叶酸靶向双载药纳米粒的制备与应用的制作方法
本发明涉及一种叶酸靶向双载药纳米粒的制备方法与应用。
背景技术:
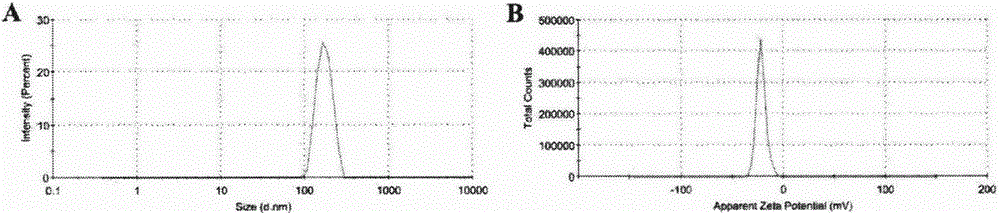
:癌症是一种严重威胁人类健康的疾病,在癌症的临床治疗中,化学治疗是最常用的手术方法。近几十年,癌症的化学治疗取得了显著的进步,癌症患者的生存指数得以明显提高,但现今临床常用的抗肿瘤药物,如阿霉素、紫杉醇、吉西他滨等,对肿瘤组织缺乏选择性或靶向性,在杀伤癌细胞的同时亦不可避免的损伤机体正常组织及细胞,引起毒副反应,限制了此类化疗药物的临床应用。随着纳米技术的发展,纳米材料常常被用作载体输送药物到肿瘤部位,这些纳米材料包括脂质体、聚合物胶束、碳纳米管等。由于叶酸基元功能化的纳米粒子可以特异的通过叶酸受体介导的胞吞效应增强肿瘤细胞对这种复合药物制剂的摄取,同时,叶酸修饰的复合聚合物纳米粒子,是肿瘤靶向给药的潜力材料。因此,使用纳米载体靶向输送抗癌药物或蛋白抑制剂,能够实现对肿瘤的高效低毒治疗。技术实现要素:针对上述面临的问题,本发明的目的在于提供一种叶酸靶向载药纳米粒的制备方法。该纳米粒以叶酸为靶向材料,水溶性树状大分子为载体,聚谷氨酸负载紫杉醇与吉西他滨,通过正负电荷相互吸引自组装成纳米粒。纳米粒中的叶酸与肿瘤细胞表面的叶酸受体结合,使纳米粒通过胞吞作用进入肿瘤细胞,在肿瘤细胞内酯键断裂释放药物,从而抑制肿瘤细胞增值,促使肿瘤细胞凋亡。本发明的另一目的在于提供一种上述叶酸靶向载药纳米粒在抗肿瘤方面的应用。本发明提供了如下技术方案:一种叶酸靶向载药纳米粒的制备。主要分为以下步骤:1)合成第三代树状大分子(pld);2)合成聚合物聚谷氨酸(pga);3)将抗癌药物紫杉醇(ptx)与吉西他滨(gem)通过酯键连接到聚谷氨酸(pga)上,得到化合物pga-ptx、pga-gem;4)将叶酸(fa)端基的羧基活化并与3-氨基丙醇反应,得到端基为羟基的化合物fa-oh,进而通过酯键连接到聚谷氨酸(pga)上,得到化合物pga-fa;5)在水溶液中,pld显正电荷,pga-ptx、pga-gem、pga-fa显负电荷,通过正负电荷自组装形成纳米粒。在本发明的技术方案中,步骤1)中第三代树状大分子聚耐氨酸pld的合成路线:式i-1与己二胺在缩合剂作用下得到式i-2,式i-2在三氟乙酸作用下生成式i-3为第一代树状大分子产物。式i-3与式i-1在缩合剂作用下得到式i-4,式i-4在三氟乙酸作用下生成式i-5为第二代树状大分子产物。式i-5与式i-1在缩合剂作用下得到式i-6,式i-6在三氟乙酸作用下生成第三代树状大分子pld。所用缩合剂为1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(缩写为edci),1-羟基苯并三氮唑(缩写为hobt),n,n-二异丙基乙胺(缩写为dipea)。在本发明的技术方案中,步骤2)中化合物聚谷氨酸(pga)的合成路线:式ii-1在光气的作用下得到式ii-2,式ii-2用正己胺为引发剂引发其开环聚合得到式ii-3,式ii-3在碱性条件下脱去苄基,得到pga。所述的碱选自碳酸钾、碳酸铯、氢化钾、氢化钠、氢氧化钾、氢氧化钠。在本发明的技术方案中,步骤3)中pga-ptx合成路线:pga与ptx在缩合剂的作用下,脱水缩合得到pga-ptx。在本发明的技术方案中,步骤3)中pga-gem合成路线:pga与gem在缩合剂的作用下,脱水缩合得到pga-gem。在本发明的技术方案中,步骤4)中pga-oh合成路线:将fa端基的羧基活化后,与3-氨基丙醇反应得到端基为羟基的fa-oh。在本发明的技术方案中,步骤4)中pga-fa合成路线:pga在缩合剂的作用下,与fa-oh端基的羟基脱水缩合得到pga-fa。在本发明的技术方案中,步骤5)中的纳米粒的制备,如表一所示。表一将样品pld,pga-fa,pga-ptx和pga-gem分别配制到0.5mg/ml,根据各比例分别加入不同量的样品,静置过夜,通过静电自组装成纳米粒。次日用马尔文动态光散射仪检测不同比例的水合粒径跟电位,结果如上表所示。在本发明纳米粒中,优选为比例1.5∶1的纳米粒进行下一步实验。因为其粒径相比于其他比例最小,为173.2nm,并且其电位为-21.6mv,如图一所示。用透射电镜观察其形态,如图二所示,形态大小均一并且不粘连。本发明纳米粒有益效果体现在:(1)所用载体为亲水性材料,制备的纳米粒具有高的生物相容性。(2)本发明所提供的纳米粒采用静电自组装的方式,高效,成本低,易操作。(3)本发明所提供的纳米粒采用叶酸为靶向材料,特异识别肿瘤细胞,从而实现更低的用药剂量和更好的治疗效果。(4)本发明所提供的纳米粒负载了紫杉醇与吉西他滨这两种抗癌药物,采用纳米粒释放紫杉醇,解决紫杉醇水溶性差的问题。(5)本发明所提供的纳米粒进入细胞后,细胞内吞形成胞内体,酯键会缓慢断裂,从而释放紫杉醇与吉西他滨。附图说明图1为马尔文动态光散射仪检测的ptx-gem-fa纳米粒的粒径与电位。图2为ptx-gem-fa纳米粒透射电镜图像。图3为紫杉醇,吉西他滨与叶酸的标准曲线。图4为紫杉醇与吉西他滨在ph5.5与ph7.4中的释放曲线。图5为动物实验中肿瘤增长率与小鼠体重变化。具体实施方式第三代树状大分子pld一种优选的合成路线。第一代1-3合成步骤:将式i-1(1000mg,1.89mmol)和hobt(640mg,4.74mmol)溶解于dcm(50ml)中,在-10℃下反应10min,加入edci(908mg,4.74mmol)反应30min,加入正己胺(109mg,0.945mmol),反应10min后加入dipea(734mg,5.67mmol)。反应20min后升至室温反应过夜。tlc检测反应,分别用10%的盐酸溶液(2×20ml),5%的nahco3溶液(2×20ml)和饱和食盐水(20ml)洗涤,ch2cl2层用无水na2so4干燥,减压蒸馏除溶剂,得到白色粉末状化合物i-2。式i-2称取800mg,加入2ml三氟乙酸,-10℃下反应30min后室温反应2h,加入大量三氟乙酸搅拌10min,抽滤,滤饼干燥得到化合物为第一代产物i-3。第二代i-5合成步骤:将式i-1(2.322g,4.4mmol)和hobt(891mg,6.6mmol)溶解于dcm(50ml)中,在-10℃下反应10min,加入edci(1.265g,6.6mmol)反应30min,加入i-3(911mg,1.1mmol),反应10min后加入dipea(854mg,6.6mmol)。反应20min后升至室温反应过夜。tlc检测反应,分别用10%的盐酸溶液(2×20ml),5%的nahco3溶液(2×20ml)和饱和食盐水(20ml)洗涤,ch2cl2层用无水na2so4干燥,减压蒸馏除溶剂,得到白色粉末状化合物i-4。式i-4称取1.1g,加入3ml三氟乙酸,-10℃下反应30min后室温反应2h,加入大量三氟乙酸搅拌10min,抽滤,滤饼干燥得到化合物为第二代产物i-5。第三代pld合成步骤:将式i-1(3.523g,6.7mmol)和hobt(1.121g,8.3mmol)溶解于dcm(50ml)中,在-10℃下反应10min,加入edci(1.591g,8.3mmol)反应30min,加入i-5(1.5g,0.83mmol),反应10min后加入dipea(1.074mg,8.3mmol)。反应20min后升至室温反应过夜。tlc检测反应,分别用10%的盐酸溶液(3×20ml),5%的nahco3溶液(3×20ml)和饱和食盐水(2×20ml)洗涤,ch2cl2层用无水na2so4干燥,减压蒸馏除溶剂,得到白色粉末状化合物i-6。1hnmr(400mhz,dmso-d6)δ7.98-7.59(-conh,m,15h),δ6.82-6.63(-conh,m,15h),4.16-3.82(-ch,s,14h),2.90(-ch2,dd,j=30.8,22.9hz,32h),1.48(-ch2,d,j=25.7hz,28h),1.36(-ch3,t,144h),1.33-1.28(-ch2,m,32h),1.22(-ch2,d,j=6.6hz,32h).式i-6称取1.1g,加入5ml三氟乙酸,-10℃下反应30min后室温反应2h,加入大量三氟乙酸搅拌10min,抽滤,滤饼干燥得到第三代产物pld。1hnmr(400mhz,d2o)δ4.58-4.11(-conh,m,14h),4.04-3.82(-ch,m,6h),3.31-3.02(-ch2,m,16h),3.00-2.84(-ch2,m,16h),2.05-1.69(-ch2,m,28h),1.66-1.43(-ch2,m,32h),1.40-1.11(-ch2,m,32h).化合物pga的合成路线:将式ii-1(5g,211mmol)溶解在100毫升无水四氢呋喃中,搅拌30min后,加入光气(6.25g,21.06mmol),50℃反应3小时。加入大量预冷的正己烷搅拌10min,抽滤得到化合物ii-2。将上步所得化合物ii-2(4.97g,18.89mmol)和正己胺(25.48g,0.25mmol)溶解在25mldmf中,35℃反应3天。加入大量预冷的甲基叔丁基醚搅拌10min,抽滤得到白色粉末为化合物ii-3。1hnmr(400mhz,dmso-d6)δ7.94(-conh,s,1h),7.29(-ph,dt,j=35.4,13.0hz,250h),5.20-4.79(-ch2,m,100h),3.91(-ch,s,50h),3.04(-ch2,m,2h),2.18(-ch2,m,100h),2.03(-ch2,m,100h),1.40(-ch2,m,2h),1.21(-ch2,m,6h),0.80(-ch3,t,3h).n=50.其中,聚合度n选自45-70,优选为n=50。取上步所得化合物ii-3(5g,22.62mmol)溶解在15ml甲醇中,称取3.6g氢氧化钠(90mmol)溶解在2ml水中,溶清后加入反应液中,室温反应5小时。后处理加入10ml乙醇,在转速9000rmp下离心10min得到沉淀,用乙醇反复洗涤沉淀3-4次,调节ph至7-8。将离心后的沉淀加入5ml盐酸ea搅拌10min,再次离心,将沉淀用真空干燥箱干燥得到聚谷氨酸(pga)。1hnmr(400mhz,dmso-d6)δ12.28(-cooh,s,50h),8.13(-conh,dd,j=133.4,54.2hz,50h),4.27-3.82(-ch,m,50h),3.43(-ch2,d,j=7.0hz,2h),2.25(-ch2,m,100h),2.00-1.68(-ch2,m,100h),1.37(-ch2,m,2h),1.23(-ch2,m,6h),0.84(-ch3,t,3h).n=50.其中,聚合度n选自45-70,优选为n=50。化合物pga-ptx合成路线:将化合物pga(400mg,3.3mmol)溶于4ml二甲基亚砜,加入edci(632mg,3.3mmol)和dmap(200mg,1.64mmol),搅拌30min后加入ptx(80mg,0.0937mmol),室温反应24h后,反应液用透析袋(mwco=1000d)透析48h得到pga-ptx。1hnmr(400mhz,dmso)δ7.89(-ph,d,50h),7.68(-ph,d,50h),7.33(-ph,d,100h),5.27(-ch,s,50h),4.48(-ch,s,50h),4.07(-ch2,m,100h),3.55(-ch,s,50h),3.14(-ch,d,j=24.2hz,50h),2.95(-ch2,m,100h),2.67(-ch2,m,100h),2.32-1.33(-ch2,m,j=100.7hz,8h),1.02(-ch3,t,j=37.1hz,3h).m=50.化合物pga-gem合成路线:将化合物pga(400mg,3.3mmol)溶于4ml二甲基亚砜,加入edci(632mg,3.3mmol)和dmap(200mg,1.64mmol),搅拌30min后加入gem(80mg,0.3mmol),室温反应24h后,反应液用透析袋(mwco=1000d)透析48h得到pga-gem。1hnmr(400mhz,dmso)07.45(-ph,m,100h),5.99(-ch,d,j=55.8hz,50h),4.49(-ch,s,50h),4.09(-ch,d,j=49.9hz,50h),3.64-3.42(-ch2,m,2h),3.14(-ch2,d,j=30.1hz,2h),2.89(-ch2,d,j=55.6hz,2h),2.61(-ch2,d,j=55.9hz,100h),2.42-2.17(-ch2,m,100h),1.92-1.24(-ch2,m,8h),0.97(-ch3,t,3h).m=50.化合物pga-oh合成路线:称取fa(1.065g,2.413mmol)溶于20ml二甲基亚砜中,加入nhs(277mg,2.413mmol),edc(694mg,3.62mmol)搅拌1h后,加入0.5mlet3n,避光搅拌36h。后处理加入300ml甲基叔丁基醚∶丙酮=7∶3搅拌20min,抽滤后将滤饼用100ml甲基叔丁基醚∶丙酮=7∶3多次洗。抽滤,滤饼真空干燥得到化合物fa-nhs。将上步所得化合物fa-nhs(200mg,0.372mmol)和3-氨基丙醇(27.9mg,0.372mmol)溶于5ml二甲基亚砜中,室温搅拌24h。后处理加入大量甲基叔丁基醚∶正己烷=7∶3(v/v)搅拌10min,抽滤,将滤饼干燥得到淡黄色粉末fa-oh。化合物pga-fa合成路线:将化合物pga(200mg,1.65mmol),edci(316.86mg,1.65mmol),dmap(201.95mg,1.65mmol)溶在4ml二甲基亚砜中,搅拌30min。加入fa-oh(66mg,0.13mmol)搅拌24小时。将反应液用透析袋(mwco=1000d)透析48h得到产物pga-fa。1hnmr(400mhz,dmso)δ7.90(-ph,d,50h),7.48(-ph,d,50h),6.66(-ph,d,j=61.6hz,100h),4.49(-ch,m,50h),4.09(-ch2,m,50h),3.50(-ch,s,50h),2.96(-ch2,d,j=27.8hz,100h),2.67(-ch2,m,100h),2.26(-ch2,d,j=29.6hz,50h),2.14-1.61(-ch2,m,8h),0.94(-ch3,t,j=25.7hz,3h).m=50.纳米粒的配制:pld端基大量的氨基在水溶液中显正电荷,pga-ptx、pga-gem、pga-fa端基大量的羧基在水溶液中显负电荷,通过正负电荷相互吸引,自组装形成纳米粒。含叶酸纳米粒ptx-gem-fanps的配制:优选比例为1.5∶1(负电荷:正电荷):用移液枪取pga-fa,pga-ptx和pga-gem各300μl,加入600μlpld水溶液,得到1.5ml的澄清透明溶液。通过正负电荷的相互吸引,静置过夜自组装成纳米粒。不含叶酸纳米粒ptx-gemnps的配制:优选比例为1.5∶1(负电荷:正电荷):用移液枪取pga-ptx和pga-gem各300μl,加入400μlpld水溶液,得到1ml的澄清透明溶液。通过正负电荷的相互吸引,静置过夜自组装成纳米粒。样品固含量的测定:称重玻璃瓶5次并计算平均值,取1ml样品(如pga-ptx)放入玻璃瓶中,在真空干燥箱中干燥至恒重。然后称量瓶子重量5次并计算平均值,得到样品固体含量。标准曲线的绘制:用甲醇制备不同浓度的fa,ptx和gem溶液,使用紫外分光光度计测量吸光度。叶酸检测波长为283nm,紫杉醇检测波长为230nm,吉西他滨检测波长为269nm,根据各浓度对应的紫外吸光值绘制标准曲线,如图三所示。根据标准曲线计算样品中药物的浓度。载药量与接枝率的计算公式:载药量=样品中药物浓度/样品固含量;接枝率=药物的摩尔浓度/聚合物摩尔浓度表二样品固含量(mg/ml)载药量(%)接枝率(%)pga-fa6.9410.63.1pga-gem7.26.83.6pga-ptx7.8518.63.5纳米粒的释放方法如下:紫杉醇与吉西他滨的释放在pbs缓冲液中进行,分别配制ph5.5与ph7.4的pbs缓冲液,各加入0.5%的吐温80,充分混匀后待用。将5mgptx-gem-fanps溶解在2.5mlpbs中,装入透析袋(mwco=3.5kda),用夹子将两端密封,放入30mlpbs中,在37℃下搅拌。以特定的时间间隔从pbs中取出1ml释放介质,取样后,立即将等体积的新鲜pbs加入释放介质中。从ptx-gem-fanps释放的药物浓度表示为透析袋外药物浓度与纳米粒中总药物的累积百分比。通过高效液相色谱(hplc)分析测量ptx和gem的浓度并绘制释放曲线,如图四所示。结果表明,前10小时药物快速释放,而纳米粒在ph5.5中包封的药物比在ph7.4中更容易释放,因为酯键在酸性条件下更易断裂,然后释放药物。另一方面,紫杉醇的释放在两个ph条件下均比吉西他滨释放的慢,这说明疏水性紫杉醇诱导降解动力学程度较低。本发明纳米粒的体外抗肿瘤活性检测方案如下:本实验所用的检测液为cck-8,所用细胞为4t1细胞。实验体系为110μl,其中含有细胞混悬液90μl,检测液为10μl,药物(纳米粒溶液)10μl。具体实验过程如下:1、药物配制:本实验配制了三种样品,分别是含有叶酸纳米粒ptx-gem-fanps,不含有叶酸纳米粒ptx-gemnps和纯药(ptx∶gem=1∶1,v/v)。将两种纳米粒分别配制成药物含量为15.4μg/ml,用移液枪吸取100μl加入5.033ml蒸馏水得到0.3μg/ml浓度的药物,稀释得到0.2μg/ml,0.1μg/ml,0.08μg/ml,0.06μg/ml,0.04μg/ml,0.02μg/ml,0.01μg/ml浓度的药物,最后一个浓度为0不加药。称取紫杉醇1mg,吉西他滨1mg,加入2mldmso溶解得到浓度1mg/ml的纯药。用移液枪吸取100μl加入3.233mldmso得到药物浓度为30μg/ml。用移液枪吸取30μg/ml浓度的药物100μl加入900μg无血清的rpm11640培养基得到0.3μg/ml浓度的药物,稀释得到0.2μg/ml,0.1μg/ml,0.08μg/ml,0.06μg/ml,0.04μg/ml,0.02μg/ml,0.01μg/ml浓度的药物,最后一个浓度为0不加药。上述三种样品加入细胞混悬液时,均取10μl加入到90μl细胞混悬液,即最终药物浓度梯度为0.03μg/ml,0.02μg/ml,0.01μg/ml,0.008μg/ml,0.006μg/ml,0.004μg/ml,0.002μg/ml,0.001μg/ml。2、细胞混悬液配制:细胞分别计数后,稀释配制4t1为1×104个/孔。3、反应体系制备:96孔荧光酶标板中每孔加入细胞悬液90μl,孵育24h,然后每孔中加入10μl待测样品,使用纯药为阳性对照药,孵育24h。反应完毕后,每孔加入10μl检测液(cck-8),孵育2-3h,450nm荧光酶标(bmglabtechpolarstaroptimamicroplatereader)检测吸光度。4、数据处理:根据公式计算,存活率=(实验组od值-空白组od值)/(对照组od值-空白组od值)×100%,计算各个浓度的细胞存活率。实验结果表明,本发明的叶酸靶向载药纳米粒能够有效抑制4t1细胞生长,并且效果与纯药相当。出现这一结果可能是由于纯药中的紫杉醇与吉西他滨直接作用于癌细胞,而纳米粒中紫杉醇与吉西他滨需要通过酯键断裂从纳米粒中缓慢释放出来,缓慢释放的过程可能导致了细胞的延迟吸收。本发明纳米粒的体内抗肿瘤活性研究方案如下:6-8周龄的balb/c雌性小鼠12只,体重20±2g,随机分为四组,每组三只。每只小鼠左侧腋下接种4t1细胞,细胞浓度为1×106/100μl,每只注射100μl,待肿瘤体积长到80mm3左右开始尾静脉注射给药,给药剂量为每只10mg/kg。第一组注射ptx-gen-fanps,第二组注射ptx-gemnps,第三组注射纯药,第四组注射生理盐水做空白对照。每两天给一次药,给药时间为两周。每隔一天用游标卡尺量小鼠肿瘤体积并称重,肿瘤体积计算公式为:v=((长×宽2)/2。15天后将小鼠处死,分离出肿瘤。实验结果由图五所示,纯药与两组纳米粒都较为明显的抑制了肿瘤的生长。纯药的抑制效果优于没有叶酸靶向的纳米粒ptx-gemnps,有叶酸靶向的纳米粒ptx-gem-fanps的肿瘤抑制效果优于纯药。然而纯药组小鼠体重下降相比于其他三组较多,说明纯药含有较大的毒副作用,而纳米粒生物相容性好,对小鼠没有较大的伤害。本发明的纳米粒有叶酸修饰的能明显增加抗肿瘤效果,叶酸受体在癌细胞表面的高表达使得靶向纳米粒更多的被细胞摄取,因此,ptx-gem-fanps可以增加药物在肿瘤部位的富集,增强治疗效果,减少全身毒副作用。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1