基于葫芦脲的水凝胶的制作方法
本案涉及2016年10月14日提交的gb1617469.0和2017年5月30日提交的gb1708589.5,这两者的内容以其整体通过引证结合于本文中。
本发明涉及基于葫芦脲交联超分子网络的水凝胶,以及由包含葫芦脲和具有葫芦脲的客体的可聚合单体的可聚合组合物制备这种水凝胶的方法。
背景技术:
一些本发明人先前已经描述了超分子水凝胶,其基于葫芦脲cb[8]与具有cb[8]的客体官能团的聚合物的超分子配位。
appel等人(j.am.chem.soc.2010,132,14251)描述了由包含cb[8]的组合物与具有cb[8]的客体的低分子量聚合物配位而形成的水凝胶。超分子水凝胶在基于cb[8]与两个客体的三元配合物的聚合物之间具有分子内和分子间的非共价交联。研究了水凝胶的流变性质。
在wo2013/124654中报道的后续工作中,一些本发明人描述了由包含cb[8]的组合物和具有cb[8]的客体的高分子量聚合物的配位而形成水凝胶。这项工作还描述了用于递送的水凝胶保持组分。这种水凝胶由包含cb[8]和具有cb[8]的客体的低分子量或高分子量聚合物的组合物的配位形成。用于递送的组分可以包含于组合物中以在配位期间引入,或组分可以后续添加。这项工作也报道于appeletal.(j.am.chem.soc.,2012,134,11767)中。
us2012/0103615描述了一些发明人在开发用于例如井筒(wellbore)流体的增粘聚合物方面的早期工作。这些增粘聚合物基于包含葫芦脲如cb[8]和聚合物链上提供的客体基团的可聚合组合物的配位。该文献中的系统与appel等人和wo2013/124654描述的体系非常相似。
发明人先前报道的水凝胶是有用的,但其用途却受限于其相对弱的机械性能。为了获得改进的机械特性,期望更高程度的交联。然而,增加cb[8]的使用量受阻于这种葫芦脲在也包含用于水凝胶网络的聚合物的组合物中的有限溶解度。本领域中描述的水凝胶体系将cb[8]添加到包含具有cb[8]的客体的聚合物的高粘度组合物中。这种混合物并不是理想的,因为它们限制了可以制备的结构的类型,并且在大规模使用时在操作混合物时可能会出现问题。
例如,在appeletal.(2010)中通过在10%应变下的动态振荡流变学分析了水凝胶材料的流变性质,观察到损耗模量(g″)在较高频率下主导储能模量(g')。因此,损耗模量在20rad/s或更大的频率下是主导的。在较低频率下,储能模量是主导的。因此,g'和g″不是线性的,并且在振荡流变学中并不平行。
wo2013/124654中报道的水凝胶通常具有在动态频率扫描实验中在0.1至100rad/s的范围上测量的小于1,000pa的储能和损失模量,和在振幅扫描实验中在0.1%至10%应变的范围上测量的小于1,000pa的储能和损失模量。
因此具有改进的机械性能的水凝胶是期望的。
大多数合成水凝胶是脆性的,因此是用于模拟类似于生物组织的生物材料(无论其柔软和湿润性质如何)的较差候选。然而,最近关于替代超分子水凝胶体系的研究表明,通过引入可以有效地改善机械强度以及韧性、阻尼和疲劳抗性的牺牲键,可以生产非常坚韧的材料以用作结构生物材料,如软骨。然而,设计具有高韧性和极高可拉伸性的柔软材料仍然是一个挑战。
本工作提供了与早期工作中描述的那些无论是制备水凝胶的方法还是水凝胶产品的结构和特征方面都不同的替代水凝胶。
技术实现要素:
总体上,本发明提供了由包含葫芦脲和具有葫芦脲的客体的第一单体的水性可聚合组合物获得的或可获得的超分子水凝胶。用于本发明的示例性葫芦脲是葫芦[8]脲(cb[8]),其适用于制备基于三元主体-客体配合物的超分子水凝胶。
本发明人已经发现这些水凝胶具有优异的机械物理化学性能,并且与一些本发明人先前描述的葫芦脲水凝胶相比,这些性能得以改进。
超分子水凝胶是由包含葫芦脲和具有葫芦脲的客体的第一单体的水性可聚合组合物获得的或可获得的。因此,水凝胶通过原位聚合而制备。在葫芦脲主体及其客体存在下制备聚合物允许形成具有高度链缠结的水凝胶,这是由先前报道的制备葫芦脲水凝胶的方法不可获得的性质。这种缠结可以在不需要高度浓缩的可聚合组合物的情况下实现。然而,为了获得水凝胶的期望的物理性质,还必须使用其中单体浓度不是太稀的可聚合组合物。因此,总单体浓度cmon通常为至少0.5m。链缠结有助于材料的有益性能。在水凝胶产品中提供超分子交联的主体-客体配合物也可以预形成于可聚合组合物中。
使用可聚合组合物制备水凝胶允许使用比先前报道的更高有效浓度的葫芦脲主体和葫芦脲客体。因此,这允许形成具有更高程度的动态交联的水凝胶。超分子键合的形成不受超分子键在聚合物之间形成的先前水凝胶制剂所见的问题的影响。在这些现有技术方法中,高水平的非共价交联通过聚合物链缠结、客体官能化聚合物的有限溶解度、聚合物混合物中的低葫芦脲溶解度和起始聚合物混合物的高粘度等来防止。
此外,由可聚合组合物制备水凝胶,为水凝胶的变化提供了许多选择,因为葫芦脲主体和第一单体以及其客体都容易变化,并且可以使用各种各样的替代主体和客体,提供了改变产品水凝胶性能的可能性。另外的试剂如另外的可聚合共聚单体可以添加到可聚合组合物中,提供改性水凝胶的进一步选择。现有技术的水凝胶的改性受限于聚合物预先构建的事实,因此变化的选择会受到一些限制。
本发明的水凝胶可以由高含水量制备,如80至95wt%的水。这些水凝胶具有很大的可拉伸性(例如,拉伸至其原始长度的45倍,如其原始长度的100倍),并具有非常高的断裂能量(例如,高达2,100j·m-2)。水凝胶还具有高收缩应力(例如,高达0.5mpa),并且它们能够比水凝胶抬升质量大得多的物体(例如,500倍质量,如2,000倍质量)。本发明的水凝胶在宽范围的应变和频率值范围内具有高储能和损耗模量(例如,1,000pa或更大,如10,000pa或更大)。
在本发明的第一方面中,提供了包含葫芦脲和具有葫芦脲的客体的第一单体的可聚合组合物。可聚合组合物可以是水性的可聚合组合物。可聚合组合物中的总单体浓度cmon可以为至少0.5m。
在本发明的第二方面中,提供了一种由包含葫芦脲和具有葫芦脲的客体的第一单体的水性可聚合组合物获得的或可获得的水凝胶。
本发明的水凝胶含有聚合物的超分子网络。聚合物通过非共价键互连或内连,并且可选地,聚合物也通过共价键互连或内连。网络可以基于葫芦脲与聚合物上提供的客体分子的三元或二元非共价配合物。
在本发明的第三方面中,提供了一种制备水凝胶的方法,方法包括聚合包含葫芦脲和具有葫芦脲的客体的第一单体的水性可聚合组合物。聚合可以在温和温度和环境温度下进行。
可聚合组合物可以包含一种或多种(如一种)与第一单体共聚的共聚单体。共聚单体不需要具有葫芦脲的客体,但可选地其可以具有葫芦脲的客体。通常,共聚单体不含有与葫芦脲形成主体-客体配合物的官能团。然而,具有与葫芦脲形成主体-客体配合物的官能团的共聚单体是可以存在。存在于可聚合组合物中的单体可以至少部分溶于水。单体可以是亲水性单体。
组合物可以含有第二可聚合共聚单体。第二可聚合共聚单体不同于第一单体。第二可聚合共聚单体可以不具有葫芦脲的客体。
葫芦脲可以是具有形成三元配合物的能力的葫芦脲。葫芦脲可以是cb[8],包括其变体和衍生物。
超分子水凝胶可以含有葫芦脲与两个客体的三元配合物。通常,三元配合物中的客体是相同的。然而,在其他实施方式中,三元配合物中的客体可以是不同的。
在一个实施方式中,水凝胶保持用于递送的组分,如药物。本文中,用于递送的组分可以在聚合之前提供于可聚合组合物中。在聚合过程期间,组分被引入水凝胶网络中。
在本发明的进一步的方面中,提供了包含本发明的水凝胶的粘合剂。
在本发明的又进一步的方面中,提供了一种生物材料,如组织、骨或软骨生物材料,其包含本发明的水凝胶。
本发明还提供了水凝胶用于治疗方法。
在本发明的进一步的方面中,提供了由本发明的可聚合组合物的聚合获得的或可获得的聚合物。
在本发明的另一个方面中,提供了用于制备聚合物的方法,方法包括聚合本发明的可聚合组合物,如本发明的第一方面的组合物的步骤。
本发明的这些和其他方面和实施方式以下将更详细地描述。
附图说明
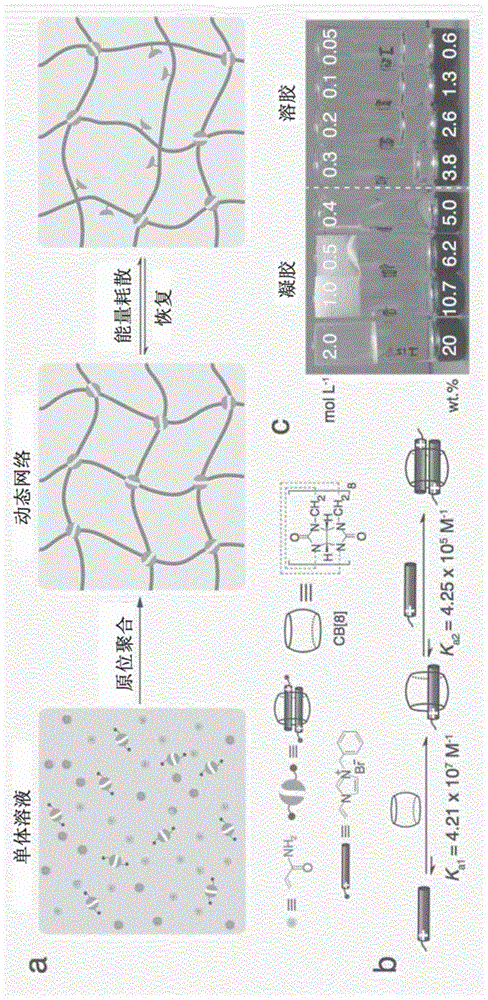
图1显示了(a)是用于制备超分子聚合物网络的原位聚合反应的示意图;(b)是在1:2结合模式之后逐步形成cb[8]和客体分子之间的三元主体-客体配位的示意图;和(c)不同的总单体浓度(mol/l)或质量分数(wt%)下的超分子聚合物水凝胶的照片,以及对于每个样品粘度的倒置小瓶展示。
图2显示了(a)由具有总单体浓度0.5m(底部线),1m(中部线)和2m(顶部线)的可聚合组合物制备的水凝胶在形变速率100mm/min下的单轴拉伸行为。(b)由具有总单体浓度2m的可聚合组合物制备的水凝胶在50(底部)至1,000mm/min(顶部)范围内的不同形变速率下的单轴拉伸行为。注意到所有样品均达到拉伸机的最大应变而未断裂;(c)由具有总单体浓度2m的可聚合组合物制备的水凝胶在施加和卸除各种应变的循环内的单轴拉伸行为。形变速率为100mm/min;和(d)由具有总单体浓度2m的可聚合组合物制备的水凝胶的单轴拉伸行为。施加应变后立即(底部曲线在40mm/mm处),在8mm/mm的应变施加和卸除的第一循环后(底部曲线在6mm/mm处),水凝胶样品在应力后室温下自修复30min(顶部曲线在40mm/mm处)。正常运行曲线(从上数第二个曲线在40mm/mm处)与30min后自修复样品的高度重叠表明,通过先前断裂的超分子键连的重新形成,网络几乎完全自我修复。在所有图中,应力(mpa)的变化随应变(mm/mm)的变化一起显示。
图3显示了(a)原始水凝胶和自愈合的水凝胶在25℃下在断裂后愈合1、2、5和12小时的应力-应变曲线(12小时后原始样品和自愈合样品在32mm/mm的应变下未断裂,最大应变可以通过拉伸机实现)。显示的插图是哑铃形聚合物网络在以λ=0,(i)和λ=22,(ii)的形变比拉伸时自愈合后的照片,其中由圆圈标记的部分表示缺口部分,并且允许在25℃下自愈合2小时的水凝胶在断裂前可以承受22mm/mm的拉伸应变。应力(mpa)的变化随着应变(mm/mm)的变化显示;(b)断裂应力(mpa)、断裂应变(表示为拉伸长度/原始长度的倒数)以及自愈合比率(自愈合的样品与原始样品的拉伸测试的功之比,原始样品在45mm/mm应变下的功取为100%);(c)两个超分子聚合物网络的荧光显微图像,一个用ritc(红色)化学标记,另一个用fitc(绿色)化学标记。两个聚合物凝胶膜(5cm(宽)×5mm(长)×50mm(厚))放置一起(接触表面:5mm×50mm),间隙约50mm(黑色区域,i),在室温下自愈合2小时(ii)和5小时(iii)。浅黄色区域的外观证实了接触区域的自我修复,以及(d)显示了水凝胶样品在20℃(i),40℃(ii)和在20℃,40℃和70℃之间的一系列步进加热和冷却上的连续步进应变测量的结果(高振幅振荡参数:应变,γ=500%,角频率,ω=10rads-1;低振幅振荡参数:γ=0.5%,角频率,ω=10rads-1)。在所有实验中,原始水凝胶是由具有总单体浓度2m的可聚合组合物制备的非共价水凝胶。
图4显示了(a)超分子聚合物网络作为导体在与电池连接时点亮led灯泡的视觉展示,在样品拉伸至应变λ=10.5之前(i)和之后(ii),恢复到其原始状态长度(iii),以及使用自愈合样品(12h,(iv)(1))作为导体,在拉伸至应变λ=10.5之前((iv)(2))和之后(iv):(b)用连接到万用表(电压通道)的网络样品在压缩(约30%形变)之前(i)和压缩时以及压缩去除和电压信号在18秒内恢复到原始值(iii)的压力传感的视觉展示;(c)电阻率(ω·m)和相对电阻(r/r0)随拉伸应变变化的变化(相对于拉伸的样品的长度与未拉伸的样品的长度的比值表示);(d)电压(mv)随压缩应变变化(%)的变化;和(e)在2%的压缩应变(约0.35kpa的应力)下循环压缩/卸压缩循环期间的电压变化(mv),每个循环的间隔为60s。在所有实验中,原始水凝胶是由具有总单体浓度2m的可聚合组合物制备的非共价水凝胶。
图5显示了通过溶解-冷冻干燥过程(下部)和(顶部)对过量的金刚烷胺(ada)渗析以去除cb[8],然后冷冻干燥后获得的聚合物的两个1hnmr谱(500mhz,298k)。5mol%的分数通过信号积分估算确认,与5mol%的化学计量进料比一致。
图6显示了(a)来自超分子聚合物水凝胶的获得的聚合物(除去了cb[8])的水性gpc迹线,提供了重均分子量(mw)为2.53mda(mw/mn=1.9),这属于自由基聚合的典型mw范围;和(b)通过粘度计测量的相对于聚合物溶液(去除cb[8])的比粘度的对数-对数绘图,以及数据点在-2至1(0.01至10g/l)和1至1.9(10至80g/l)范围内的线性绘图,提供1.42(26.3g)处的交叉点,即重叠浓度。
图7显示了以总单体浓度2.0m制备的水凝胶的杨氏模量(mpa)随形变速率(mm/min)变化的变化。此处,杨氏模量的定量增加据估计当形变速率从10(0.017mpa)增加到2,000mmmin-1(0.42mpa)时增加约24倍。
图8显示了具有拉伸变化的以总单体浓度2.0m制备的水凝胶的透射率(%)随波长(nm)变化的变化,其中原始厚度为2mm的膜在将两端固定到uv/vis谱固定器上时,在单轴拉伸之下分别拉伸至5和15的应变,记录样品的透射率,填充milli-q水的比色皿作为空白。
图9显示了(a)用不同的总单体浓度制备的水凝胶在动态室温振幅扫描中(从0.1%到1000%应变,10rads-1)g'和g″的模量(pa)随着应变变化(%)的变化(pa);(b)显示了用不同总单体浓度制备的水凝胶的g'和g″的模量(pa)随频率扫描中的频率(rad/s)变化(从0.1s-1到100s-1)的变化;(c)用不同总单体浓度制备的水凝胶的粘度(pa.s)随应变速率(1/s)变化的变化;和(d)用不同总单体浓度制备的水凝胶的粘度(pa.s)随时间变化的变化,其中使用的总单体浓度为0.3、0.4、0.5、1.0和2.0m,并且应力弛豫函数是从线性形变方案(在0.1s内γ=10%,t=20℃)中得到的g(t)。
图10显示了(a)g'和g″的模量(pa)和tan(δ)随动态室温振幅扫描在的应变(%)变化(0.1%至1000%应变,10rads-1)的变化;(b)g'和g″的模量(pa)和tan(δ)随频率扫描中频率(rad/s)变化(0.01至100rads-1,0.5%应变)的变化;和(c)在cb[7](c-i)和无cb[n](c-ii)存在下受控的超分子水凝胶样品(流动流体)的照片。
图11显示了仿生模块化多级超分子双网络设计的概念图,其中(a)由超分子物理主体-客体交联(蓝色圆圈)和永久交联(粉色圆圈)组成的模块化双网络的示意图。三组分cb[8]三元配合物化学计量比为1:2;(b)肌联蛋白(titin)结构的典型部分,具有约200至300个重复免疫球蛋白(ig)样结构域,和为多级组装肌肉机械提供核心功能的模块化三级折叠结构域蛋白;(c)含有由葫芦[8]脲介导的主体-客体配位保持的多环的双网络(从(a)中的阴影区域提取的结构),超分子相互作用的机械诱导的解离,能量耗散的基本机制,以及卸除和松弛时主体-客体配位的重新形成的示意图;(d)在17的应变下拉伸时的无缺口的双网络样品的条带的照片(用两个载玻片和刚性夹具固定,样品尺寸:4cm(l)×1mm(w)×1.5mm(t));(e)和在17的应变下的缺口样品的伸长率(样品尺寸:4cm(长)×1mm(宽)×1.5mm(t),缺口尺寸2cm)。(当网络样品形变时,应变由两个夹具之间的形变除以原始长度定义);和(f)显示维持200g的重量的双网络(4cm(长)×2mm(宽)×1.5mm(长))的能力的图像。聚合物双网络以1.0m的cmon制备。
图12显示了(a)使用10至1000mmmin-1范围内的不同初始形变速率的超分子聚合物双网络的单轴拉伸行为。每个测试通过拉动哑铃形样品至断裂而进行;和(b)双网络的断裂应力、断裂应变和杨氏模量的形变速率依赖性。聚合物双网络以1.0m的cmon制备。
图13显示了(a)对超分子聚合物双网络的样品进行施加和卸除不同应变的循环(形变速率为100mmmin-1);(b)样品在8×(原始长度)的应变下通过循环拉伸测试进行的不同等待时间下的恢复;(c)滞后比(下一个滞变回线与第一次循环运行的面积比)和残余应变对于等待时间的依赖性;(d)在8×(原始长度)的应变下加载和卸除的第一次循环后(粉红色曲线)立即(蓝色曲线)的样品的拉伸测试,以及使自修复的样品在室温下静置30min(绿色曲线)的样品的拉伸测试。原始样品曲线(黑色曲线)与30分钟后自修复样品(绿色曲线)之间的高水平重叠表明通过超分子主体-客体缀合的重新形成的机械性质的完全恢复。聚合物双网络以1.0m的cmon制备。
图14显示了(a)来自动态室温频率扫描(0.01至200rads-1,0.5%应变)的超分子双网络的g'、g″和tan(δ)值;(b)温度扫描(5至80℃,间隔为5℃,60rads-1,0.5%应变)。(c)超分子双网络的主曲线(0至60℃,0.5%应变,10rads-1);和(d)样品在40℃下的连续步进应变测量(高振幅振荡参数:应变(γ)=500%,角频率(ω)=10rads-1;低振幅振荡参数:γ=0.5%,ω=10rads-1)。
图15显示了1mm的1-苄基-3-乙烯基咪唑鎓溶液向0.05mm的cb[8]溶液(均在10mmpbs缓冲液中,ph7,25℃下制备)的itc滴定。对于2:1结合检测到高结合常数1.8×1013m-2,熵7.63kjmol-1(δh1)和7.60kjmol-1(δh2)。结合能与肌联蛋白内的蛋白质折叠相当,证实了材料作为仿生物的用途。
图16是拉伸期间双网络的uv/vis透射谱图,以及可见光范围(400至700nm)内的完全100%透明度。
图17显示了动态室温振幅扫描实验(0.1%至1000%,10rads-1)中超分子双网络的g'、g″(pa)和tan(δ)值随应变(%)的变化。
图18显示了双网络在20℃(a)和70℃(b)下以10rads-1在0.5%和500%应变之间的连续步进应变测量中g'和g″(pa)随时间的变化。对于20和70℃的大应变扫描可以观察到一次模量的迅速降低,并且检测到快速自修复性能,与cb[8]介导的超分子相互作用的动态本质一致。无论大或小应变扫描,样品的模量在20℃下保持稳定,表明稳定性良好。然而,对于70℃,由于主体-客体相互作用的动态本质和更高的解离动力学,模量保持下降。
图19是一对图表,显示了力(n)随拉伸距离(mm)变化的变化;其中(a)是双网络的撕裂测试的代表性力-伸展曲线;和(b)显示了双网络的无缺口(蓝色)和缺口(红色)样品的力-伸展曲线。
图20显示了由具有总单体浓度为0.3m(顶部线,100rad/s)至2m(底部线,100rads-1)的可聚合组合物制备的五种水凝胶的tan(δ)随频率(rad/s)变化的变化。水凝胶具有超分子非共价网络。在所有实验中,原始水凝胶是非共价水凝胶。
图21显示了(a)用作根据本发明的实施方式的可聚合组合物中的第二单体的示例性单体(顶行),cb[8]的结构,以及第一单体溴化1-苄基-3-乙烯基咪唑鎓与cb[8]的三元配合物的示意图(第二行);(b)由含有可聚合单体的可聚合组合物与cb[8]主体形成水凝胶的示意图,其中水凝胶具有由cb[8]主体和溴化1-苄基-3-乙烯基咪唑鎓的非共价三元配合物组装的动态网络;和(c)由溴化1-苄基-3-乙烯基咪唑鎓与(a)(顶行)中所示的每种单体一起制备的超分子水凝胶网络的照片,以及其粘度的相应的倒置小瓶展示。注:丙烯酰胺(aam)、n-异丙基丙烯酰胺(nipam),甲基丙烯酸2-(二甲基氨基)乙酯含有(dmaema)、丙烯酸(aa)、溴化1-乙烯基-3-乙基咪唑鎓(viet)、3-[2-(甲基丙烯酰氧基)乙基](二甲基)铵-1-丙磺酸盐(mps)、聚(乙二醇)甲基丙烯酸酯(pegma)、二甲基丙烯酰胺(dma)和n-羟乙基丙烯酰胺(heam)。
图22显示了基于heam的cb[8]水凝胶经由动态室温(a)振幅扫描(10-1%至103%应变,10rads-1);和(b)频率扫描(10-2至102rads-1,1%应变)的g'和g″(模量,pa)的变化。
图23显示了基于heam的cb[8]水凝胶在(a)采用0至80℃之间的加热和冷却处理的热稳定性测试中;和(b)在20℃下网络样品的连续步进应变测量(高振幅振荡参数:应变γ=500%,角频率ω=10rads-1,低振幅振荡参数:γ=0.5%,ω=10rads-1)的g'和g″(模量,pa)的变化。
图24显示了基于aam的cb[8]水凝胶经由动态室温(a)振幅扫描(10-1%至103%应变,10rads-1);和(b)频率扫描(10-2至102rads-1,1%应变)的g'和g″(模量,pa)的变化。
图25显示了基于n-异丙基丙烯酰胺(nipam)的cb[8]水凝胶经由动态室温(a)振幅扫描(10-1%至103%应变,10rads-1);和(b)频率扫描(10-2至102rads-1,1%应变)的g'和g″(模量,pa)的变化。
图26显示了基于甲基丙烯酸2-(二甲基氨基)乙基酯的cb[8]水凝胶经由动态室温(a)振幅扫描(10-1%至103%应变,10rads-1);和(b)频率扫描(10-2至102rads-1,1%应变)的g'和g″(模量,pa)的变化。
图27显示了基于丙烯酸(aa)的cb[8]水凝胶经由动态室温(a)振幅扫描(10-1%至103%应变,10rads-1);和(b)频率扫描(10-2至102rads-1,1%应变)的g'和g″(模量,pa)的变化。
图28显示了基于溴化1-乙烯基-3-乙基咪唑鎓(viet)的cb[8]水凝胶经由动态室温(a)振幅扫描(10-1%至103%应变,10rads-1);和(b)频率扫描(10-2至102rads-1,1%应变)的g'和g″(模量,pa)的变化。
图29显示了基于3-[2-(甲基丙烯酰氧基)乙基](二甲基)铵-1-丙磺酸盐(mps)的cb[8]水凝胶经由动态室温(a)振幅扫描(10-1%至103%应变,10rads-1);和(b)频率扫描(10-2至102rads-1,1%应变)的g'和g″(模量,pa)的变化。
图30显示了基于聚(乙二醇)甲基丙烯酸酯(pegma)的cb[8]水凝胶经由动态室温(a)振幅扫描(10-1%至103%应变,10rads-1);和(b)频率扫描(10-2至102rads-1,1%应变)的g'和g″(模量,pa)的变化。
图31显示了基于二甲基丙烯酰胺(dma)的cb[8]水凝胶经由动态室温(a)振幅扫描(10-1%至103%应变,10rads-1);和(b)频率扫描(10-2至102rads-1,1%应变)的g'和g″(模量,pa)的变化。
具体实施方式
在本发明人的早期工作之一中,基于葫芦脲的超分子水凝胶通过具有适合的用于葫芦脲主体的客体官能团的聚合物的超分子配位形成。因此,在制备聚合物后,仅将聚合物暴露于葫芦脲主体。这些方法的实例陈述于appeletal.(2010和2012)和wo2013/124654中。
在本发明的工作中,提供了一种可聚合组合物,其中葫芦脲主体与用于制备聚合物的单体一起存在。单体具有与葫芦脲主体形成配合物的客体官能团。因此,在单体聚合之前和期间可以允许主体-客体配位,从而形成超分子水凝胶。因此,该方法不同于appeletal.(2010和2012)和wo2013/124654先前描述的方法。
发现与通过聚合物配位而制备的那些水凝胶相比,通过本发明方法制备的水凝胶具有改变的流变性质。
本发明人认为,先前制备水凝胶的方法受阻于葫芦脲主体分子在用于制备水凝胶的聚合物混合物中的有限溶解度。该溶解度限制阻止了具有非常高的交联度的水凝胶形成。还认为葫芦脲的有限溶解度主要是由于现有技术的水凝胶结构内形成的不均匀性所致。
本发明人认为,本发明的超分子水凝胶的韧性归因于两种机制的协同作用:通过聚合物链缠结的裂缝桥接(即,阻止裂缝生长的裂缝尖端),和通过超分子葫芦脲介导的主体-客体配位的解离的能量耗散。水凝胶内超分子交联的解离是水凝胶结构的内部损伤,并且随后通过主体-客体配合物的重新形成而愈合这种损伤。
因此,本发明的水凝胶是极其韧性的并具有极其可拉伸的性质,可以用作模型系统以探索形变和能量耗散的机制,并且水凝胶可以用于需要韧性和可拉伸材料的应用,如生物材料和可穿戴电子设备中。本发明的水凝胶还提供了具有相对较弱的机械性能的迄今已知的葫芦脲水凝胶的替代物。
本发明的水凝胶具有改进的拉伸韧性,且易于调变可聚合组合物中的组分的能力提供了改变(如增强)材料性质如改进高吸水性而不牺牲强度和韧性的机会。水凝胶内存在的动态物理结合促进了可加工性和自我修复。
本案的水凝胶可以通过原位聚合可聚合组合物而制备。水凝胶形成其中可聚合组合物的容器的形状。具有期望形状的水凝胶可以通过简单选择聚合容器而制备。
发明人已经发现,通过在水凝胶结构中引入补充性价键,可以进一步改性水凝胶的性质。因此,交联剂(交联单体,本文中称为第四单体)可以另外存在于可聚合组合物中。这种水凝胶被称为水凝胶双网络,因为它们包含了聚合物分子之间的非共价和共价连接两者
在wo2013/124654中关于水凝胶的早期工作中,发明人之一考虑在水凝胶结构内形成共价键。然而,其工作实施例描述了以超分子配合物中的非共价键合为代价形成共价键。本发明人最初制备了具有由cb[8]与两个蒽客体的三元配合物形成的分子间和分子内交联的超分子水凝胶,每个蒽客体都与聚合物分子连接。辐照水凝胶导致配合物内的蒽基团的分子间环化,从而形成通过配合物的共价连接。
相反,本案的水凝胶保持动态非共价连接,其与抗断裂性、能量耗散和抗疲劳性相关,并还保持永久的共价连接,其维持了网络的形状并赋予材料弹性。
song等人最近描述了由n-异丙基丙烯酰胺和在与两个各自与丙烯酸酯部分共价连接的phe-gly-gly基团的三元配合物中的cb[8]的水性混合物制备的微凝胶产品的制备。混合物的聚合提供了分散于水相中的微凝胶。song等人的工作与本案中的工作有区别。通常,本案的水凝胶由其中总单体浓度cmon较高,如至少0.5m的可聚合组合物制备。相反,song等人描述了总单体浓度为21mm的水性组合物的使用。通过本发明的方法制备的水凝胶具有高机械强度和高度链缠结,而这在由具有低总单体浓度如低于0.5m的可聚合组合物制备水凝胶的情况下是不可获得的。
song等人描述的微凝胶较小,并具有直径400至900nm(取决于温度)。相反,本发明的水凝胶可以在宏观尺度上,例如,以μm和mm尺度制备。
song等人没有公开微凝胶的流变性质。因此,没有报告储存或损失模量值,没有报告应力研究,也没有报告tan(δ)值。也没有表明微凝胶可以或将会用于生物材料应用或用作粘合剂。song等人没有证明或暗示微凝胶具有自我修复特性。
水凝胶双网络也可以在由可聚合组合物,以在组合物中包含合适的交联剂的单步骤中形成。在早期报道的工作中,如wo2013/124654,有必要首先制备具有合适客体官能团的聚合物,然后使用这些聚合物形成超分子水凝胶,并随后最终在该水凝胶内产生共价交联。
与常规的共价交联的材料相比,水凝胶双网络具有增加的拉伸强度、韧性和弹性。水凝胶的行为类似于结构蛋白的那些,其利用类似的作用机制,主要通过模块域的力诱导的解折叠和可逆重折叠实现期望的机械性质和功能。双网络水凝胶是高度的可伸缩的和韧性的,并且显示出高效的能量耗散,并可以在松弛时通过主体-客体三元配合物的重新形成而完全自我修复。动态cb[n]主体-客体相互作用和一种材料中共价交联的这种组合允许构建仿生超分子材料,用于很多应用,包括人造肌肉、软骨置换和组织工程、传感器、药物递送系统、可穿戴电子装置、用于生物分离的微致动器和基质。
水凝胶双网络有利地相当于结构蛋白肌联蛋白,本发明人将其视为适于吸收能量的代表性生物材料。骨骼肌蛋白质肌联蛋白(3mda,1mm长)主导肌肉的机械强度和韧性。通过单分子力谱研究,先前已经揭示了肌联蛋白通过力诱导的二级分子内相互作用的断裂而吸收能量的能力。在展开肌联蛋白之后重新折叠诱导形状恢复,使其成为人工适应性材料设计的感兴趣的模型。
本文描述的水凝胶双网络由永久和暂时的非共价相互作用构成,非常类似于肌联蛋白的拓扑结构。在这方面,水凝胶提供了对迄今描述的仿生系统的改进。例如,guan等人报道了一种引入超分子四联氢键合部分,2-脲基-4[1h]-嘧啶酮(upy)作为模块单元的仿生系统,模拟免疫球蛋白(ig)样结构域。然而,除非使用分段两亲性upy-栓系聚合物,否则基于upy的材料难以引入水性体系中。水溶液中如此形成的纤维可以交联并产生瞬时超分子网络。
song等人没有描述具有双网络的水凝胶。由该研究组描述的微凝胶中的聚合物链仅通过超分子非共价配合物连接。
wo2015/103125描述了葫芦脲使用形成聚合物膜。本文中,葫芦脲与用于与也含有烯基基团的有机单体共聚的可聚合官能团如烯基基团共价连接。含有葫芦脲的单体仅用于形成共价连接,并且没有公开由可聚合有机单体提供的葫芦脲和客体分子之间形成的非共价配合物。此外,wo2015/103125中描述的可聚合组合物含有有机溶剂,并且没有暗示在水中可以制备聚合物以提供水凝胶产物。
类似地,cn104086691描述了提供有可聚合官能团的葫芦脲的用途。这种可聚合单体由单官能cb-oh与4-苄基氯的反应获得。随后使含cb的单体与共聚单体反应而提供聚合物产物。葫芦脲用于容纳含咪唑鎓化合物,然而,该化合物不是可聚合单体,并且其不包含在由改性的葫芦脲与4-乙烯基苄基氯的聚合形成的聚合物的主链内。在本案中,聚合物分子通过基于葫芦脲的主体-客体配合物非共价连接。这些并不存在于cn104086691的聚合物产品中。
cn105061783广泛地描述了可聚合组合物,其包含主体,如环糊精或葫芦脲,以及可聚合单体1-乙烯基咪唑和丙烯酸羟丙酯。使组合物聚合而得到聚合物产物。cn105061783的作者称其产品为自我修复凝胶。由可聚合组合物和凝胶产物中的环糊精或葫芦脲化合物形成的连接并没有清楚描述,并且没有提供分析数据。
本案的聚合物通过基于葫芦脲的主体-客体配合物非共价连接。此处,聚合物上的客体分子容纳于超分子配合物中的葫芦脲主体的空腔中,从而在聚合物链之间形成非共价连接。聚合物的带有客体的部分衍生自在前体可聚合组合物中提供有合适客体的可聚合单体。主体-客体配合物未在cn105061783中明确描述。此外,认为cn105061783中描述的单体不能形成主体-客体配合物,因为它们不具有合适的官能团。认为聚合物产物在葫芦脲和由单体提供的咪唑基团之间含有非共价结合。然而,在存在这种结合的情况下,这只能是葫芦脲门户(portal)和咪唑基团之间的结合。这在本案的意义上并不是配合物,其中葫芦脲在其腔中容纳一个或多个客体。
不希望受理论束缚,本发明人认为由1-乙烯基咪唑单体提供的咪唑基基团不能延伸到葫芦脲的空腔中,因为其直接连接到形成聚合物产物主链的乙烯基可聚合基团上。在本案的情况下,发明人举例说明了含咪唑鎓的可聚合单体1-苄基-3-乙烯基咪唑鎓与cb[8]一起的使用。此处,苄基基团位于葫芦脲主体的空腔中,而咪唑鎓基团位于门户处(参见图5)。
在本案的情况下,由超分子聚合物产物的nmr谱分析看出主体-客体配合物的存在。如本领域技术人员已知的,主体-客体配合物的存在可以通过其他方法,如uv-vis光谱证实。
cn105061783中的可聚合组合物显然是在无溶剂的情况下制备和使用。相反,在本发明的优选实施方式中,可聚合组合物是用于制备水凝胶产品的水性组合物。
xu等人最近在本案优先权日之后发表的出版物描述了由cb[8]和可聚合单体制备的超分子水凝胶。这项工作源自与以上描述的song等人相同的团队。此处,水性可聚合组合物由与fggea和丙烯酰胺(aam)配位的cb[8]制备。该组合物的聚合产生超分子水凝胶。作者证实,提高组合物中丙烯酰胺的量,因此提高组合物中的总单体浓度,会改进储能和损失模量,以及配合物的剪切模量。如预期的,水凝胶可以通过引入竞争性粘合剂,此处是美金刚胺(memantine)来破坏,以破坏cb[8]超分子配合物。
xu等人的工作因此支持本案中描述和要求授权保护的早期工作。
可聚合组合物
本发明提供了一种可聚合组合物,包含葫芦脲和第一可聚合单体,其中第一可聚合单体具有适合与葫芦脲一起参与主体-客体配合物的客体官能团。可聚合组合物可以是水性组合物,并且水性组合物可以用于制备本发明的水凝胶。在其他方面中,可聚合组合物可以用于制备聚合物。本文中,组合物不必是水性组合物。通常,可聚合组合物中的总单体浓度为至少0.5m。
第一单体为超分子网络中的主体-客体配合物的形成提供客体。因此,存在于可聚合组合物中的客体量以及因此存在的第一单体的量,连同葫芦脲主体的量,将决定网络中的交联水平。通常,第一单体与其他不参与主体-客体配合物的形成的可聚合单体一起提供。
如下面进一步详细讨论,在可聚合组合物中可以提供第三单体,并且这种单体还可以具有适合与葫芦脲一起参与主体-客体配合物的客体官能团。第三单体与第一单体是不同的。
葫芦脲可以与第一可聚合单体和/或第三单体(存在的情况下)中的一个或两个客体形成配合物。因此,可聚合组合物可以含有三元主体-客体配合物或二元主体-客体配合物,其中客体由可聚合组合物中的单体提供。
至少一些葫芦脲与可聚合组合物中提供的一种或多种单体客体配位。例如,至少5%,如至少10%,如至少20%,如至少25%,如至少50%,如至少75%,如至少95%的葫芦脲与可聚合组合物中提供的一种或多种客体配位。基本上所有的可聚合组合物中的葫芦脲可以处于客体-主体配合物中。
可聚合组合物可以包含一种或多种另外的可聚合单体,如以下进一步的详细讨论的。这些另外的可聚合单体的每一种都不同于第一可聚合单体。
第二单体可以存在于可聚合组合物中,用于与第一单体聚合。第二单体不同于第一单体。第二单体不具有适合与葫芦脲一起参与主体-客体配合物的客体官能团。通常,第二单体作为产物聚合物的结构元素提供,该聚合物形成水凝胶产物中的延伸的网络的基础。
通常,第二单体存在于可聚合组合物中。第二单体也可以过量于第一单体存在。即使当第一单体的量与第二单体的量相比相对较低时,本发明的水凝胶也具有优异的流变性质。因此,交联的量非常高不是必要的。
相对于第一单体的摩尔量,第二单体可以以等摩尔量或更高的量存在。
第二单体可以以第一单体的摩尔量的2倍或更多倍,5倍或更多倍,10倍或更多倍,或15倍或更多倍的摩尔量存在。
第一单体也应该以在水凝胶内提供足够的非共价交联的水平存在,以提供韧性和可拉伸性的期望的物理特性。因此,第二单体可以以第一单体的摩尔量的至多25倍,至多30倍,至多50倍,或至多100倍的摩尔量存在。
在工作实施例中,第二单体以第一单体的95摩尔当量至5摩尔当量使用,即第二单体以第一单体摩尔量的19倍的摩尔量存在。
在可聚合组合物中可以提供第三单体,其中第三单体不同于第一单体,并也不同于第二单体(在存在的情况下)。第三单体具有适合与葫芦脲一起参与主体-客体配合物的客体官能团。客体官能团可以与第一单体的客体官能团相同,或可以不同。在前一种情况下,当客体官能团相同时,第三单体的剩余部分不同于第一单体。此处,第一和第三单体的客体可以一起形成三元配合物,并还可以形成单独的配合物。因此,由含有这些单体的组合物形成的水凝胶可以具有配合物的混合物。
当第三单体具有与第一单体不同的客体官能团时,第一和第三单体可以一起使用以形成三元配合物,例如,其中配合物中的客体是不同的(称为客体与葫芦脲主体的1:1:1配合物)。合适的混合客体配对描述于以下的葫芦脲客体部分中。另外或可替代地,第一和第三单体的单体客体可以形成单独的配合物,其中每种配合物中的客体是相同的(客体与主体的2:1配合物),并且由含有这种单体的组合物形成的水凝胶因此可以具有配合物的混合物。
当存在第三单体时,其可以以大于或小于,或等于第一单体的摩尔量的摩尔量存在。
通常,仅当可聚合组合物中存在第二单体时才存在第三单体。此处,相对于第三单体的摩尔量,第二单体可以以等摩尔量或更多的量存在。
第二单体可以以第三单体摩尔量的2倍或更多倍,5倍或更多倍,10倍或更多倍,15倍或更多的摩尔量存在。
第三单体也应该以在水凝胶内提供足够的交联的水平存在,以提供期望的韧性和可拉伸性的物理特性。因此,第二单体可以以第三单体摩尔量的至多20倍,至多25倍,至多30倍,至多50倍,或至多100倍或更多的摩尔量存在。
可替换地,相对于第一单体和第三单体的总摩尔量(在这两者都存在于可聚合组合物中的情况下),第二单体可以以等摩尔量或更多的量存在。
第二单体可以以第一单体和第三单体的总摩尔量的2倍或更多倍,5倍或更多倍,10倍或更多倍,15倍或更多的摩尔量存在。
第一和第三单体也应该以在水凝胶内提供足够的交联的水平存在,以提供期望的韧性和可拉伸性的物理特性。因此,第二单体可以以第一单体和第三单体的总摩尔量的至多20倍,至多25倍,至多30倍,至多50倍或至多100倍的摩尔量存在。
在可聚合组合物中可以提供第四单体,其中第四单体是交联单体(也称为交联剂)。例如,单体是多官能单体,如双官能单体。除了由葫芦脲配合物提供的非共价交联之外(由第一单体和如果存在的第三单体提供),第四单体可以用于聚合物之间提供额外的共价交联。
在存在交联剂的情况下,交联剂通常以相对于第一单体和如果存在的第二单体的摩尔量,如相对于第一单体的量相对低的摩尔量提供。本发明人已经发现,在其中共价交联水平相对低的体系中可以获得具有优异的抗断裂性和抗疲劳性的水凝胶。因此,水凝胶中的非共价交联可以超过共价交联,因此第一单体的量通常超过第四单体。
相对于第四交联单体的摩尔量,第一单体可以以等摩尔量或更多的量存在。
第一单体可以以第四单体的摩尔量的至少2倍,至少5倍或至少10倍的摩尔量存在。
第四单体应该以在水凝胶内提供将会提供抗断裂性和抗疲劳性的期望物理特性的水平的共价交联的摩尔量存在。因此,第一单体可以以第四单体的摩尔量至多20倍,至多25倍,至多50倍,至多100倍或至多150倍的摩尔量存在。
在工作实施例中,交联单体以第一单体的0.05摩尔当量至5摩尔当量使用,因此第一单体以交联单体的摩尔量100倍的摩尔量存在。
在本案中,发明人已经制备了仅具有非共价交联的水凝胶,以及具有非共价和共价交联两者的水凝胶(后者称为双网络)。向水凝胶中添加共价交联为水凝胶提供了结构完整性,并还赋予水凝胶一定程度的弹性。因此,在要求水凝胶产品具有非共价和共价交联两者时提供第四单体。
额外的共价键合为水凝胶提供了有用的额外特性,并补充了那些具有非共价键合的水凝胶可获得的益处。
认为非共价超分子主体-客体相互作用同时发挥多种机械功能。例如,非共价相互作用通过牺牲键断裂增强材料的抗断裂性,从而使网络增韧。超分子配合物的存在都通过产生高内部摩擦而增强能量耗散。在动态系统中,断裂的键可以重新形成,从而在损坏后使材料愈合。以这种方式增强了抗疲劳性。
可选地,葫芦脲是第五可聚合单体内的基团。此处,葫芦脲与第五单体的可聚合官能团直接或间接共价键合。在优选的实施方式中,葫芦脲不是可聚合单体内的基团。因此,优选地可聚合组合物中不存在第五可聚合单体。
其他可聚合单体可以提供于组合物中。例如,还可以在可聚合组合物中提供包含可检测标记的单体。以这种方式,水凝胶可以引入标记,例如,用于检测和分析水凝胶产品。
标记的类型没有特别限制,并可以包括,例如,荧光或发光标记。在本案的工作实施例中,荧光单体用于可聚合组合物中以提供荧光水凝胶。本发明人已经使用荧光标记跟踪在接触的水凝胶之间的动态连接的形成,导致接触水凝胶片的一体化。
可聚合组合物可以包含旨在控制聚合反应和/或改性产品水凝胶性质的其他组分。
可聚合组合物还可以包括催化剂或聚合引发剂以控制或引发聚合反应。
可聚合组合物可以通过在例如水中简单混合组分而制备。可以控制添加顺序以控制组合物的后期聚合。
在可聚合组合物中,葫芦脲通常处于与第一单体的客体的非共价配合物中。这可以是二元配合物或三元配合物。此处,当聚合反应进行时,第一单体在与葫芦脲配位的同时反应,并且第一单体化合物也可以通过配合物与另一种第一单体化合物非共价连接。配合物可以继续在从第一单体化合物延伸的生长的聚合物链之间提供连接,并且最终配合物存在于最终水凝胶产品中,在形成的聚合物之间提供非共价连接。
水性可聚合组合物通常具有至少0.2m,至少0.3m,至少0.5m,至少1.0m,至少1.5m,至少2.0m的总单体浓度cmon。本发明人发现,当总单体浓度相对较高时,例如,当浓度至少为1.0m,最优选至少2.0m时,水凝胶的机械性能是最有用的。在这些较高浓度下,获得的水凝胶中的链缠结较高,而这为水凝胶的机械强度提供了有益的贡献。
水性可聚合组合物通常具有至多5.0m,至多10.0m或至多20m的总单体浓度cmon。
总单体浓度可以是在由具有选自以上提供的值的下限和上限量的范围内的量。例如,总单体浓度可以处于0.5至5.0m的范围内。
总单体浓度是指可聚合组合物中存在的可聚合单体的总摩尔量,如第一至第五单体的量之和。
葫芦脲和第一单体,在与其它可聚合单体一起存在的情况下,不需要大量存在。当动态交联水平(由葫芦脲配合物提供的非共价键的量)相对较低时,本发明的水凝胶保持其拉伸和愈合性质。因此,如果存在的话,第三单体也不需要大量存在。
水性可聚合组合物通常具有至少0.005m,至少0.01m,至少0.02m,至少0.05m,至少0.1m的葫芦脲浓度。
水性可聚合组合物通常具有至多0.5m,至多1.0m或至多2.0m的葫芦脲浓度。
葫芦脲的浓度可以存在于具有选自以上给出的值中的上限和下限的范围内。例如,葫芦脲可以以0.01至0.5m的浓度存在。
在葫芦脲旨在形成三元超分子配合物的情况下,其通常以第一单体和如果存在的第三单体的摩尔量的约一半存在。本文中,葫芦脲可以相对于总单体量以至少0.25摩尔%,至少0.5摩尔%,至少1.0摩尔%,至少2.5摩尔%的量存在。葫芦脲可以以相对于总单体量至多5.0摩尔%,至多7.5摩尔%,至多10摩尔%,至多12.5摩尔%,或至多25摩尔%的量存在。
葫芦脲可以以具有选自以上提供的值的上限和下限量的范围内的量存在。例如,葫芦脲可以以0.5摩尔%至5摩尔%的量存在。
在葫芦脲旨在形成二元超分子配合物的情况下,其通常以与第一单体和如果存在的第三单体大约相同的摩尔量存在。本文中,葫芦脲可以相对于总单体量以至少0.5摩尔%,至少1摩尔%,至少2摩尔%,至少5摩尔%的量存在。葫芦脲可以以相对于总单体量至多10摩尔%,至多15摩尔%,至多20摩尔%,至多25摩尔%,或至多50摩尔%的量存在。
葫芦脲可以以具有选自以上提供的值的上限和下限量的范围内的量存在。例如,葫芦脲可以以1至10摩尔%范围内的量存在。
可聚合组合物可以进一步包含催化剂或聚合引发剂。
相对于总单体量cmon,催化剂或聚合引发剂可以以至少0.01摩尔%,至少0.05摩尔%,至少0.1摩尔%,至少0.5摩尔%的量存在。
相对于总单体量cmon,催化剂或聚合引发剂可以以至多1摩尔%,至多2摩尔%或至多5摩尔%的量存在。
催化剂或聚合引发剂可以以具有选自以上提供的值的上限和下限量的范围内的量存在。例如,催化剂或聚合引发剂可以以0.01至1摩尔%的量存在。
单体
可聚合组合物含有一种或多种单体,这些单体适于在聚合反应中反应以生成聚合物产物。
在优选的实施方式中,组合物包含两种或更多种单体。具有客体官能团的第一单体旨在在水凝胶产品中提供非共价交联。第一单体通常与旨在为水凝胶材料提供结构主链的第二单体一起存在。可选地,可以存在第四单体,这种单体是交联剂,其旨在在产物中提供共价交联。
单体没有特殊限制,只要该单体含有适合参与聚合反应的官能团。单体可以适用于在逐步增长或链增长条件下聚合。单体可以适用于在自由基、阳离子或阴离子(加成)聚合条件下聚合。单体通常适用于水性条件下的聚合反应。
单体可以适合参与自由基聚合反应。在本案的工作实施例中,自由基聚合用于制备水凝胶产品。
在一个实施方式中,适用于可聚合组合物的单体包括碳-碳双键或三键,最优选碳-碳双键。碳-碳双键或三键可以存在于单体的末端,因此单体如第一或第二单体可以具有乙烯基(乙烯)基团,如烯丙基基团,或乙炔基(乙炔)基团,如炔丙基基团。碳-碳双键或三键可以用于在聚合反应中进行反应。
其他官能团可以用于代替碳-碳双键或三键,如用于形成聚酰胺的酸和胺官能团等。
通常,适用于本案的替代官能团是其中可以在水中实现聚合而提供水凝胶产品的那些。实例包括单体与硫醇和烯烃如乙烯基官能团之间的迈克尔加成反应;与酸官能团或酸的活化形式如酸酐和醇或胺官能团的酯化和酰胺化反应单体;使用具有环氧基团和醇、胺或羧基官能团的单体的开环反应;具有醛和羟基或羧基官能团的单体之间的席夫碱反应;和点击反应,例如,在具有叠氮化物和炔烃官能团的单体之间的点击反应。
在可聚合组合物中存在两种或更多种不同单体的情况下,单体具有在聚合反应中彼此反应的官能团。不同的单体可以具有相同的官能团(例如,乙烯基官能团)或可以具有不同的官能团(例如,一种单体上的氨基官能团,另一种单体上的羧基官能团)。
第一单体通常是乙烯基单体,其中客体是乙烯基基团的取代基。合适的客体将在下面进一步详细描述。在本案的工作实施例中,第一单体是1-苄基-3-乙烯基咪唑鎓。
适用于可聚合组合物的单体,如用作第二单体的那些,可以是丙烯酸类或丙烯酰胺类单体。本文中的实例包括丙烯酸酯和丙烯酰胺单体,烷基丙烯酸酯单体如甲基丙烯酸酯单体和烷基丙烯酰胺如甲基丙烯酰胺单体,和(烷基)丙烯酸酯单体如(甲基)丙烯酸酯,和(烷基)丙烯酰胺如(甲基)丙烯酰胺单体。
在一个实施方式中,丙烯酸类单体是较不优选的,例如,优选的是第一单体和如果存在的第三单体不是丙烯酸类单体,如第一和如果存在的第三单体不包括丙烯酸酯基团(-oc(o)chch2)。然而,丙烯酸类单体仍可以用于本发明,并可用作第二单体,用于制备具有期望的流变性质的水凝胶。本案中的工作实施例显示基于非环酸和其他丙烯酸酯的单体作为第二单体的用途。
可聚合组合物中存在的单体可以至少部分溶于水。单体可以是亲水性单体。
单体可以具有至少1g/l,如至少5g/l,如至少10g/l,如至少15g/l的在水中的溶解度。
水溶解度信息可以从单体的商业供应商提供,或可以通过实验方法,如本领域技术人员已知的那些测定。溶解度可以在25℃下测定。本案的工作实施例使用了甲基丙烯酸酯单体,且单体甲基丙烯酸甲酯在25℃下具有约15g/l的溶解度。
优选地,第一单体是乙烯基单体。如本文所示,客体分子可以与乙烯基基团连接,并且该单体可以参与聚合反应而形成超分子水凝胶。
第二单体可以是乙烯基单体,且特别是取代的乙烯基单体,如被酰胺基团取代的那些,如丙烯酰胺单体,或酯基团取代的那些,如丙烯酸酯单体,或杂芳基基团取代的那些,如咪唑鎓单体。
优选地,第二单体是丙烯酰胺类单体,如丙烯酰胺。
另外或可替代地,第二单体是基于(烷基)丙烯酰胺的单体,基于(烷基)丙烯酸酯的单体,如基于(烷基)丙烯酸酯的单体,或芳基乙烯基单体,如1-乙烯基-3-乙基咪唑鎓单体。
(烷基)丙烯酰胺单体的实例包括(烷基)丙烯酰胺、烷基(烷基)丙烯酰胺如二甲基丙烯酰胺(dma),二乙基丙烯酰胺、n-异丙基酰胺(nipam)和n-叔丁基丙烯酰胺;和羟烷基丙烯酰胺,如n-羟乙基丙烯酰胺(heam)、n-[三(羟甲基)甲基]丙烯酰胺和2-羟丙基甲基丙烯酰胺;磺酰基烷基(烷基)丙烯酰胺如2-丙烯酰胺基-2-甲基-1-丙磺酸;氨基烷基(烷基)丙烯酰胺如n-(3-氨基丙基)甲基丙烯酰胺;
(烷基)丙烯酰胺单体可以是丙烯酰胺单体。
(烷基)丙烯酸酯单体的实例包括丙烯酸、(烷基)丙烯酸烷基酯如甲基丙烯酸2-乙基己酯和甲基丙烯酸乙酯;(烷基)丙烯酸氨基烷基酯如甲基丙烯酸2-(二甲基氨基)乙酯(dmaema)和甲基丙烯酸2-(二异丙基氨基)乙酯;(烷基)丙烯酸羟烷基酯如甲基丙烯酸2-羟乙酯;(烷基)丙烯酸(羟基)杂烷基酯如甲基丙烯酸聚(乙二醇)酯(pegma)、乙二醇甲基醚甲基丙烯酸酯和甲基丙烯酸2-乙氧基乙基酯;磺酰基(杂)烷基(烷基)丙烯酸酯如3-[2(甲基丙烯酰氧基)乙基](二甲基)铵-1-丙磺酸盐(mps)和3-磺丙基甲基丙烯酸酯。
(烷基)丙烯酸酯单体可以是烷基丙烯酸酯单体,如甲基丙烯酸酯单体。
芳基乙烯基单体的实例包括杂芳基乙烯基单体,如咪唑单体,如1-乙烯基咪唑和1-乙烯基-3-乙基咪唑鎓(viet)。
本案中的工作实施例证实了以下作为可聚合组合物中的第二单体的用途:丙烯酰胺(aam),n-异丙基丙烯酰胺(nipam),甲基丙烯酸2-(二甲基氨基)乙酯(dmaema),丙烯酸(aa),溴化1-乙烯基-3-乙基咪唑鎓(viet),3-[2(甲基丙烯酰氧基)乙基](二甲基)铵-1-丙磺酸盐(mps),甲基丙烯酸聚(乙二醇)酯(pegma),二甲基丙烯酰胺(dma)和n-羟乙基丙烯酰胺(heam)。
可聚合组合物可以含有多种具有葫芦脲的客体官能团的单体,如可聚合组合物可以含有第一单体以及第三单体。本文中,每种单体的客体官能团可以与葫芦脲形成非共价配合物。
在其他实施方式中,第一单体可以与第二单体一起提供于可聚合组合物中,其中第二单体具有能够在不存在第一单体的情况下与葫芦脲形成配合物,但当第一单体不存在时不如此的官能团。在这种组合物中,第一单体客体对葫芦脲的结合亲和力比第二单体官能团对葫芦脲的结合亲和力更大,如大得多。因此,配合物形成可以主要在葫芦脲和第一单体客体之间。
本案中的工作实施例包括含有1-苄基-3-乙烯基咪唑鎓作为第一单体且1-乙烯基-3-乙基咪唑鎓(viet)作为第二单体的可聚合组合物。苄基-咪唑鎓部分表现出比第二单体的乙基咪唑鎓基团强得多的与葫芦脲的结合表现出,因此配合物形成主要(如果并非完全)是葫芦脲和第一单体之间。
在一个实施方式中,可聚合组合物中的单体是盐。因此,单体具有离子基团和该离子基团的反离子。离子基团可以不作为单体的可聚合官能团的一部分存在。因此,离子基团并非旨在参与聚合反应。单体中存在的离子基团可以在聚合后与合适的反离子一起存在于水凝胶产物中。
优选地,第一单体是盐,其最优选地,离子基团提供于第一单体的客体上。当存在第三单体时,其可以是盐,并且其也可以具有提供于第三单体的客体上的离子基团。
在本案的工作实施例中,用于在水凝胶产品中提供共价交联的第四单体是n,n'-亚甲基双丙烯酰胺。交联剂的使用在本领域是常规的,并且可以使用其他双官能(或多官能)单体代替n,n'-亚甲基双丙烯酰胺。
第一单体的客体被连接,如共价直接连接或共价间接连接至参与聚合反应的可聚合基团,如乙烯基基团。合适的客体将在下面进一步详细描述。
优选的是可聚合组合物含有第一单体和第二单体。如上文解释的,第一单体与葫芦脲一起在水凝胶产物中提供非共价连接。本发明人发现,在非共价交联的水平相对较低的情况下可以制备强水凝胶。因此,可聚合组合物中存在的第一单体的量也可以相对较低。因此,为水凝胶产品提供结构主链的第二单体的量相对较大。
所指的总单体量是指可聚合组合物中所有可聚合单体的总摩尔量,如第一单体和第二单体可选地与如果存在的第三和第四单体一起以及任何其他如果存在的可聚合单体的总量。
相对于总单体量,第一单体可以以至少0.5摩尔%,至少1摩尔%,至少2摩尔%,至少5摩尔%的量存在。
相对于总单体量,第一单体可以以至多10摩尔%,至多15摩尔%,至多20摩尔%,至多25摩尔%,或至多50摩尔%的量存在。
第一单体可以以具有选自以上给出的值的上限和下限量的范围内的量存在。例如,第一单体可以以1摩尔%至10摩尔%的量存在。
优选地,第二单体存在于可聚合组合物中。通常,第二单体相对于第一单体大幅过量存在。
相对于总单体量,第二单体可以以至少50摩尔%,至少60摩尔%,至少70摩尔%,至少80摩尔%,至少90摩尔%,至少95摩尔%的量存在。
相对于总单体量,第二单体可以以至多96摩尔%,至多98摩尔%,或至多99摩尔%的量存在。
第二单体可以以具有选自以上给出的值的上限和下限量的范围内的量存在。例如,第二单体可以以90摩尔%至99摩尔%的量存在。
如果存在,第三单体通常将会以与第一单体等摩尔量存在。
第三单体的客体被连接,如共价直接连接或共价间接连接至参与聚合反应的可聚合基团,如乙烯基基团。合适的客体将在下面进一步详细描述。
第一和第三单体的总摩尔量可以选自上面对第一单体提供的值。
因此,相对于总单体量,第一和第三单体的总摩尔量可以为至少0.5摩尔%,至少1摩尔%,至少2摩尔%,至少5摩尔%。
因此,相对于总单体量,第一和第三单体的总摩尔量可以为至多10摩尔%,至多15摩尔%,至多20摩尔%,至多25摩尔%,或至多50摩尔%。
第一和第三单体可以以具有选自以上给出的值的上限和下限量的范围内的量存在。例如,第一和第三单体可以以1至10摩尔%范围内的总量存在。
可以考虑具有用于形成配合物的客体的组合物中存在的单体的总摩尔量来选择组合物中存在的葫芦脲的量。当水凝胶将含有三元配合物的情况下,组合物中存在的葫芦脲的摩尔量将是具有用于形成配合物的客体的组合物中存在的单体的总摩尔量(其可以是存在的第一单体的量,或可以是存在的第一和第三单体的总摩尔量)的约50%。
在水凝胶含有二元配合物的情况下,组合物中存在的葫芦脲的摩尔量将是具有用于形成配合物的客体的组合物中存在的单体的总摩尔量(其可以是存在的第一单体的量)的等摩尔量。
第一和/或第三单体可以具有一个或多个,如两个葫芦脲的客体。通常,第一和第三单体仅具有一个客体。
在存在的情况下,第四单体可以以相对低的量存在。如前解释的,第四单体在水凝胶产品中提供共价连接。本发明人发现,可以制备其中共价交联的水平相对较低的有用的水凝胶。因此,可聚合组合物中存在的第四单体的量也可以相对较低。同样,为水凝胶产品提供结构主链的第二单体的量相对较大。
第四单体可以在可聚合组合物中以相对低的量存在。
相对于总单体量,第四单体可以以至少0.01摩尔%,至少0.05摩尔%,至少0.1摩尔%,至少0.5摩尔%的量存在。
相对于总单体量,第四单体可以以至多1摩尔%,至多2摩尔%或至多5摩尔%的量存在。
第四单体可以以具有选自以上给出的值的上限和下限量的范围内的量存在。例如,第四单体可以以0.01摩尔%至1摩尔%范围内的量存在。
在一些实施方式中,第五可聚合单体存在于可聚合组合物中,其中第五单体包含葫芦脲。葫芦脲被连接,如共价直接连接,或共价间接连接至可以参与聚合反应的可聚合基团,如乙烯基基团。通常,第五单体以第一单体量的等摩尔量使用。
第一和第五单体的总摩尔量可以选自以上对于第一单体给出的值。
因此,相对于总单体量,第一和第五单体的总摩尔量可以为至少0.5摩尔%,至少1摩尔%,至少2摩尔%,至少5摩尔%。
因此,相对于总单体量,第一和第五单体的总摩尔量可以为至多10摩尔%,至多15摩尔%,至多20摩尔%,至多25摩尔%,或至多50摩尔%。
第一和第五单体可以以具有选自以上给出的值的上限和下限量的范围内的量存在。例如,第一和第五单体可以以1摩尔%至10摩尔%范围内的总量存在。
可聚合组合物中的单体可以具有至少50,至少100或至少200的分子量。
可聚合组合物中的单体可以具有至多500,至多1,000,至多1,500或至多2,000的分子量。
提及单体还可以包括提及低聚物,如可以由少量单体的反应形成的低聚物。低聚物具有至多50个重复单体单元,如至多20个重复单元,如至多10个重复单元,如至多5个重复单元,如至多2个重复单元(二聚体)。本发明人认识到,在小分子量原料与葫芦脲主体一起用于聚合反应中的情况下,可获得本发明的优点。
在一个实施方式中,提及单体仅是指单体,而不包括低聚物形式。
配合物
水凝胶是通过超分子手铐(handcuff)保持在一起的聚合物网络。形成这种超分子手铐的配合物基于容纳一个客体(二元配合物)或两个客体(三元配合物)的葫芦脲。葫芦脲与每种客体形成非共价键。本发明人先前已经证实,葫芦脲的配合物易于形成,并可以用于聚合物结构单元之间提供稳固的非共价键。在葫芦脲和第一单体的客体之间可以形成配合物,并且配合物的形成要耐受单体内的许多官能团。
认识到其腔内容纳一个或多个客体的主体,该配合物可以被称为包含配合物。如本文描述的配合物可以有别于其中葫芦脲通过葫芦脲的门户非共价结合于另一化合物的排布。本文中,其他化合物不是由葫芦脲主体配位,因为化合物不是葫芦脲腔内的客体。
在本案的方法中,配合物的形成是指葫芦脲的空腔接收一个或多个客体,从而形成主体-客体配合物。
主体-客体配合物的形成如图1中示意性地示出,其中清楚地显示了葫芦脲在腔内容纳有一个或多个客体。
在本发明的各种实施方式中,聚合物网络通过聚合物之间共价交联的存在而补充。当在可聚合组合物中包含交联剂如双官能单体时,在聚合过程期间会形成这种交联。
如上指出,葫芦脲与一个或两个客体的配合物是非共价连接,其交联和/或互连聚合物而形成材料的超分子网络。
在配合物在葫芦脲腔内包含两个客体的情况下,配合物的缔合常数ka为至少103m-2,至少104m-2,至少105m-2,至少106m-2,至少107m-2,至少108m-2,至少109m-2,至少1010m-2,至少1011m-2,或至少1012m-2。
在葫芦脲容纳两个客体分子的情况下,客体分子可以是相同的或它们可以是不同的。能够容纳两个客体分子的葫芦脲也可以与单个客体形成稳定的二元配合物。认为三元主体-客体配合物的形成是通过中间二元配合物进行的。在本发明的水凝胶中,可以存在在客体分子和葫芦脲之间形成的二元配合物。二元配合物可以被认为是尚未与另一个客体分子形成非共价结合的部分形成的三元配合物。
在一个实施方式中,水凝胶是具有多个配合物的网络,其中每个配合物包含容纳一个客体分子的葫芦脲。
在配合物在葫芦脲腔内包含一个客体的情况下,该配合物的缔合常数ka为至少103m-1,至少104m-1,至少105m-1,至少106m-1,至少107m-1,至少108m-1,至少109m-1,至少1010m-1,至少1011m-1,或至少1012m-1。
在一个实施方式中,客体是能够形成具有104至107m-1范围内的缔合常数的配合物的化合物。
在一个实施方式中,配合物的形成是可逆的。客体与葫芦脲主体分离,从而切断与聚合物的连接或交联,可以称为解配位。按照这种方式,水凝胶可以被认为是动态材料。当水凝胶暴露于应力时,认为配合物会解离,并稍后在愈合过程中重新缔合。
配合物解配位而分离一个或多个客体可以响应于外部刺激而发生,包括,例如,竞争客体化合物、光、氧化剂或还原剂、电化学电势和温度变化等。本文描述的工作还研究了水凝胶响应机械应力,如剪切应力和轴向应力的能力,以及材料在受到应力后响应和修复的能力。
用于基于cb[8]的网络的解配位的竞争性客体是金刚烷胺(ada)。竞争客体可以过量于网络的聚合物上存在的客体的量(摩尔量)使用。在一个实施方式中,竞争性客体具有比配合物的客体更高的缔合常数。
在其他实施方式中,通过配合物中客体的氧化或还原实现网络的解配位,并因此实现水凝胶的解配位。客体的氧化态的变化可以使用化学氧化剂或还原剂或施加电化学电势实现。如本文描述的,包含紫精如甲基紫精的配合物可以通过用还原剂如连二亚硫酸盐处理而解配位。
在一个实施方式中,解配位反应是可逆的。因此,水凝胶可以转化为低粘度的解配位形式,然后视情况恢复至高粘度的水凝胶形式,其可以与原始水凝胶相同或不同。
水凝胶
水凝胶是截留并储存大量水的三维交联聚合物网络。鉴于它们与软生物组织的相似性和可变的机械性能,即从柔软而脆弱到硬而坚韧,它们在各种生物医学和工业应用中越来越重要。
本发明的水凝胶是保持水的三维交联聚合物网络。在本案中,网络是由葫芦脲主体与具有在第一单体上提供的合适的客体分子官能团的配位可获得的或获得的。因此,水凝胶包含这种网络和截留的水。
本发明的水凝胶可以是由本发明的水性可聚合组合物获得的或可获得的。
水凝胶由可聚合组合物中的可聚合单体的聚合而形成。水凝胶形成还可以包括葫芦脲主体与组合物内的单体上提供的客体官能团之间的一定程度的超分子(非共价)结合。
认为当葫芦脲主体与可聚合组合物中的单体混合时,葫芦脲将与可聚合单体的客体形成配合物,从而用于非共价连接组合物中的单体。在聚合期间,可以形成另外的非共价连接,并且考虑到超分子配合物的可逆性质,在反应条件下,可以存在主体和客体一定程度的解离和重新缔合。
在一个实施方式中,水凝胶的水含量为至少90wt%,至少95wt%,至少97wt%,至少98wt%,至少99wt%,至少99.5wt%。在水含量为这样的量的情况下,本发明人已经发现,水凝胶可以用例如相当于水凝胶体积的体积的水稀释,并且网络结构保持原位。应当注意的是,水凝胶的相当大的稀释可以降低水凝胶的机械性能。
在一个实施方式中,水凝胶中存在的聚合物的总量为至多20wt%,至多10wt%,至多7.0wt%,至多5.0wt%,至多2.5wt%,至多2.0wt%,至多1.5wt%,至多1.0wt%,至多0.5wt%或至多0.4wt%。
在一个实施方式中,水凝胶中存在的聚合物的总量为至少0.05wt%,至少0.1wt%,至少0.2wt%,至少0.3wt%。
在一个实施方式中,水凝胶中存在的聚合物的总量处于选自以上实施方式的最小和最大值的范围内。例如,存在的聚合物的总量在0.3至2.0wt%的范围内。
聚合物的总量可以对应于可聚合反应中包含的可聚合单体的总量,假设所有可聚合单体在聚合反应中被消耗。
在制备水凝胶期间,水性可聚合组合物中单体的浓度和量可以选择为使得在水凝胶产品中提供期望的重量的聚合物。
在聚合中形成的聚合物的分子量没有特别限制。本案的工作实施例显示了如何测定水凝胶样品中聚合物的平均分子量。
在一个实施方式中,水凝胶中的聚合物具有至少50kda,至少100kda,至少500kda,至少1.0mda或至少2.0mda的分子量,如重均mw或数均分子量mn。
在一个实施方式中,水凝胶中的聚合物具有至多5.0mda,至多10mda,至多50mda或至多100mda的分子量,如重均mw或数均分子量mn。
水凝胶中的聚合物可以具有选自以上提供的值的下限和上限量的范围内的平均分子量。例如,水凝胶可以具有500kda至10mda范围内的平均分子量。优选地,分子量平均值是指重均分子量,如在本案的工作实施例中测定的重均分子量。
水凝胶通常具有制备其的反应容器的形状和尺寸。本发明的水凝胶可以容易地在宏观尺度上,如μm、mm或cm尺度上制备。
水凝胶可以具有至少1μm,至少5μm,至少10μm,至少20μm,至少50μm,至少100μm,至少500μm,至少1mm,至少5mm,至少1cm,至少100cm的最大尺寸。
song等人已经描述了微凝胶的制备。这些微凝胶具有约400至约900nm范围的平均最大尺寸,其取决于微凝胶样品的温度。
在一个实施方式中,水凝胶在稳定的剪切测量中具有至少10,至少100,至少500,至少1,000或至少10,000pas的粘度。
在一个实施方式中,水凝胶在稳定剪切测量中具有至多15,000,至多20,000,至多50,000pas的粘度。
在一个实施方式中,水凝胶具有100至15,000pa·s,如1,000至15,000pa·s范围内的粘度。
粘度值可以是在低剪切速率下,例如,在0.1至0.51/s,例如,0.1至0.31/s范围内的剪切速率下记录的值。粘度值可以是25℃下在稳定剪切测量中记录的值。在本案的工作实施例中描述了这种测量,并且示例性结果如图9中所示。
应该注意的是,wo2013/124654中描述的水凝胶在0.1至0.51/s范围内的剪切速率下具有更低的粘度值(参见,例如,wo2013/124654中的图16(a)和(b))。
水凝胶的储能模量值(g')可以是25℃下由应变振幅扫描测量记录的值,并是在0.01%至100%范围内,例如0.1%至10%范围内的应变值下取得的值。角频率可以是10或60rad/s,如10rad/s。
在一个实施方式中,水凝胶具有至少10pa,至少100pa,至少500pa,至少1,000pa,至少2,000pa,至少5,000pa,或至少10,000pa的储能模量g'(来自应变振幅扫描测量)。
在一个实施方式中,水凝胶具有至多20,000,至多30,000pa,至多50,000pa,至多100,000pa,或至多500,000pa的储能模量g'(来自应变振幅扫描测量)。
在一个实施方式中,水凝胶具有其中最小和最大量选自以上实施方式的范围内的储能模量(来自应变振幅扫描测量)。例如,储能模量处于100至30,000pa,例如,1,000至30,000pa的范围内。
应该注意的是,wo2013/124654中报道的储能模量值g'(来自应变振幅扫描测量)显著低于本案的水凝胶所记录的那些。参见例如wo2013/124654的图9(a),其中所报道的储能模量值在0.1%至10%的应变范围内不大于1,000pa。
储能模量的大小将决定水凝胶的预期用途。具有高储能模量,如在应变范围0.1%至10%上的应变振幅扫描测量中高于1,000pa的材料,非常适合用于生物材料应用,如软骨置换。此处,水凝胶的韧性和机械性能与原始生物材料的非常匹配。具有较低储能模量,如在应变范围0.1%至10%上的应变振幅扫描测量中1,000pa或更低的材料不适合这些应用,但它们可以用作替代软组织的一般材料。本发明的水凝胶具有有用的储能模量值,如高储能模量值,使其适合用作生物材料。
可替换地,水凝胶的储能模量值可以是25℃下由频率扫描测量记录的值,并是在0.1至100rad/s范围内,例如在0.1至10rad/s范围内,例如0.1,1.0或10rad/s的频率值下获得的值。应变可以为0.5%或1%。
在本案的工作实施例中,频率扫描实验通常在0.5%或1%应变下进行。预期在这些不同的应变下记录的频率扫描数据将是相似的,并可能仅由于样品操作和测量条件的差异而不同(并且在不同应变下测试的相同水凝胶中也观察到这种情况:例如,参见图9和24,其显示了在不同批次中制备并在0.5%或1%应变下测试的的相同水凝胶的频率扫描数据)。
在一个实施方式中,水凝胶具有至少10pa,至少100pa,至少500pa或至少1,000pa的储能模量(来自频率扫描测量)。
在一个实施方式中,水凝胶具有至多5,000pa,至多10,000pa,至多50,000pa,或至多100,000pa的储能模量(来自频率扫描测量)。
在一个实施方式中,水凝胶具有其中最小和最大量选自上述实施方式的范围内的储能模量(来自频率扫描测量)。例如,储能模量处于10至10,000pa,例如,100至10,000pa,例如,500至10,000pa的范围内。
应该注意的是,wo2013/124654中报道的储能模量值g'(来自频率扫描测量)显著低于对本案的水凝胶记录的那些。参见例如wo2013/124654的图9(c),其中报道的储能模量值在0.1至10rad/s的频率范围内不大于1,000pa。
水凝胶的损耗模量值(g″)可以是25℃下由应变振幅扫描测量记录的值,并是在0.1%至100%范围内,例如,在0.1%至10%范围内,例如,0.1%,1.0%或10%的应变值下获得的值。
在一个实施方式中,水凝胶具有至少10pa,至少100pa,至少500pa,至少1,000pa,或至少2,000pa的损耗模量(来自应变振幅扫描测量)。
在一个实施方式中,水凝胶具有至多5,000pa,至多10,000pa,至多50,000pa,或至多100,000pa的损耗模量(来自应变振幅扫描测量)。
在一个实施方式中,水凝胶具有其中最小和最大量选自上述实施方式的范围内的损耗模量(来自应变振幅扫描测量)。例如,损耗模量处于100至10,000pa,例如,500至10,000pa,例如,1,000至10,000pa的范围内。
wo2013/124654中对于应变振幅扫描报道的损耗模量值显著低于对本案的水凝胶所记录的那些。参见例如wo2013/124654的图9(a),其中报道的储能模量值不大于1,000pa,并更接近于100pa。
损耗模量通常用于描述网络系统的粘度,并且损耗模量越高表示系统具有越高的能量耗散能力,因此网络可以被视为具有那些具有低损耗模量的材料并不享有的特定韧性。本案的水凝胶具有有用的损耗模量值。耗散能量的能力与非共价配合物的解离有关,非共价配合物能够作为动态自我修复过程的一部分而重新形成。
可替换地,水凝胶的损耗模量值可以是25℃下由频率扫描测量记录的值,并且是在0.1至100rad/s范围内,例如,在0.1至10rad/s的范围内,例如,0.1,1.0或10rad/s的频率值下获得的值。
在一个实施方式中,水凝胶具有至少1pa,至少10pa,至少100pa,至少500pa,或至少1,000pa的损耗模量(来自频率扫描测量)。
在一个实施方式中,水凝胶具有至多5,000pa,至多10,000pa,或至多50,000pa的损耗模量(来自频率扫描测量)。
在一个实施方式中,水凝胶具有其中最小和最大量选自上述实施方式的范围内的损耗模量(来自频率扫描测量)。例如,损耗模量处于1至5,000pa,例如,100至5,000pa,例如,1,000至5,000pa的范围内。
wo2013/124654中对于频率扫描报告的损耗模量值显著低于对本案的水凝胶所记录的那些。参见例如wo2013/124654的图9(c),其中报道的储能模量值不大于1,000pa,并更接近于100pa。
在一个实施方式中,储能模量和/或损耗模量值在0.1%至100%,0.1%至10%或1%至10%的应变范围内基本相同。本发明人发现本发明的水凝胶具有极宽的线性粘弹性区域,例如高达100%。例如在100%或更大的应变值下,水凝胶开始显示与线性粘弹性的偏差。
在一个实施方式中,对于25℃下通过频率扫描测量分析的水凝胶,损耗模量对于0.1至100rad/s范围内的任何频率值都不大于储能模量。因此,水凝胶的储能模量是主导的。
在一个实施方式中,对于25℃下通过的应变振幅扫描测量分析的水凝胶,损耗模量对于0.1%至100%应变范围内的任何应变值都不大于储能模量。因此,水凝胶的储能模量是主导的。
在一个实施方式中,(在频率扫描实验中)储能和损耗值随着频率变化的变化在0.01至1rad/s的范围内基本相同。因此,可以说储能和损耗模量是平行的。模量值的平行性质在频率扫描实验中是明显的,其中频率(以rad/s计)和模量(以pa计)均以对数标度表示。这如本案的图9(b)中所示。
在一个实施方式中,储能和损耗值随着应变变化(在应变振幅扫描实验中)的变化在0.01%至100%的范围内基本相同。因此,可以说储能和损耗模量是平行的。模量值的平行性质在应变振幅扫描实验中是明显的,其中应变(以%计)和模量(以pa计)均以对数标度表示。这如本案的图9(a)中所示。
在记录应变振幅扫描测量的情况下,频率可以设置为1或10rad/s。
在记录频率扫描测量的情况下,应变振幅可以设定为1%,5%或10%应变。技术人员应选择适合于研究的材料的应变值。技术人员应理解的是,应变值选择为使频率扫描在材料的线性粘弹性区域中进行。
tan(δ)值可以是25℃下由应变振幅扫描测量记录的值,并且是在0.1%至10%范围内的应变值下取得的值。角频率可以为10或60rad/s。
在一个实施方式中,水凝胶具有至少0.1,至少0.2,或至少0.4的tan(δ)值(来自应变振幅扫描测量)。
在一个实施方式中,水凝胶具有至多0.5,至多0.1,或至多2.0的tan(δ)值(来自应变振幅扫描测量)。
在一个实施方式中,水凝胶具有其中最小和最大量选自上述实施方式的tan(δ)值(来自应变振幅扫描测量)。例如,tan(δ)值处于0.1至0.5,例如,0.2至0.4的范围内。
在一个实施方式中,tan(δ)值在0.1%至100%,0.1%至10%或1%至10%的应变范围内基本相同。
本发明人已经发现本发明的水凝胶具有高弹性并且记录了在频率扫描测量中测量的约0.3的tan(δ)值,。
本发明的水凝胶可以被拉伸而不显著损失光学透明度。
在一个实施方式中,在150%、200%、300%或500%的应变下的水凝胶具有例如在400nm、450nm、500nm、550nm或600nm的波长下测量80%或更大,例如,90%或更大的透射率。透射率值是相对于100%应变下水凝胶的透射率。
在一个实施方式中,水凝胶是一种导电水凝胶,如离子导电水凝胶。此处,水凝胶聚合物是具有合适的反离子的离子聚合物。在工作实施例中,显示水凝胶据在不同的应变下导电。水凝胶聚合物提供有带正电荷的咪唑鎓客体和溴反离子。
例如在0,1,2.5或4的应变下测量的水凝胶的电阻率可以为至少0.002ω.m,至少0.005ω.m,至少0.01ω.m,至少0.05ω.m,至少0.1ω.m,至少0.5ω.m。
例如在0,1,2.5或4的应变下测量的水凝胶的电阻率可以是至多1ω.m,至多2ω.m,至多5ω.m,或至多10ω.m。
应变可以表示为样品的拉伸长度与其未拉伸长度之间的比率(如mm/mm)。
在应变下水凝胶的电阻(r),相对于无应变下的电阻(r0)测量,例如,在1、2.5或4的应变下测量,可以为至多40,至多50,或至多100。电阻表示为r/r0。
水凝胶的电阻,例如,在1,2.5或4的应变下测量,可以为至少1,至少2,至少5,或至少10。
电阻率和电阻都可以在室温下测定。
本发明的水凝胶在形变时具有优异的重塑特性。因此,在扰动后重新组装水凝胶时可以重新获得原始水凝胶的原始粘度性质。
水凝胶响应应力而响应和重塑的能力可以根据连续步进应变实验测定。可以使水凝胶样品,例如在恒定温度如20℃下在多个循环中经受应变。本案的水凝胶能够在重复的应变循环中保持其储能和损耗模量值,然后自修复,而没有明显的模量损失。
因此,在应变和自修复循环后水凝胶的储能和/或损耗模量值可以为水凝胶在应变前的值的至少90%,至少95%,或至少98%。应变实验可以在10rad-1下在0.5%至500%的应变下进行。储能和/或损耗模量值可以在一个、两个、三个或四个应变和修复循环之后测量。在应变和自修复循环后双网络水凝胶的恢复如本案的图18(a)中所示。
如预期的,由于主体-客体相互作用的动态性质和更高的解离动力学,在70℃下自修复的能力较差,并且在重复循环中损耗和储能模量降低。
在一个实施方式中,在至少一个形变和重塑的循环,如两个形变和重塑的循环之后,水凝胶的流变性质保持基本相同。流变性质可以是选自粘度、储能模量、损耗模量和tan(δ)的一种或多种性质。
水凝胶对形变循环的响应证实了基于葫芦脲的网络可逆形成强水凝胶结构的强度。
葫芦脲
本发明的水凝胶利用了葫芦脲与一个或两个客体的非共价配合物。在本领域中,术语葫芦脲和葫芦[n]脲可以互换使用。
在优选的实施方式中,葫芦脲作为主体存在于具有两个客体的三元配合物中,其中客体提供于聚合物上,聚合物可以是相同的聚合物分子(从而提供分子内结合)或不同的聚合物分子(从而提供分子间结合)。通常,三元配合物中的客体是相同的,但不是必需的。
这种水凝胶可以由包含葫芦脲和具有葫芦脲的客体官能团的(第一)可聚合单体,并可选地进一步包含具有葫芦脲的客体官能团的第二可聚合单体的可聚合组合物聚合而形成。
然而,本发明不限于具有三元配合物的那些水凝胶,并且水凝胶可以包含基于二元配合物的形成的非共价交联。此处,葫芦脲作为主体存在于具有一个客体的二元配合物中,其中客体提供于聚合物上且葫芦脲与聚合物共价连接,并且这些可以是相同的聚合物分子(从而提供分子内结合)或不同的聚合物分子(从而提供分子间结合)。
这种水凝胶可以由包含葫芦脲和具有葫芦脲的客体官能团的(第一)可聚合单体的可聚合组合物聚合而形成,其中葫芦脲与第二可聚合单体共价结合。
因此,能够形成三元配合物和二元配合物的葫芦脲可以用于本发明中。葫芦脲的领域中有许多适用于形成这种配合物的实例。
本发明人之一先前已经证实葫芦脲作为超分子手铐而使用存在于一种或多种聚合物上的客体分子连接和/或交联聚合物的用途。这种配合物的形成连接各个聚合物分子,从而形成材料网络。这种网络与水一起形成水凝胶。
最近的工作表明,葫芦脲化合物具有高的体外和体内生物相容性并具有极低的毒性(参见uzunovaetal.,org.biomol.chem.2010,8,2037-2042)。因此,当与无毒聚合物组分一起使用时,本发明的水凝胶也适用于生物系统。
葫芦脲可以能够形成三元配合物。例如,cb[8]能够形成三元配合物。cb[10]和cb[12]化合物也是如此。
葫芦脲可以能够形成二元配合物。例如,cb[7]能够形成三元配合物。cb[8]与合适的客体分子也是如此。
葫芦脲可以是cb[8]、cb[10]或cb[12]化合物。
在一个实施方式中,葫芦脲是cb[8]化合物。
提及的葫芦脲化合物是指其变体和衍生物。
cb[8]的变体可以包括具有一个或多个结构类似于甘脲的重复单元的结构。重复单元可以包括乙基脲单元。在所有单元都是乙基脲单元的情况下,变体是半葫芦脲。变体可以是半葫芦[12]脲(如下所示,也参见lagonaetal.,angew.chem.int.ed.2005,44,4844)。
提供了葫芦脲衍生物并用于本文描述的方法中。葫芦脲的衍生物是具有一个、两个、三个、四个或更多个取代的甘脲单元的结构。取代的葫芦脲化合物可以由以下结构表示:
其中:
n是至少为5的整数;
并且对于每个甘脲单元
每个x为o、s或nr3,并且
-r1和-r2各自独立地选自-h和以下的可选取代的基团:-r3,-oh,-or3,-cooh,-coor3,-nh2,-nhr3和-n(r3)2,其中-r3独立地选自c1-20烷基、c6-20碳芳基(carboaryl)和c5-20杂芳基,或其中-r1和/或-r2是-n(r3)2,两个-r3一起形成c5-7杂环;或-r1和r2一起是与尿嘧啶骨架一起形成c6-8碳环的c4-6亚烷基。
在一个实施方式中,甘脲单元之一是取代的甘脲单元。因此,对于n-1个甘脲单元,-r1和-r2各自独立地为h。
在一个实施方式中,n为5、6、7、8、9、10、11或12。
在一个实施方式中,n为5、6、7、8、10或12。
在一个实施方式中,n为8、10或12。
在一个实施方式中,n为8。
在一个实施方式中,n为7。
在一个实施方式中,每个x为o。
在一个实施方式中,每个x为s。
在一个实施方式中,r1和r2各自独立地为h。
在一个实施方式中,对于每个单元,r1和r2中一个是h,而另一个独立地选自-h和以下的可选取代的基团:-r3,-oh,-or3,-cooh,-coor3,-nh2,-nhr3和-n(r3)2。在一个实施方式中,对于一个甘脲单元,r1和r2之一是h,而另一个独立地选自-h和以下的可选取代的基团:-r3,-oh,-or3,-cooh,-coor3,-nh2,-nhr3和-n(r3)2。在该实施方式中,剩余的甘脲单元使得r1和r2各自独立地为h。
优选地,-r3是c1-20烷基,最优选c1-6烷基。c1-20烷基基团可以是直链和/或饱和的。每个基团-r3可以独立地是未取代或取代的。优选的取代基选自:-r4,-oh,-or4,-sh,-sr4,-cooh,-coor4,-nh2,-nhr4和-n(r4)2,其中-r4选自c1-20烷基、c6-20碳芳基和c5-20杂芳基。取代基可以独立地选自-cooh和-coor4。
在一些实施方式中,-r4与-r3不同。在一些实施方式中,-r4优选是未取代的。
在-r1和/或-r2是-or3、-nhr3或-n(r3)2的情况下,则r3优选是c1-6烷基。在一些实施方式中,r3被取代基-or4、-nhr4或-n(r4)2取代。每个-r4是c1-6烷基并其本身优选地被取代。
适用于本发明的是与可聚合单体共价连接的葫芦脲。这在上面的描述中被称为第五单体。通常,葫芦脲在位置r1和/或r2处通过一个或多个甘脲单元与可聚合单体连接。例如,jungetal.(biomacromoelcules2014,15,708)描述了在r1和r2处由烯丙基醚基团官能化的葫芦脲的使用。
在优选的实施方式中,葫芦脲不与可聚合单体共价连接。
应该注意的是,直到形成引入单体的聚合物,这些单体不用于形成非共价配合物。聚合物形成后,使聚合物结合的葫芦脲与适于形成非共价配合物的客体接触。
葫芦脲客体
如上所述,客体是能够与葫芦脲形成主体-客体配合物的化合物。因此,术语配位是指主体-客体配合物的建立。客体提供于单体内,如第一和第三单体内。
在本发明的一些实施方式中,主体-客体配合物是三元配合物,其包含葫芦脲主体和第一客体和第二客体。通常,此类配合物基于cb[8]及其变体和衍生物。优选的是第一客体和第二客体是相同的。
原则上,任何具有合适结合亲和力的化合物(如上述配合物的讨论中提到的那些)都可以用于本发明的方法中。使用的化合物可以基于被认为与葫芦脲的腔相互作用的部分的尺寸来选择。这些部分的尺寸可以足够大以允许仅与较大的葫芦脲形式配位。
在可聚合单体内,葫芦脲客体被连接,如共价直接连接或共价间接连接至参与聚合反应的可聚合基团,如乙烯基基团。
在一个实施方式中,客体并未经由酯键与可聚合基团连接。本发明的水凝胶可以用于其中相对不稳定的酯官能团将会不是有益的应用中。
葫芦脲客体分子在本领域是熟知的。适用的客体化合物的实例包括wo2009/071899、jiaoetal.(jiaoetal.,org.lett.2011,13,3044)、jiaoetal.(jiaoetal.,j.am.chem.soc.2010,132,15734),rauwaldetal.(rauwaldetal.,j.phys.chem.2010,114,8606)和wo2011/077099中描述的那些。
本发明人先前已经研究了当这些分子与另一基团(如聚合物或小有机化合物)连接时以及当这些分子未连接时客体分子的配位作用。使用等温量热法已经证明,客体与另一基团的附连不会导致客体的结合常数降低。因此,对聚合物空间位阻的结合没有可观察到的影响。
在本发明的一个实施方式中,葫芦脲是cb[8]并且客体是适于与该主体形成三元配合物的分子。
在优选的水凝胶中,葫芦脲的配合物是由两个客体形成的三元配合物,其中每个客体是相同的。这种水凝胶可以由仅具有一种具有客体官能团的单体的可聚合组合物形成。水凝胶也可以由具有一种以上的可聚合单体的可聚合组合物形成,其中每种单体具有相同的客体,而单体的剩余部分在单体之间是不同的。
水凝胶还可以含有三元配合物,其中第一和第二客体是不同的。这种水凝胶由含有具有客体官能团的第一单体和具有不同客体官能团的另外的单体,如第三单体的可聚合组合物形成。在该配合物中,一个客体可以是富电子客体,而一个客体分子可以是缺电子的。
客体可以是离子的,如阳离子或阴离子的,并可以存在合适的反离子。客体中存在的离子基团也可以与反离子一起存在于水凝胶产物中。有利地,单体如客体内的离子官能团的存在提供了具有离子导电性的水凝胶。可以用于提供离子导电性的客体类型的实例是下面描述的阳离子有机氮杂环。
在一个实施方式中,客体是离子液体。通常,这种客体适合与cb[7]形成配合物。然而,它们也可以在二元配合物中与cb[8]形成配合物,或在三元配合物与另一种小客体分子或溶剂一起形成配合物(参见jiaoetal.org.lett.2011,13,3044)。离子液体通常是阳离子有机氮杂环。
因此,客体可包含阳离子有机氮杂环,其可以是芳族氮杂环(杂芳基)或非芳族氮杂环。本文中,客体通常包含阳离子有机氮杂环的反阴离子。氮杂芳基优选是氮c5-10杂芳基基团,最优选氮c5-6杂芳基基团,其中下标是指一个或多个环中的原子总数,包括碳和氮原子。非芳族氮杂环优选是氮c5-6杂环,其中下标是指一个或多个环中的原子总数,包括碳和氮原子。氮杂环的环中的氮原子是季铵化的。
反阴离子可以是卤素离子,优选溴离子。适合使用的其他反阴离子是产生可溶于水的配合物的那些。
客体优选选自由以下组成的组:咪唑鎓部分;吡啶鎓部分;喹啉鎓部分;嘧啶鎓部分;吡咯鎓部分;季吡咯烷部分;和季铵。
优选地,客体是咪唑鎓部分或包含咪唑鎓部分。特别优选的客体是1-烷基-3-烷基咪唑鎓,其中烷基基团是可选地取代的。
其中烷基基团未取代的1-烷基-3-烷基咪唑鎓化合物特别适合与cb[7]形成配合物。
其中烷基基团未取代的1-烷基-3-烷基咪唑鎓化合物特别适合与cb[6]形成配合物。
其中烷基基团被芳基(优选萘基)取代的1-烷基-3-烷基咪唑鎓化合物特别适合与cb[8]形成配合物。
1-烷基和3-烷基取代基可以相同或不同。优选地,它们是不同的。
在一个实施方式中,3-烷基取代基是甲基,并优选是未取代的。
在一个实施方式中,1-烷基取代基是乙基或丁基,并各自优选是未取代的。
在一个实施方式中,可选的取代基是芳基,优选c5-10芳基。芳基包括碳芳基和杂芳基。芳基基团包括苯基、萘基和喹啉基。
在一个实施方式中,本文描述的烷基基团是直链烷基基团。
每个烷基基团独立地是c1-6烷基基团,优选c1-4烷基基团。
芳基取代基本身可以是另一个1-烷基-3-取代的咪唑鎓部分(其中烷基基团连接于环的3-位)。
在另一个实施方式中,化合物优选包含吡啶鎓部分。
上述的离子液体分子特别适用于形成二元主体-客体配合物。包含两个离子液体分子作为葫芦脲主体内的客体的配合物也包括于本发明中。
葫芦脲可以能够形成二元和三元配合物。例如,先前已经注意cb[6]化合物与短链1-烷基-3-甲基咪唑鎓客体分子形成三元配合物,而更长链的1-烷基-3-甲基咪唑鎓客体分子与葫芦脲主体形成二元配合物。
用于本发明的优选客体是h+x-形式,其中h+是以下阳离子中的一种,
并且x-是合适的反阴离子,如上定义。优选的反阴离子是卤素阴离子,优选br-。
在优选的实施方式中,阳离子a或阳离子b可以用于与cb[7]或cb[6]形成配合物。
在优选的实施方式中,阳离子c、d或e可以用于与cb[8]形成配合物,例如,阳离子c用作本工作中的客体。
阳离子a和b可以分别称为1-乙基-3-甲基咪唑鎓和1-丁基-3-甲基咪唑鎓。
阳离子d和e可以称为1-萘基甲基-3-甲基咪唑鎓,其中d是1-萘-2基甲基-3-甲基咪唑鎓,且e是1-萘-1基甲基-3-甲基咪唑鎓。
可替代地或另外,客体可以是或含有式(i)的咪唑鎓盐:
其中x-是反阴离子;
r1独立地选自h和饱和的c1-6烷基;
r2独立地是可以可选地含有一个或多个双键或三键的c1-10烷基,并可以可选地被选自-o-、-s-、-nh-和-b-的杂原子中断,并且可以可选地被取代。
在一个实施方式中,x-独立地选自由以下组成的组:cl-,br-,i-,bf4-,pf6-,oh-,sh-,hso4-,hco3-,ntf2-,c2n5o4-,alcl4-,fe3cl12-,no3-,nmes2-,meso3-,sbf6-,prcb11h11-,aucl4-,hf2-,no2-,ag(cn)2-和nicl4-。在一个实施方式中,x-选自cl-、br-和i-。
在一个实施方式中,r1选自h和直链饱和c1-6烷基。
在一个实施方式中,r2是可以可选地含有一个或多个双键的直链c1-10烷基,并可以可选地被选自-o-、-s-、-nh-和-b-的杂原子中断,并且可以可选地被取代。
在一个实施方式中,r2是可以可选地含有一个或多个双键的直链c1-10烷基,并且可以是可选地取代的。
在一个实施方式中,在存在双键或三键的情况下,其可以与咪唑鎓部分共轭。可替换地,双键或三键可以不与咪唑鎓部分共轭。
在一个实施方式中,可选的取代基独立地选自由以下组成的组:卤素,可选取代的c5-20芳基,-or3,-ocor3,=o,-sr3,=s,-br3,-nr3r4,-nr3cor3,-n(r3)conr3r4,-coor3,-c(o)r3,-c(=o)sr3,-conr3r4,-c(s)r3,-c(=s)sr3和-c(=s)nr3r4,其中r3和r4各自独立地选自h和可选地取代的饱和c1-6烷基、c5-20芳基和c1-6亚烷基-c5-20芳基,或者r3和r4可以一起形成可选地被基团-r3取代的可选地饱和的5元、6元或7元杂环。
在一个实施方式中,可选的取代基独立地选自由以下组成的组:卤素,可选取代的c5-20芳基,-or3,-ocor3,-nr3r4,-nr3cor3,-n(r3)conr3r4,-coor3,-c(o)r3和-conr3r4,其中r3和r4如上定义。
每个c5-20芳基基团可以独立地选自c6-20碳芳基基团或c5-20杂芳基基团。
c6-20碳芳基基团的实例包括苯基和萘基。
c5-20杂芳基基团的实例包括吡咯(pyrrole)(唑(azole))(c5)、吡啶(pyridine)(吖嗪(azine))(c6)、呋喃(furan)(呋喃(oxole))(c5)、噻吩(thiophene)(噻吩(thiole))(c5)、噁唑(c5)、噻唑(c5)、咪唑(1,3-二唑)(c5)、吡唑(1,2-二唑)(c5)、哒嗪(1,2-二嗪)(c6)和嘧啶(1,3-二嗪)(c6)(例如,胞嘧啶,胸腺嘧啶,尿嘧啶)。
每个c5-20芳基优选选自可选取代的苯基、萘基和咪唑鎓。
每个c5-20芳基基团可选地被取代。可选的取代基独立地选自卤代基、c1-6烷基、-or3、-ocor3、-nr3r4、-nr3cor3、-n(r3)conr3r4、-coor3、-c(o)r3和-conr3r4,其中r3和r4如上定义。
在一个实施方式中,每个c5-20芳基基团可选地被c1-6烷基取代。
在c5-20芳基基团是咪唑鎓的情况下,其优选在氮上被基团r1取代(从而形成季氮)。
式(i)的结构包含在1-位上具有取代基r2和在3-位上具有取代基r1的咪唑鎓部分。式(i)的结构可以可选地在2-、4-或5-位上进一步被基团ra取代,其中ra具有与r1相同的含义。
视情况,上述实施方式可以以任何组合进行组合。
葫芦脲客体可以是或包含下表中的结构:
其中该结构可以是盐,包括合适的质子化形式。在一个实施方式中,客体分子是cb[8]的客体分子。
在一个实施方式中,客体分子是,或衍生自,或包含以上表格中的结构a1-a43、a46或b1-b4。
在一个实施方式中,客体分子是,或衍生自,或包含以上表格中的结构a1、a2或a13。
在一个实施方式中,客体分子是,或衍生自,或包含结构b1。
另外,客体分子是,或衍生自,或包含金刚烷、二茂铁或环辛烷(包括双环[2.2.2]辛烷)。其由moghaddam等人描述(参见j.am.chem.soc.2011,133,3570)。
适合使用的其他客体分子包括芘、二苯并呋喃和芴及其衍生物。衍生物可以是其中芳环原子被杂原子如氮取代的化合物。另外或可替代地,衍生物可以是环原子上被如卤素、烷基、羟基、氨基、烷氧基的基团取代的化合物。
在一些实施方式中,第一和第二客体分子形成一对,其可以在葫芦脲的腔内相互作用而形成稳定的三元主体-客体配合物。可以使用任何适合葫芦脲的腔内的客体对。在一些实施方式中,客体分子对可以形成包含富电子和缺电子化合物的电荷转移对。第一和第二客体分子中的一种充当电子受体,而另一个充当ct对中的电子供体。例如,第一客体分子可以是充当电子受体的缺电子分子,且第二客体分子可以是充当电子供体的富电子分子,或反之亦然。在一个实施方式中,葫芦脲是cb[8]。
合适的电子受体包括4,4'-联吡啶鎓衍生物,例如,n,n'-二甲基二吡啶鎓基乙烯,及其相关受体,如基于二氮杂芘和二氮杂菲的那些。包括烷基紫精的紫精化合物特别适用于本发明。烷基紫精化合物的实例包括n,n'二甲基-4,4'-联吡啶鎓盐(也称为百草枯(paraquat))。
合适的电子供体包括富电子芳族分子,例如,1,2-二羟基苯、1,3-二羟基苯、1,4-二羟基苯、四硫富瓦烯、萘如2,6-二羟基萘和2-萘酚、吲哚和芝麻酚(3,4-亚甲二氧基苯酚)。多环芳族化合物通常可以用作本发明中合适的电子给体。这些化合物的实例包括蒽和并四苯。
在一个实施方式中,客体是蒽。在一个实施方式中,客体是肉桂酸。
氨基酸,如色氨酸、酪氨酸和苯丙氨酸,可以适合用作电子供体。可以使用其末端包含这些氨基酸的肽序列。例如,可以使用包含氨基酸序列n-wgg-c、n-ggw-c或n-gwg-c的供体。
在一个实施方式中,客体是色氨酸或苯丙氨酸。在一个实施方式中,客体是苯丙氨酸。
在一些实施方式中,客体分子是一对化合物,例如,第一和第二客体分子,其中该对中的一种是如上表中列出的a化合物(例如,a1,a2,a3等),且该对中的另一个是如上表中所示的b化合物(例如,b1,b2,b3等)。在一个实施方式中,a化合物选自a1-a43和a46。在一个实施方式中,b化合物是b1。
其他合适的客体分子包括肽如wgg(bush,m.e.etal.,j.am.chem.soc.2005,127,14511-14517)。
富电子客体分子可以与任何缺电子的cb[8]客体分子成对。如本文描述的,适合使用的合适客体分子对例如第一和第二客体分子的实例可以包括:
紫精和萘酚;
紫精和二羟基苯;
紫精和四硫富瓦烯;
紫精和吲哚;
甲基紫精和萘酚;
甲基紫精和二羟基苯;
甲基紫精和四硫富瓦烯;
甲基紫精和吲哚;
n,n'-二甲基二吡啶鎓基乙烯和萘酚;
n,n'-二甲基二吡啶鎓基乙烯和二羟基苯;
n,n'-二甲基二吡啶鎓基乙烯和四硫富瓦烯;
n,n'-二甲基二吡啶鎓基乙烯和吲哚;
2,7-二甲基二氮杂芘鎓和萘酚;
2,7-二甲基二氮杂芘鎓和二羟基苯;
2,7-二甲基二氮杂芘鎓和四硫富瓦烯;和
2,7-二甲基二氮杂芘鎓和吲哚。
具体地,如本文描述的适用的合适客体分子对可以包括2-萘酚和甲基紫精、2,6-二羟基萘和甲基紫精以及四硫富瓦烯和甲基紫精。
在一个实施方式中,客体对是2-萘酚和甲基紫精。
在一个实施方式中,客体对是指适合与cb[8]形成三元配合物的客体分子对。
组分
本发明的水凝胶可以保持组分。水凝胶适合于储存组分,并且该组分可以随后根据需要在选定的位置释放。
本发明的一些发明人先前已经描述了保持组分的水凝胶,以及水凝胶将组分递送至目标位置的用途。参见,例如,wo2013/124654,其内容通过引证结合于本文中。
应该理解的是,所指由水凝胶保持的组分并非是指溶剂分子。例如,组分不是水或有机溶剂。因此,该组分是在可以存在于水凝胶内的溶剂之外提供。
还应该理解的是,所指的组分不是指葫芦脲,或用于制备水凝胶的单体,或可聚合组合物的其他组分,或由葫芦脲与适当官能化的单体配位形成的中间产物,或在聚合反应期间形成的中间产物。此外,组分并不受特别限制。
在一个实施方式中,组分具有至少100,至少200,至少300,至少1,000,至少5,000(5k),至少10,000(10k),至少15,000(15k),至少20,000(20k),至少50,000(50k),至少100,000(100k)或至少200,000(200k)的分子量。
本发明人先前已发现超分子水凝胶可以有效地保持和递送如生物活性组分的组分至一定位置。组分可以在其储存于水凝胶中的整个期间以及在其随后递送至所需位置之后保持活性。
在一个实施方式中,组分是治疗化合物。
在一个实施方式中,组分是生物分子或包含生物分子,如多核苷酸(例如,dna和rna),多肽或多糖。
在一个实施方式中,组分是聚合物分子,包括生物聚合物,如上述的那些生物分子。
在一个实施方式中,组分是细胞。
在一个实施方式中,组分具有可检测的标记。可检测标记可用于量化和/或定位组分。标记可以用于测定水凝胶中含有的组分的量。
在一个实施方式中,可检测标记是发光标记。在一个实施方式中,可检测标记是荧光标记或磷光标记。
在一个实施方式中,可检测标记是可见标记。
在一个实施方式中,荧光标记是罗丹明或荧光素标记。
在一个实施方式中,组分是多肽,如蛋白质。蛋白质可以是血清白蛋白或溶菌酶。前者的实例包括牛血清白蛋白。
在一个实施方式中,组分是颗粒。颗粒可以是金属颗粒。
在一个实施方式中,组分选自由以下组成的组:毒性分子(如神经毒剂和重金属),激素,除草剂,杀虫剂,抗体,病原体(如病毒),佐剂,凝胶,纳米颗粒(包括金属或非金属颗粒),聚合物(包括合成和天然聚合物),催化剂(有机,无机和有机金属),粘合剂和密封剂。
在另一个实施方式中,组分可以是递送载体,其可以装载用于递送的试剂,并且试剂可以选自上面列出的组分。例如,递送载体可以是胶囊、胶束或脂质体。
水凝胶内组分的存在可以使用可以区分网络材料和组分的合适分析技术进行检测。这些技术是本领域技术人员所熟知的。
组合物和水凝胶的制备方法
如上描述的,本发明的可聚合组合物可以通过化合物的简单混合物,例如,在水中容易地制备。
在一个方面中,本发明提供了制备可聚合混合物的方法,方法包括将葫芦脲与可聚合单体混合的步骤,如将葫芦脲和可聚合单体与水混合在一起。
可聚合组合物的组分可以全部一起添加,或组分可以逐步添加。例如,葫芦脲主体可以与第一单体和可选的第三单体(在存在的情况下)混合于例如水中,从而形成葫芦脲与单体客体的配合物。然后可以将可聚合组合物的其余组分加入混合物中。通常,可聚合组合物中的总单体浓度cmon为至少0.5m。
优选地,可聚合组合物包含葫芦脲与由可聚合组合物中的单体提供的一种或多种客体的配合物。例如,葫芦脲形成与由第一单体提供的两个客体的三元配合物。在第三单体存在于可聚合组合物中的情况下,可以存在与由第一和第三单体提供的客体的三元配合物,以及与仅由第一单体和仅第三单体提供的客体的配合物。
在可聚合组合物中保持于配合物中的葫芦脲的量可以是存在的葫芦脲的总量的至少5%,如至少10%,至少20%,至少50%,至少60%,至少70%,至少80%,至少90%,或至少95%。
可聚合组合物内的配合物的形成可以通过标准光谱法检测。例如,发明人通过us/vis光谱和nmr谱等观察到配合物形成。
聚合反应可以通过存在于可聚合组合物中的聚合引发剂和/或通过刺激如光或热引发。在本案的工作实施例中,描述了聚合光引发剂,并且这种光引发剂通过可聚合组合物的uv照射而活化。聚合反应的条件适合相对于聚合反应中存在的单体选择。
聚合反应可以在室温下进行,例如,在10至25℃,如15至20℃的温度下进行。
因此,本发明的水凝胶可以在温和和环境条件下制备。
本发明的方法允许制备具有任何期望的形状的水凝胶。水凝胶材料的形状由用于聚合的反应容器确定。因此,适当选择反应容器形状是形成期望的水凝胶设计所需的全部。通常,水凝胶以宏观尺度制备,因此水凝胶的最大尺寸处于μm、mm或cm范围内。因此选择的反应容器适合于形成具有这种尺寸的水凝胶。
当然,具有期望的形状和期望的尺寸的产品可以由更大的水凝胶块,例如通过将该大块切割成期望的的形状和尺寸而制成。
水凝胶还可以通过组装多个水凝胶块制备。本案表明,本发明的水凝胶具有自愈合性能,这是由超分子网络中的非共价交联的动态解离和形成所致。这种动态特性还允许将单块的水凝胶材料放至一起并连接至一起。本案中的工作实施例显示了两个单独的水凝胶块融合成整体水凝胶形式的融合。在工作实施例中对水凝胶愈合过程提供的流变学研究表明,给予足够的愈合时间,愈合的水凝胶可以重新获得原始水凝胶形式的有益机械性质。
本发明的水凝胶可以保持组分。组分可以在水凝胶形成期间引入水凝胶中,或可以将组分添加到预先形成的水凝胶中,然后破坏而使之将组分并入其中。
在水凝胶形成期间将组分引入水凝胶中的情况下,组分仅需要在水凝胶形成之前混合入可聚合组合物中。超分子网络在组分周围形成,从而提供保持组分的水凝胶。
因此,本发明的可聚合组合物,如水性可聚合组合物,可以进一步包含组分。水性可聚合组合物的聚合将提供含有组分的水凝胶。
该组分不参与聚合反应。该组分可以不具有聚合所需的单体的官能团。例如,组分可以不包含碳-碳双键或三键,因为其在自由基聚合反应中可以是反应性的。
可替换地,制备保持组分的水凝胶的方法包括以下步骤:提供本发明的水凝胶,并在组分存在下搅拌该水凝胶,从而将组分并入水凝胶中。搅拌步骤可以是水凝胶的机械搅拌或破坏。
本发明的超分子配合物是可逆的,并且被破坏的配合物可以重新形成,如本案的工作实施例中所示。
水凝胶的用途
由于其低毒性和高含水量,本文描述的水凝胶可以用作医学应用中的材料。当适当加载有组分时,本发明的水凝胶可以用于将该组分递送至目标位置。
在本发明的进一步的方面中,提供了一种生物材料,其包含本发明的水凝胶或本发明的聚合物。生物材料用作位于动物如哺乳动物,如人之上或之内的结构元件。生物材料可以是植入物。生物材料可以作为组织、软骨或骨替代材料提供。
在本发明的又一个进一步的方面中,提供了一种电子装置,如传感器,其包含本发明的水凝胶或聚合物。如本文描述的,本发明的水凝胶可以是离子导电的,并且水凝胶可以作为电子装置的导电特征提供。更一般而言,水凝胶材料具有任何可以为电子装置提供结构的有益的机械性能。本发明的聚合物可以具有使其以类似的方式使用的类似性质。
本发明人已经发现,本发明的水凝胶具有优异的粘合性能。因此,本发明还提供了水凝胶作为粘合剂的用途。本发明的聚合物也可以用作粘合剂。在另一方面中,提供了包含本发明的水凝胶或聚合物的粘合剂。
本发明人先前已发现,保持于本发明的葫芦脲水凝胶中的组分可以在选定的位置从水凝胶中释放。因此,在本案中,提供了将组分递送至一定位置的方法,该方法包括以下步骤:
(i)如本文描述的提供保持组分的水凝胶;
(ii)使水凝胶在目标位置可用;
(iii)从水凝胶中释放组分。
在一个实施方式中,目标位置是体内位置。因此,水凝胶可以置于受试者中或受试者上的目标位置。受试者可以是哺乳动物,如人或啮齿动物,如大鼠或小鼠。
在这个实施方式中,组分可以是用于治疗或预防疾病的治疗药物。因此,包含治疗化合物的水凝胶适用于治疗人体或动物体的方法。
在其他实施方式中,水凝胶适于将组分递送至离体或体外的位置。
本发明人先前已发现,改变水凝胶组成可以用于改变组分从水凝胶中释放的时机。此处,时机可以是指组分释放的速率,并且另外计时机可以是指该速率随时间的变化。因此,可聚合组合物中组分的绝对和相对量的变化可以用于改变组分从水凝胶中的释放曲线。
在一个实施方式中,释放组分而无需对水凝胶施加任何外部刺激。因此,水凝胶可以置于期望的位置,并简单地允许组分从水凝胶中浸出。
在其他实施方式中,组分的释放可以与网络的至少部分解配位相关。配合物的解配位而使一个或多个客体与主体分离,可以响应于外部刺激而发生,包括例如竞争性客体化合物、光、氧化剂或还原剂、电化学电势和温度变化等。可以诱导这种解配位以在水凝胶中提供保持于水凝胶中的组分可以通过的额外的或更大的孔隙。解配位还可以用于破坏整个网络并导致水凝胶的分解。因此,通过向水凝胶施加外部刺激可以引发解配位。
在上述方法和用途中,本发明的聚合物可以用于代替本发明的水凝胶。
其他优选
上述实施方式的每种相容性组合在此明确地公开,如同每种组合被单独和明确地叙述一样。
鉴于本公开,本发明的各个进一步的其他方面和实施方式对于本领域技术人员而言是显而易见的。
“和/或”在本文中使用时,将被视为两个指定的特征或组件一个具有或不具有另一个指定的特征或组件的每个的具体公开。例如,“a和/或b”将被视为(i)a、(ii)b和(iii)a和b中的每个的具体公开,就像每个在本文中单独列出一样。
除非上下文另有规定,否则上述特征的描述和定义不限于本发明的任何具体方面或实施方式,并且同样适用于所描述的所有方面和实施例。
本发明的某些方面和实施方式现在将通过示例并参考上述附图进行举例说明。
实验和结果
材料
除非另有说明,否则本工作所用的化学品,包括丙烯酰胺、n,n'-亚甲基双丙烯酰胺、乙烯基咪唑、1-金刚烷胺、苄基溴、irgacure2959,都购自sigma-aldrich,并且未进行进一步纯化而使用。按照先前的报道合成葫芦[7]脲和葫芦[8]脲(schermanetal,chem.commun.,2010,46,2007-2009;kimetal.,j.am.chem.soc.,2000,122,540-541)。
仪器和测量
1hnmr(500mhz)谱使用brukeravancebb500记录。化学位移以ppm(δ)报告。
光照射在具有365nm波长灯(功率密度:4.8mwcm2)的光反应器上进行。
所指的室温是指通常在20℃或25℃下进行实验。如果没有给出温度数字,则可以假设温度为20℃或25℃。
本工作中使用的仪器仪表和测量结果如下。
粘度测试
聚合物溶液粘度使用具有悬浮液位计泡(levelbulb)的玻璃schott-gerateubbelohde微粘度计并使用pvs1测量装置测量,并使用dlk10恒温器单元(均由lauda制造)将微型粘度计恒温于25℃的pv15水浴中。测量的溶液是在mill-q(18mw)h2o中浓度为0.01至80gl-1。
拉伸测试
样品的室温(20℃下)机械测试在配备有5-n测力传感器的拉伸hounsfield机上以预定的十字头速度进行,并且对每种情况测试五个样品。
拉伸测试在配备有5-n测力传感器的拉伸hounsfield机上进行。实验在室温下以100mmmin-1的形变速率进行。为了避免夹具附近的系统性失效,使用符合iso4661-1标准的哑铃形模切机切割样品,其中样品的缩小部分具有12mm×2mm×2mm的尺寸。由直至断裂的拉伸应力-应变曲线之下的面积计算断裂时的延伸功wb(jm-3),该参数表征每单位体积样品断裂所需的功。在压缩测试中,具有圆柱形状(直径20mm,初始厚度5mm)的样品放置于涂有硅油的金属板上以减小摩擦。加载速度通常设定为0.5mmmin-1。
itc测试
itc滴定实验在microcalinc.的vpitc上在25℃下于磷酸钠缓冲液(ph7,10mm)中进行。水性gpc装置由串联连接至shimadzuspd-m20a突出二极管阵列检测器、wyattdawnheleos多角度光散射检测器和wyattoptilabrex折射率检测器的shodexohpaksb柱组成。
在典型的实验中,主体分子(cb[8])以0.05mm的浓度加载到样品池中,并将客体分子(1-苄基-3-乙烯基咪唑鎓)以高20倍的浓度(1mm)装入注射器中。滴定由20次连续注射2-10ml构成,注射间隔至少60秒。在曲线拟合之前从数据集中移除第一个数据点。
稀释热通过将客体溶液滴定到缓冲溶液中检查,并从归一化的焓中减去,即使它们在所有情况下都相对较小。数据采用origin7.0软件使用一组位点模型(sitemodel)分析。
撕裂测试
撕裂测试使用配备有5-n测力传感器的拉伸hounsfield机进行以表征空气中的韧性。厚度约2mm(w)的样品切割成标准jis-k62521/2尺寸(50mm×7.5mm;初始缺口长度20mm)。将测试片的两个臂夹紧,然后以100mmmin-1拉动上臂,并记录撕裂力f。撕裂能量t按照t=2f/w以恒定的撕裂力f计算,其中w是样品的厚度。
纯剪切测试
纯剪切试验也用于表征韧性。使用缺口和无缺口的两种不同样品测量撕裂能量t。使用矩形样品(20mm(宽度)×40mm(长度))×1.02mm(厚度)。
使用剃刀刀片切割长度为20mm的初始缺口。将测试片在两侧夹住,两个夹具之间的距离固定为8mm(l0)。上夹具以100mmmin-1拉动,下夹具固定。记录样品的力-长度曲线,并由t=u(lc)/(a0×b0)计算撕裂能量,其中u(lc)是施加的力对无缺口样品在临界拉伸距离lc下作的功,且lc是裂纹开始在缺口样品中扩展时两个夹具之间的距离。
裂纹扩展的开始由照相机记录的视频图像确定。本发明人已经证实,撕裂测试和纯剪切测试对于相同种类样品给出了一致的值。
流变学测试
使用discoveryhybrid流变仪(dhr)-2,来自tainstruments的配备有设定为各种温度的水浴的受控应力混合流变仪,进行流变学测试。所有测量均使用20mm和40mm平行板几何结构,间隙500nm进行。使用厚度为0.5mm且直径为20mm的圆盘形样品,并由供应商提供的溶剂捕集器(solventtrap)包围,以减轻测量期间的溶剂(水)损失,并使用tainstrumentstrios软件分析结果。通常,流变测试在20℃或25℃下进行,除了温度扫描实验之外,其温度为例如5℃到80℃不等(如以下讨论的)。
动态振荡应变振幅扫描测量以10rads-1的频率进行。动态振荡频率扫描测量在1%应变振幅下,在0.01至100rads-1或0.01至200rads-1进行,且温度频率扫描测试在5至80℃以5℃的速率采用1%的剪切应变和60rad/s的剪切频率下进行。
松弛测试采用在0.01s(剪切速率10s-1)内施加的10%的最大应变振幅(γ0)进行,然后记录松弛模量g(t)随时间的变化。
样品的连续步进应变测量在20℃、40℃和70℃下以高振幅振荡(参数:应变(γ)=500%,角频率(ω)=10rads-1)和低振幅振荡(参数:γ=0.5%,ω=10rads-1)进行。
透光率
对于整个可见光范围,使用uv/vis分光光度计测量水凝胶的光学透射谱。使用具有milli-q水的石英比色皿(18mw,在550nm处100%透射率)作为参照以降低来自折射率不匹配的反射。
聚合物分子量
通过粘度显著增加的聚合物溶液的重叠浓度(c*)进行非共价水凝胶的重均分子量(mw)的粗略估计。通过milli-q溶液中的粘度测量确定的c*值为约26.3g/l,对应于约0.32m的单体摩尔浓度(不包括cb[8]部分)。由于c*与重复单元尺寸a(约0.2nm)、平均聚合度n以及聚合物链的卷绕尺寸r有关,遵循以下方程:
假设聚合物处于θ态,则r为约an1/2,并且
因此,聚合度n为约24,500,并且相应的分子量为约2.1mda,其仍属于来自自由基聚合的典型mw范围的聚合物。如通过流变学测量观察和证实,在0.4m单体浓度(重量分数为33g/l)下获得凝胶网络,而对于0.3m,获得流动溶胶溶液。因为缠结浓度仅比重叠浓度高几倍,因此,聚合物链缠结涉及那些超分子聚合物水凝胶,与cb[8]介导的主体-客体相互作用一起有助于极端韧性和可拉伸性。
单体合成
1-苄基-3-乙烯基咪唑鎓的合成
在0℃下在10分钟内将苄基溴(50mmol)滴加到乙烯基咪唑(50mmol)的乙醚(100ml)溶液中。反应在室温下再保持16小时。滤出反应粗产物,得到灰色粉末,然后用乙醚洗涤,并真空干燥至恒重(产率约100%)。
1hnmr(500mhz,d2o,298k,d,ppm):10.68(s,1h,-n+-ch-n-),7.17-7.91(m,7h,芳烃),7.22(dd,1h,-n-ch=ch2),5.89(dd,1h,-n-ch=ch-h反式),5.52(s,2h,ph-ch2-n+-),5.16(dd,1h,-n-ch=ch-h顺式)。
水凝胶合成-非共价交联
将预定量的aam(95摩尔当量)、1-苄基-3-乙烯基咪唑鎓(5摩尔当量)和cb[8](2.5摩尔当量)溶解于h2o中。用氮气吹扫30min后,向单体中加入光引发剂(irgacure2959,0.03摩尔当量)。将反应混合物转移到预先密封的实验室制玻璃模具中,然后暴露于uv照射2h。
以0.05m至2.0m的总单体浓度(cmon),包括0.05、0.1、0.2、0.3、0.4、0.5、1.0和2.0m,制备水凝胶。
在室温下剧烈搅拌5天后,将超分子网络样品(用2.0m的cmon制备;直径1cm,厚度0.5cm)完全溶解于水(~50ml)中。在冷冻干燥聚合物溶液后获得含有cb[8]的聚合物。为了除去cb[8],将聚合物溶液相对ada溶液渗析一周,然后相对milli-q水渗析并冷冻干燥。在cb[8]解配位之前和之后通过1hnmr分析样品。经由质子信号积分比证实约5摩尔当量的具有客体的单体,与用于网络合成所用的5摩尔当量单体一致。
质子信号在这两个谱之间的化学位移,由苄基和咪唑鎓基团中的质子(e',f',g'和h'的峰)产生,也证实了聚合物侧基团与cb[8]主体分子的配位作用,以及cb[8]在相对作为竞争性客体的ada渗析后的完全去除。nmr谱如图5中所示。
水凝胶合成-与荧光标记的非共价交联
使用上述用于合成具有非共价交联的水凝胶的方法制备具有非共价交联的荧光标记的水凝胶。向可聚合组合物中加入若丹明丙烯酰胺或荧光素丙烯酰胺(0.002摩尔当量),并将总单体浓度保持于2.0m。制备了两种水凝胶样品,一种具有罗丹明标记,另一种具有荧光素标记。这两种样品用于愈合测试,如图3(c)中所示。
水凝胶合成-双网络
通常,将预定量的aam(94.95摩尔当量)、1-苄基-3-乙烯基咪唑鎓(5摩尔当量)、cb[8](2.5摩尔当量)和mba(0.05摩尔当量)溶解于milli-qh2o(18mw)中。用氮气吹扫30min后,将光引发剂(irgacure2959,0.03摩尔当量)加入到单体溶液中。将反应混合物转移到实验室制玻璃模具中,然后暴露于uv照射(4.8mwcm-2)2h。
总单体浓度(cmon)为1.0m。与非共价网络一样,可以改变总单体浓度以调节水凝胶产品的性质。
结果—非共价网络
水凝胶由包含主体(葫芦[8]脲)、具有客体分子(1-苄基-3-乙烯基咪唑鎓)的单体和单体丙烯酰胺的可聚合组合物制备,其提供水凝胶产物中的主骨架。动态cb[8]介导的主体-客体配位作用充当了在形变下断裂并消散所施加的能量的牺牲键,其可以进一步重新形成,导致宏观超分子网络的自我修复。
如上描述的,在倒置小瓶测试中采用0.05至2.0m范围内的总单体浓度(cmon)聚合并检查了一系列超分子聚合物网络(结果显示于图1(c)中)。随着单体浓度(cmon)的变化,观察到粘度的明显增加:在0.2和0.3m下粘性流动,浓度为0.5m及以上时网络稳定。这表明聚合物链之间的主体-客体相互作用在高浓度下稳定以形成稳定的,甚至没有化学交联的超分子水凝胶。
据证实,在网络中没有共价交联,因为它们在5天后可以完全溶解于milli-q水(25℃,18mω)中,其可以在冷冻干燥后产生蓬松/无定形聚合物,以及随后在水中重新溶解后再次形成超分子聚合物网络(结果未显示)。对凝胶化的强烈浓缩效应归因于聚合物链的缠结。来自样品的分子量的重叠浓度的粗略估计支持该论点(参见以上的“聚合物分子量”讨论)。当添加少量化学交联剂时胶凝化临界cm向较低值迁移的事实也支持这一论点(参见以下描述的双网络系统)。
图6(a)显示了在相对过量ada溶液(1mm)渗析后冷冻干燥的聚合物的gpc迹线。此处,通过nmr分析证实,通过竞争性形成ada配合物,从聚合物中完全除去了cb[8]分子。
在mill-q(18mω)水中制备浓度范围为0.01至80g/l的聚合物溶液,并使用玻璃的schott-gerteubbelohde微粘度计在25.0℃下测量聚合物溶液的粘度。生成两组线性图以在交叉点处给出临界缠结浓度(参见图6(b))。
超分子聚合物网络的拉伸应力-应变曲线(图2(a))显示,cmon的增加显著提高了机械性能。在低单体浓度(例如,cmon=0.5m)下合成的凝胶不显示明显的屈服点,表明链缠结较弱,而在较高浓度下合成的凝胶(例如,cmon=2.0m)显示出明显的屈服点以及显著增强的模量。显然,机械强度增强是由于高浓度下强链缠结,并结合链间的cb[8]介导的主体客体配位作用。这也通过采用cb[7]或无cb[n]的对照证实,其只能产生高粘性流体,模量低2个数量级(参见图10)。
此外,超出某一应变观察到的明显屈服应该来自主体客体配合物的内部断裂。值得注意的是,这种类型的超分子聚合物网络可以拉伸超过原始尺寸的45倍(由拉伸机可获得的最大拉伸),而在拉伸测试期间没有观察到断裂(结果未显示)。简单的举重测试表明,网络的带可以容易地抬升网络本身重量500倍的重量(结果未显示)。
拉伸行为的形变率依赖性也证实了上述物理图像(图1(a))。超分子聚合物水凝胶的强度和杨氏模量随着形变速率增加而显着增加(从10mm/min下的0.016mpa到2000mm/min下的0.42mpa,为24倍)指示整个形变速率范围内的较不稳定的主体-客体相互作用的动态和可逆特征,其特征在传统的共价网络中并不存在(图2(b))。这种粘弹性特征有助于超分子网络的高冲击吸收率和韧性。应该注意的是,尽管形变速度从10mm/min增加到2,000mm/min,但在拉伸比为45(研究中使用的拉伸机器可达到的最大比率)之前没有观察到断裂。
为了进一步证明超分子网络的粘弹性,发明人进行了循环拉伸测试以观察形变及其恢复的滞变。如图2(c)所示,在将样品加载到预定的拉伸应变时观察到明显的屈服,并且在卸除到零应力时观察到大的滞变。在零应力下观察到残余的应变。屈服揭示了凝胶的结构变化,即耗散能量的超分子主体-客体配合物的解离。残余应变和滞变表明在卸除时间内结构变化得以保留。在一定的等待时间后,残余应变减小到零。
如图2(d)所示,在8的应变下的循环拉伸测试后立即在拉伸测试中检测模量和应力的降低。然而,在等待时间约30分钟时,样品恢复所有的机械性能,这由拉伸曲线与原始曲线的重叠证明。这表明即使没有共价交联,由于网络内主体-客体配位的广泛分布,在大形变下没有发生链滑动。此外,cb[8]介导的主体-客体配合物充当了可逆牺牲键,其会动态断裂和重新形成,为这种新型粘弹性超分子聚合物网络提供高韧性。
图7显示了水凝胶样品(在1.0m的cmon下制备)的杨氏模量随形变速率变化的变化。葫芦脲主体客体相互作用的存在赋予网络显著的粘弹性,因此在较高的形变速率下观察到较高的模量值。这可以与由于材料的弹性本质而显示出对形变率的依赖性的共价交联水凝胶系统相反。
虽然最近已经开发了几种基于物理缔合的高强度和韧性的凝胶(参见sunetal.nature;haqueetal.;tanakaetal.),但其大多数仅具有部分自我恢复能力,可能是由于加载时共价键的一些永久损坏所致。因此,刚性和脆性的物理结合与柔软和延性主链的共价键之间的微妙平衡对于坚韧的水凝胶是至关重要的。
水凝胶在600至900nm范围内具有相对高的可见光透射率(参见图8,采用1.0m的cmon制备的厚度为2mm的膜样品在拉伸前)。可见光范围内的透光率是适中的,并且这在拉伸时,例如,当样品被拉伸5次或15次时会增加(图8)。
传统的共价水凝胶太脆而不能承受压缩、切片或伸长(参见aidaetal.;yanetal.)。然而,由于存在cb[8]介导的超分子主体客体配位而耗散能量,超分子网络具有高弹性和可拉伸性,表现如同橡胶一样。网络样品非常有弹性,可以承受高压缩或高水平形变,如弯折、扭曲、打结和大范围拉伸。
此外,网络还可以承受用刀片的切片,留下痕迹,这可以在几分钟内快速愈合(结果未显示)。根据提出的化学/拓扑网络结构,认为用这些超分子相互作用构建的超分子聚合物网络具有相对均匀且大的长度。如果不施加负荷,它们表现出自然的卷绕状态。它们可以在应力下伸长直到它们变直,并当应力移除时可以恢复到最低能量的卷绕状态。因此,应力可以有效地被耗散于那些主体-客体超分子交联的解离。
超分子聚合物网络也是非常缺口不敏感的:具有片状的原始超分子网络可以拉伸至其原始长度的25倍而不会断裂,而在累积了所有强烈的应力的玻璃或金属夹具附近观察到后续的断裂。当向凝胶切入缺口然后将其拉伸时,缺口显著变钝并在拉伸率分别为13.8和15.5之前保持稳定。在临界施加的拉伸之后,在缺口的前部开始断裂,并且快速通过整个样品。在临界施加的拉伸(10的应变)下,通过将金属球落于由圆形夹具固定的凝胶膜上,显示大的、可恢复的形变。在击中膜时,球大幅拉伸膜并随后反弹回来。在振动衰减后,膜保持完整,振动并恢复其初始平坦构型。然而,具有更大动能的球会导致膜在大形变后断裂。
用哑铃形样品进行室温自愈比率测试,将样品切成两块并随后使二者接触一系列不同的自愈时间。愈合比率,定义为破坏愈合结点所需的功与通过拉伸形变破坏原始样品所需的功之间的比率,在室温下愈合1小时后达到35%。愈合结点可以承受非常大的应力(约为原始应力的40%,参见图3(a)),形变比为约28,随后愈合的部分断裂(参见图3(a)(ii)和(iii),圆圈表示断裂的位置)。
12小时的较长自愈合时段可以导致材料几乎完全自我修复。这种高自愈合效率也与先前报道的基于水凝胶结合(cordieretal.;whiteetal.;chenetal.;wangetal.;teeetal.)、金属配体缀合(burnworthetal.)、静电相互作用(sunetal.nat.mater.)和主体-客体相互作用(nakahataetal.)等的自愈系统相当。
此外,通过荧光标记的水凝胶的荧光显微镜成像也观察到未切割表面之间的粘附。水凝胶如前制备,向可聚合组合物中加入少量红色或绿色标记的单体(0.002摩尔当量荧光单体至95摩尔当量的aam),然后将其聚合而得到红色和绿色标记的水凝胶。使水凝胶接触至一起,并荧光成像。对应于红色发光(ritc)和绿色发光(fitc)的重叠的黄色区域的出现证实了接触区域的自我修复。
离子导体通常具有比大多数电子导体更高的电阻率。然而,当需要高拉伸性和透光率时,离子导体具有比如银纳米线、单壁碳纳米管、石墨烯和氧化铟锡的电子导体更低的薄层电阻(参见,keplingeretal.)。
图4(a)表明超分子聚合物网络即使在水凝胶沿其长度拉伸高达五倍时的导电性足以为led灯供电(参见,图4(a)(ii))。此外,由于长度增加和相交表面积减小,一旦拉伸四倍,网络的电阻增加两倍(图4(c))。然而,网络的电阻率(1至15)×103ω.m)比海水的电阻率(约0.2ω.m)高约两个数量级。
在压缩下,测量电压看起来增加,在初始压缩范围(0至5%)内显著增加(参见图4(d))。还研究了循环压缩/卸压循环期间的电压变化。结果如图4(e)所示。
图20显示了在不同cmon,包括0.3、0.4、0.5、1.0和2.0m下制备的一系列非共价水凝胶的频率扫描实验中tan(δ)的变化。实验在室温下进行。据观察,在较低的总单体浓度cmon下制备的水凝胶仅在较高频率下表现出类固体特征。在较高的总单体浓度下制备的那些水凝胶在整个扫描范围内具有较高的网络稳定性。
结果—双网络
如上描述的,在与cb[8]以2:1方式配位(ka1=4.21×107m-1,ka2=4.25×105m-1,参见图15)时充当非共价超分子交联剂的可聚合客体(1-苄基-3-乙烯基咪唑鎓)用少量的化学交联剂n,n'-亚甲基双丙烯酰胺(mba,2摩尔当量的动态cb[8]交联)和亲水单体丙烯酰胺聚合,产生所谓的水性双网络(参见图11(a))。双网络易于在室温下水性介质中制备。
cb[8]介导的主体-客体相互作用(物理交联)沿聚合物链形成动态的“环”(图11(c))。主体-客体配合物的力诱导解离,类似于肌联蛋白结构域的解折叠(图11(b)),可以有效地耗散能量并赋予系统显著的韧性。
2:1的cb[8]苄基-咪唑鎓配合物的断裂需要7.6kcalmol-1(δh2,参见图15),其与蛋白解折叠能量(免疫球蛋白结构域为约4kjmol-1;参见fongetal.)相当。在这种水凝胶中,系统中存在的化学交联不仅改进了机械性能,而且还增加了整个网络的弹性,类似于肌联蛋白和肌球蛋白的作用,其是负责肌肉收缩的两种运动蛋白。
水凝胶具有高水平的透明度,并在压缩时既坚韧且耐久。实际上,使用刀片也不能轻易地切割网络,并且在移除切割刀片后表现出至其原始形状的迅速恢复。这种双网络足够坚固且可以轻松承受自身重量的500倍(图11(f))。感兴趣的是,网络迅速地各向同性收缩,在冷冻干燥时不会在凝胶表面发生任何断裂;此外,其可以在水中吸收高达自身重量的500倍。
为了测试材料的可拉伸性,将8.0mm(长)×40mm(宽)×1.5mm(t)的样品常规拉伸至其原始尺寸的17至25倍(图11(d))而没有断裂。在拉伸期间,双网络在可见光范围(400至700nm)内保持优异的透明度(参见图16)。
此外,缺口样品(具有20mm(w)的缺口,总宽度的一半)达到7的应变,表现出显著的缺口不敏感性。在拉伸期间,缺口显著变钝并保持稳定(图11(e)),直至达到临界应变。为了确定材料消散能量的能力,进行了落球测试。当金属球(16g,直径2cm,下落高度45cm)落于样品膜上时,观察到大的可恢复的形变。在击中膜时,球在弹回之前将材料大幅拉伸:膜保持完整,振动并恢复到其初始的扁平构型,成功地消散了所有能量。
为了确定双网络在大形变下的非线性和粘弹性行为,在各种初始形变速率(10至1,000mmmin-1)下对哑铃形样品进行单轴拉伸研究。拉伸应力-应变曲线(标称应力相对标称应变)如图12(a)所示。显然,机械性能受形变速率的强烈影响,并且在特定应变以上在所有情况下观察到屈服,该特定应变随着更高的形变速率而降低。屈服可归因于主体-客体配合物在拉伸时在分子水平上的连续解离,导致能量吸收。
值得注意的是,杨氏模量从4.6kpa(10mmmin-1)至77.5kpa(1,000mmmin-1)增加了约17倍(参见图12(b))。材料的断裂应力和断裂应变在较低的形变速率(10和50mmmin-1)下变化轻微,然而,它们在较高的速率下降低(参见图12(b))。这通过在低速拉伸期间cb[8]主体-客体相互作用的动态解离和重新形成,其进一步维持了断裂应力和应变来解释。然而,在较高的拉伸速率(>100mm/min)下,双网络失去其韧性可能是因为主体-客体配位在解离后没有足够的时间重新形成,而使应力在断裂尖端处扩展。双网络的这种粘弹性特征类似于gong及其同事报道的聚两性电解质水凝胶(参见sunetal.;以及各种luoetal.的出版物)。
进行循环拉伸测试以研究主体-客体相互作用作为可逆牺牲键的动态本质,类似于肌联蛋白内可逆蛋白折叠。每个应变实验中的形变速率为100mmmin-1。图13(a)显示了不同应变(1,2,4和8)下的四个拉伸-回缩循环,显示了显著的类塑性形变和显著的滞变。因此,证实了超分子体系通过非共价超分子交联的牺牲性断裂的有效能量耗散,其是所有高韧性材料的重要特征。此外,在收缩过程(去除负荷)期间,残余应变逐渐减小(图13(b)),这可以归因于由共价交联产生的弹性和主体-客体相互作用的重新形成两者。
50%的所观察到的滞变在5秒内恢复(图13(c)),而剩余的50%在慢得多和复杂得多的过程后恢复。如果在连续循环之间施加120秒等待,则材料没有显示出任何明显的塑性形变。在双网络经受其原始长度8倍的循环应变之后,其略微弱于原始材料,然而,在室温下30分钟的等待时间之后其完全自我修复(图13(d))。
双网络表现出宽的线性粘弹性区域,对图17所示的坚韧水凝胶是典型的,临界屈服应变为80%,高于该值时刚性网络开始断裂(g″>g')。角频率为10rad/s,且实验在室温下进行。由于cb[8]主体-客体相互作用的可逆性,双网络是剪切敏感的,并且随着角频率(ω)增加表现出高粘弹性模式。
频率依赖性的粘弹性模量清楚地证实了双网络的弹性行为,高于ω=1rads-1时g'>g″(图14(a)),验证了各种应变速率下的拉伸测试(图12(a))。对于图14(a)所示的工作,应变为0.5%,并且测量在室温下进行。
双网络的g'恒定直至40℃,然后随着温度的升高逐渐降低(图14(b))。相反,g″作为温度的函数继续增加并且在45℃附近达到峰值,表明双网络的软化,源于cb[8]主体-客体配合物的热诱导的解离。tan(δ)达到最大值时双网络的软化温度约为70℃,高于此温度时材料完全熔融,这远高于聚两性电解质水凝胶的软化温度(参见sunetal.)。由于稳定双网络的整体结构的共价交联,在整个研究的温度范围内没有检测到明显的凝胶-溶胶转变。对于图14(b)所示的工作,应变为0.5%,而角频率为60rads-1。
图14(c)使用20℃下参照的时间-温度叠加(tts)描绘双网络的流变学主曲线。数据在10-3至103rads-1的频率范围内良好叠加,得到185kjmol-1的活化能(ea),与128pnnm(1kjmol-1=1.6pnnm,即80kjmol-1)的典型肌联蛋白的蛋白解折叠活化能相当(参见,例如,riefetal.和reifetal.)。对于图14(c)所示的工作,应变为0.5%,而角频率为10rad/s。
在20℃(图18(a))和40℃(图14(d))下都观察到大振幅振荡分解后其机械性能的快速恢复。在施加大振幅振荡力(γ=500%,ω=10rads-1)时,g'从5,000pa降至600pa,导致产生类液态(tanδ=g″/g'为约2.0)。然而,当振幅减小(γ=0.5%,ω=10rads-1)时,g'立即恢复其类固态初始值(tanδ=0.5),类似于肌肉蛋白质在外部机械刺激下的可逆解折叠-折叠过程。
图19显示了力随着双网络水凝胶样品拉伸距离变化的变化。在第一个实验中,对双网样品进行撕裂测试。代表性的力延伸曲线如图19(a)所示。此处,撕裂能量(t)由恒定拉伸力f按照t=2f/w计算。对于双网络样品,t为630jm-2。
在第二个实验中,对缺口和未缺口双网络样品进行拉伸。结果如图19(b)所示。由于用于样品的撕裂和拉伸测试的性质,图19(a)和(b)中记录的值不同。
lc是缺口样品的裂缝开始扩展的拉伸距离,且u(lc)是当力被施加到未缺口样品上直到样品达到拉伸距离lc时所做的功。撕裂能量(t)计算为(t)=u(lc)/(a0×b0)(其中a0和b0分别表示为样品宽度和厚度)。由该方法(rivlin-thomas纯剪切测试)获得的断裂能量为750jm-2,这与根据撕裂测试(630jm-2)获得的750jm-2的值相当。此外,这种韧性接近于软骨(1,000jm-2),进一步有望使用这种双网络作为再生组织工程的结构生物材料。
工作实施例—双网络
发明人观察并记录了圆柱形双网络(原始直径10mm,高度5mm)压缩至薄膜(直径29mm,高度约0.59mm,压缩应变约88.2%)的高压缩,在3分钟内该材料恢复到其原始尺寸。
发明人观察并记录了经受落球测试时双网络的形变。在测试中,当金属球(16g,直径2cm,下落高度45cm)落于双网络的膜(2mm厚度)上时,材料表现出大的形变。在击中膜时,球大幅拉伸膜并随即反弹回来。在振动衰减并且冲击能量完全耗散后,膜保持完整,振动并恢复到其初始的平坦构型。
其他实验和结果
发明人已经制备了另外的水凝胶以突出本发明的优点。水凝胶由1-苄基-3-乙烯基咪唑鎓与选自aam、dma、nipam、heam、aa、dmaema、viet和mps的第二单体一起制备。
按照与上述的非共价水凝胶类似的方式制备水凝胶,其中1-苄基-3-乙烯基咪唑鎓和第二单体的水性混合物与cb[8]和光引发剂一起使用。这在下面进一步详细描述。
材料
单体如丙烯酰胺(aam)、二甲基丙烯酰胺(dma)、n-异丙基丙烯酰胺(nipam)、n-羟乙基丙烯酰胺(heam)、丙烯酸(aa)、甲基丙烯酸2-(二甲基氨基)-乙基酯(dmaema)、聚(乙二醇)甲基丙烯酸酯(pegma)、溴化1-乙烯基-3-乙基咪唑鎓(viet)和3-[2-(甲基丙烯酰氧基)乙基](二甲基)铵-1-丙磺酸盐(mps)购自sigma-aldrich。除非另有说明,否则工作中使用的所有化学品均未经进一步纯化而使用。
如上描述的合成溴化1-苄基-3-乙烯基咪唑鎓。
仪器
在具有uv灯(365nm,功率密度为4.8mwcm-2)的光反应器上进行光照射。
使用discoveryhybrid流变仪(dhr)-2,来自tainstruments的配备有设定为各种温度的水浴的受控应力混合流变仪,进行流变学测试。制备具有0.5mm的厚度和20mm的直径的圆盘形样品,并且这些由供应商提供的溶剂捕集器包围,以减轻测量期间的溶剂(水)损失,并使用tainstrumentstrios软件分析结果。
以10rads-1的频率进行动态振荡应变振幅扫描。动态振荡频率扫描测量在1%应变振幅,0.01至100rads-1下进行,且温度频率扫描测试在剪切应变为1%且剪切频率为60rads-1下,以5℃的间隔0至80℃的渐变进行。
振幅扫描、频率扫描和步进应变实验在20℃的温度下进行。
在下面描述的温度稳定性测试中,处理温度以5℃的间隔升高。每个测试温度之间的渐变相对较快。典型的测试包括在每个测试温度下约5分钟的测试样品处理。
水凝胶合成—非共价交联
通常,对于基于丙烯酰胺(aam)的水凝胶网络,将预定量的1-苄基-3-乙烯基咪唑鎓(5摩尔当量)、cb[8](2.5摩尔当量)和丙烯酰胺(95摩尔当量)溶解于milli-qh2o(2ml,18mω)中。用氮气吹扫30分钟后,将光引发剂irgacure2959(0.03摩尔当量)加入到单体溶液中。单体溶液暴露于uv照射(4.8mwcm-2)下6小时。
基于其他单体的水凝胶网络,其中aam被替代单体代替,按照相同的策略制备。使用的单体是dma、nipam、heam、aa、dmaema、viet和mps。每种单体在水凝胶制剂中以与aam相同的mol%使用。
如此获得的样品直接用于测量。
流变学测试
为了探测超分子水凝胶网络的网络动力学,以及量化其机械性质,进行了水凝胶产品的流变学表征。
基于heam的水凝胶网络(固体分数为12wt%,cb[8]交联为2.5mol%)的代表性的应变依赖性振荡流变学(图22(a))显示出在高应变下在结构失效之前的宽线性粘弹性区域(应变为0.1-550%),表明宽的加工区域和剪切稀化行为。与先前的具有中等机械性能的cb[8]水凝胶(即平台模量为101-103pa)相比(参见appeletal.2010;tanetal.),g'>104pa的水凝胶网络可以容易地通过调节总固体分数和/或动态交联度而获得。
g'和g″的频率依赖性证实了超分子网络的弹性行为,因为g'在施加的整个频率范围(10-2-102rads-1,图22(b))内是主导的。在之前的研究中,当g'和g″在振荡频率扫描中交叉时估算临界松弛时间(β),低于其时水凝胶网络是弹性的(ω>β),但是随着粘性组分主导(ω<β)而变为粘性流体(参见appeletal.2010;tanetal.)。虽然此处的cb[8]水凝胶网络的β不能在实验时间尺度(10-2-102rads-1)内测定,但时间-温度叠加(tts)原理可以在更宽的角频率范围(10-3-103rads-1)内延伸b值(参见appeletal.2010;tanetal.)。由于动态交联数量更多,固体质量分数更高(>10wt%)且聚合物链缠结更多,因此对于这些cb[8]水凝胶网络获得了振荡扰动后更缓慢的松弛(即,更高弹性),由整个角频率范围内具有g'>g″的更高模量证实。
记录了利用上述不同单体的所有其他水凝胶网络的振荡动态流变学扫描,并且结果显示于图24至31中。因此,由dma、nipam、heam、aa、dmaema、viet和mps连同1-苄基-3-乙烯基咪唑鎓一起制备的水凝胶在振幅扫描和频率扫描条件下测试,并记录储能和损耗模量。
基于heam的水凝胶网络的温度依赖性流变行为记录至高达80℃(图23(a)),高于该温度,在平行板几何结构中来自cb[8]水凝胶网络的水的蒸发可能带来问题。在整个温度范围内,g'在40℃之前是稳定的,之后网络样品开始失去其机械性能。随着温度升高的机械性能降低可以直观地归因于在升高的温度下伴随的主体-客体配合物的解离动力学的增加。该观察进一步证明了三元配合物负责3d网络及其相应的粘弹性。在随后的冷却过程中,网络样品逐渐恢复其初始机械强度,这在先前的水凝胶系统中未观察到(参见tanetal.),标志着高的热稳定性和可逆性。
天然系统的愈合开裂的能力总是涉及通过牺牲键的能量耗散机制,牺牲键可以在破坏整个瞬态支架之前动态地断裂和重新形成。作为牺牲键引入动态相互作用,可以有效地增加整体粘弹性耗散,而且赋予其在失效后恢复其初始强度的能力(参见gemertetal.;degreefetal.;和liuetal.)。
通过流变学步进-应变测量研究了基于heam的水凝胶网络的微观自愈合行为,其中在高应变(加工和注射软材料的关键参数)下诱导中尺度断裂。如图23(b)所示,高应变振荡扫描(γ=500%)导致产生类液体结构(g″/g'为约2.0),其中水凝胶样品易于流动。然而,当网络经受随后的低振幅应变扫描(γ=0.5%)时,g'在应力诱导流动后的几秒内立即恢复到其准固态的初始值(tanδ约0.5)。感兴趣的是,在同时断裂和重新形成的几个循环内,恢复的速度和程度几乎相同,突出了这种动态交联网络的可逆和强健的本质。因此,主体-客体配合物的可逆解离/缔合可以有效地延缓网络失效,并确保惊人的韧性。
如以上工作实施例中描述的,在基于丙烯酰胺的cb[8]网络的拉伸测试中也观察到类似的水凝胶网络的宏观自愈合。
cb[8]水凝胶网络表现出高达2,000jm-2的韧性(来自rivlin-thomas纯剪切测试),这与软骨的(1,000jm-2)相当。因此,这些坚韧的水凝胶网络作为用于再生组织工程的结构生物材料显示出巨大的潜力,类似于gong及其同事报道的双网络(参见nonoyamaetal.)和li及其同事报道的石墨烯的水凝胶(参见luetal.)。
此外,由于具有不受限于溶液粘度的一系列单体浓度的单体前体溶液的适用性,这种合成策略允许在宽范围的机械强度下灵活制备水凝胶网络。例如,通过调整总固体分数和交联度可以获得具有中等强度(1至1,000pa)的水凝胶网络。
这种控制使其具有用作体内药物递送平台的可模制和可注射的水凝胶的潜在用途,类似于langer及其同事报道的聚合物/颗粒水凝胶(参见appeletal.,2015)。
除了受这些网络的卓越的机械性能启发的应用外,选择的水凝胶网络中的移动离子可以实现导电通路的渗透,从而扩展其作为高度可拉伸的离子导体和压力传感器的潜力。这些柔韧而坚韧的透明材料的固有离子传导性开启了离子/生物和离子/电子系统之间构建稳固界面的可能性,类似于suo,zhao及其同事所展示的藻酸盐(aliginate)的网络(参见keplinger等人;yuk等人)。
引入带正电单体如viet、带负电单体如aa、两性离子单体(例如,mpc)和其他ph-敏感单体(例如,dmaema和aa)或热敏单体(例如,nipam,dmaema和pegma)也可以丰富这些具有额外特性的超分子水凝胶网络。
参考文献:
本说明书中提及的所有文献通过引证以其全部内容结合于本文中。
aidaetal.science2012,335,813
appeletal.j.am.chem.soc.2010,132,14251
appeletal.j.am.chem.soc.2012,134,11767
appeletal.nat.commun.2015,6,6295
burnworthetal.nature2011,472,334.
chenetal.nat.chem.2012,4,467
cn104086691
cn105061783
cordieretal.nature2008,451,977
degreefetal.nature2008,453,171
fongetal.j.mol.biol.1996,264,624
haqueetal.macromolecules2011,44,8916
keplingeretal.science2013,341,984
kimetal.j.am.chem.soc.2000,122,540
liuetal.macromol.chem.phys.2016,217,319
luetal.adv.mater.2016,28,4025
luoetal.acsmacrolett.2015,4,961
luoetal.ady.mater.2015,27,2722
luoetal.macromo/ecules2014,47,6037
luoetal.macromolecules2016,49,2750-2760
nakahataetal.nat.commun.2011,2,511
nonoyamaetal.adv.mater.2016,28,6740
riefetal.biophys.j.1998,75,3008
rietetal.phys.rev.lett.1998,81,4764
schermanetal.chem.commun.2010,46,2007
songetal.acsmacrolett.2016,5,1084
sunetal.nat.mater.2013,12,932
sunetal.nature2012,489,133
tanetal.polym.chem.2015,6,7652
tanakaetal.j.phys.chem.b2005,109,11559
teeetal.naturenanotechnology2012,7,825
us2012/0103615
vangemertetal.macromol.chem.phys.2012,213,234
wangetal.nat.chem.2013,5,1042
whiteetal.na山re2001,409,794
wo2013/124654
wo2015/103125
xuetal.acsappl.mater.interfaces2017,9,11368
yanetal.chem.soc.rev.2012,41,6042
yuketal.nat.mater.2016,15,190。
- 还没有人留言评论。精彩留言会获得点赞!