糖皮质激素受体促进剂樟芝酸A在抑制乳腺癌肿瘤发生与转移中的应用的制作方法
本发明有关于一种抑制乳癌肿瘤发生与转移的糖皮质激素受体促进剂,尤指涉及一种不论是合成或天然的糖皮质激素受体(glucocorticoidreceptor,gr)促进剂皆可诱导微小核糖核酸-708(microrna-708,mir-708)表现,进而抑制乳癌肿瘤发生与转移,特别指透过转录活化糖皮质激素受体α(grα)活化mir-708造成乳癌细胞增殖减少及诱导细胞周期停滞,主要是透过转录抑制nf-κb及其靶基因cox-2与cmyc的作用。
背景技术:
乳癌是全世界女性中最常见的恶性肿瘤与导致癌症相关死亡的主要原因,每年约有130万例乳癌新病例被诊断出来,估计有50万人死于乳癌[1],根据国际癌症研究机构(internationalagencyforresearchoncancer,iarc)的报告,乳癌占所有癌症的11.88%,是全球第二大最常见的恶性肿瘤[2],这些统计结果显示乳癌的检测、诊断与治疗仍是目前急需改善的重要议题。
糖皮质激素(glucocorticoids,gcs)是属于皮质类固醇的一种,主要功能与代谢、存活、增殖及细胞分化相关,作用途径大部分是通过糖皮质激素受体(glucocorticoidreceptor,gr)。糖皮质激素受体是一种转录因子,属于核受体超家族成员[3],糖皮质激素与糖皮质激素受体结合的皮质类固醇,几乎存在于所有脊椎动物细胞中[4],合成糖皮质激素,如地塞米松(dexamethasone,dex)、倍他米松(betamethasone)、强的松(prednisone)与甲泼尼龙(methylprednisolone)被广泛用作抗发炎药物。事实上,在许多癌症治疗中,地塞米松常被用在化学治疗之前的预防药物,用以防止超敏反应[5]。越来越多的体外与体内研究发现,地塞米松具有抑制前列腺癌[6]、黑色素瘤[6]与部分恶性淋巴癌肿瘤生长并诱导癌症细胞凋亡的作用[7]。最近的一项研究显示,糖皮质激素受体拮抗剂米非司酮(mifepristone)可增加三阴性乳癌(triplenegativebreastcancer,tnbc)细胞对紫杉醇的敏感性,但是若只用米非司酮处理三阴性乳癌,则对癌细胞存活率无显著影响[8],此外,在异种移植肿瘤动物实验发现,以地塞米松预先治疗动物,可显著加强紫杉醇对肿瘤的抑制效果[9]。迄今为止,没有其他前瞻性研究探讨糖皮质激素促进剂对乳癌肿瘤生长、癌症干细胞分化与转移的影响。因此,需要进一步研究厘清糖皮质激素在乳癌治疗中的确切作用与应用。
糖皮质激素促进剂诱导的细胞凋亡与肿瘤生长似乎涉及多个信号传导途径的复杂过程,包含藉由核因子活化b细胞κ轻链增强子(nuclearfactorkappa-light-chain-enhancerofactivatedbcells,nf-κb)的转录抑制机制与转录活化诱导细胞凋亡的相关基因,包括bim1进而负调控存活、增殖与细胞周期调控相关基因,如cox-2、cmyc及cyclind1[7]。nf-κb是rel家族的一个转录因子,在生存、增殖、分化、发炎及免疫方面扮演相当重要的功能。在大多数乳癌肿瘤中经常发现若不断地活化nf-κb,nf-κb的活化失调会导致p50、p52、p65、crel及relb等蛋白质持续性定位于细胞核内,进而透过抗凋亡基因的上调,破坏细胞增殖与死亡间的平衡[10]。癌症干细胞(cancerstemcells,cscs)或肿瘤初始干细胞(tumorinitiatingstem-likecells,tics)是肿瘤起始细胞的一小部分,其能够自我更新并抵抗各种化学治疗药物。癌症干细胞已在多种癌症中发现,包括脑癌、乳癌、卵巢癌、前列腺癌、胰脏癌与大肠癌[11]。乳癌干细胞的自我更新行为涉及wnt/β-catenin蛋白、notch、hedgehog与nf-κb等数个讯号传递途径[12],其中,nf-κb特别受到关注,因为它是大多数乳癌肿瘤中常见的特征,nf-κb的转录活性在乳癌干细胞自我更新中发挥重要作用[13]。然而,目前尚未有研究阐明gr促进剂引发的抑制nf-κb与乳癌的干细胞分子机制。
微小核糖核酸(micrornas,mirna)经常于植物、动物与病毒基因组被发现,是一种内源性、非编码的小rna分子,长度约为22个核苷酸,其主要功能为抑制rna与基因转录后调控作用。mirna可直接与转录本的3'非转录区(3′utr)结合,在各种生物过程[14]包括癌细胞增殖、转移、癌症干细胞分化与其他生物功能中抑制转录作用及/或诱导mrna降解[15]。mirna的发现为癌症研究提供了新见解。在2005年,iorio等人首先发表了mirna对乳癌的调控[16],他们发现29种mirna在乳房肿瘤组织与正常乳腺组织表现上的差异。mir-708是一种新发现的mirna,可抑制许多类型的癌症,包括肾细胞癌、肝癌、卵巢癌、前列腺癌、乳癌、多形性胶质母细胞瘤及尤文氏肉瘤。然而,在少数癌症类型中,mirna是具有致癌性的mirna,特别是在膀胱癌、大肠直肠癌及某些肺癌中[17]。最近的一项研究报导,在慢性淋巴细胞性白血病中,mir-708的表观遗传抑制藉由直接作用于ikkβ而增强nf-κb信号传导[18]。接着在2015年,ma等人研究发现,在三阴性乳癌细胞中强制表达mir-708,可透过作用于离胺酸特异性组织蛋白去甲基化酶(lysine-specifichistonedemethylase,lsd1)进而抑制三阴性乳癌细胞的增殖与侵袭能力[19]。然而,只有少数研究着眼于mir-708在乳癌中的功能,而且相关的分子作用机制尚有一大部分未被探讨。
合成的gr促进剂被广泛用在化疗期间的辅助治疗,而天然存在的gr促进剂或植物固醇则很少被研究。植物固醇在发炎与免疫系统中是极具潜力的调节剂[20],本案申请人之前曾发现从药用真菌牛樟芝中分离得到的固醇植物化学物质樟芝酸a(antcina,ata)具有模拟gr促进剂作用,可抑制肺细胞的发炎反应,这种效应与合成的gr促进剂(如可的松(cortisone)与地赛米松)很相似[21]。此外,关于乳癌中通过gr促进剂介导的信号传导途径对mirna的调节知之甚少。故,一般无法符合使用者于实际使用时所需。
技术实现要素:
本发明的主要目的在于,克服已知技术所遭遇的上述问题,并提供一种糖皮质激素受体促进剂樟芝酸a在抑制乳腺癌肿瘤发生与转移中的应用,不论是合成或天然的gr促进剂皆可诱导mir-708表现,进而抑制乳癌肿瘤发生与转移,透过转录活化糖皮质激素受体α(grα)活化mir-708造成乳癌细胞增殖减少及诱导细胞周期停滞,其主要是透过转录抑制nf-κb及其靶基因cox-2与cmyc的作用。
本发明的次要目的在于,提供一种藉由gr促进剂樟芝酸a来诱导mir-708表现,以有效抑制人类乳癌细胞增殖、细胞周期进程以及肿瘤干细胞表现与癌细胞转移。
本发明的另一目的在于,提供一种使用糖皮质激素受体促进剂或mir-708转染处理细胞,以显著抑制ikkβ的表现,也抑制nf-κb活性及其下游目标基因,包括cox-2、cmyc、cyclind1、mmp-2、mmp-9、cd24及cd44的表现,并提高参与癌细胞增生、细胞周期进程、转移及肿瘤干细胞标记蛋白p21cip1与p27kip1的表现量。
本发明的再一目的在于,提供一种用gr促进剂樟芝酸a来治疗动物,可显著抑制肿瘤生长以及肿瘤重量与体积,从而以一新型治疗方式应用于乳癌治疗。
为达以上目的,本发明所采用的技术方案是:一种糖皮质激素受体促进剂樟芝酸a在抑制乳腺癌肿瘤发生与转移中的应用,其中,该应用是指将樟芝酸a与乳腺癌细胞反应后,透过转录活化grα活化mir-708,抑制该乳腺癌细胞的增生并诱发细胞周期停滞,而藉由该樟芝酸a的讯号传递诱发mir-708的表现后抑制nf-κb活性,以及抑制调控其下游目标基因cox-2与cmyc的表现。
于本发明上述实施例中,该应用更包含抑制肿瘤干细胞表现。
于本发明上述实施例中,该应用更包含抑制nf-κb下游目标基因cyclind1、mmp-2、mmp-9、cd24及cd44的表现。
于本发明上述实施例中,该乳腺癌细胞包含非恶性人类乳腺癌细胞株(mcf-7)与高度恶性人类乳腺癌细胞株(mda-mb-231)。
本发明同时提供糖皮质激素受体促进剂樟芝酸a在制备抑制乳腺癌肿瘤发生与转移药物中的应用。
于本发明上述实施例中,该药物还包括医药上可接受的载体或赋形剂,该载体或赋形剂基于施用类型来选择,并且可以包括但不限于局部的载体或赋形剂、静脉注射、皮内注射、口服或皮下的载体或赋形剂。
于本发明上述实施例中,该药物以1~20μm的浓度施用。
于本发明上述实施例中,该樟芝酸a从牛樟芝子实体中分离。
于本发明上述实施例中,该樟芝酸a根据高效液态层析仪(hplc)与质谱仪(1h-nmr)分析,其纯度高于99%。
附图说明
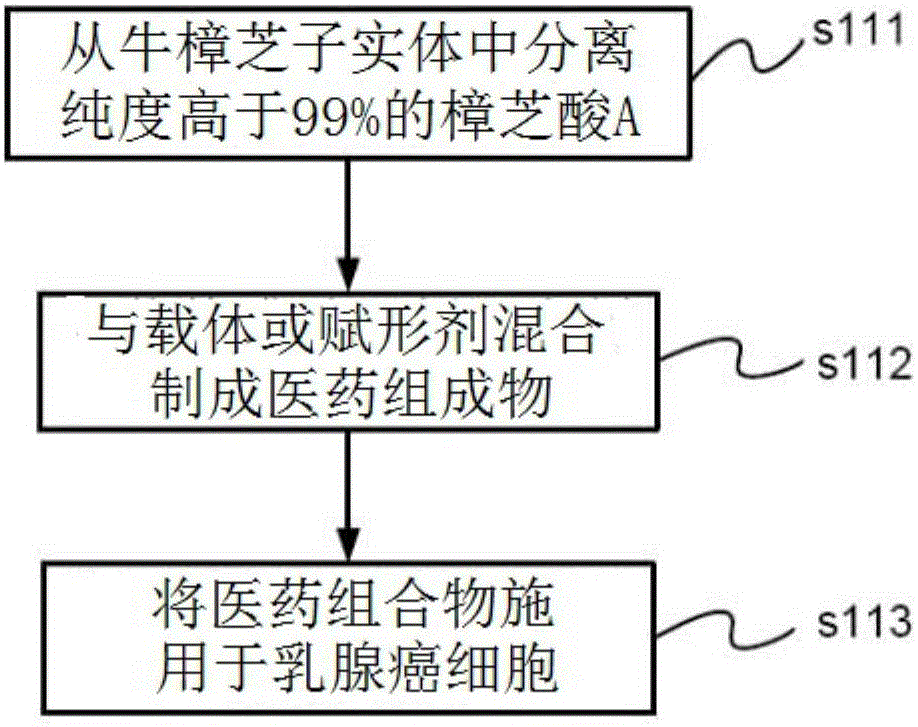
图1是本发明投药ata调节乳癌肿瘤发生与转移的流程示意图。
图2是本发明的gr促进剂通过grα信号活化乳癌细胞中的mir-708示意图;
其中,a为定量rt-pcr分析mir-708在正常、非恶性及恶性人类乳癌细胞中的基因表现;b为pre-mir-708转染72小时后在乳癌细胞中mir-708的表现;c为乳癌细胞处理dex(1μm)及ata(10μm)72小时后mir-708的表现,对照为未进行dex和ata处理;d为mir-708在乳癌细胞经转染杂乱sirna(scsirna)或grα特异性sirna(sigrα)后,利用dex或ata处理72小时的表现,以未进行dex和ata处理作为对照;e为乳癌细胞用米非司酮(mifepristone)预处理2小时后再用dex或ata处理72小时,测量mir-708的表现,前一个对照为未进行dex和ata及米非司酮处理,后一个对照表示仅进行米非司酮处理;f为用放线菌酮(cycloheximide,chx,10μm)预处理乳癌细胞12小时后再用dex或ata处理72小时测定mir-708表现,利用u6基因计算标准化mir-708的含量,对照为未进行dex和ata及chx处理;g为用dex或ata处理乳癌细胞48小时后grα的荧光素酶活性(rlu,相对荧光素酶单位),对照为未进行dex和ata处理;h为用dex或ata处理48小时后,通过免疫荧光测量乳癌细胞中grα的细胞定位,dapi用于细胞核染色,对照为未进行dex和ata处理;merge指dapi及grα两种染色图合并;i为用dex或ata处理48小时后细胞质与细胞核中grα的蛋白质表现,β-actin与laminb1蛋白分别是细胞质与细胞核组的内部对照,对照为未进行dex和ata处理。
图3是本发明投予dex或ata对乳癌细胞存活率的影响示意图;
其中,a是将mcf-7细胞与ata(1~20μm)或dex反应24~72小时;b是将mda-mb-231细胞与递增浓度的ata(1~20μm)或dex(0.1~10μm)一起反应24~72小时。
图4是本发明使用流式细胞仪分析annexinv/pi染色乳癌细胞凋亡情况的示意图。
图5是本发明mirna-708/grα讯号途径抑制乳癌细胞的增生示意图;
其中,a为投予dex或ata处理乳癌细胞72小时后,使用市售细胞增生试剂测定乳癌细胞增生作用;b为用pre-mir-708转染乳癌细胞72小时后,乳癌细胞增生情况,以未用pre-mir-708转染为对照;c为dex或ata处理乳癌细胞72小时后,使用pi染色后藉由流式细胞仪测量细胞周期分布;d为pre-mir-708转染乳癌细胞72小时,以未用pre-mir-708转染为对照;e为dex或ata处理乳癌细胞72小时后,利用西方墨点分析细胞周期调控蛋白的表现;f藉由免疫墨点法分析cdk抑制剂的蛋白质表现,β-actin蛋白作为内部控制。a、c、e及f中以未进行dex或ata处理为对照。
图6是本发明利用台盼蓝排除试验评估细胞增生情况的示意图;
其中,a和b分别为使用pre-mir-708暂时转染乳癌细胞mcf-7及mda-mb-231,并在24、48及72小时后测量细胞的增生情况;c和d分别为乳癌细胞mcf-7及mda-mb-231使用dex或ata处理24~72小时后的活细胞数量。
图7是本发明mirna-708/grα讯号传递途径调控体外与体内肿瘤的生长示意图。
其中,a为将乳癌细胞接种到6孔培养板上,每孔含有200个细胞,细胞沉降后,使用dex或ata处理14天,以未进行dex或ata处理为对照。观察肿瘤球形成并计数,直方图中显示每孔中肿瘤球的数量;b为使用pre-mir-708转染细胞14天后测定肿瘤球的数量,以未用pre-mir-708转染为对照;c为利用scsirna或sigrα转染乳癌细胞后再以dex或ata处理14天,检测肿瘤球形成情况,对照表示未进行dex或ata处理;d和e为利用膜蛋白萃取试剂萃取细胞膜蛋白,接着利用免疫墨点法分析cd44与cd24的蛋白质表现,α-tubulin作为内部控制,d中以未进行dex或ata处理为对照,e中以未用pre-mir-708转染为对照;f为利用dex或ata处理人类乳癌肿瘤异种移植小鼠7周,以未进行dex或ata处理为对照;g为在治疗结束时(10周)时测量的肿瘤体积;h为在治疗结束时(10周)时测量的肿瘤重量;i为肿瘤组织学检查显示的肿瘤细胞坏死情况;j为肿瘤组织学检查显示的肿瘤细胞凋亡图(h&e染色;放大率100×),g、h、i及j以未进行dex或ata处理为对照。
图8是本发明gr促进剂在体外试验可抑制乳癌细胞的转移示意图。
其中,a为在存在或不存在tgf-β的情况下,用dex或ata处理乳癌细胞24小时后,利用伤口愈合试验分析细胞迁移情形,在直方图中显示肿瘤细胞迁移的倍数增加;b为在存在或不存在tgf-β之情况下,用dex或ata处理乳癌细胞24小时后,通过transwell试验法测定肿瘤细胞侵袭作用,在0小时与24小时使用倒置显微镜拍摄照片(放大倍率10倍);c为使用特异性抗体藉由西方墨点法分析mmp-2与mmp-9的蛋白质表现;d为通过定量q-pcr测定mmp-2与mmp-9的mrna表现;e为将乳癌细胞转染scsirna或sigrα后与dex或ata反应24小时,测定乳癌细胞迁移,直方图中显示了肿瘤细胞迁移的百分比。a、b、c、d中以在存在或不存在tgf-β的情况下未进行dex或ata处理为对照。
图9是本发明mirna-708/grα讯号途径具有专一调控乳癌细胞中的ikkβ表现作用示意图。
其中,a为mir-708的目标位点落在ikbkb(ikkβ)3'utr的位置;b为pre-mir-708转染细胞48小时后乳癌细胞中ikkβ的启动子活性,以未用pre-mir-708转染为对照;c为用pre-mir-708转染细胞48小时后,乳癌细胞中ikk亚基(subunits)的mrna表现,以未用pre-mir-708转染为对照;d为用dex或ata处理细胞48小时后,乳癌细胞中ikk亚基的mrna表现,以未进行dex或ata处理为对照;e为利用西方墨点法分析ikkα与ikkβ蛋白表现,以未进行dex或ata处理为对照;f为将细胞转染scsirna或sigrα后与dex或ata反应48小时,分析ikkβmrna的表现,以未转染scsirna或sigrα为对照;g为将细胞转染scsirna或sigrα后与dex或ata反应48小时,ikkα与ikkβ的蛋白表现,以未转染scsirna或sigrα为对照。
图10是本发明mirna-708抑制乳癌细胞中nf-κb的活性示意图。
其中,a为pre-mir-708转染细胞48小时后乳癌细胞中nf-κb的荧光素酶活性,以未用pre-mir-708转染为对照;b为用dex或ata处理48小时后乳癌细胞中nf-κb的荧光素酶活性,以未进行dex或ata处理为对照;c为用pre-mir-708转染细胞48小时后,细胞质与细胞核中nf-κb的蛋白表现,gapdh与laminb1分别作为细胞质与细胞核的内部对照,以未用pre-mir-708转染为对照;d为dex或ata处理细胞48小时后,细胞质与细胞核中nf-κb的蛋白表现,gapdh与laminb1分别作为细胞质与细胞核的内部对照,以未进行dex或ata处理为对照;e为用scsirna或sigrα转染细胞后与dex或ata反应48小时的nf-κb荧光素酶活性,以未用scsirna或sigrα转染为对照;f为细胞转染sirna或sigrα后与dex或ata反应48小时,细胞质与细胞核中nf-κb的蛋白表现,以未用scsirna或sigrα转染为对照;g为肿瘤异种移植物中nf-κb的免疫组织化学染色,以未进行dex或ata处理为对照;h为细胞转染pre-mir-708转染48小时后,乳癌细胞中i-κbα的蛋白质表现及其磷酸化蛋白表现,以未用pre-mir-708转染为对照;i为用dex或ata处理细胞48小时后,乳癌细胞中i-κbα的蛋白质及其磷酸化蛋白表现,以未进行dex或ata处理为对照。
图11是本发明gr促进剂藉由活化mir-708抑制nf-κb下游目标基因示意图。
其中,a为pre-mir-708转染细胞48小时后,乳癌细胞中cox-2与cmyc的mrna表现,以未用pre-mir-708转染为对照;b为乳癌细胞经dex或ata处理48小时后,利用西方墨点法分析cox-2与cmyc蛋白表现,以未进行dex或ata处理为对照;c为乳癌细胞经pre-mir-708转染细胞48小时后,细胞中cox-2与cmyc蛋白的表现,以未用pre-mir-708转染为对照;d为人类乳癌肿瘤异种移植动物对照组、dex或ata处理组的cox-2免疫组织化学染色;e为细胞经scsirna或sigrα转染再与dex或ata反应48小时后cox-2mrna的表现,以未经dex或ata处理为对照。
具体实施方式
本发明为糖皮质激素受体促进剂樟芝酸a在抑制乳腺癌肿瘤发生与转移中的应用,是针对乳癌具有抗肿瘤与抗转移作用。本发明揭示了樟芝酸a(antcina,ata)抑制乳线癌肿瘤发生与转移的作用并阐明了该作用的分子机制。
请参阅图1所示,是本发明投药ata调节乳癌肿瘤发生与转移的流程示意图。如图所示:了解将ata作为医药组成物给药以抑制乳癌肿瘤发生与转移。在该流程中,如第一步骤s111,来自牛樟芝的材料,分离ata。在一些实施例中,ata是从牛樟芝子实体中分离。在一些实施例中,ata的纯度为99%。
在第二步骤s112中,ata与医药上可接受的载体或赋形剂混合制成医药组成物。医药上可接受的载体或赋形剂可以基于施用类型来选择,并且可以包括但不限于局部的载体或赋形剂、或者是静脉注射、皮内注射、口服或皮下的载体或赋形剂。
第三步骤s113涉及将乳癌细胞投予有效量的医药组成物以抑制乳癌细胞的增生与转移。在至少一个实施例中,该医药组成物允许与非恶性人类乳癌细胞株(mcf-7)及高度恶性人类乳癌细胞株(mda-mb-231)以任何方式进行处理。在一些实施例中,将医药组成物注射于腹腔以处理乳癌细胞,并且医药上可接受的载体或赋形剂是允许易于施用的二甲基亚砜(dimethylsulfoxide,dmso)或其他物质。
在其他实施例中,该有效量为1μm至20μm的浓度,包括但不限于:2μm、3μm、4μm、5μm、6μm、7μm、8μm、9μm、10μm、11μm、12μm、13μm、14μm、15μm、16μm、17μm、18μm及19μm。在其他实施例中,该有效量为10μm至20μm。
本发明将进一步藉由下面的实施例来做说明,但应明了的是,该些实施例仅为说明之用,而不应被视为本发明实施上的限制。
制备例:材料与方法
一、化学药剂:
如前所述,ata是从牛樟芝子实体中分离出来,其方法如之前已发表的研究所述[21]。根据高效液态层析仪(hplc)及质谱仪(1h-nmr)分析证实ata的纯度高于99%。dmem培养基(dulbecco'smodifiedeagle'smedium)购自invitrogen(carlsbad,ca)。rpmi培养基(roswellparkmemorialinstitute)、最低必需培养基(minimumessentialmedium)、胎牛血清(fetalbovineserum,fbs)、丙酮酸钠(sodiumpyruvate)、青霉素(penicillin)、链霉素(streptomycin)、地塞米松(dexamethasone,dex)、噻唑蓝(3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazoliumbromide,mtt)、4',6-二脒基-2-苯基吲哚(4',6-diamidino-2-phenylindole,dapi)及环己酰亚胺(cycloheximide)皆购自sigma-aldrich(st.louis,ca)。本发明中使用的抗体列于表一。所有其他化学品均为试剂级或hplc级,购自merck(darmstadt,germany)或sigma-aldrich。
表一本发明使用的抗体
二、方法:
细胞培养、样品处理及转染
人类正常乳腺上皮细胞株(mcf-10a)与乳癌细胞株mcf-7及mda-mb-231皆购自中国台湾新竹的台湾生物资源保存和研究中心(bcrc)。利用dmem/ham's-f12、mem与rpmi培养基(该三种细胞培养基均含有10%fbs、1.5g/l碳酸氢钠与100u/l青霉素及链霉素)分别培养mcf-10a、mcf-7及mda-mb-231三种细胞,将细胞培养在37℃、5%二氧化碳(co2)的培养箱中。利用地赛米松(dex)(1μm)或ata(10μm)分别处理细胞24~72小时。另一方面,利用lipofectamine2000(invitrogen)转染小分子干扰核糖核酸(smallinterferingrna,sirna)或微小核糖核酸(microrna,mirna)模拟物至细胞中。每次转染使用20nmpre-mirna或sirna。利用20ng/ml的肿瘤坏死因子-α(tumornecrosisfactor-α,tnf-α)刺激细胞。对照组为分别含有0.1%dmso的dmem/ham's-f12、mem与rpmi培养基。
细胞增生及细胞周期分析
利用市售细胞增生测定试剂(celltiteraqueousonesolutioncellproliferationassay,promega)进行细胞增生试验。细胞周期试验则使用流式细胞仪(beckmancoulterfc500)进行分析。简言之,将上述培养的乳癌细胞以1×105个细胞/孔的细胞数分别培养在6孔培养盘中,加入pre-mir-708或dex与ata至培养盘中处理72小时。接着收集细胞,在1500×g下离心3分钟,利用95%冰乙醇固定细胞,在-20℃下反应过夜;用pbs洗涤细胞,再以1500×g离心3分钟,然后加入含有1mlpi/tritonx-100(20μg/mlpi、0.1%tritonx-100与2.5μg/mlrnase)的染剂,在冰上反应30分钟。利用流式细胞仪分析细胞的dna含量,至少分析10000个细胞数资料。细胞周期的分析则是使用cxp软件(beckmancoulter)进行分析。
本发明另利用台盼蓝排除试验(trypanblueexclusionassay)评估细胞增生情况,将上述增生后的乳癌细胞分别以5x104个细胞/孔接种到6孔细胞培养盘中,培养过夜后,将细胞使用pre-mir-708转染,或使用dex或ata处理24~72小时,接着以0.2%台盼蓝进行细胞染色,再利用细胞计数器计数染色的细胞。活细胞的百分比为计算每孔中活细胞占总细胞的比率。细胞增生比率为样品处理组的活细胞数量相对于对照组的活细胞量。细胞凋亡检测
根据制造商的说明书(bdbiosciences,sanjose,ca),用annexinv-fitc/pi凋亡检测试剂进行annexinv-fitc/pi染色测定。简言之,将染色后的5×105个细胞/培养盘接种于10cm培养盘中,培养过夜后,将细胞暴露于dex或ata24小时,接着用pbs洗涤两次并使用不含乙二胺四乙酸(ethylenediaminetetraaceticacid,edta)的0.25%胰蛋白酶处理细胞,在1500xg下离心5分钟后收集细胞。然后,将细胞悬浮于500μl含有1μlannexinv-fitc及5μlpi的结合缓冲液中,在黑暗中反应5分钟后,藉由流式细胞仪直接分析染色的细胞。使用cxp软件(beckmancoulter)采集与分析数据。
癌症球型细胞形成试验
乳癌细胞的肿瘤球型细胞形成测定,先加入200μl基底膜基质(matrigel,bdbiosciences,sanjose,ca)至6孔培养盘中,并使其在37℃聚合15分钟。接着将mcf-7与mda-mb-231细胞(5×104个细胞/孔)分别放入1ml的mem或dmem培养基(培养基中含有10%基底膜基质及10%fbs)中。将培养盘放在37℃反应使细胞完全沉降,然后加入含有5%matrigel的mem或dmem细胞培养基取代原有的培养基。接着在细胞加入dex或ata或赋形剂(0.1%dmso)培养15天;每三天更换一次含有基底膜基质的新鲜培养基。在第15天进行细胞拍照分析。
体外癌细胞迁移及侵入试验
将mcf-7或mda-mb-231细胞以1×105个细胞/孔接种到12孔培养盘并插入无细胞的硅板间隙(ibidigmbh,martinsried,德国)。在单层细胞形成后,取出插入板,用pbs洗涤细胞,然后加入dex或ata处理细胞24小时。为了诱导mcf-7细胞迁移,加入转化生长因子-β1(transforminggrowthfactor-β1,tgf-β1)至mcf-7tgf-β细胞培养基(为含20ng/mltgf-β1但不含fbs的dmem培养基)中。在0与24小时,对迁移之细胞进行拍照(100倍放大)以监测细胞向间隙区域的迁移,并计算间隙区域的闭合程度。为了测定肿瘤细胞侵袭作用,利用24孔通透性(transwell)培养盘进行细胞侵袭试验,将基底膜基质(10μl,0.5mg/ml;bdbioscience,losangeles,ca)覆盖于8-μm聚碳酸酯膜滤器上,并将1x105个细胞接种到膜滤器上,加入200μl含有dex或ata的无血清培养基,每组分别三重复。在培养装置之下室加入750μl完全生长培养基(completegrowthmedium,为含10%的fbs的dmem或rpmi培养基),将细胞培养在37℃反应24小时,观察细胞迁移现象。将培养装置之下室的侵入细胞利用冰甲醇固定15分钟,再用pbs洗涤3次,接着使用吉姆萨染剂(giemsastain)进行细胞染色,然后用pbs洗涤,最后使用光学显微镜(200x放大率)拍照细胞,以手动计数定量侵入细胞。针对基因沉默作用,则在实验之前,先用杂乱(scrambled)的sirna或sigrα转染细胞。
免疫荧光染色
免疫荧光染色方法如之前已发表的研究所述[22]。简言之,将mcf-7与mda-mb-231细胞(1x104细胞/孔)接种在8孔nunclab-tek玻璃腔室载玻片中,(thermofisherscientific),并加入dex与ata,或微小核糖核酸-708(microrna-708,mir-708)模拟物反应72小时,接着去除培养基后,利用4%多聚甲醛固定细胞15分钟,接着使用0.1%tritonx-100透化10分钟,然后用含有10%fbs的pbs洗涤并进行阻隔作用,再加入配制于1.5%fbs特异性一级抗体反应过夜,之后将细胞与带有异硫氰酸荧光素(fitc)的二级抗体(配制于6%牛血清白蛋白(bovineserumalbumin,bsa))反应1小时。然后利用1μg/ml4',6-二脒基-2-苯基吲哚(dapi,cellsignalingtechnology)染色5分钟后用pbs洗涤,最后使用荧光显微镜(moticelectricgroup)以40×倍率进行观察。
蛋白质萃取及免疫墨点法按照制造商的使用手册指示,分别利用放射性免疫沉淀分析(radioimmunoprecipitationassay,ripa)裂解缓冲液、或细胞核与细胞质萃取试剂(thermofisherscientific)、或跨膜蛋白萃取试剂(fivephotonbiochemicals,sandiego,ca)进行细胞蛋白质萃取,并利用bio-rad蛋白质测定试剂(bio-radlaboratories,hercules,ca)测定蛋白质含量。接着利用7~12%十二烷基硫酸钠聚丙烯酰胺凝胶电泳(sodiumdodecylsulfate-polyacrylamidegelelectrophoresis,sds-page)分离蛋白质样品(60μg)后转渍到聚偏二氯乙烯膜上(polyvinylidenechloride,pvdc),再加入特异性一级抗体反应过夜,然后加入带有辣根过氧化物酶(horseradishperoxidase-conjugated)的山羊抗兔或抗小鼠抗体反应2小时,最后加入化学冷光(enhancedchemiluminescence,ecl)西方墨点试剂(millipore,billerica,ma),并使用vlchemi-smart3000(viogenebiotek,sunnyvale,ca)侦测冷光讯号。
定量实时聚合酶链锁反应(quantitativereal-timepcr,qrt-pcr)
首先使用trizol试剂(thermofisherscientific)萃取细胞总rna。用nanovueplus分光亮度计(gehealthcarelifesciences,chicago,il)定量rna浓度。利用定量实时聚合酶链锁反应侦测系统和软件(appliedbiosystems,fostercity,ca)进行实时聚合酶链锁反应。先利用superscriptiii反转录酶试剂(invitrogen)将rna反转录产生第一链cdna,接着将正向与反向引子(primer,10μm)、荧光试剂sybrgreenmastermix(appliedbiosystems)加入cdna中混合均匀,再透过实时聚合酶链锁反应定量目标基因之表现量,mrna定量分析利用gapdh基因表现量进行标准化,而mirna定量分析则用u6基因表现量标准化。每个基因的引子序列详列于表二。
表二qrt-pcr引子序列
冷光试验
利用荧光素酶报告基因测定试剂(gal4reporterkit,bpsbiosciences,sandiego,caandcignalnf-κbpathwayreporterassaykit,quiagen,chatsworth,ca)测定糖皮质激素受体α(grα)与nf-κb的转录活性。在三端非转译区(3'utr)报告基因试验中,将pgl3-ikkβ3'utr及renilla荧光连同或不连同pre-mir-708转染乳癌细胞,分别在处理或不处理dex与ata情况下,最后利用双荧光素酶报告基因试验系统(promega)进行测定。引子序列详列于表三。
表三
动物及肿瘤细胞接种
异种移植动物肿瘤模式是使用四周龄之雌性无胸腺裸鼠(balb/c-nu购自中国台湾的台北乐斯科(biolasco)公司),饲养于无特殊病原的动物房中,提供囓齿动物饲料(orientalyeast,tokyo,japan)与随意饮水,并给予12小时光照及12小时黑暗的光照周期,所有动物实验均按照“实验动物管理和使用指南”与中国台湾有关动物保护的法律进行,并经实验动物伦理委员会批准。使用细胞代数小于15的细胞进行肿瘤细胞接种试验,将mcf-7与mda-mb-231细胞(200μl基质凝胶中含有1×106个细胞)以皮下注射接种到裸鼠的右后腹。将小鼠分成6组,每组5只小鼠,当平均肿瘤体积达到50mm3时(细胞接种后约3周),开始投予药物,将dex与ata配制于pbs缓冲液中,总共有四个给药组,包含mcf-7_dex、mcf-7_ata、mda-mb-231_dex及mda-mb-231_ata,以腹腔注射dex(1mg/kg)或ata(10mg/kg),每周投药两次,而对照组包含mcf-7_pbs与mda-mb-231_pbs,仅投予pbs缓冲液。整个实验期为10周,其中给药期为7周。10周后牺牲小鼠并取出肿瘤,将肿瘤组织拍照并称重,然后放入4%多聚甲醛中进行固定。利用卡尺测量肿瘤体积,每连续3天测量一次,肿瘤体积计算公式为:(长度×宽度2×高度×1/2)。为了监测药物毒性,每3天测量一次动物体重。
组织切片及免疫组织染色分析
将肿瘤组织包埋在石蜡中并切成3mm厚的切片,并用苏木精-伊红染色(h&estain)后利用光学显微镜进行观察,检测分析存活的肿瘤细胞、坏死及细胞凋亡。为了检验肿瘤标志物的表现,将组织切片用cox-2与nf-κb特异性抗体进行免疫组织染色,并在室温下反应2小时,最后加入二级检测抗体与realenvision侦测试剂(dakocytomationa/s,丹麦)进行反应。
统计分析
实验数据皆以平均值±标准偏差表示。所有实验数据皆利用统计分析软件graphpadprism版本6.0(graphpadsoftware,lajolla,ca)进行分析,使用单因素方差分析(one-wayanova)进行统计分析,然后用dunnett检验进行多重比较,p值小于0.05表示具有显著统计差异。
结果
糖皮质激素受体(glucocorticoidreceptor,gr)促进剂透过grα信号活化人类乳癌细胞中的mir-708
请参阅图2所示,是本发明的gr促进剂通过grα信号活化乳癌细胞中的mir-708示意图。如图所示:为了厘清mir-708在正常人类乳腺与乳癌细胞中的表现差异,本发明利用正常乳腺上皮细胞株(mcf-10a)、非恶性人类乳癌细胞株(mcf-7)及高度恶性人类乳癌细胞株(mda-mb-231)进行试验。数据以三次独立实验的平均值±标准偏差表示。*p<0.05、**p<0.01及***p<0.001表示对照组(control/con)与pre-mir-708转染或dex及ata处理组相比具有显著差异。
由图2a的rt-pcr分析结果显示,与mcf-10a(11.11倍)相比,mcf-7(2.43倍)与mda-mb-231(0.43倍)中mir-708表现显著低于正常人类乳腺细胞。此外,进一步比较非转移性mcf-7细胞与高度转移性mda-mb-231细胞,在mda-mb-231细胞中几乎观察不到mir-708的表现。再者,由图2b的rt-pcr结果显示,暂时性转染mir-708前驱物会造成mcf-7与mda-mb-231细胞过度表现mir-708。ma等人之前的一项研究发现[19],透过暂时性转染mir-708前驱物,使mda-mb-231细胞过度表现mir-708可抑制乳癌细胞增生与侵袭,然而抑制mir-708表现则会增加mda-mb-231细胞生长与转移。另外,最近一项研究发现,在卵巢癌转移过程中,mir-708的上调与糖皮质激素受体活化反应相关[23]。因此,本发明假设grα媒介的讯号传递途径可能参与调控乳癌中mir-708表现。为了验证此项假设,将乳癌细胞分别与dex及ata反应72小时,藉由图2c的rt-pcr测定细胞中mir-708的mrna表现可知,如同本发明所预期的,dex与ata显著增加了mir-708的表现量。然而如图2d所示,在grα沉默的乳癌细胞中,几乎观察不到dex与ata引发的mir-708表现增加,而在以grα药物抑制剂米非司酮处理的乳癌细胞中也观察到类似的结果,由图2e所示这些结果显示grα在诱导mir-708表现中扮演相当重要的功能。为了进一步测试从头合成(denovosynthesis),利用蛋白质合成抑制剂(chx)处理乳癌细胞后,用dex与ata处理72小时,由图2f的rt-pcr分析结果显示,经chx预处理的乳癌细胞中,dex与ata仍可增加细胞mir-708的表现量。这些结果显示,dex与ata诱导的grα媒介的mir-708转录是不需要从头蛋白质合成。
先前研究已发现糖皮质激素(glucocorticoids,gc)刺激后,mir-708的四个潜在启动子区域。其中具有cpg岛的启动子区域-3(在哺乳动物中高度相似)在卵巢癌细胞中具有超过40倍之启动子活性[23]。因此,本发明假设启动子区域-3或许可作为乳癌细胞中mir-708的启动子区域。本发明建构了携带启动子区域-3的报告质体,并利用荧光素酶报告基因测定法分析grα的转录活性。如图2g所示,dex与ata分别使mcf-7细胞中荧光素酶活性显著增加至8.2倍与6.5倍,mda-mb-231细胞中荧光素酶活性显著增加4.9倍与4.8倍,显示grα的转录活性参与了调控gc刺激下mir-708转录作用。接下来本发明试图测试gc媒介之grα入核作用,首先利用dex与ata处理乳癌细胞48小时,然后透过免疫荧光分析侦测grα在细胞间定位。结果如图2h所示,在对照组的细胞中,grα的基本表现主要存在于细胞质中,在细胞核中几乎观察不到,而用dex与ata处理后,显著提高乳癌细胞核中grα的表现量。西方墨点法(westernblot)分析进一步证实了类似的结果,如图2i所示,用dex与ata处理的细胞,其细胞核中grα蛋白表现显著增加,而细胞质中grα蛋白表现则减少,且两者之间成比例,因此gc诱导的grα转录活化,系在其入核后之一连串的反应事件。
mirna-708/grα途径可抑制乳癌细胞之增生
接下来请参阅图3所示,是本发明投予dex或ata对乳癌细胞存活率的影响示意图。如图所示:为了评估gr促进剂在乳癌细胞中的功能,本发明将dex与ata处理干细胞并分析其增生状况。另外,为了探讨gr促进剂对乳癌细胞的作用,将mcf-7与mda-mb-231细胞分别与不同剂量的dex(0.1、1、2.5、5及10μm)与ata(1、5、10及20μm)反应24、48及72小时,比较组则是0.1%的dmso,然后使用mtt与mts试验分析细胞生长与增生,其中图3a是将mcf-7细胞与ata(1~20μm)或dex反应24~72小时;以及图3b是将mda-mb-231细胞与递增浓度的ata(1~20μm)或dex(0.1~10μm)一起反应24~72小时。以上通过mtt比色测定评估细胞存活率,数据以三次独立实验的平均值±标准偏差表示。经mtt结果显示,与对照组(control)和比较组相比,细胞经dex与ata处理后,其生长曲线急剧下降且具有剂量与时间相关性。而且,使用dex与ata处理72小时,即使在低剂量的dex与ata(1μm)下,细胞数量也显著减少。
请参阅图4所示,是本发明使用流式细胞仪分析annexinv/pi染色乳癌细胞凋亡情况的示意图。如图所示:接着利用annexin-v/pi染色测定细胞数量的减少是否与细胞凋亡相关,数据以三次独立实验的平均值±标准偏差表示。经流式细胞仪分析结果显示,与对照组(未进行dex或ata处理)相比,dex或ata处理组中凋亡细胞没有显著增加。因此,本发明假设gr促进剂可能诱导乳癌细胞中的生长停滞或衰老,进而造成细胞数目减少。
接着请参阅图5及图6所示,是分别为本发明mirna-708/grα讯号途径抑制乳癌细胞的增生示意图、及本发明利用台盼蓝排除试验评估细胞增生情况的示意图。本发明利用mts的细胞增殖试验证实这个假设。图5的数据以三个独立实验的平均值±标准偏差表示。**p<0.01表示对照组与pre-mir-708转染或dex及ata处理组相比具有显著差异。图6a和图6b分别为使用pre-mir-708暂时转染乳癌细胞mcf-7及mda-mb-231,并在24、48及72小时后测量细胞的增生情况,对照为未使用pre-mir-708转染;图6c和图6d分别为乳癌细胞mcf-7及mda-mb-231使用dex或ata处理24~72小时后,藉由台盼蓝排除分析测定活细胞的数量,以未进行dex或ata处理为对照(control)。数据以三次独立实验的平均值±标准偏差表示。*p<0.05、**p<0.01及***p<0.001表示对照组与pre-mir-708转染或dex及ata处理组相比具有显著差异。
由图5a的mts测定的结果显示dex与ata显著降低了细胞数目,表示gr促进剂具有抑制乳癌细胞增生的作用。为了进一步确认gr促进剂抑制细胞增生是否藉由mir-708的作用,将乳癌细胞转染pre-mir-708前驱物使其过度表现mir-708,然后测定细胞增生情况,如图5b所示,细胞过度表达mir-708可显著降低存活细胞的比率,显示gr促进剂抑制乳癌细胞的增生是透过mir-708的作用。并且,由图6在平行(parallel)台盼蓝排除分析中也观察到类似的结果。
本发明进一步利用流式细胞仪分析细胞周期进程,以确认gr促进剂是否透过引起细胞周期停滞来抑制细胞增生。由图5c的结果显示,dex使mcf-7与mda-mb-231在细胞周期g2的细胞群增加,显示细胞都停滞在g2-m过渡期。相较之下,ata处理后的细胞,g0/g1期细胞群的比例显著增加,显示ata使细胞停滞在g1-s转换期。亦如图5d所示,将乳癌细胞转染pre-mir-708也进一步验证此结果,与dex结果一致,pre-mir-708的转染在使mcf-7与mda-mb-231细胞停滞在g2-m转换期。为了进一步证实这些结果,透过免疫墨点法侦测细胞周期调控蛋白的表现,像是周期蛋白(cyclins)、细胞周期蛋白依赖型激酶(cyclin-dependentkinases,cdks)及cdk抑制因子(cdkinhibitors,ckis)。图5e的实验结果强烈支持上述观点,dex与ata显著抑制调控g1-s过渡调节蛋白,特别是cyclind1、cdk4与cdk6,此外,ata显著抑制cyclinb及cdk1。然而,cyclina与cycline的蛋白表现则不受dex或ata影响。有趣的是,图5f显示投药dex或ata处理都显著上调mcf-7与mda-mb-231细胞中的p21cip1与p27kip1的蛋白表现,而p21cip1与p27kip1是经常涉及生长停滞与细胞衰老的蛋白质。
mirna-708/grα途径抑制乳癌细胞中的癌症干细胞表现
接下来请参阅图7所示,是本发明mirna-708/grα讯号传递途径调控体外与体内肿瘤的生长示意图。如图所示:进行gr促进剂是否抑制肿瘤球形细胞的形成测试,这是癌症干细胞的一个特征。数据以三次独立实验的平均值±标准偏差表示。*p<0.05、**p<0.01及***p<0.001表示对照组(control/con)与pre-mir-708转染或dex及ata处理组相比具有显著差异。
首先将乳癌细胞在处理或不处理dex与ata的情况下在三维微环境中培养14天,然后计数在三维培养条件下形成肿瘤球形细胞的数量,由图7a显示mcf-7与mda-mb-231细胞分别在7天与5天后形成肿瘤球形细胞。在未处理药物的情况下,mcf-7细胞每孔形成5-7个肿瘤球形细胞,而在dex与ata处理下,未发现任何肿瘤球形细胞的形成。另一方面,在未处理药物的情况下,mda-mb-231细胞每孔形成15-22个肿瘤球形细胞,而dex与ata的细胞则分别显著减少形成5-7个肿瘤球与3-7个肿瘤球。这些结果显示gr促进剂可有效地抑制乳癌细胞中肿瘤球的形成。为了进一步探讨mir-708参与gr促进剂的抑制作用,利用pre-mir-708转染细胞后测定肿瘤球形成,如图7b所示,与未处理的对照组相比,mir-708过度表现明显减少了mcf-7与mda-mb-231细胞中肿瘤球的数量。本发明还由图7c发现在grα沉默细胞中,dex或ata无法抑制肿瘤球体形成,此结果显示gr促进剂的体外抗肿瘤作用是藉由mir-708/grα讯号串联反应。此外,图7d的西方墨点法结果显示,dex与ata显著抑制调控mcf-7与mda-mb-231细胞中的cd44与cd24蛋白表现。同样地,图7e显示pre-mir-708的转染显著抑制调控乳癌细胞中的cd44与cd24蛋白表现,显示dex与ata或pre-mir-708抑制肿瘤球体形成与抑制调控cd44与cd24表面蛋白标志具有强烈相关性。
gr促进剂在异种移植动物模型中抑制乳癌肿瘤生长
根据上述体外致癌性测试结果显示gr促进剂具有成为抗肿瘤试剂的潜力。为了在体内验证这些效果,将mcf-7与mda-mb-231细胞接种到雌性无胸腺小鼠的右侧皮下,建立乳癌异种移植模式,在肿瘤大小达到50mm3后,以腹腔注射dex与ata进行投药,每周两次。如图7f及图7g所示,在mcf-7肿瘤对照组的小鼠,在第五周后肿瘤大小从40mm3增加至450mm3,在mda-mb-231肿瘤对照组小鼠,则从60mm3增加至2700mm3。然而,dex与ata投药的小鼠则显著减缓了肿瘤的生长,mcf-7肿瘤体积分别减少到170±37mm3与110±28mm3,mda-mb-231肿瘤则分别减少至330±117mm3与437±136mm3。另外,图7h显示mcf-7对照组的肿瘤重量为2.1±0.5g,mda-mb-231对照组的肿瘤重量为3.7±1.3g,而dex与ata处理下mcf-7肿瘤重量为0.6±0.2g与0.15±0.02g,dex与ata处理下mda-mb-231的肿瘤重量分别为1.5±0.5g与1.1±0.5g。此外,ata处理的小鼠体重略有降低(体重减少不到10%),但并无显著的副作用影响。由图7i的组织切片结果显示对照组肿瘤组织仅有些微的坏死现象,而dex与ata处理组均发现mcf-7与mda-mb-231两个肿瘤组织中有大量坏死区域。图7j的组织化学染色也显示对照组肿瘤组织并无发现显著的细胞核浓缩及凋亡小体的产生,而在mcf-7与mda-mb-231两个肿瘤组织中,dex与ata则增加细胞核浓缩及凋亡小体的产生,这样的结果与上述细胞凋亡试验结果互相矛盾,dex与ata在体外不会诱导乳癌细胞的凋亡。
gr促进剂抑制乳癌细胞之转移
请参阅图8所示,是本发明gr促进剂在体外试验可抑制乳癌细胞的转移示意图。如图所示:为了进一步测试gr促进剂抑制癌细胞转移的活性,利用dex与ata处理乳癌细胞,进行癌细胞迁移与侵入试验分析。由于mcf-7细胞转移能力差,因此在培养基中加入tgf-β1诱导mcf-7细胞(mcf-7tgf-β)进行转移。数据以三次独立实验的平均值±标准偏差表示。#p<0.001表示对照组与tgf-β1处理组相比具有显著差异。**p<0.01及***p<0.001表示单用tgf-β1处理组与dex或ata处理组具有显著差异。δp<0.001表示单用sigrα转染与dex或ata处理组具有显著差异。
如图8a所示,与未处理药物的对照组细胞相比,在tgf-β1处理的细胞中细胞迁移程度显著增加,而用dex或ata处理的细胞,其细胞间隙的闭合比例明显较低,显著降低了tgf-β1诱导的细胞迁移。同样的,gr促进剂的dex与ata也显著降低了乳癌细胞的迁移能力。接下来,利用通透性培养盘侵袭试验(trans-wellinvasionassay)测试gr促进剂是否能够抑制tgf-β1诱导mcf-7细胞的侵袭,图8b显示tgf-β1组侵袭细胞的数量显著高于对照组,然而,在dex或ata处理下,则减少mcf-7侵入细胞的数目。此外,gr促进剂也显著降低了mda-mb-231细胞的侵袭作用。接下来,为了证实gr促进剂抑制癌细胞迁移与侵袭作用可能与mmps的抑制调控有关,因此检测mmp-2与mmp-9的基因表现。正如本发明所预期,图8c显示在mcf-7tgf-β与mda-mb-231细胞中,dex与ata均显著降低了mmp-2与mmp-9的mrna表现,此外,由图8d可见dex与ata也显著抑制mmp-2与mmp-9蛋白质表现。为了进一步验证grα在乳癌细胞中调控转移的作用,先将mcf-7tgf-β与mda-mb-231细胞转染grα特异性sirna,使grα沉默,接着再进行迁移试验测定dex与ata的抗转移作用,如图8e所示,dex或ata显著降低了scsirna转染细胞的肿瘤细胞迁移,但在grα沉默的细胞中,dex则无法抑制mcf-7tgf-β与mda-mb-231细胞的迁移,有趣的是,ata可部分抑制grα沉默细胞中的肿瘤细胞迁移,显示ata可能可以透过调节其他讯号途径抑制肿瘤细胞转移。
mirna-708/grα途径具有专一调控乳癌细胞中之ikkβ表现的作用
请参阅图9所示,是本发明mirna-708/grα讯号途径具有专一调控乳癌细胞中的ikkβ表现作用示意图。如图所示:为了鉴定调节乳癌增生的mir-708的标靶基因,利用三种标靶预测算法(miranda、targetscan及mirtarbase)搜寻与mir-708相互作用的mrna,三种预算法都显示mir-708可以作用于nf-κb活化激酶β(ikkβ)基因,而且ikk-nf-κb途径也被认为是乳癌发生的重要因素,也是治疗乳癌的潜在标靶[24]。数据以三次独立实验的平均值±标准偏差表示。*p<0.05及**p<0.01表示对照组与pre-mir-708转染或dex与ata处理组具有显著差异。δp<0.001表示单用sigrα转染组与dex或ata处理组具有显著差异。
图9a显示了ikbkb(inhibitorofnuclearfactorkappabkinasesubunitbeta)抑制剂与mir-708之间预测的结合位点,为了评估mir-708与ikkβ的直接作用,图9b将ikkβ的3'utr区域克隆到萤火虫荧光素酶下游的报告质体中,在mir-708转染mcf-7与mda-mb-231后,荧光素酶活性分别下降至对照组的38.7%与40.1%,这些结果强烈显示出ikkβ是mir-708的直接标靶。接着本发明分析乳癌细胞中ikkα、ikkβ及ikkγ等ikk亚基的mrna表现,图9c显示当mcf-7与mda-mb-231细胞过度表现mir-708时,会导致细胞中ikkβmrna表现显著降低,而ikkα与ikkγ的mrna表现则不受pre-mir-708转染的影响。同样地,图9d显示gr促进剂的dex或ata显著抑制调控ikkβmrna的表现,而ikkα与ikkγ则不受影响。这些结果强烈显示,gr促进剂引起的ikkβ的减少是藉由mir-708的作用。图9e免疫墨点结果也显示,dex与ata不仅减少ikkβ的蛋白表现,还抑制其磷酸化作用,而dex或ata则不影响ikkα蛋白表现及其磷酸化。为了进一步检测grα参与ikkβ调控,图9f及图9g是用grαsirna暂时转染乳癌细胞后,测定ikkβ蛋白与mrna的表现,在grα抑制(knockdown)的细胞中,dex与ata都不能抑制调控ikkβmrna与蛋白质的表现,然而ata在grα沉默的mda-mb-231细胞中,仍可显著抑制ikkβ磷酸化,显示ata抑制ikkβ磷酸化系部分grα非依赖性的。根据以上结果推测,gr促进剂可活化mir-708,而ikkβ是mir-708的直接作用标的。
mirna-708抑制乳癌细胞中nf-κb的活性
目前已经发现nf-κb信号在许多癌细胞中可调节许多基因之转录表现,而这些基因通常和促进细胞存活与增生、抑制细胞凋亡、及诱导癌症干细胞分化与肿瘤细胞转移[13,25]相关。最近一项研究[18]证实了mir-708的表观遗传学沉默增强了慢性淋巴细胞白血病(cll)中之nf-κb信号。请参阅图10所示,是本发明mirna-708抑制乳癌细胞中nf-κb的活性示意图。如图所示:鉴于mir-708在细胞增殖、肿瘤发生与转移的功能,本发明测试了mir-708是否透过nf-κb调控癌细胞。为了诱导mcf-7细胞中的nf-κb信号传导,利用tnf-α(10ng/ml)预处理细胞,并将tnf-α刺激的mcf-7细胞表示为mcf-7tnf。数据以三次独立实验的平均值±标准偏差表示。**p<0.01表示对照组与pre-mir-708转染或dex与ata处理组具有显著差异。
图10a的荧光素酶报告基因活性试验结果显示,乳癌细胞中mir-708的表现会导致nf-κb转录活性的显著降低。此外,图10b显示gr促进剂的dex或ata显著抑制乳癌细胞中nf-κb的转录活化。接下来,本发明测试了pre-mir-708处理下nf-κb的核输出作用。图10c的西方墨点法结果显示在对照组细胞中,nf-κb主要表达在细胞核中,而在pre-mir-708转染的细胞中则发现细胞质中的nf-κb表现增加。同样地,图10d显示dex与ata明显抑制nf-κb入核作用,其细胞核中的nf-κb表现较低。先前研究指出,诱发糖皮质激素受体表现可抑制hela细胞中的nf-κb活性[26]。因此,本发明推测gr促进剂抑制调控ikkβ表现在乳癌细胞中抑制nf-κb活性可能扮演重要的功能,如图10e所示,dex与ata显著抑制了scsirna转染的细胞中的nf-κb活性,而dex与ata无法抑制grα沉默细胞中的nf-κb活性。图10f的免疫墨点法结果显示,在grα沉默的细胞中几乎观察不到dex与ata抑制nf-κb入核作用。为了进一步探讨gr促进剂对nf-κb调节作用,将异种移植皮下肿瘤组织进行免疫组织化学染色,如图10g所示,在对照组的肿瘤组织中观察到nf-κb阳性的免疫染色的细胞,然而,在dex与ata组中nf-κb阳性免疫染色细胞的数量则显著降低。
ikkphosphorylatesnf-κb-boundiκbs(inhibitor-κbs)在iκbn-末端调节结构域内的两个保守丝氨酸上磷酸化,iκb蛋白降解是藉由磷酸化与泛素化(ubiquitination)依赖途径作用,且是nf-κb活化的关键步骤。iκb磷酸化主要依赖于ikk复合物的ikkβ催化亚基[10]。因此,测量了nf-κb之上游蛋白i-κbα的表现,如图10h所示,在对照组中几乎没有观察到i-κb的表现,而在pre-mir-708转染的细胞中则观察到i-κb表现增加,然而,在对照组或pre-mir-708转染细胞中都未观察到i-κb的磷酸化表现。图10i显示dex与ata处理后的细胞i-κb蛋白表现与处理前也无明显差异。
gr促进剂藉由活化mir-708抑制nf-κb下游目标基因
最近研究指出,人类乳癌细胞异常表现cox-2与cmyc蛋白,而且在乳癌患者中cox-2与cmyc表现提升具有预后价值[27,28]。请参阅图11所示,为本发明gr促进剂藉由活化mir-708抑制nf-κb下游目标基因示意图。如图所示:为了测定mir-708是否会影响nf-κb讯号途径下游基因的转录活性,将细胞处理tnf-α诱发nf-κb下游基因cox-2与cmyc的表现后进行测试。数据以三个独立实验的平均值±标准偏差表示。**p<0.01表示对照组与pre-mir-708转染或dex与ata处理组具有显著差异。#p<0.05表示scsirna组与sigrα组有显著差异。δp<0.001表示单用sigrα转染组与dex或ata处理组具有显著差异。
由图11a和图11c的结果发现过度表现mir-708会造成cox-2与cmyc基因(图a)与蛋白质(图c)的表现受到抑制。图11b亦显示gr促进剂的dex与ata处理的细胞中也观察到类似的结果。此外,在图11d的动物实验中,与未处理药物的对照组相比,dex与ata组的肿瘤组织中cox-2阳性细胞的数量明显减少。为了评估grα活化对cox-2表现的影响,将dex与ata处理grα沉默细胞后进行rt-pcr与西方墨点法分析。由图11e的rt-pcr分析结果显示,与对照组相比,grα沉默的细胞中cox-2基因表现显著增加,然而,dex与ata则部分抑制grα沉默细胞中cox-2的表现。
讨论
mir-708在许多癌症中是属于最抑制调控的mirna之一,包括肾细胞癌、前列腺癌、成胶质细胞瘤、肝癌、卵巢癌及乳癌[17,29]。相比之下,mir-708高度表现在非小细胞肺癌、儿童常见前体b细胞急性淋巴细胞白血病、及膀胱癌[17,29]。先前研究指出,经常在大多数转移性肿瘤细胞观察到mir-708表现,然而,到目前为止,几乎没有太多研究探讨它在肿瘤发生中的作用。ryu等人[30]研究指出,藉由polycomb抑制mir-708会造成乳癌中钙诱导之细胞迁移进而促进癌症转移,并由ma等人研究[19]证明强制表现mir-708可藉由抑制lsd1表现进而抑制乳癌细胞的增生与侵袭。由于mir-708在物种间具高度相似性,且在特定组织中有不同的表现,因此本发明一开始测试一组非致瘤性、致瘤性(非转移性)及致瘤性(转移性)乳癌细胞的mir-708表现差异,结果显示mir-708在非致瘤性mcf-10a细胞中表现量较高,而mir-708在转移性癌细胞的表现又显著低于非转移性癌细胞。此外,本发明发现透过转染mir-708前驱物在细胞中强制表达mir-708,可显著抑制乳癌细胞增生与体外致癌性,此结果与其他研究中观察到乳癌细胞[19]或其他癌细胞[31,33]结果很类似。实验结果也发现mir-708相似物可造成细胞周期停滞进而抑制细胞增生,然而,在转移与非转移癌细胞中之细胞周期停滞在不同阶段,在mcf-7细胞中,mir-708相似物可将细胞停滞在g1-s转换期,在mda-mb-231细胞则停滞在g2-m转换期。进一步分析细胞周期调节蛋白也发现mir-708相似物在两种细胞系中均诱导p21cip1与p27kip1的表现。整体来说,本发明的研究结果显示mir-708在乳癌中具有抗肿瘤的作用。
越来越多证据显示,mirna具有调控各种真核生物体中胚胎干细胞与组织干细胞的特性。近期研究发现mirna提供了调控癌症干细胞的机制的新方向。shimono等人[34]发现在乳癌干细胞中,有8个mirna被选择性地抑制调控并位于三个mirna簇,包含mir-200c-141、mir-200b-200a-429及mir-183-96-182,而其中两个是mir-200簇(mir-200c-141与mir-200b-200a-429)。bmi1是一种调节干细胞自我更新的调控因子,其表现受到mir-200c的调控,显示抑制mir-200家族在维持干细胞功能中系至关重要的[35]。此外,活化调控let-7家族mirnas目标激因h-ras与hmga2,会抑制乳癌干细胞的自我更新与分化[36]。这一证据显示,乳癌干细胞特异性mirna在调控自我更新能力、致癌性及转移中具有相当重要的功能。在本发明中,选择高侵略性的乳癌细胞株mda-mb-231来测试gr促进剂或mir-708对癌症干细胞的潜在作用。mda-mb-231系属于er-/pr-/her-1-三阴性乳癌细胞(triplenegativebreastcancer,tnbc),且表现高比例cd44+/cd24-细胞群。由于mda-mb-231细胞具有高cd44+细胞群,可作为乳癌干细胞的良好模型。为了研究gr促进剂与mir-708过度表现对乳癌干细胞的影响,本发明藉由肿瘤球形成试验分析癌症干细胞的表现,结果发现gr促进剂或转染mir-708相似物皆显著降低肿瘤球形成与cd44+表现,显示gr促进剂可藉由活化mir-708抑制乳癌细胞中癌症干细胞的表现。
先前研究指出,使用gr促进剂治疗癌症展现了具有争议的结果。合成的gr促进剂-地赛米松(dex),其透过增加雌激素磺基转移酶与降低肿瘤组织中的雌二醇来抑制小细胞肺癌及乳癌肿瘤生长[37]。然而,另一项研究发现,糖皮质激素受体拮抗剂-米非司酮合并化疗使用可有效抑制三阴性乳癌细胞的致癌性并诱导细胞凋亡,然而单独使用米非司酮治疗则对三阴性乳癌细胞无效[38]。lin等人最近一项研究指出,gr促进剂dex可藉由作用在rap1b进而活化mir-708来抑制卵巢癌转移[23]。因此,本发明假设gr促进剂具有活化调控mir-708的作用,并且可能具有抑制乳癌细胞之致癌性潜力。在这里,本发明发现合成之gr促进剂dex,或天然gr促进剂-樟芝酸a(ata)都可藉由诱导mir-708表现抑制乳癌细胞之增生与癌细胞群落形成,以及造成细胞周期停滞。有趣的是,本发明还观察到不论是合成的或天然的gr促进剂都不会诱导乳癌细胞凋亡,此结果与先前在急性淋巴细胞白血病中观察到的现象互相矛盾[39],此外,dex或ata皆显著降低裸鼠皮下mcf-7与mda-mb-231乳癌肿瘤的生长、肿瘤体重与体积。根据以上结果显示gr促进剂具有治疗乳癌的潜力。
在人类乳癌中,nf-κb途径是过度活跃的,并提供了一种有利于生存的功能。nf-κb讯号途径会受到许多不同因子的刺激而活化,从细胞激素、生长因子讯号传导到识别病原体产物或dna损伤与致癌压力等[40]。目前研究发现药理学抑制剂具有增加乳癌细胞凋亡以及抑制体内肿瘤生长的作用[41,42]。nf-κb讯号途径的活化是通过其上游激酶ikkα与ikkβ活化后,促进蛋白酶体降解i-κbα/β,使nf-κb进入细胞核随后转录活化其下游基因[40]。许多活化nf-κb的讯号因子汇集在ikkβ上,包括生长因子、发炎细胞激素、内毒素及病毒感染等,活化之nf-κb不仅参与免疫与发炎反应,还参与调控细胞黏附、血管生成、细胞自噬、能量代谢、衰老及诱导细胞增生与存活[43],因此,不论在实验模式或癌症病患中发现nf-κb参与癌症的发生与发展并不特别令人感到惊奇。在这项研究中,本发明预测mir-708会作用在nf-κb之上游激酶ikkβ,荧光素酶报告基因试验结果发现mir-708会直接结合在ikkβ之3'utr区域,此外,过度表现mir-708或gr促进剂的处理皆可显著降低ikkβ蛋白表现以及nf-κb转录活性,另外,gr促进剂显著降低了人类乳癌异种移植小鼠的肿瘤组织中的nf-κb表现。其他研究也发现mir-708在慢性淋巴细胞白血病中具有抑制nf-κb的功能[18],此外,近期研究指出,乳腺上皮细胞nf-κb会调控乳癌干细胞的自我更新[13],nf-κb的药理学抑制剂可有效减少癌症干细胞自我更新作用与抑制异种移植肿瘤生长[44]。因此,本发明认为gr促进剂或mir-708相似物抑制癌症干细胞与cd44+细胞群的作用可能与抑制nf-κb讯号传递有关,这些结果证实mir-708与nf-κb会透过密切相互作用调控乳癌细胞生长与癌症干细胞表现。
cox-2高度活化是人类上皮肿瘤的共同特征,包括大肠直肠癌、乳癌及前列腺癌[45]。在乳癌中,cox-2的表现会调控肿瘤生长、侵袭及转移,或许可作为预测乳癌预后的标志物[28]。目前已经发现数种cox-2抑制剂可抑制肿瘤癌细胞增生并诱导细胞凋亡[46]。此外,流行病学研究发现,长期暴露于非类固醇消炎药(nsaid)可抑制cox-2诱发产生前列腺素,并且降低多种癌症发病率,包括乳癌[47],因此,临床上已将cox-2抑制剂与抗癌药物合并使用[48]。另一方面,cmyc也是在大多数癌症中高度表现的致癌基因,包括淋巴瘤、黑色素瘤、骨髓性白血病、胃肠道癌、前列腺癌及乳癌[49]。先前研究发现,在转殖基因小鼠的乳腺上皮细胞中藉由诱导系统引发c-myc过度表现导致浸润性乳癌的形成,然而抑制cmyc的活性则会使肿瘤持续消退[50]。为了探讨cox-2与cmyc在预防癌症中的作用以及与抗癌药物的组合,本发明实验评估了gr促进剂对乳癌细胞中cox-2与cmyc表现的影响,rt-pcr与免疫墨点分析结果显示gr促进剂或mir-708相似物可显著抑制调控mcf-7与mda-mb-231细胞中cox-2与cmyc基因与蛋白质表现,此外,在动物实验中,gr促进剂也显著抑制mcf-7或mda-mb-231细胞异种移植肿瘤中的cox-2表现。
本发明首次证明,合成或天然来源的gr促进剂可透过grα活化mir-708,进而抑制乳癌细胞的增生并诱发细胞周期停滞。而藉由gr促进剂活化的讯号传递诱发mir-708的表现后抑制nf-κb活性,以及抑制调控其下游目标基因cox-2与cmyc的表现。根据本发明实验结果推测gr促进剂或其下游mirna-708具有应用在抑制乳癌肿瘤发生的潜力。
糖皮质激素经常与化疗合并使用在乳癌治疗中当作缓和治疗。最近研究发现,糖皮质激素治疗诱导卵巢癌细胞中mir-708的表现进而抑制肿瘤细胞的转移,如果在乳癌细胞中强制表达mir-708可抑制肿瘤细胞增殖与转移。此外,在慢性淋巴细胞性白血病中,nf-κb的上游激酶ikkβ受mir-708调控,然而,在乳癌细胞中糖皮质激素诱导之mir-708之谱系及其下游nf-κb之标的皆尚待确定。在本发明中,发现用合成的糖皮质激素dex或天然的糖皮质激素相似物ata,处理人类乳癌细胞mcf-7与mda-mb-231后,通过活化grα显著增加mir-708表现。藉由gr促进剂诱导mir-708表现可有效抑制人类乳癌细胞增殖、细胞周期进程以及肿瘤干细胞表现与癌细胞转移。另外,gr促进剂或mir-708转染处理细胞,皆显著抑制ikkβ的表现,也抑制nf-κb活性及其下游目标基因,包括cox-2、cmyc、cyclind1、mmp-2、mmp-9、cd24及cd44的表现,并提高参与癌细胞增生、细胞周期进程、转移及肿瘤干细胞标记蛋白p21cip1与p27kip1的表现量。人类乳癌细胞异种移植模式试验发现,用gr促进剂治疗动物,可显著抑制肿瘤生长以及肿瘤重量与体积。整体来说,本实验结果有力推测gr促进剂诱发mir-708表现与抑制nf-κb下游信号传导,可成为一新型治疗方式应用于乳癌治疗。
综上所述,本发明的糖皮质激素受体促进剂樟芝酸a在抑制乳腺癌肿瘤发生与转移中的应用,可有效改善现有技术的种种缺点,不论是合成或天然的糖皮质激素受体(gr)促进剂皆可诱导mir-708表现,进而抑制乳癌肿瘤发生与转移,透过转录活化糖皮质激素受体α(grα)活化mir-708造成乳癌细胞增殖减少及诱导细胞周期停滞,主要是透过转录抑制nf-κb及其靶基因cox-2与cmyc的作用,进而使本发明的产生能更进步、更实用、更符合使用者所须,确已符合发明专利申请的要件,依法提出专利申请。
但以上所述,仅为本发明的较佳实施例而已,当不能以此限定本发明实施的范围。故,凡依本发明申请专利范围及发明说明书内容所作的简单的等效变化与修饰,皆应仍属本发明专利涵盖的范围内。
参考文献:
[1]1.jemala,brayf,centermm,ferlayj,warde,formand.globalcancerstatistics.cacancerjclin2011;61:69-90.
[2]2.ferlayj,soerjomatarami,dikshitr,esers,mathersc,rebelom,etal.cancerincidenceandmortalityworldwide:sources,methodsandmajorpatternsinglobocan2012.intjcancer2015;136:e359-e86.
[3]3.schoneveldojlm,gaemersic,lamerswh.mechanismsofglucocorticoidsignalling.biochimbiophysacta2004;1680:114-28.
[4]4.beckerde.basicandclinicalpharmacologyofglucocorticosteroids.anesthprog2013;60:25-32
[5]5.chenfc,wanglh,zhengxy,zhangxm,zhangj,lilj.meta-analysisoftheeffectsoforalandintravenousdexamethasonepremedicationinthepreventionofpaclitaxel-inducedallergicreactions.oncotarget2017;8:19236-43.
[6]6.mooney,ryuyk,leegh.dexamethasoneinhibitsinvivotumorgrowthbythealterationofbonemarrowcd11b(+)myeloidcells.intimmunopharmacol2014;21:494-500.
[7]7.link-t,wangl-h.newdimensionofglucocorticoidsincancertreatment.steroids2016;111:84-8.
[8]8.skormn,wonderel,kocherginskym,goyala,hallba,caiy,etal.glucocorticoidreceptorantagonismasanoveltherapyfortriple-negativebreastcancer.clincancerres2013;19:10.1158/078-0432.ccr-12-3826.
[9]9.suim,chenf,chenz,fanw.glucocorticoidsinterferewiththerapeuticefficacyofpaclitaxelagainsthumanbreastandovarianxenografttumors.intjcancer2006;119:712-7.
[10]10.karinm,caoy,gretenfr,lizw.nf-kappabincancer:frominnocentbystandertomajorculprit.natrevcancer2002;2:301-10.
[11]11.matsuiwh.cancerstemcellsignalingpathways.medicine2016;95:s8-s19.
[12]12.dandawatepr,subramaniamd,jensenra,anants.targetingcancerstemcellsandsignalingpathwaysbyphytochemicals:novelapproachforbreastcancertherapy.semincancerbiol2016;40-41:192-208.
[13]13.shostakk,chariota.nf-κb,stemcellsandbreastcancer:thelinksgetstronger.breastcancerres2011;13:214.
[14]14.huangy,shenxj,zouq,wangsp,tangsm,zhanggz.biologicalfunctionsofmicrornas:areview.jphysiolbiochem2011;67:129-39.
[15]15.crocecm.causesandconsequencesofmicrornadysregulationincancer.naturerevgenet2009;10:704-14.
[16]16.ioriomv,ferracinm,liucg,veronesea,spizzor,sabbionis,etal.micrornageneexpressionderegulationinhumanbreastcancer.cancerres2005;65:7065-70.
[17]17.monteleonenj,lutzcs.mir-708-5p:amicrornawithemergingrolesincancer.oncotarget2017;8:71292-316.
[18]18.baerc,oakescc,ruppertas,clausr,kim-wannersz,mertensd,etal.epigeneticsilencingofmir-708enhancesnf-kappabsignalinginchroniclymphocyticleukemia.intjcancer2015;137:1352-61.
[19]19.mal,mas,zhaog,yangl,zhangp,yiq,etal.mir-708/lsd1axisregulatestheproliferationandinvasionofbreastcancercells.cancermed2016;5:684-92.
[20]20.aldinir,micuccim,ceveninim,fator,bergaminic,nannic,etal.antiinflammatoryeffectofphytosterolsinexperimentalmurinecolitismodel:prevention,induction,remissionstudy.plosone2014;9:e108112.
[21]21.chenyc,liuyl,lify,changci,wangsy,leeky,etal.antcina,asteroid-likecompoundfromantrodiacamphorata,exertsanti-inflammatoryeffectviamimickingglucocorticoids.actapharmacolsin2011;32:904-11.
[22]22.senthilkk,gokilavm,maujl,lincc,chufh,weicc,etal.asteroidlikephytochemicalantcinmisananti-agingreagentthateliminateshyperglycemia-acceleratedprematuresenescenceindermalfibroblastsbydirectactivationofnrf2andsirt-1.oncotarget2016;7:62836-61.
[23]23.linkt,yehym,chuangcm,yangsy,changjw,sunsp,etal.glucocorticoidsmediateinductionofmicrorna-708tosuppressovariancancermetastasisthroughtargetingrap1b.natcommun2015;6:5917.
[24]24.kendellenmf,bradfordjw,lawrencecl,clarkks,baldwinas.canonicalandnon-canonicalnf-κbsignalingpromotesbreastcancertumor-initiatingcells.oncogene2014;33:1297-305.
[25]25.rinkenbaughal,baldwinas.thenf-κbpathwayandcancerstemcells.cells2016;5:16.
[26]26.nelsong,wildegj,spillerdg,kennedysm,raydw,sullivane,etal.nf-kappabsignallingisinhibitedbyglucocorticoidreceptorandstat6viadistinctmechanisms.jcellsci2003;116:2495-503.
[27]27.xuj,cheny,olopadeoi.mycandbreastcancer.genecancer2010;1:629-40.
[28]28.janad,sarkardk,gangulys,sahas,sag,mannaak,etal.roleofcyclooxygenase2(cox-2)inprognosisofbreastcancer.indianjsurgoncol2014;5:59-65.
[29]29.sevenm,karatasof,duzmb,ozenm.theroleofmirnasincancer:frompathogenesistotherapeuticimplications.futureoncol2014;10:1027-48.
[30]30.ryus,mcdonnellk,choih,gaod,hahnm,joshin,etal.suppressionofmirna-708bypolycombgrouppromotesmetastasesbycalcium-inducedcellmigration.cancercell2013;23:63-76.[31]31.jangjs,jeonhs,sunz,aubrymc,tangh,parkch,etal.increasedmir-708expressioninnsclcanditsassociationwithpoorsurvivalinlungadenocarcinomafromneversmokers.clincancerres2012;18:3658-67.
[32]32.guop,lanj,gej,nieq,maoq,qiuy.mir-708actsasatumorsuppressorinhumanglioblastomacells.oncolrep2013;30:870-6.
[33]33.leisl,zhaoh,yaohl,cheny,leizd,liukj,etal.regulatoryrolesofmicrorna-708andmicrorna-31inproliferation,apoptosisandinvasionofcolorectalcancercells.oncollett2014;8:1768-74.
[34]34.shimonoy,mukohyamaj,nakamurasi,minamih.micrornaregulationofhumanbreastcancerstemcells.jclinmed2016;5:2.[35]35.shimonoy,zabalam,chorw,lobon,dalerbap,qiand,etal.downregulationofmirna-200clinksbreastcancerstemcellswithnormalstemcells.cell2009;138:592-603.
[36]36.yuf,yaoh,zhup,zhangx,panq,gongc,etal.let-7regulatesselfrenewalandtumorigenicityofbreastcancercells.cell2007;131:1109-23.
[37]37.gongh,jarzynkamj,coletj,leejh,wadat,zhangb,etal.glucocorticoidsantagonizeestrogensbyglucocorticoidreceptor–mediatedactivationofestrogensulfotransferase.cancerres2008;68:7386.
[38]38.skormn,wonderel,kocherginskym,goyala,hallba,caiy,etal.glucocorticoidreceptorantagonismasanoveltherapyfortriple-negativebreastcancer.clincancerres2013;19:6163-72.
[39]39.laanee,panaretakist,pokrovskajak,buentkee,corcoranm,
[40]40.fusellaf,seclìl,bussoe,krepelovaa,moisoe,roccas,etal.theikk/nf-κbsignalingpathwayrequiresmorganatodrivebreastcancermetastasis.natcommun2017;8:1636.
[41]41.keanemm,rubinsteiny,cuellom,ettenbergsa,banerjeep,naumm,etal.inhibitionofnf-kappabactivityenhancestrailmediatedapoptosisinbreastcancercelllines.breastcancerrestreat2000;64:211-9.
[42]42.connellyl,barhamw,onishkohm,sherrillt,chodoshla,blackwellts,etal.inhibitionofnf-kappabactivityinmammaryepitheliumincreasestumorlatencyanddecreasestumorburden.oncogene2011;30:1402-12.
[43]43.xiay,shens,vermaim.nf-kappab,anactiveplayerinhumancancers.cancerimmunolres2014;2:823-30.
[44]44.songl,liul,wuz,liy,yingz,linc,etal.tgf-betainducesmir-182tosustainnf-kappabactivationingliomasubsets.jclininvest2012;122:3563-78.
[45]45.xuxc.cox-2inhibitorsincancertreatmentandprevention,arecentdevelopment.anticancerdrugs2002;13:127-37.
[46]46.basugd,pathangeylb,tindertl,gendlersj,mukherjeep.mechanismsunderlyingthegrowthinhibitoryeffectsofthecyclo-oxygenase-2inhibitorcelecoxibinhumanbreastcancercells.breastcancerres2005;7:r422-35.
[47]47.harrisre,chlebowskirt,jacksonrd,friddj,ascenseojl,andersong,etal.breastcancerandnonsteroidalanti-inflammatorydrugs:prospectiveresultsfromthewomen'shealthinitiative.cancerres2003;63:6096-101.
[48]48.bundrednj,barnesnlp.potentialuseofcox-2–aromataseinhibitorcombinationsinbreastcancer.brjcancer2005;93:s10-s5.
[49]49.millerdm,thomassd,islama,muenchd,sedorisk.c-mycandcancermetabolism.clincancerres2012;18:5546-53.
[50]50.d'cruzcm,guntherej,boxerrb,hartmanjl,sintasathl,moodyse,etal.c-mycinducesmammarytumorigenesisbymeansofapreferredpathwayinvolvingspontaneouskras2mutations.natmed2001;7:235-9.
序列表
<110>台湾利得生物科技股份有限公司
<120>糖皮质激素受体促进剂樟芝酸a在抑制乳腺癌肿瘤发生与转移中的应用
<130>003
<160>25
<170>siposequencelisting1.0
<210>1
<211>23
<212>dna
<213>人工序列()
<400>1
cggcggaaggagcttacaatcta23
<210>2
<211>15
<212>dna
<213>人工序列()
<400>2
gtgcagggtccgagg15
<210>3
<211>23
<212>dna
<213>人工序列()
<400>3
ctcgcttcggcagcacatatact23
<210>4
<211>22
<212>dna
<213>人工序列()
<400>4
acgcttcacgaatttgcgtgtc22
<210>5
<211>21
<212>dna
<213>人工序列()
<400>5
cagccatttacctggcatgag21
<210>6
<211>22
<212>dna
<213>人工序列()
<400>6
gagggtcccaattcaacatcaa22
<210>7
<211>23
<212>dna
<213>人工序列()
<400>7
acctgaaagtgtcagctgtatcc23
<210>8
<211>18
<212>dna
<213>人工序列()
<400>8
ggatgctgtgccagacct18
<210>9
<211>22
<212>dna
<213>人工序列()
<400>9
acgtactgggcgaagagtctcc22
<210>10
<211>20
<212>dna
<213>人工序列()
<400>10
gacgtcacctgggctttcac20
<210>11
<211>20
<212>dna
<213>人工序列()
<400>11
ctcatcgcagatgcctggaa20
<210>12
<211>25
<212>dna
<213>人工序列()
<400>12
ttcaggtaataggcacccttgaaga25
<210>13
<211>20
<212>dna
<213>人工序列()
<400>13
acgcacgacgtcttccagta20
<210>14
<211>21
<212>dna
<213>人工序列()
<400>14
ccacctggttcaactcactcc21
<210>15
<211>20
<212>dna
<213>人工序列()
<400>15
gaatggggtgatgagcagtt20
<210>16
<211>19
<212>dna
<213>人工序列()
<400>16
cagaagggcaggatacagc19
<210>17
<211>19
<212>dna
<213>人工序列()
<400>17
ccaccagcagcgactctga19
<210>18
<211>20
<212>dna
<213>人工序列()
<400>18
gcagaaggtgatccagactc20
<210>19
<211>19
<212>dna
<213>人工序列()
<400>19
tcctggtatgacaacgaat19
<210>20
<211>19
<212>dna
<213>人工序列()
<400>20
ggtctctctcttcctcttg19
<210>21
<211>21
<212>dna
<213>人工序列()
<400>21
ccgagauguuagcugaaautt21
<210>22
<211>23
<212>dna
<213>人工序列()
<400>22
aaggagcuuacaaucuagcuggg23
<210>23
<211>21
<212>dna
<213>人工序列()
<400>23
uucuccgaacgugucacgutt21
<210>24
<211>30
<212>dna
<213>人工序列()
<400>24
gtacactagtttgtctgcacactggaggtc30
<210>25
<211>32
<212>dna
<213>人工序列()
<400>25
gtacacgcgttggaacccagtatgagtgtttg32
- 还没有人留言评论。精彩留言会获得点赞!
- 荧光定量RT‑PCR检测促甲状腺激素受体基因mRNA表达的试剂盒及其使用方法与制造工艺
- 神经肽Corazonin和其受体基因及在桔小实蝇特异性控制剂中的应用的制造方法与工艺
- 具有促凋亡活性的人IgG1衍生的抗体的制造方法与工艺
- 使用抗STEAP1抗体和免疫缀合物的方法与制造工艺
- 包含热弹性感觉剂和TRPA1受体抑制剂的皮肤接合剃刮助剂的制造方法与工艺
- 一种基于分子动力学模拟筛分黄酮类化合物抗雄活性方法与制造工艺
- 人雌激素受体α亚型单克隆抗体杂交瘤细胞株、单克隆抗体、制备方法、应用与制造工艺
- 一种抗高盐梳型微嵌段疏水缔合聚合物及其制备方法与制造工艺
- 水源性高碘地区人群甲状腺自身抗体筛查分析方法及应用与制造工艺
- 一种促甲状腺激素受体抗体化学发光免疫测定试剂盒及其制备方法与制造工艺