抗HCV联合用药物的制作方法
本发明涉及化合物1和化合物2在制备抗hcv联合用药物中的应用。
背景技术:
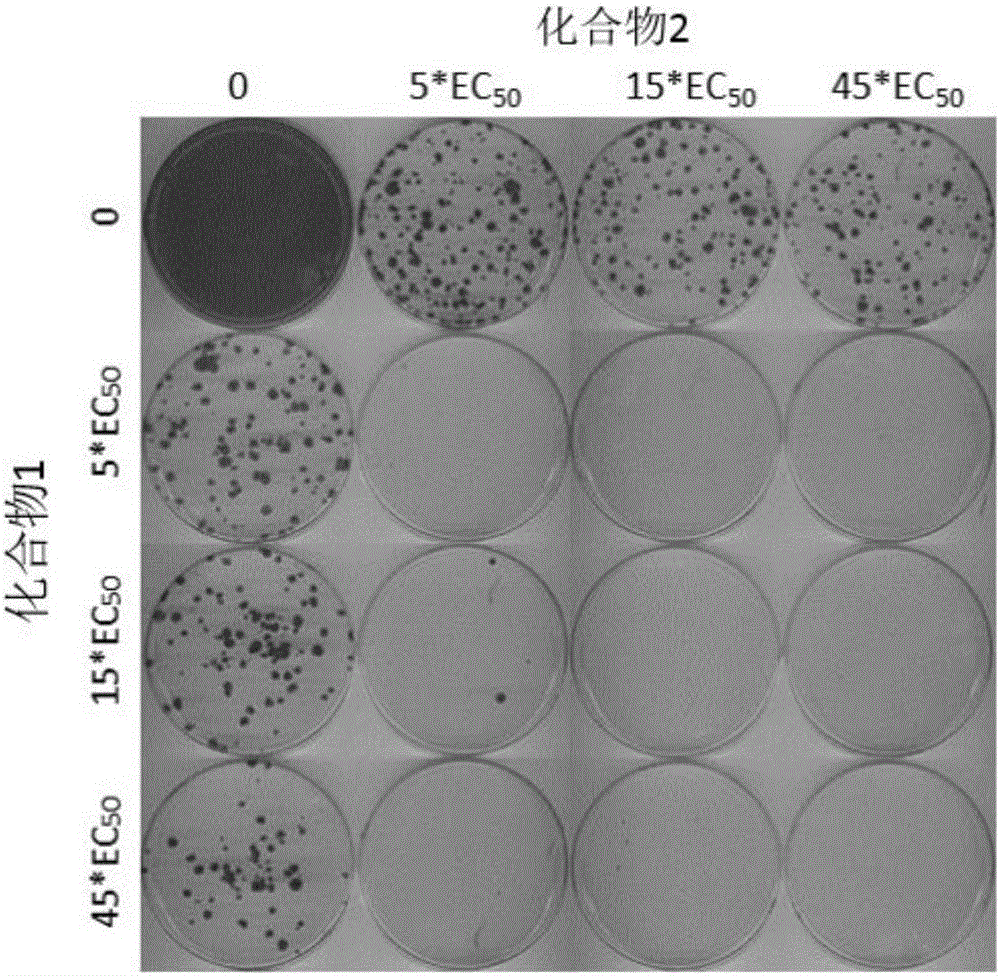
:hcv是主要的人类病原体之一,估计全球慢性hcv感染者约1.7亿,为人免疫缺陷病毒1型感染人数的5倍。慢性hcv感染者会发展成严重的进行性肝病,包括肝硬化和肝细胞癌。因此,慢性hcv感染是全球患者因肝病而死亡的主要原因。目前,标准的慢性hcv感染疗法是釆用α-干扰素和利巴韦林与近两年来批准的其中的一个直接作用抗病毒(daa)药物的联合用药。疗效虽较之前的α-干扰素和利巴韦林联合用药明显提高,但仍对部分慢性hcv感染者无效,而且病毒可产生抗药性。加之α-干扰素和利巴韦林有明显的副反应。因此,新的有效的治疗慢性hcv感染的药物是目前迫切所需的。hcv是单链正链rna病毒。属黄病毒科(flaviviridaefamily)单独的一个属内。黄病毒科的所有成员都是含正链rna基因组的有包膜病毒粒子,该基因组通过单个不间断开放阅读框(orf)的翻译,编码所有已知的病毒特异性蛋白。hcv基因组的核苷酸和所编码的氨基酸序列存在相当多的异质性。已经鉴定出至少6个主要的基因型,50多个亚基因型。hcv的主要基因型在全球的分布不同,虽然进行了大量基因型对发病机制和治疗作用的研究,但仍不清楚hcv遗传异质性的临床重要性。hcvrna基因组长度约为9500个核苷酸,具有单个开放阅读框,编码单个约3000个氨基酸的多聚蛋白。在感染细胞中,该多聚蛋白在多个位点上被细胞蛋白酶和病毒蛋白酶切割,产生结构和非结构(ns)蛋白。就hcv而言,成熟非结构蛋白(ns2、ns3、ns4a、ns4b、ns5a和ns5b)的形成是通过两种病毒蛋白酶实现的。一般认为第一种(ns2)是金属蛋白酶,在ns2-ns3接点进行切割;第二个蛋白酶是包含在ns3(本文中亦称为ns3蛋白酶)n端区域的丝氨酸蛋白酶,它介导ns3下游所有的后续切割,在ns3-ns4a切割位点为顺式,在ns4a-ns4b、ns4b-ns5a、ns5a-na5b位点则为反式。ns4a蛋白似乎有多种功能,起ns3蛋白酶辅因子的作用,并可能协助ns3和其他病毒复制酶组分进行膜定位。ns3蛋白还显示出核苷三磷酸酶和rna解旋酶活性。ns4b和ns5a两个蛋白的功能尚不完全清楚,但对hcv的复制起着重要的作用。ns4b是一个穿膜蛋白,参与病毒复制复合体的形成。ns5a是一个磷酸化蛋白,参与病毒rna的复制和病毒颗粒的形成。ns5b(亦称hcv聚合酶)是参与hcv基因组rna复制的依赖于rna的rna聚合酶。wo2013095275、wo2012122716、cn102863428a等文献各自报道了一系列作为hcv抑制剂的化合物,其活性、溶解性等方面的效果有待改善。技术实现要素:本发明提供了化合物1和化合物2在制备抗hcv联合用药物中的应用。定义和说明本发明的化合物可以通过本领域技术人员所熟知的多种合成方法来制备,包括下面列举的具体实施方式、其与其他化学合成方法的结合所形成的实施方式以及本领域技术上人员所熟知的等同替换方式,优选的实施方式包括但不限于本发明的实施例。本发明所使用的溶剂可经市售获得。本发明采用下述缩略词:aq代表水;hatu代表o-(7-氮杂苯并三唑-1-基)-n,n,n',n'-四甲基脲六氟磷酸盐;edc代表n-(3-二甲基氨基丙基)-n'-乙基碳二亚胺盐酸盐;m-cpba代表3-氯过氧苯甲酸;eq代表当量、等量;cdi代表羰基二咪唑;dcm代表二氯甲烷;pe代表石油醚;diad代表偶氮二羧酸二异丙酯;dmf代表n,n-二甲基甲酰胺;dmso代表二甲亚砜;etoac代表乙酸乙酯;etoh代表乙醇;meoh代表甲醇;cbz代表苄氧羰基,是一种胺保护基团;boc代表叔丁基羰基是一种胺保护基团;hoac代表乙酸;nacnbh3代表氰基硼氢化钠;r.t.代表室温;o/n代表过夜;thf代表四氢呋喃;boc2o代表二-叔丁基二碳酸酯;tfa代表三氟乙酸;dipea代表二异丙基乙基胺;socl2代表氯化亚砜;cs2代表二硫化碳;tsoh代表对甲苯磺酸;nfsi代表n-氟-n-(苯磺酰基)苯磺酰胺;ncs代表1-氯吡咯烷-2,5-二酮;n-bu4nf代表氟化四丁基铵;iproh代表2-丙醇;mp代表熔点;lda代表二异丙基胺基锂;edci代表碳化二亚胺;hobt代表1-羟基苯并三唑;pd(dppf)cl2代表[1,1'-双(二苯基膦)二茂铁]二氯化钯。化合物经手工或者软件命名,市售化合物采用供应商目录名称。附图说明图1:化合物2与化合物1联合用药对耐药丙型肝炎病毒复制子产生的抑制效果。在g418存在的条件下,丙型肝炎病毒gt1b复制子稳转细胞由化合物2或tmc435单独或联合处理21天。复制子细胞集落由结晶紫染色显示。具体实施方式参考例1:片段bb-63-a和bb-63-b合成路线:步骤1:化合物bb-63-1的合成将2,5-二氢呋喃(7g,99.9mmol)和4-甲基苯亚磺酸钠(18.2g,1.1.9mol)溶于400ml的水和400ml的二氯甲烷的混合溶剂中,分批加入碘(25.9g,101.9mol),室温下搅拌过夜。加入400ml的二氯甲烷,萃取得到有机相,分别用饱和碳酸氢钠水溶液(200ml),饱和的亚硫酸氢钠水溶液(50ml),饱和食盐水(100ml)洗涤,有机相用无水硫酸钠干燥,减压除去溶剂得到目标化合物bb-63-1(油状物粗品,35g,100%)。步骤2:化合物bb-63-2的合成将化合物bb-63-1(35g,99.9mmol)溶于乙腈(500ml),加入三乙胺(21ml,149.8mol),室温下搅拌2小时。减压旋干溶剂,残留物用乙酸乙酯(500ml)稀释,分别用1mhcl(50ml),饱和碳酸氢钠水溶液(50ml),食盐水(50ml)洗涤,有机相用无水硫酸钠干燥,减压除去溶剂得到目标化合物bb-63-2(淡黄色固体,15g,收率:67%)。1h-nmr(400mhz,cdcl3)δ:7.89-7.82(m,2h),7.35-7.33(m,2h),6.77(s,1h),4.77-4.69(m,4h),2.44(s,3h).步骤3:化合物bb-63-3的合成在冰浴条件下,将化合物bb-63-2(7g,31.2mmol)和异腈乙酸乙酯(5.32g,46.8mmol)的四氢呋喃(50ml)溶液缓慢滴加到nah(3.12g,78.1mmol)的四氢呋喃(100ml)悬浊液中,0℃下搅拌1小时,缓慢升至室温继续搅拌2小时。反应液用甲醇(20ml)淬灭,减压除去溶剂,残留物进行硅胶柱层析分离(淋洗剂:etoac/pe=1/2),得到目标化合物bb-63-3(灰色固体,2g,收率:35%)。lcmsm/z:182.1[m+h]+步骤4:化合物bb-63-4的合成将化合物bb-63-3(2g,11mmol)溶于四氢呋喃(50ml),加入nis(2.98g,13.3mmol),反应液室温搅拌下过夜。减压除去溶剂,用乙酸乙酯(100ml)稀释,饱和食盐水(50ml)洗涤,有机相用无水硫酸钠干燥,减压除去溶剂,残留物进行硅胶柱层析分离(淋洗剂:etoac/pe=1/5),得到目标化合物bb-63-4(淡黄色固体,1.5g,收率:44%)。1hnmr(400mhz,cdcl3)δ:4.87(s,2h),4.52(s,2h),4.1(t,j=7.1hz,2h),1.23(q,j=6.8hz,3h).lcmsm/z:308.1[m+h]+步骤5:化合物bb-63-5的合成将化合物bb-63-4(1.5g,4.88mmol)溶于二氯甲烷(20ml),依次加入boc2o(1.28g,5.86mmol),三乙胺(1.48g,14.7mmol),室温下搅拌3小时。减压旋干溶剂,残留物进行硅胶柱层析分离(淋洗剂:etoac/pe=1/6),得到目标化合物bb-63-5(无色固体,4g,收率:70%)。lcmsm/z:353.2[m+h-55]+步骤6:化合物bb-63-6的合成将化合物bb-63-5(1.4g,3.44mmol)和对溴苯硼酸(0.76g,3.78mmol)溶于1,4-二氧六环/水=5/1(100ml),依次加入pd(dppf)cl2(0.3g,0.344mmol)和碳酸钠(1.1g,10.3mmol),反应液在氮气保护下,100℃反应4小时。反应液用乙酸乙酯(100ml)稀释,加水(30ml),萃取得到有机相用无水硫酸钠干燥,减压除去溶剂,残留物进行硅胶柱层析分离(淋洗剂:etoac/pe=1/2),得到目标化合物bb-63-6(白色固体0.7g,收率:46%)。lcmsm/z:379.9[m-56+h]+步骤7:化合物bb-63-7的合成将koh(1.3g,22.9mmol)溶于乙二醇(50ml),加热回流1小时,再加入化合物bb-63-6(2g,4.58mmol),继续加热回流1小时。冷却至室温后倒入冰水(100ml)中,用二氯甲烷(2×50ml)萃取,合并有机相用无水硫酸钠干燥,减压除去溶剂得到目标化合物bb-63-7(褐色固体,0.5g,收率41%)。1hnmr(400mhz,cdcl3)δ:8.41(br,1h),7.41-7.39(m,2h),7.05-7.03(m,2h),9.48(s,1h),4.98(s,2h),4.84(s,2h).lcmsm/z:263.9[m+h]+步骤8:化合物bb-63-8的合成在冰浴条件下,将三氯氧磷(1.74g,11.4mmol)逐滴加入化合物bb-63-7(0.5g,1.89mmol)和2-吡咯烷酮(0.81g,9.47mmol)的1,2-二氯乙烷(50ml)的溶液中,室温反应2小时。反应结束后倒入饱和的醋酸钠溶液(50ml)中,在0℃下,用10m的koh水溶液调至水相的ph为11,用二氯甲烷(3×50ml)萃取,合并有机相用无水硫酸钠干燥,减压除去溶剂得到目标化合物bb-63-8(褐色固体,0.3g,收率:48%)。lcmsm/z:333.0[m+h]+步骤9:化合物bb-63-9的合成将化合物bb-63-8(0.3g,0.91mmol)溶于二氯甲烷/meoh(1:1,20ml),加入nabh4(0.34g,9.06mmol),反应液加热回流过夜。反应液用水(10ml)淬灭,用二氯甲烷(2×50ml)萃取,合并有机相用无水硫酸钠干燥,减压除去溶剂得到目标化合物bb-63-9(褐色固体,0.1g,收率:33%)。lcmsm/z:335.0[m+h]+步骤10:化合物bb-63-a和bb-63-b的合成将化合物bb-63-9(100mg,0.3mmol)溶于二氯甲烷(5ml),依次加入化合物bb-2-6(63mg,0.36mmol),hatu(0.17g,0.45mmol)和三乙胺(91mg,0.9mmol),室温下搅拌2小时。加入水(20ml),用二氯甲烷(3×50ml)萃取,合并有机相用无水硫酸钠干燥,减压除去溶剂,残留物经硅胶柱层析分离(淋洗剂:etoac/pe=1/3)得到目标化合物bb-63-a(淡黄色固体,50mg,收率:34%)和目标化合物bb-63-b(淡黄色固体,50mg,收率:34%)。lcmsm/z:490.1[m+h]+参考例2:片段bb-21合成路线:步骤1:化合物bb-2-6的合成将l-缬氨酸(100g,751mmol)加入氢氧化钠溶液(2mol/l,535ml)中。冰浴冷却至5℃以下,滴加氯甲酸甲酯(118.13g,1.25mmol),并室温搅拌过夜。tlc检测反应完毕后,冰浴冷却至5℃以下,滴加浓盐酸调节ph值至5左右,收集析出的固体,用水(100ml)洗涤,干燥后得到目标化合物bb-2-6(白色固体,141g,产率98.2%)。产品无需纯化,直接应用于下一步。1hnmr(cdcl3400mhz):δ5.19(d,j=8.8hz,1h),4.32(dd,j=8.8hz,j=4.4hz,1h),3.71(s,3h),2.26-2.18(m,1h),1.01(d,j=7.2hz,3h),0.94(d,j=6.4hz,3h).步骤2:化合物bb-21-1的合成将edc·hcl(26.3g,136.9mmol),n-moc-l-缬氨酸(bb-2-6,17.6g,92.05mmol),二异丙基乙胺(35.4g,274.4mmol)溶于无水二氯甲烷(500ml),室温搅拌10分钟后加入化合物bb-8(参考例8,30g,102.7mmol),并在氮气保护下室温搅拌过夜。tlc检测反应完毕后,加入水(20ml)淬灭反应,有机相用10%的盐酸洗涤至ph值为5~6,然后再用饱和食盐水(100ml)洗涤,无水硫酸钠干燥。过滤后,滤液减压除去溶剂得到化合物bb-21-1(灰色泡沫状固体,35g,收率76%)。产品无需纯化,直接应用于下一步。msm/z:449.0[m+h]+.步骤3:化合物bb-21的合成室温下,将化合物bb-21-1(80g,178mmol),双联频哪醇硼酸酯(90g,354mmol)溶于二氧六环(600ml),在氮气保护下加入醋酸钾(35g,357mmol)和pd(dppf)cl2(13g,1.78mmol)。反应液在氮气保护下加热至90℃并搅拌过夜。tlc检测反应完毕后冷却至室温。过滤后,滤液旋干溶剂,残留物经硅胶柱层析(石油醚/乙酸乙酯=20:1→8:1)得到目标化合物bb-21(灰色固体,70g,收率80%)。msm/z:519.1[m+na]+.实施例1:化合物1合成路线:步骤1:化合物1的合成将化合物bb-63-a(30mg,61.2mmol)和化合物bb-21(36mg,73.4mmol)溶于thf/dmf/h2o=1:1:1(6ml),依次加入pd(dppf)cl2(5mg,6.1mmol)和碳酸钠(19mg,183.5mmol),反应液在氮气保护下,100℃反应4小时。反应液用乙酸乙酯(20ml)稀释,加水(10ml),萃取得到有机相用无水硫酸钠干燥,减压除去溶剂,残留物经制备分离得到目标化合物1(白色粉末,10mg,收率:19%)。1hnmr(400mhz,cdcl3)δ:10.79(br,1h),10.41(br,1h),10.01(br,1h),7.78-7.72(m,1h),7.57-7.44(m,5h),7.24-7.20(m,3h),5.64-5.59(m,2h),5.46-5.39(m,2h),5.01(s,2h),4.93-4.86(m,2h),4.47-4.41(m,2h),3.94-3.49(m,8h),3.11-3.08(m,1h),2.51-2.34(m,1h),2.27-1.90(m,8h),1.12-0.86(m,12h)。lcmsm/z:780.3[m+h]+。实施例2化合物11-1的制备步骤1:化合物9-1的合成将化合物8-1(1.6g,5.36mmol)溶于正己烷(30ml),然后依次加入双联嚬哪醇硼酸酯(1.497g,5.896mmol),二(1,5-环辛二烯)二-μ-甲氧基二铱(i)(0.216g,0.32mmol),4,4’-二叔丁基-2,2’-联吡啶(0.144g,0.536mmol),反应物在氮气保护下回流反应2小时。反应液旋干,然后快速过柱(淋洗剂,乙酸乙酯/石油醚=1/10)得到目标化合物9-1(白色固体,2.1g,收率:92%)。ms(esi)m/z:343[m-82+h]+。步骤2:化合物10-1的合成将化合物9-1(0.30g,0.707mmol)溶于甲醇(8ml),然后加入溴化铜(0.316g,1.41mmol),该反应液升温至50℃反应20小时。冷却后,反应液旋干得粗产品,快速过柱(淋洗剂,乙酸乙酯/石油醚=1/10)得到目标化合物10-1(灰色固体,0.22g,收率:82%)。ms(esi)m/z:377[m+h]+,379[m+h+2]+。步骤3:化合物11-1的合成将化合物10-1(0.160g,0.424mmol)溶于氮,氮-二甲基甲酰胺(3ml),然后加入cucn(0.152g,1.7mmol),该反应液在160℃反应4小时,冷却后,加入乙酸乙酯(50ml),有机相用食盐水(20ml)洗,无水硫酸钠干燥,过滤,浓缩得粗产品,再用薄层硅胶板分离(展开剂,乙酸乙酯/石油醚=1/3)得到目标化合物11-1(灰色固体,0.040g,收率:29%)。ms(esi)m/z:324[m+h]+。实施例3化合物19的制备合成路线:步骤1:化合物14的合成将化合物13(500g,2.27mol)溶于浓盐酸(1250ml)和水(1250ml),然后一小时内在25-30℃下滴加亚硝酸钠(469.89g,6.81mol)溶于水(1000ml)的溶液。反应物在25℃搅拌1小时。tlc(石油醚:乙酸乙酯=1:1)显示反应完全。反应物倒入乙酸乙酯(2l),分层,水层用乙酸乙酯萃取(1lx2)。合并有机相,用饱和食盐水1l洗涤,无水硫酸钠干燥,过滤,浓缩得到目标化合物14(黄色油状物,560g,收率:94.58%)没有纯化直接用于下一步反应。ms(esi)m/z:250[m+h]+。1hnmr(400mhz,chloroform-d)δ7.37-7.36(m,5h),5.18(s,2h),4.29-4.28(m,2h),3.83-3.82(m,2h),3.76(t,j=5.2hz,2h),3.53(t,j=5.2hz,2h)。步骤2:化合物15的合成将化合物14(1.00kg,4.01mol)溶于甲醇(10l),然后加入氯化铵(1.50kg,28.08mol),再于一小时内分批加完锌粉(1.05kg,16.05mol)。反应液加热至55℃反应2小时。tlc(石油醚:乙酸乙酯=4:1)显示原料转化完全。反应物用硅藻土过滤,滤液旋蒸浓缩,残留物溶于二氯甲烷10l,混合物再次用硅藻土过滤。滤液浓缩得粗产品15(浅黄色油状物,950g)直接用于下一步反应。ms(esi)m/z:236[m+h]+。步骤3:化合物17的合成将化合物16(415.64g,2.91mol)和二异丙基乙胺(626.21g,4.85mol)溶于二氯甲烷(10l),然后在-65℃下于一小时内慢慢滴加化合物15(570g,2.42mol)溶于2l二氯甲烷的溶液。反应物在-65℃反应2小时。lcms显示反应完成。反应混合物用水洗(4lx2),1m的稀盐酸洗(4l),饱和食盐水洗涤(4l),无水硫酸钠干燥,过滤,浓缩得粗产品17(浅黄色固体,700g)。将粗产品17(600g,1.76mol)溶于二氯甲烷(1.2l)中,搅拌下于一小时内慢慢滴加石油醚(1.2l)。析出物过滤收集,得到粗产品17(500g)。该白色固体产品再次溶于二氯甲烷(1l)中,于一小时慢慢滴加石油醚(1l)。析出物过滤收集,得到目标化合物17(白色固体,300g,结晶收率:49.87%,反应、结晶总收率:42.50%)。另外,合并重结晶的母液,浓缩得到粗产品(300g,纯度:60%)可通过重复如上重结晶进一步提纯。1hnmr(400mhz,chloroform-d)δ7.39-7.32(m,5h),5.88(br,1h),5.13(s,2h),4.35(t,j=5.2hz,2h),3.69(t,j=5.2hz,2h),3.64(t,j=4.8hz,4h),2.81-2.80(m,4h)。ms(esi)m/z:342[m+h]+。步骤4:化合18的合成将化合物17(40g,117.03mmol)溶于乙腈(400ml),然后在25℃下加入碳酸铯(38.13g,117.03mmol),该反应物在60℃搅拌4小时。lcms显示反应完全。反应液过滤掉碳酸铯,乙腈洗涤(100ml*3),滤液浓缩旋干。残留物溶于乙酸乙酯(1l),酸洗(1nhcl,120ml*2),饱和食盐水洗涤(120ml*2),有机相无水硫酸钠干燥,过滤,滤液浓缩旋干得白色固体粗品直接用于下一步反应。收率:35.5g。ms(esi)m/z:306[m+h]+。1hnmr(400mhz,chloroform-d)δ7.36-7.31(m,5h),5.12(s,2h),4.33(t,j=7.2hz,2h),3.65-3.59(m,6h),3.0-2.85(m,4h)。步骤5:化合物19的合成将化合物18(34.5g,112.99mmol)溶于甲醇(700ml),然后加入pd/c(3.45g),在氢气(50psi)保护下室温(25℃)搅拌18小时,lcms显示原料反应完全。过滤pd/c,滤饼用甲醇洗(100mlx2),滤液浓缩旋干,所得粗品化合物(19.0g)直接用于下一步。1hnmr(400mhz,chloroform-d)δ4.32(t,j=8hz,2h),3.64(t,j=8hz,2h),3.01-2.93(m,8h)。实施例4化合物2的制备合成路线2:步骤1:化合物6-1的合成将化合物4(250g,929.3mmol)溶于氮,氮-二甲基甲酰胺(2l),然后加入碳酸钾(256.88g,1.86mol),缓慢加入溴乙酸乙酯(310.39g,1.86mmol)。反应物在25℃反应1小时再加热到90-95℃,搅拌11小时。反应液变成褐色,lcms检测反应完全,降温至25℃后过滤,滤液倒入6l冷水中,搅拌2小时,过滤,空气中干燥得到目标化合物6-1(灰褐色固体,收率:69.27%)直接用于下一步合成。ms(esi)m/z:337[m+h]+,339[m+h+2]+。1hnmr(400mhz,cdcl3)8.02(s,1h),7.81(s,1h),7.52(s,1h),4.46(q,j=7.19hz,2h),1.44(t,j=7.03hz,3h).步骤2:化合物8-1的合成将化合物6-1(250g,741.64mmol),环丙基硼酸(76.45g,889.97mmol),磷酸钾(393.57g,1.85mol),醋酸钯(15.00g,66.81mmol),三环己基膦(20.80g,74.16mmol)溶于甲苯(2l)和水(500ml),然后换气,再在氮气保护下加热至100℃反应12小时。hplc显示反应完全。冷却后分液,水相用乙酸乙酯(1lx2)萃取,合并后有机相用食盐水洗(500mlx2),有机相用硫酸钠干燥,过滤浓缩得到粗产物,过柱分离(淋洗剂,乙酸乙酯/石油醚=1/20)得到目标化合物8-1(棕黑色油状物,210g,收率:93.99%)。ms(esi)m/z:299[m+h]+。1hnmr(400mhz,cdcl3)7.54(s,1h),7.49(s,1h),7.42(s,1h),4.47-4.42(m,2h),2.07-2.02(m,1h),1.44-1.41(m,3h),1.06-1.02(m,2h),0.77-0.73(m,2h)。步骤3:化合物11-1的合成将化合物8-1(200.00g,670.56mmol)溶于无水四氢呋喃(2.0l),在-40℃氮气保护下加入二氯化镁(2,2,6,6-四甲基哌啶)锂盐溶液(1.5m,893.33ml),在-40℃下搅拌2小时,然后加入4-甲苯磺酰腈(182.27g,1.01mol)的无水四氢呋喃(500ml)溶液,在-40℃至-10℃条件下继续搅拌1小时。tlc(石油醚/乙酸乙酯=20/1)显示产物形成。反应液用120ml水淬灭。乙酸乙酯(5l)加入到上述体系中,过滤,滤液浓缩,所得残留物经柱层析纯化(展开剂:石油醚/乙酸乙酯=30/1)得目标化合物11-1(白色固体,90g,收率:41.52%)。1hnmr(400mhz,meod)δ7.76(s,1h),7.66(s,1h),4.53(q,j=7.2hz,2h),2.21~2.17(m,1h),1.46(t,j=7.2hz3h),1.14~1.11(m,2h),0.86~0.84(m,2h)。步骤4:化合物12的合成将化合物11-1(405.00g,1.25mol)溶于四氢呋喃(3.00l),然后将水合氢氧化锂(105.14g,2.51mol,2.00eq)和水(1.25l)加入到上述的溶液中,在25℃的条件下搅拌2小时,tlc(pe:ea=30:1)检测原料完全消失。溶剂四氢呋喃在减压下旋蒸除去,水相加2nhcl调ph值至4~5。乙酸乙酯(2.0l*3)萃取有机相合并水洗(500ml*3),无水硫酸钠干燥减压浓缩得浅黄色粗品。将混合溶剂(石油醚:乙酸乙酯=30:1,600ml)加入到上述的粗品中搅拌打浆,然后砂芯漏斗过滤得浅黄色固体,在旋蒸下干燥得浅黄色纯品化合物12(300.00g,收率:79.67%yield)。ms(esi)m/z:296(m+h)+。1hnmr(400mhz,meod)δ7.75(s,1h),7.65(s,1h),2.23~2.20(m,1h),1.35~1.13(m,2h),0.87~0.86(m,2h)。步骤5:化合物2的合成将化合物19(5.0g,29.21mmol)溶于dmf(80ml),然后在25℃下依次加入化合物12(8.0g,27.10mmol),hatu(12.36g,32.52mmol),diea(10.51g,81.30mmol)。该反应液在25℃下搅拌1.5小时。lcms显示原料反应完全。反应结束后加入120ml水,沉淀物过滤,固体加甲醇(120ml)搅拌15分钟,过滤,真空干燥得到白色固体目标化合物2(7.4g,收率:60.89%)。ms(esi)m/z:449[m+h]+。1hnmr(400mhz,chloroform-d),δ7.62(s,1h),7.50(s,1h),4.36(t,j=8.0hz,2h),3.97(m,4h),3.67(t,j=8.0hz,2h),3.16-3.13(m,4h),2.12-2.06(m,1h),1.15-1.11(m,2h),0.81-0.79(m,2h)。实验例1:体外评价实验目的:用hcv基因型1a(hcv-1a)和1b(hcv-1b)稳转复制子(replicon)细胞测定抗hcv化合物的ec50和cc50值。基因型1a复制子来源为h77克隆,含有k1691r,k2040r和s2204i适应性突变。基因型1b复制子来源为con1克隆,含有e1202g,t1280i和k1846t适应性突变。背景介绍:hcv1a(hcv-1a)和1b(hcv-1b)基因型亚基因组复制子系统含有相关hcv基因亚型非结构蛋白基因,g418抗性基因neo和荧光素酶基因,使得hcv相关蛋白及荧光素酶可在细胞中稳定表达。通过检测荧光素酶基因的表达高低可以确定hcv复制子的复制水平的高低。因此,该系统作为体外筛选抗hcv化合物活性的模型。实验材料:hcv复制子细胞系:hcv-1a和hcv-1b细胞。细胞培养液:dmem(invitrogen,cat.#11960077)培养液,加10%胎牛血清(fbs,sigma,cat.#12003c)和1%双抗(青霉素5000iu/ml,链霉素10mg/ml,hyclone,cat.#sv30010)。胰酶(invitrogen,cat.#25200072)。pbs(invitrogen,cat.#10010023)。台盼蓝(invitrogen,cat.#15250061)。celltiter-fluor(promega,cat.#g6082)。bright-glo(promega,cat.#e2650)。co2培养箱,thermo240i。multidrop自动分液器,thermo。pod810plateassembler全自动微孔板预处理系统,labcyte。scepterhandheldautomatedcellcounter手持式自动细胞计数器,millipore。microplatespectrophotometer微孔板分光光度计,moleculardevice。实验步骤和方法:a)化合物溶液制备、稀释和加样:将化合物粉剂溶解于100%dmso。然后对化合物以5倍稀释6个点,用echo声波移液设备(echoliquidhandler)加到细胞板中。保证dmso终浓度为0.5%。每个化合物做双复孔。最高起始浓度为100,10或1nm,5倍稀释,6个点。b)细胞培养(hcv-1a或hcv-1b复制子细胞):1)吸掉细胞培养的培养上清,用10mlpbs洗细胞。2)加入预热过的胰酶到洗过的细胞培养瓶中,旋转培养瓶使胰酶均匀覆盖培养瓶底部。放到37℃,5%co2培养箱中消化。3)每个t150培养瓶用10-15ml培养液悬浮细胞,吸取0.1ml用台盼蓝溶液稀释2倍计数。4)用培养液稀释细胞到8×104/ml,用自动分液器(thermoscientific)将稀释好的细胞加入到含化合物的96孔板(greiner,cat.#655090)(100μl/孔,8000cells/孔)。置于37℃,5%co2培养箱培养3天。细胞对照孔:不加化合物,只含0.5%dmso。5)加化学发光底物celltiter-fluor到细胞孔,孵育30分钟后用化学发光检测系统envison(exat405nmandreadat515nm)检测信号。根据发光数据分析化合物对hcv复制子细胞活性的影响,并用于计算cc50值。6)然后加荧光素酶发光底物bright-glo,温孵5分钟后用化学发光检测系统envison检测(波长>700nm)荧光素酶活性;根据荧光素酶数据分析化合物的抗hcv抑制活性并用于计算ec50值。c)数据处理和分析:采用graphpadprism软件对抑制百分比(inh﹪)数据进行非线性拟合分析得到ec50或cc50值。实验结果见表1:表1hcv复制子细胞ec50/cc50测试结果注:ec50表明分子的体外抗丙肝病毒的活性,ec50小于1um即代表化合物具有体外活性。活性区间:a(0.001nm~0.1nm。cc50的数值表明分子的体外毒性的大小,数值越大毒性越小。结论:化合物1具有优异的体外抗丙肝病毒活性。实验例2:体外细胞活性测试实验目的:用hcv基因型1a(hcv-1a)和1b(hcv-1b)稳转复制子(replicon)细胞测定抗hcv化合物的ec50和cc50值。基因型1a复制子来源为h77克隆,含有k1691r,k2040r和s2204i适应性突变。基因型1b复制子来源为con1克隆,含有e1202g,10t1280i和k1846t适应性突变。背景介绍:hcv1a(hcv-1a)和1b(hcv-1b)基因型亚基因组复制子系统含有相关hcv基因亚型非结构蛋白基因,g418抗性基因neo和荧光素酶基因,使得hcv相关蛋白及荧光素酶可在细胞中稳定表达。通过检测荧光素酶基因的表达高低可以确定hcv复制子的复制水平的高低。因此,该系统作为体外筛选抗hcv化合物活性的模型。实验材料:hcv复制子细胞系:hcv-1a和hcv-1b细胞。细胞培养液:dmem(invitrogen,cat.#11960077)培养液,加10%胎牛血清(fbs,sigma,cat.#12003c)和1%双抗(青霉素5000iu/ml,链霉素10mg/ml,hyclone,cat.#sv30010)。胰酶(invitrogen,cat.#25200072)。pbs(invitrogen,cat.#10010023)。台盼蓝(invitrogen,cat.#15250061)。celltiter-fluor(promega,cat.#g6082)。bright-glo(promega,cat.#e2650)。co2培养箱,thermo240i。multidrop自动分液器,thermo。pod810plateassembler全自动微孔板预处理系统,labcyte。envisionmultilabelplatereaders微孔板分光光度计,perkineelmer。实验步骤和方法:a)化合物溶液制备、稀释和加样:将化合物粉剂溶解于100%dmso。然后对化合物以5倍稀释6个点。用echo声波移液设备(echoliquidhandler)5加到细胞板中。dmso终浓度为0.5%。每个化合物做双复孔。b)细胞培养(hcv-1a或hcv-1b复制子细胞):1)吸掉细胞培养的培养上清,用10mlpbs洗细胞。2)加入预热过的胰酶到洗过的细胞培养瓶中,旋转培养瓶使胰酶均匀覆盖培养瓶底部。放到37℃,5%co2培养箱中消化。3)每个t150培养瓶用10-15ml培养液悬浮细胞,吸取0.1ml用台盼蓝溶液稀释2倍计数细胞。4)用培养液稀释细胞到8×104/ml,用自动分液器(thermoscientific)将稀释好的细胞加入到含化合物的96孔板(greiner,cat.#655090)(100μl/孔,8000cells/孔)。置于37℃,5%co2培养箱培养3天。细胞对照孔:不加化合物,只含0.5%dmso。5)加化学发光底物celltiter-fluor到细胞孔,孵育30分钟后用化学发光检测系统envison(exat405nmandreadat515nm)检测信号。根据发光数据分析化合物对hcv复制子细胞活性的影响,并用于计算cc50值。6)然后加荧光素酶发光底物bright-glo,温孵5分钟后用化学发光检测系统envison检测(波长>700nm)荧光素酶活性;根据荧光素酶数据分析化合物的抗hcv复制子抑制活性并用于计算ec50值。c)数据处理和分析:采用graphpadprism软件对抑制百分比(inh﹪)数据进行非线性拟合分析得到ec50或cc50值。实验结果见表2:表2hcv复制子细胞ec50/cc50测试结果化合物ec50(1b)ec50(1a)cc50化合物2aa>3000注:a(0.1nm~100nm)。结论:化合物2具有优异的体外抗丙肝病毒活性。实验例3:化合物1与化合物2联合用药对丙型肝炎病毒复制子耐药产生的影响研究1.试验目的联合应用抗病毒药物的目的除了增强药效而外,另一重要目的是增强抑制耐药病毒的出现。本实验旨在评价化合1与化合物2联合用药时对抑制耐药病毒出现的效果。2.实验材料2.1受试药物药物名称:化合物1与化合物2。配制方法:化合物溶于100%dmso制备成10mm的母液,保存于氮气柜中。2.2细胞株丙型肝炎病毒基因型1b复制子细胞hcv-1b在含10%胎牛血清、2mm谷氨酰胺、100u/ml青霉素、100μg/ml链霉素和250μg/mlg418的dmem培养液中培养。2.3试剂及仪器结晶紫(sigma,货号c3886)。扫描仪(惠普,型号scanjetg4050)。3.试验方法实验将50000个hcv-1b复制子细胞种于10cm平皿后,化合物2和化合物1各取三个浓度,分别为12nm,36nm,108nm和0.05nm,0.15nm,0.45nm(约5×,15×和45×ec50),进行组合加入含有复制子细胞的平皿,每3或4天换一次含化合物的新鲜培养液,直至耐药复制子细胞集落的出现。hcv-1b复制子细胞在无化合物或单一化合物的培养液中培养作为参照。复制子细胞经化合物处理3周后出现细胞集落。复制子细胞集落用1%结晶紫染色。4.结果为了进一步评价化合物2联合用药的效果,检测了化合物2与ns5a蛋白抑制剂化合物1联合应用后对耐药复制子产生的抑制效果。如果化合物能够抑制复制子在细胞中复制,则细胞在g418存在的条件下不能生存。如果复制子细胞含耐药复制子,则复制子可以在细胞中复制,同时复制子细胞对g418表现抗药性,因而当含药物敏感复制子的细胞在g418的压力下死亡后,含有耐药复制子的细胞则分裂复制并进而形成集落。实验结果见图1。化合物2或化合物1单独用药时,耐药复制子细胞克隆出现的数量与药物剂量成反比,即药物剂量越高,耐药复制子细胞克隆数越少。化合物2与化合物1联合使用时,出现的耐药复制子细胞克隆数量均比任一药物单独作用时显著减少,如化合物2与化合物1在5xec50浓度,单独应用时复制子细胞集落分别为120和90,但当二者联合用药时,无耐药复制子细胞集落。因而实验结果表明化合物2与化合物1联合用药可明显增强抑制丙型肝炎病毒复制子耐药的产生。表3化合物2与化合1联合用药细胞集落数5.结论研究结果表明化合物2与化合物1联合用药后可以增强抑制耐药丙型肝炎病毒复制子的产生,因而进一步支持化合物2可以与化合物1在临床进行联合用药。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1