31,aib32]pnpy通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例24
[d-trp34]-npy-peg-dspe材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-peg-dspe、nhs、edc混合活化2小时后加入1摩尔比的[d-trp34]-npy,在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
[d-trp34]-npy-peg-dspe与mpeg-dspe复合的负载阿霉素胶束的制备
(1)取[d-trp34]-npy-peg-dspe与mpeg-dspe质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为57.29%,释放速率明显比ph7.4的条件下为14.56%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为7.156微克/毫升,非靶向胶束组的半数致死量为11.825微克/毫升,结果表明,靶分子[d-trp34]-npy通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例25
[cpp1-7,pnpy19-23,ala31,aib32,gln34]hpp-peg-dspe材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-peg-dspe、nhs、edc混合活化2小时后加入1摩尔比的[cpp1-7,pnpy19-23,ala31,aib32,gln34]hpp,在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
[cpp1-7,pnpy19-23,ala31,aib32,gln34]hpp-peg-dspe与mpeg-dspe复合的负载阿霉素胶束的制备
(1)取[cpp1-7,pnpy19-23,ala31,aib32,gln34]hpp-peg-dspe与mpeg-dspe质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为52.37%,释放速率明显比ph7.4的条件下为17.29%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为8.246微克/毫升,非靶向胶束组的半数致死量为11.825微克/毫升,结果表明,靶分子[cpp1-7,pnpy19-23,ala31,aib32,gln34]hpp通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例26
npy(3-36)-peg-dspe材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-peg-dspe、nhs、edc混合活化2小时后加入1摩尔比的npy(3-36),在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
npy(3-36)-peg-dspe与mpeg-dspe复合的负载阿霉素胶束的制备
(1)取npy(3-36)-peg-dspe与mpeg-dspe质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为51.43%,释放速率明显比ph7.4的条件下为19.27%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为8.716微克/毫升,非靶向胶束组的半数致死量为11.825微克/毫升,结果表明,靶分子npy(3-36)通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例27
npy(22-36)-peg-dspe材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-peg-dspe、nhs、edc混合活化2小时后加入1摩尔比的npy(22-36),在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
npy(22-36)-peg-dspe与mpeg-dspe复合的负载阿霉素胶束的制备
(1)取npy(22-36)-peg-dspe与mpeg-dspe质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为52.73%,释放速率明显比ph7.4的条件下为18.24%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为8.426微克/毫升,非靶向胶束组的半数致死量为11.825微克/毫升,结果表明,靶分子npy(3-36)通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例28
[asn6,pro34]pnpy-dspe-pla材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-dspe-pla、nhs、edc混合活化2小时后加入1摩尔比的[asn6,pro34]pnpy,在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
[asn6,pro34]pnpy-dspe-pla与dspe-pla复合的负载阿霉素胶束的制备
(1)取[asn6,pro34]pnpy-dspe-pla与dspe-pla质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为85.43%,释放速率明显比ph7.4的条件下为24.47%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为5.127微克/毫升,非靶向胶束组的半数致死量为10.348微克/毫升,结果表明,靶分子[asn6,pro34]pnpy通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例29
[asn6,pro34]pnpy-pla材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-pla、nhs、edc混合活化2小时后加入1摩尔比的[asn6,pro34]pnpy,在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
[asn6,pro34]pnpy-pla与pla复合的负载阿霉素胶束的制备
(1)取[asn6,pro34]pnpy-pla与pla质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为81.49%,释放速率明显比ph7.4的条件下为22.49%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为4.826微克/毫升,非靶向胶束组的半数致死量为9.167微克/毫升,结果表明,靶分子[asn6,pro34]pnpy通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例30
[asn6,pro34]pnpy-plga材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-plga、nhs、edc混合活化2小时后加入1摩尔比的[asn6,pro34]pnpy,在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
[asn6,pro34]pnpy-pla与plga复合的负载阿霉素胶束的制备
(1)取[asn6,pro34]pnpy-plga与plga质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为87.46%,释放速率明显比ph7.4的条件下为19.13%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为3.157微克/毫升,非靶向胶束组的半数致死量为7.462微克/毫升,结果表明,靶分子[asn6,pro34]pnpy通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例31
[asn6,pro34]pnpy-dspe材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-dspe、nhs、edc混合活化2小时后加入1摩尔比的[asn6,pro34]pnpy,在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
[asn6,pro34]pnpy-dspe与dspe复合的负载阿霉素胶束的制备
(1)取[asn6,pro34]pnpy-dspe与dspe质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为85.16%,释放速率明显比ph7.4的条件下为20.43%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为4.597微克/毫升,非靶向胶束组的半数致死量为8.146微克/毫升,结果表明,靶分子[asn6,pro34]pnpy通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例32
[asn6,pro34]pnpy-peg材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-peg、nhs、edc混合活化2小时后加入1摩尔比的[asn6,pro34]pnpy,在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
[asn6,pro34]pnpy-peg与peg复合的负载阿霉素胶束的制备
(1)取[asn6,pro34]pnpy-peg与peg质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为87.16%,释放速率明显比ph7.4的条件下为18.43%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为8.127微克/毫升,非靶向胶束组的半数致死量为12.496微克/毫升,结果表明,靶分子[asn6,pro34]pnpy通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例33
[asn6,pro34]pnpy-peg-pcl材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-peg-pcl、nhs、edc混合活化2小时后加入1摩尔比的[asn6,pro34]pnpy,在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
[asn6,pro34]pnpy-peg-pcl与peg-pcl复合的负载阿霉素胶束的制备
(1)取[asn6,pro34]pnpy-peg-pcl与peg-pcl质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为71.46%,释放速率明显比ph7.4的条件下为23.89%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为8.726微克/毫升,非靶向胶束组的半数致死量为13.459微克/毫升,结果表明,靶分子[asn6,pro34]pnpy通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例34
[asn6,pro34]pnpy-peg-pei材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-peg-epi、nhs、edc混合活化2小时后加入1摩尔比的[asn6,pro34]pnpy,在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
[asn6,pro34]pnpy-peg-epi与peg-pei复合的负载阿霉素胶束的制备
(1)取[asn6,pro34]pnpy-peg-pei与peg-epi质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为74.46%,释放速率明显比ph7.4的条件下为21.89%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为6.197微克/毫升,非靶向胶束组的半数致死量为10.589微克/毫升,结果表明,靶分子[asn6,pro34]pnpy通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
实施例35
[asn6,pro34]pnpy-p123材料的合成
步骤如下:
在室温下将摩尔比为1:1:1.5的cooh-p123、nhs、edc混合活化2小时后加入1摩尔比的[asn6,pro34]pnpy,在室温下搅拌48小时后,用分子量为5000的透析袋透析48小时后冻干保存。
[asn6,pro34]pnpy-p123与p123复合的负载阿霉素胶束的制备
(1)取[asn6,pro34]pnpy-p123与p123质量比为1:80的混合物共25毫克溶于2毫升丙酮,加入到1毫升阿霉素(1毫克每毫升)的丙酮溶液中混合均匀;
(2)将步骤(1)得到的混合溶液逐滴加入到2毫升去离子水中,搅拌5分钟,在37摄氏度下旋转蒸发去除丙酮;
(3)将步骤(2)中得到的溶液过0.22微米的亲水滤膜后得到胶束。
胶束中的药物在ph5.0条件下72小时释放率为84.82%,释放速率明显比ph7.4的条件下为19.26%,表明,该实施例中的胶束具有ph响应,在肿瘤的微环境中(ph5.0-6.5)能够加速药物的释放,从而提高药物在肿瘤部位的浓度,提高抗肿瘤效果。
与非靶向胶束相比,靶向胶束对乳腺癌瘤癌细胞具有较好的抑制效果,靶向胶束组的半数致死量为6.846微克/毫升,非靶向胶束组的半数致死量为11.279微克/毫升,结果表明,靶分子[asn6,pro34]pnpy通过特异性的与肿瘤细胞神经肽y多肽受体结合,提高靶向胶束在肿瘤部位的聚集浓度,从而提高抑瘤效果。
此外,实施例中所用的丙酮、乙醇也可替换成包括但不限于:蒸馏水、甲醇、二氯甲烷、三氯甲烷、二氯亚砜、n,n-二甲基甲酰胺。
在胶束的制备中,实施例所用的旋转蒸发法、搅拌挥发法也可替换成下组(包括但不限于):化学结合法、空白胶束载药法、透析法、乳化法。
实施例中所用的阿霉素、多西他赛,也可替换成下组(包括但不限于):顺铂、长春新碱、去甲长春碱、紫杉醇、丝裂霉素、长春地辛。
在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
- 还没有人留言评论。精彩留言会获得点赞!