细胞坏死抑制剂TAK-632及其作为药物的用途的制作方法
本发明属于医药技术领域,更具体地,本发明涉及细胞坏死抑制剂tak-632及其作为药物的用途。
背景技术:
程序性细胞坏死是一种由死亡受体介导的、caspase蛋白水解酶非依赖性的细胞死亡方式。当细胞凋亡被阻断时,tnf-α和tnf-α家族的受体成员,toll样受体3和4以及α干扰素的受体在被相应的配体激活后能诱发细胞发生程序性坏死。
研究表明,蛋白激酶ripk1和ripk3形成复合体并激活mlkl蛋白,mlkl多聚化后直接定位细胞膜上并导致细胞膜破裂是诱发程序性细胞坏死的关键机制。由于细胞内容物的释放,程序性细胞坏死会引起体内大量炎症细胞浸润从而诱发严重的炎症反应。目前的研究表明,程序性细胞坏死作为新的一类细胞程序性死亡方式,在缺血性损伤、神经退行性疾病、恶性肿瘤、病毒感染和免疫性疾病等多种疾病的病理生理过程中起重要作用。
因此,鉴定并发现程序性细胞坏死的抑制剂,对于程序性细胞坏死相关疾病的临床治疗具有十分重要的意义。
技术实现要素:
本发明的目的在于提供细胞坏死抑制剂tak-632及其作为药物的用途。
在本发明的第一方面,提供式(i)所示化合物或其异构体、溶剂合物或前体,或它们的药学上可接受的盐的用途,用于制备抗细胞坏死的药物;
在一个优选例中,所述的式(i)所示化合物或其异构体、溶剂合物或前体,或它们的药学上可接受的盐的药物作用靶点为ripk1/ripk3,通过抑制ripk1/ripk3激酶活性,从而抑制细胞坏死。
在本发明的另一方面,提供式(i)所示化合物或其异构体、溶剂合物或前体,或它们的药学上可接受的盐的用途,用于制备ripk1/ripk3抑制剂。
在本发明的另一方面,提供式(i)所示化合物或其异构体、溶剂合物或前体,或它们的药学上可接受的盐的用途,用于制备ripk1/ripk3激酶紊乱、过度激活或过度相互作用相关疾病的药物。
在一个优选例中,所述的ripk1/ripk3激酶紊乱、过度激活或过度相互作用相关疾病为炎症性、感染性、缺血性和退行性相关疾病或组织损伤。
在另一优选例中,所述的炎症性、感染性、缺血性和退行性相关疾病由ripk1/ripk3激酶介导,或由细胞坏死引发(通过抑制细胞坏死抑制炎症和感染性相关疾病)。
在另一优选例中,所述的炎症性、感染性、缺血性和退行性相关疾病包括:全身炎症综合征(即,脓毒血症)、组织损伤、急性胰腺炎、炎症性肠病、沙门氏菌感染、李斯特菌感染、牛痘病毒感染、缺血性心肌病、缺血性脑卒中、阿尔茨海默病、动脉粥样硬化。
在另一优选例中,所述的组织损伤包括:肝损伤(如,对乙酰氨基酚引起的肝损伤)或肝细胞坏死。
在本发明的另一方面,提供一种抑制细胞坏死的方法,包括:以式(i)所示化合物或其异构体、溶剂合物或前体,或它们的药学上可接受的盐处理细胞。
在一个优选例中,所述的抑制细胞坏死的方法是不以治疗疾病为目的的方法;如为体外细胞培养方法。
在本发明的另一方面,提供一种预防、缓解或治疗炎症性、感染性、缺血性和退行性相关疾病的方法,包括:以式(i)所示化合物或其异构体、溶剂合物或前体,或它们的药学上可接受的盐给予需要预防、缓解或治疗炎症和感染性相关疾病的对象。
本发明的其它方面由于本文的公开内容,对本领域的技术人员而言是显而易见的。
附图说明
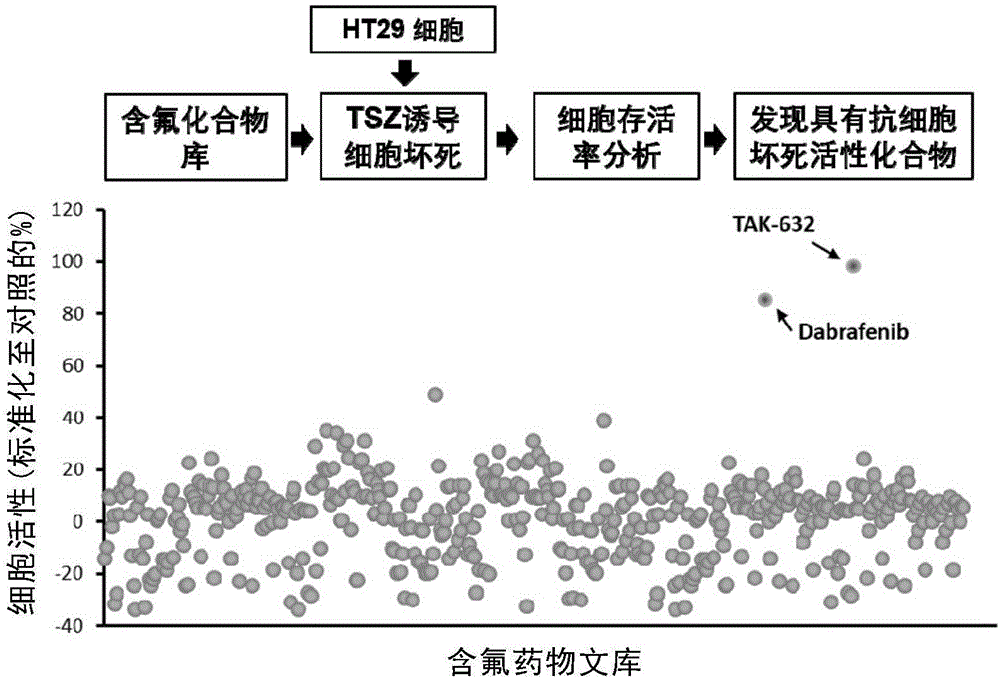
图1、利用自建含氟药物筛选抗程序性坏死抑制剂。
图2、tak-632对tsz刺激后ht-29细胞坏死的保护作用;
a.在显微镜下观察tak-632对tsz刺激后ht-29细胞的保护作用;
b.采用流式细胞术检测碘化吡啶(pi)染色后的活细胞数量;
c.采用流式细胞术方法考察tak-632的ec50;
d.tak-632对ht-29细胞的毒性检测。
图3、tak-632对不同细胞坏死抑制活性考察;
a.thp-1细胞;
b.u937细胞;
c.l929细胞;
d.j774a.1细胞。
图4、tak-632抑制rip1和rip3的磷酸化并阻止坏死小体的形成;
a.tak-632抑制坏死的作用与iκbα的相关性;
b.ht-29细胞在tak-632处理30min后,用tsz刺激不同时间点,考察ripk1,ripk3,mlkl及其磷酸化蛋白的表达;
c.小鼠l929细胞中tak-632对于ripk3和mlkl磷酸化的抑制作用;
d.tak-632抑制ripk1和ripk3在tsz坏死激活后的相互作用的检测。
图5、tak-632结合靶点确认;
a.采用分子对接的方法考察tak-632与ripk1和ripk3的相互作用;
b.采用同源的b-raf结构(pdbid:4ksp)和tak-632重构ripk3的三维结构,tak-632被对接到同源模型ripk3的atp结合口袋中;
c.药物亲和响应靶标稳定性试验(darts);
d.生物素化的tak-632对于tnf诱导的坏死的抑制作用。
图6、tak-632抑制ripk1和ripk3的激酶活性;
a.tak-632抑制ripk1的激酶活性;
b.tak-632抑制ripk3的激酶活性;
c.tak-632阻止了ripk1-ripk3的相互作用;
d.tak-632阻止了ripk3-mlkl的相互作用。
图7、tak-632在体内保护小鼠抵抗tnf诱导的全身免疫综合征(sirs)和对乙酰氨基酚(apap)诱导的肝毒性;
a~b.tak-632显著保护小鼠免于低温和死亡;
c.tak-632处理显著降低了il-6的血清水平;
d~f.tak-632预处理可显著降低对乙酰氨基酚处理小鼠的肝坏死和血浆ast水平。
具体实施方式
本发明人经过深入的研究,首次揭示一种新的抗细胞坏死抑制剂——化合物tak-632。其通过抑制ripk1/ripk3激酶活性,有效地抑制细胞坏死;其还可用于制备ripk1/ripk3激酶紊乱、过度激活或过度相互作用相关疾病的防治药物。
术语
本文所用的术语“异构体”包括:几何异构体、对映异构体、非对映异构体(如顺反异构体,构象异构体)。
本文所用的术语“溶剂合物”表示携带有溶剂分子的化合物,例如,所述的溶剂合物可以是水合物。
本发明中,术语“含有”表示各种成分可一起应用于本发明的混合物或组合物中。因此,术语“主要由...组成”和“由...组成”包含在术语“含有”中。
本发明中,“药学上可接受的”成分是适用于人和/或动物而无过度不良副反应(如毒性、刺激和变态反应)即有合理的效益/风险比的物质。
本发明中,“药学上可接受的载体”是用于将本发明的式(i)化合物、异构体、溶剂合物、前体,或它们的药学上可接受的盐传送给动物或人的药学上或食品上可接受的溶剂、悬浮剂或赋形剂。载体可以是液体或固体。
化合物tak-632
首先,提供一种如结构式(i)所示的化合物,其命名为:n-(7-氰基-6-(4-氟-3-(2-(3-三氟甲基苯基)乙酰胺基苯氧基-苯并噻唑环丙酰胺;其结构式如下:
本发明还包括上述式(i)化合物的异构体、溶剂合物、前体,或它们的药学上可接受的盐,只要它们也具有与式(i)化合物具有相同或基本相同的功能。所述的“药学上可接受的盐”是指化合物与无机酸、有机酸、碱金属或碱土金属等反应生成的盐。这些盐包括(但不限于):(1)与如下无机酸形成的盐:如盐酸、硫酸、硝酸、磷酸、二磷酸、氢溴酸;(2)与如下有机酸形成的盐,如乙酸、乳酸、琥珀酸、对甲苯磺酸、水杨酸、草酸、丁二酸、酒石酸、甲磺酸、马来酸、富马酸、或精氨酸。其它的盐包括与碱金属或碱土金属(如钠、钾、钙或镁)形成的盐,以酯、氨基甲酸酯,或其它常规的“前体药物”的形式。化合物具有一个或多个不对称中心。所以,这些化合物可以作为外消旋的混合物、单独的对映异构体、单独的非对映异构体、非对映异构体混合物、顺式或反式异构体存在。
所述的“化合物的前体”指当用适当的方法服用后,该化合物的前体在病人体内进行代谢或化学反应而转变成结构式(i)的一种化合物,或化学结构式(i)的一个化合物所组成的盐或溶液。
用途
本发明人发现,化合物tak-632靶向作用于ripk1/ripk3,通过抑制ripk1/ripk3相互磷酸化,抑制ripk1和ripk3的激酶活性,发挥抗细胞坏死活性,从而治疗ripk1/ripk3激酶紊乱、过度激活或过度相互作用相关疾病。ripk1/ripk3激酶的紊乱、过度激活或过度相互作用与多种疾病存在密切的相关性,例如文献necroptosisisactiveinchildrenwithinflammatoryboweldiseaseandcontributestoheightenintestinalinflammation.amjgastroenterol.2014feb;109(2):279-87报导了ripk1/ripk3激酶介导的坏死与炎症性肠病相关。也有不少文献报导了细胞坏死与疾病的相关性,例如,文献necroptosisisanimportantseveritydeterminantandpotentialtherapeutictargetinexperimentalseverepancreatitis.cellmolgastroenterolhepatol.2016jul;2(4):519-535报导了坏死与急性胰腺炎相关;文献typeiinterferoninducesnecroptosisinmacrophagesduringinfectionwithsalmonellaentericaserovartyphimurium.natimmunol.2012oct;13(10):954-62报道了坏死与沙门氏菌感染相关;文献liver-residentmacrophagenecroptosisorchestratestype1microbicidalinflammationandtype-2-mediatedtissuerepairduringbacterialinfection.immunity.2015jan20;42(1):145-58报道了坏死与李斯特菌感染相关;文献vacciniavirusinducesrapidnecrosisinkeratinocytesbyastat3-dependentmechanism.plosone.2014nov24;9(11):e113690报道了坏死与与牛痘病毒感染相关;文献camkiiisarip3substratemediatingischemia-andoxidativestress-inducedmyocardialnecroptosis.natmed.2016feb;22(2):175-82报道了坏死与缺血性心肌病相关;文献chemicalinhibitorofnonapoptoticcelldeathwiththerapeuticpotentialforischemicbraininjury.natchembiol.2005jul;1(2):112-9报道了坏死与缺血性脑卒中相关;文献necroptosisactivationinalzheimer'sdisease.natneurosci.2017sep;20(9):1236-1246报导了坏死与阿尔茨海默病相关;文献targetingmacrophagenecroptosisfortherapeuticanddiagnosticinterventionsinatherosclerosis.sciadv.2016jul22;2(7):e1600224报道了坏死与与动脉粥样硬化相关。
基于本发明人的新发现,本发明提供了化合物tak-632或其异构体、溶剂合物、前体,或它们的药学上可接受的盐的用途,用于制备抗细胞坏死的药物。
本发明还提供了化合物tak-632或其异构体、溶剂合物、前体,或它们的药学上可接受的盐的用途,用于预防、缓解或治疗炎症、感染性相关疾病或组织损伤全身免疫综合征。作为本发明的优选方式,所述的炎症性、感染性、缺血性和退行性相关疾病包括:全身炎症综合征、肝损伤(如,对乙酰氨基酚引起的肝损伤)、急性胰腺炎、炎症性肠病、脓毒血症、沙门氏菌感染、李斯特菌感染、牛痘病毒感染、阿尔茨海默病、缺血性心肌病、缺血性脑卒中、动脉粥样硬化。作为本发明的优选方式,所述的组织损伤包括:肝损伤、肝中毒或肝细胞坏死。
本发明还提供了化合物tak-632或其异构体、溶剂合物、前体,或它们的药学上可接受的盐的用途,用于制备ripk1或ripk3抑制剂。ripk1或ripk3作为丝氨酸/苏氨酸激酶,是介导细胞坏死性凋亡的关键元件。
基于本发明人的新发现,本发明还提供一种抑制细胞坏死的方法,所述方法包括:以化合物tak-632或其异构体、溶剂合物或前体,或它们的药学上可接受的盐处理细胞。所述的抑制细胞坏死,可以是体内状态下的抑制细胞坏死,也可以是离体状态下的抑制细胞坏死。例如,可以是在体外细胞培养物中,抑制细胞坏死,这是非治疗目的的情形。
本发明还提供了一种药物组合物。含有:(a)有效量的化合物tak-632、或其异构体、溶剂合物、前体,或它们的药学上可接受的盐;和(b)药学上可接受的载体或赋形剂。一般地,所述的药物组合物含有按照重量比例为0.001-50%的化合物tak-632或其异构体、溶剂合物、前体,或它们的药学上可接受的盐。
本发明所述的药物组合物的剂型可以是多种多样的,只要是能够使活性成分有效地到达哺乳动物机体的剂型都是可以的。比如可选自:溶液、悬浮液、片剂、胶囊、粉末、颗粒或糖浆。根据本发明的化合物所治疗的疾病类型,本领域人员可以选择方便应用的剂型。
化合物tak-632作为活性成分的有效施用剂量可随给药的模式和待治疗的疾病的严重程度而变化。然而,通常当本发明的化合物每天以约0.005-500mg/kg动物体重的剂量给予时,能得到令人满意的效果,较佳地每天以1-3次分开的剂量给予,或以缓释形式给药。可调节此剂量方案以提供最佳治疗应答。例如,由治疗状况的迫切要求,可每天给予若干次分开的剂量,或将剂量按比例地减少。
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件如j.萨姆布鲁克等编著,分子克隆实验指南,第三版,科学出版社,2002中所述的条件,或按照制造厂商所建议的条件。
实施例1、含氟化合物库筛选抗细胞坏死抑制剂
自建含氟化合物库中每个化合物用dmso溶解成10mm母液备用,人结肠癌ht-29细胞铺96孔板,每孔加入一种化合物20μm,用肿瘤坏死因子tnf-α、凋亡抑制蛋白抑制剂smacmimetic和caspase抑制剂z-vad-fmk(简称tsz)共刺激16小时后,共孵育,采用mtt法测定细胞生存率。
如图1所示,结果表明,大部分化合物都对经tsz刺激所导致的程序性细胞坏死没有保护作用,只有少量化合物呈现出理想的保护坏死作用,tak-632具有最佳的效果,同时,dabrafenib也具有很好的保护坏死效果,最近已被文献报道,证明该高通量筛选方法具有很好的选择性。
实施例2、tak-632的抗ht-29细胞坏死活性
在显微镜下观察tak-632对tsz刺激后ht-29细胞的保护作用,如图2a所示,在20μmtak-632的作用下,ht-29细胞全部存活。
采用流式细胞术检测碘化吡啶(pi)染色后的活细胞数量,如图2b所示,tak-632几乎完全保护细胞发生坏死,但是,采用ts(肿瘤坏死因子tnf-α加上凋亡抑制蛋白抑制剂smacmimetic的联合)刺激细胞发生凋亡的情况下,tak-632却不能发挥保护作用。
进一步地,采用流式细胞术方法考察了tak-632的ec50,为2.87μm(图2c)。
同时,在1~100μm的浓度下,tak-632对ht-29细胞没有毒性作用(图2d)。
实施例3、tak-632对不同细胞的坏死抑制活性
使用两种人来源的细胞系(人单核细胞thp-1,人白血病细胞u937)和两种小鼠来源的细胞系(小鼠成纤维细胞l929,小鼠单核巨噬细胞j774a.1),考察tak-632对tsz刺激后不同细胞系坏死的抑制活性。
结果如图3a~d所示,tak-632在低浓度(0.1~5μm)下能显著保护人来源和鼠来源细胞发生程序性坏死。
实施例4、tak-632可通过抑制ripk1/ripk3相互磷酸化发挥抗细胞坏死活性
ht-29细胞在tak-632(10μm)处理30min后,用tnf-α(20ng/ml)刺激不同时间点,观察iκbα蛋白的表达,结果表明iκbα蛋白的表达没有显著变化,表明tak-632抑制坏死的作用与iκbα相关的凋亡通路无关(图4a)。
ht-29细胞在tak-632(10μm)处理30min后,用tsz刺激不同时间点,考察ripk1,ripk3,mlkl及其磷酸化蛋白的表达,如图4b所示,tak-632作用2,4,6h后,可以显著抑制ripk1,ripk3和mlkl相互磷酸化蛋白的表达,但是对ripk1,ripk3和mlkl蛋白本身的表达量没有影响。
同时,本发明人在小鼠l929细胞中发现tak-632同样可以抑制ripk3和mlkl磷酸化的作用(图4c)。
进一步,采用免疫共沉淀的方法(ip),考察了tak-632能否抑制ripk1和ripk3在tsz坏死激活后的相互作用,结果表明在tsz处理4h之后,tak-632可直接抑制细胞内ripk1和ripk3的相互作用(图4d)。ripk1与ripk3是同源的两类丝氨酸/苏氨酸激酶,它们是介导细胞坏死性凋亡的关键元件,因此,tak-632通过抑制ripk1/ripk3相互磷酸化发挥抗细胞坏死活性。
实施例5、生物素标记的tak-632分子探针制备
合成tak-632分子探针,合成路线如下:
将化合物1(710mg,1.79mmol)溶于100ml甲苯中,加入戌二酸酐(409mg,3.58mmol),混合液在氮气保护下加热至130℃反应5小时,tlc显示反应完全,冷却反应液至室温,浓缩,再用50ml乙酸乙酯稀释,无水硫酸钠干燥。滤去不溶物,浓缩滤液,最后用柱层析分离(洗脱液:石油醚/乙酸乙酯=3:1)得到化合物2(582mg)。1h-nmr(dmso-d6,300mhz):δ12.71(s,1h),12.12(s,1h),11.39(s,1h),8.04(d,j=9.0hz,1h),7.46(t,j=9.5hz,1h),7.36(dd,j=6.1,3.0hz,1h),7.29–7.18(m,1h),7.15(d,j=9.0hz,1h),2.59(t,j=7.3hz,2h),2.30(t,j=7.2hz,2h),1.94–1.78(m,2h)。其中,化合物1的合成参照文献(okaniwam,etal.discoveryofaselectivekinaseinhibitor(tak-632)targetingpan-rafinhibition:design,synthesis,andbiologicalevaluationofc-7-substituted1,3-benzothiazolederivatives.jmedchem.2013,56(16):6478-6494)。
将20ml甲醇冷却至0℃后缓慢滴加氯化亚砜(44μl,0.57mmol),滴毕,在0℃下继续搅拌30分钟,加入化合物2(100mg,0.19mmol),2小时后tlc显示反应完全,直接将反应液浓缩得到甲基化产物用于下步反应。将0.5ml甲醇加入到硼氢化钠(130mg,3.45mmol)的乙醇(20ml)溶液中,再将甲基化产物(90mg,0.17mmol)在0℃下加入,并继续反应30分钟后,升至室温反应30分钟,tlc显示反应完全。混合液用乙酸乙酯稀释,5%碳酸氢钠溶液和盐水洗,有机相用无水硫酸钠干燥,滤去不溶物后浓缩滤液,柱层析分离得到化合物3(55mg,76%).1h-nmr(dmso-d6,300mhz):δ12.68(s,1h),8.00(d,j=9.0hz,1h),7.25–6.89(m,2h),6.50(dd,j=7.6,3.0hz,1h),6.27(dt,j=8.7,3.3hz,1h),5.42(s,2h),3.60(s,3h),2.59(t,j=7.3hz,2h),2.40(t,j=7.3hz,2h),1.97-1.83(m,2h)。
化合物3(20mg,0.046mmol)和2-(3-三氟甲基)苯乙酸(10mg,0.046mmol)溶于20ml吡啶中,室温加入2-(7-偶氮苯并三氮唑)-n,n,n’,n’-四甲基脲六氟磷酸酯(hatu,36mg,0.094mg)后,将混合物加热至85℃反应8小时,tlc监测显示反应完毕,浓缩反应液。剩余物用乙酸乙酯稀释,5%碳酸氢钠溶液和盐水洗,有机相用无水硫酸钠干燥,滤去不溶物后浓缩滤液,柱层析分离得到化合物4(9mg,31.8%).1h-nmr(dmso-d6,300mhz):δ12.68(s,1h),10.23(s,1h),7.99(d,j=9.0hz,1h),7.84(dd,j=6.4,3.0hz,1h),7.68(s,1h),7.51-7.61(m,,3h),7.38(dd,j=10.5,9.1hz,1h),7.08(d,j=9.0hz,1h),6.98(dt,j=8.9,3.5hz,1h),3.87(d,j=13.2hz,2h),3.60(s,3h),2.59(t,j=7.3hz,2h),2.40(t,j=7.3hz,2h),1.97-1.83(m,2h)。
化合物4(100mg,0.162mmol)溶于20mlthf/meoh/h2o(2:2:1)混合液,加入1mol/l的氢氧化钠溶液3ml。反应液在室温下反应5小时,调节混合液的ph值至2左右,析出酸化产物,过滤得到产品用于下一步。将酸化产物(83mg,0.138mmol)溶于四氢呋喃中,加入pybop(79mg,0.152mmol),n-叔丁氧羰基-1,3-丙二胺(24mg,0.152mmol),三乙胺(15.35mg,0.152mmol),室温搅拌8小时,tlc显示反应完全,滤去不溶物,浓缩滤液,柱层析得到化合物5,86mg,收率83.8%。将86mg化合物5加入5ml二氯甲烷中,在加入1ml三氟乙酸,室温反应2小时,浓缩得到氨基粗产物。将20mg氨基粗产物溶于5mldmf中,加入dipea(6mg,0.046mmol)和(+)生物素-n-琥珀酰亚胺基酯(16mg,0.05mmol),室温反应15小时,用乙酸乙酯萃取反应液,浓缩有机相,柱层析分离得到生物素标记的tak-632目标化合物,10mg,37.0%1h-nmr(dmso-d6,600mhz):δ10.17(s,1h),7.91–7.90(m,2h),7.76–7.73(m,2h),7.67(s,1h),7.60–7.59(m,2h),7.54–7.53(m,1h),7.33–7.30(m,1h),6.96(d,j=8.9hz,1h),6.89–6.87(m,1h),6.39(s,1h),6.32(s,1h),4.29–4.27(m,1h),4.12–4.09(m,1h),3.85(s,2h),3.08–3.05(m,5h),2.80–2.77(m,1h),2.56(m,1h),2.38–2.35(m,2h),2.10(t,j=7.4hz,2h),2.05(t,j=7.2hz,2h),1.84–1.79(m,3h),1.59–1.42(m,5h).hrms(esi+)m/zcalculatedforc40h41f4n8o6s2869.2521;observed869.2523(m+h+),867.2378(m-h-),891.2342(m+na+).hplc纯度:97.46%,rt=24.964min,uv254nm.
实施例6、细胞坏死抑制剂tak-632的靶点确认
采用分子对接的方法考察tak-632与ripk1和ripk3的相互作用。tak-632的三氟苯基可以占据ripk1的疏水口袋(lys45,leu90和met92)。苯氧基-苯并[d]噻唑部分可以完全插入到ripk1激酶的“开放”环中,环丙基暴露在溶剂中(图5a)。采用同源的b-raf结构(pdbid:4ksp)和tak-632重构ripk3的三维结构,tak-632被对接到同源模型ripk3的atp结合口袋中。相似的是,三氟苯基可占据ripk3疏水口袋(lys127,ala146和asn125)(图5b),环丙烷甲酰胺部分在酰胺的羰基和his-85的nh之间形成氢键。
采用药物亲和响应靶标稳定性试验(darts),依赖于靶蛋白对药物结合的蛋白酶敏感性的降低,以检测tak-632和ripk1/3激酶之间的潜在相互作用。如图5c所示,在tak-632处理的细胞的提取物中,ripk1和ripk3都可以保护0.01%pronase蛋白酶的消化作用,而在相同的样品中没有检测到mlkl的保护,提示tak-632可能与ripk1和ripk3相互作用。
通过将长链生物素替换其环丙基来合成生物素化的tak-632(图5d),发现生物素化的tak-632能够在较高浓度下抑制tnf-α诱导的坏死,为了证实tak-632和ripk1/3之间的相互作用,通过在未刺激的ht-29细胞裂解物中使用生物素化的tak-632进行pull-down实验。如图5d所示,ripk1和ripk3都被生物素化的tak-632富集,而mlkl不在同一复合物中,这表明了tak-632与ripk1和ripk3相互作用的特异性。因此,该结果证明,tak-632在tsz-未刺激的细胞中可与ripk1和ripk3直接结合。
实施例7、细胞坏死抑制剂tak-632抑制ripk1和ripk3的激酶活性
用flag-ripk1或ripk3-v5转染hek293t细胞。12h后,用tak-632不同浓度处理6h。然后通过sds-page分析细胞裂解物,并用所示抗体进行免疫印迹。本发明人发现,tak-632剂量依赖性地抑制hek293t细胞中ripk1和ripk3的自身磷酸化,表明tak-632抑制ripk1和ripk3的激酶活性(图6a和6b)。因为ripk1和ripk3的激酶活性对于ripk1-ripk3坏死体形成和ripk3-mlkl相互作用是必需的,所以本发明人进一步考虑tak-632是否阻断坏死形成和ripk3-mlkl相互作用。为此,本发明人在hek293t细胞中共表达rip3-v5和flag-ripk1或flag-mlkl,并利用tak-632进行共免疫沉淀测试。如图6c和6d所示,tak-632不仅阻断了ripk1-ripk3的相互作用,而且阻止了ripk3-mlkl的相互作用。因此,这些结果表明,tak-632可抑制两种ripk1/3的激酶活性,从而破坏ripk1-ripk3和ripk3-mlkl复合物的形成。
实施例8、细胞坏死抑制剂tak-632在体内保护小鼠抵抗tnf诱导的全身免疫综合征(sirs)和对乙酰氨基酚(apap)诱导的肝毒性
为了探索tak-632是否能够在体内保护rip激酶驱动的炎症,本发明人首先在tnf诱导的全身炎症反应综合征(sirs)模型中测试了它。静脉注射tak-632前3小时灌胃给药。注射mtnf-α,本发明人发现tak-632显著保护小鼠免于低温和死亡(图7a和b)。此外,当这些小鼠在6小时检查时,通过tak-632处理显著降低了il-6的血清水平(图7c)。因此,这些结果表明tak-632在体内保护免受tnf诱导的sirs。以前的研究表明ripk1是apap诱导肝毒性所必需的。然后本发明人研究tak-632对这个模型的组织损伤和炎症的影响。对乙酰氨基酚治疗(300mg/kg)导致显著的肝损伤,导致肝脏细胞坏死和血浆天冬氨酸转氨酶(ast)水平升高(图7d,e和f)。然而,tak-632预处理可显著降低对乙酰氨基酚处理小鼠的肝坏死和血浆ast水平(图7d,e和f)。
总的来说,本发明人的研究结果表明,tak-632可以保护ripk1/3激酶介导的组织损伤和体内炎症。
以上实验结果表明,本发明的化合物tak-632具有优异的抗细胞坏死活性,可作为ripk1/ripk3抑制剂,可抵抗tnf诱导的全身免疫综合征(sirs)和对乙酰氨基酚(apap)诱导的肝毒性,用于制备抗阿尔茨海默病、抗缺血性心肌病、抗缺血性脑卒中、抗动脉粥样硬化、抗急性胰腺炎、抗儿童炎症性肠病、抗脓毒血症、抗沙门氏菌感染、抗李斯特菌感染、抗牛痘病毒感染等炎症或感染性相关疾病的药物。
综上,因此本发明化合物及其盐类可以用于制备抗细胞坏死抑制剂。
在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
- 还没有人留言评论。精彩留言会获得点赞!
- SPNP‑I在制备Nav1.7通道工具试剂中的应用的制造方法与工艺
- SPNP‑I在制备Nav1.2通道工具试剂中的应用的制造方法与工艺
- SPNP‑IX在制备Nav1.3通道抑制剂中的应用的制造方法与工艺
- SPNP‑IX在制备Kv4.2通道工具试剂中的应用的制造方法与工艺
- SPNP‑IX在制备Kv4.3通道工具试剂中的应用的制造方法与工艺
- SPNP‑27在制备Nav1.5通道抑制剂中的应用的制造方法与工艺
- SPNP‑27在制备Nav1.3通道抑制剂中的应用的制造方法与工艺
- 一种多肽在制备Nav1.3钠通道工具试剂中的应用的制造方法与工艺
- SPKP‑XI在制备Kv4.2通道抑制剂中的应用的制造方法与工艺
- SGLT‑2糖尿病抑制剂及其中间体的制备方法与流程