一种染色质拓扑结构域边界的分析方法与流程
本发明涉及染色质结构的分析方法,具体涉及一种分析染色质拓扑结构域边界的分析方法。
背景技术:
染色质结构及其在基因调控和细胞特性中的作用引起了细胞生物学研究的广泛关注。测序和成像技术的进步进一步使人们在理解染色质结构方面取得飞速的进展。其中,染色质结构中一个最显著的特征就是在hi-c数据所观察到的染色质相互作用矩阵的对角线上具有增强的接触频率的矩形块,其最早在40kb分辨率的hi-c图(hi-cmap)中观测到,并且被命名为拓扑结构域(topologicallyassociatingdomains,tads)。拓扑结构域是连续的大片段的染色质折叠缠绕形成的三维结构,并且同一个拓扑结构域内部的染色质相互作用相对富集,而不同的拓扑结构域之间的相互作用则非常少。拓扑结构域同时也是基因组复制时机调控(replication-timingregulatoion)的稳定单元,具有重要的功能。
从结构上,不同的拓扑结构域之间会存在一个边界(contactdomainboundaries,cdbs),边界上往往有ctcf蛋白和cohesin蛋白复合体的结合。目前对于cdb的系统研究还相对较少,这也部分是由于灵敏而鲁棒(robust)的cdb检测方法还较为缺乏所导致的。现有技术中已有一些基于hi-c图的cdb拓扑结构域和cdb的计算方法,例如一维基于统计的方法,包括di、insulatonscore和topdom法,这些方法从原理上是通过计算原始染色质接触矩阵每个滑动窗口中的平均染色质相互作用频率来得出针对每个bin的一维统计量。另外还有一部分方法属于基于二维邻接矩阵的方法,其使用了原始染色质邻接矩阵中的全局信息,而并非前述的一维统计量,这些方法包括armatus、hicseq、ic-finder和arrowhead法等。
然而所有以上方法主要存在以下几个问题:计算复杂度较高,不适合应用在高分辨的hi-c矩阵;检测拓扑结构域的准确率不高,尤其是灵敏度低等。因此,有必要开发新的检测拓扑结构域的算法。
技术实现要素:
发明人经过深入研究,提供了一种新的检测拓扑结构域的方法,在本发明中该方法也被称为hicdb。该方法能够快速准确地检测和精细划分拓扑结构域,并且能够精准的寻找差异cdb。
在本发明的第一个方面中,提供了一种染色质拓扑结构域边界(cdb)的识别方法,其包括:
(1)针对至少一个条件下(例如1个、2个、3个、4个、5个、6个、7个、8个、9个、10个)的目标样本获得至少一次重复(例如1次、2次、3次、4次、5次、6次、7次、8次、9次或10次)的染色质相互作用结果;
(2)利用步骤(1)得到的结果数据获得染色质相互作用矩阵;
(3)给定窗长w,其中w为区间大小的x倍,所述区间大小优选为步骤(1)中染色质相互作用的分析方法的分辨率,优选的,所述区间大小为1kb至1mb之间,例如10kb、20kb、30kb、40kb、50kb、60kb、70kb、80kb、90kb、100kb、200kb、300kb、400kb、500kb、600kb、700kb、800kb、900kb和1mb;x优选为1-50之间的整数,例如1、2、3、4、5、6、7、8、9、10、20、30、40或50,对于位于第k和k+1个区间(bin)之间的每个位点s,计算不同窗口大小下的相对绝缘性ri(w,s),
其中,u、d和b分别表示位点s上游、下游以及中间区域的平均染色质相互作用频率,如下式计算:
(4)获得多个(例如2个、3个、4个、5个、6个、7个、8个、9个、10个、20个、30个、40个或50个)不同窗口大小下的ri值,取均值获得平均ri,如下式所示:
(5)检测步骤(4)得到的平均ri的局部峰值,优选的,利用matlab中的内置函数findpeaks来检测峰值;
(6)计算局部相对绝缘性lri值,并根据lri值确定cdb,其中lower_envelope指平均ri的下包络,通过线性插值拟合ri的局部最小峰值得到;
在一个实施方案中,其中步骤(1)中所述染色质相互作用结果是通过hi-c技术获得的,例如singlecellhi-c、dilutionhi-c、insituhi-c、dnasehi-c、capture-c和bl-hi-c等。
在另一个实施方案中,其中步骤(6)中,当lri值高于lri的截止值时,即可被确定为cdb。
在另一个实施方案中,所述lri截止值可以根据需要自行确定,或者通过下面的步骤确定:
a、根据lri值的大小对候选cdb进行排序,
b、基于步骤a所述排序依次计算富集分数es,计算公式如下式所示,其中s表示具有ctcf基序的候选cdb集合;li表示第i候选cdb;lrii表示第i候选cdb的局部相对绝缘;nhit是s中候选cdb的数量,而n表示候选cdb的总数;
c、选择在最大es处的lri作为cdb检测截止值。
在另一个实施方案中,其中步骤(2)中获得的染色质相互作用矩阵还经过kr标准化。
在本发明的第二个方面中,提供了一种差异cdb的分析或检测方法,其包括下列步骤:
i)利用第一个方面所述的方法,针对至少2个条件下(例如2个、3个、4个、5个、6个、7个、8个、9个或10个)目标样本获得各自的cdb信息;
当样本的染色质相互作用结果具有至少2次重复(例如2次、3次、4次、5次、6次、7次、8次、9次或10次)时,
ii)对于每个条件下的数据,合并位于一个区间(bin)内的cdb;随后针对不同重复进行库深度的归一化;
iii)计算每个基因组区间(bin)内不同重复的平均ri;优选的,每次重复使用kr标准化来校正样本内偏差;
iv)每个重复乘以一个用于校正文库大小差异的库深度调整因子(sizefactor),所述sizefactor为每个hi-c重复矩阵总和的平均值除以所有重复的矩阵总和;
v)应用ma归一化校正相同条件的重复之间的系统偏差;
vi)如果两个条件之间的平均ri值的差异高于所有cdb平均ri差异的90%分位数,或者其平均ri值在不同条件之间显著不同(p<0.05,t检验)同时所述差异高于所有cdb的50%分位数,则认为cdb存在差异;相反,则认为不存在差异;
当样本的染色质相互作用结果不存在重复时,进行如下操作:
ii’)对于每个条件下的数据,合并位于一个区间(bin)内的cdb;随后进行库深度的归一化;
iii’)通过交集确定差异cdb。
在本发明的第三个方面中,提供了一种染色质拓扑结构域边界(cdb)的识别系统,其包括:
输入模块:用于输入针对至少一个条件下(例如1个、2个、3个、4个、5个、6个、7个、8个、9个、10个)的目标样本获得至少一次重复(例如1次、2次、3次、4次、5次、6次、7次、8次、9次或10次)的染色质相互作用结果和/或属于依据所述结果所得到的染色质相互作用矩阵;
优选的,还包括矩阵生成模块:用于基于输入模块所输入的染色质相互作用结果生成染色质相互作用矩阵;以及
计算模块,所述计算模块具体包括:
(a)相对绝缘ri(w,s)计算器:在下述条件下:给定窗口大小w,其中w为区间大小的x倍,所述区间大小优选为用于获得步骤(1)中染色质相互作用结果的方法的分辨率,例如10kb、1mb等;x优选为1-50之间的整数,例如1、2、3、4、5、6、7、8、9、10、20、30、40或50,对于位于第k和k+1个区间(bin)之间的每个位点s,计算不同窗口大小下的相对绝缘ri(w,s),
其中,u、d和b分别表示位点s上游、下游以及中间区域的染色质相互作用频率,如下式计算:
(b)平均ri计算器:获得多个(例如2个、3个、4个、5个、6个、7个、8个、9个、10个、20个、30个、40个或50个)不同窗口大小下的ri值,取均值获得平均ri,如下式所示:
(c)候选cdb生成器:检测平均ri的局部峰值,优选的,利用matlab中的内置函数findpeaks来检测峰值;
(6)lri值计算器:计算局部相对绝缘性,并根据lri值确定cdb,其中lower_envelope指平均ri的下包络,通过线性插值拟合ri的局部最小峰值得到;
在一个实施方案中,其中步骤(1)中所述染色质相互作用结果是通过hi-c技术获得的,例如singlecellhi-c、dilutionhi-c、insituhi-c、dnasehi-c、capture-c和bl-hi-c等。
在另一个实施方案中,其中步骤(6)中,当lri值高于lri的截止值时,即可被确定为cdb。
在另一个实施方案中,其还进一步包括lri截止值确定器:并通过下面的步骤确定的所述lri截止值:
a、根据lri度量值的大小对候选cdb进行排序,
b、基于步骤a所述排序依次计算富集分数es,计算公式如下所示,其中s表示具有ctcf基序的候选cdb集合;li表示第i候选cdb;lrii表示第i候选cdb的局部相对绝缘;nhit是s中候选cdb的数量,而n表示候选cdb的总数:
c、选择在最大es处的lri作为cdb检测截止值。
在本发明的第四个方面中,提供了一种分析cdb差异的系统,其包括第三个方面所述系统中所包括的模块,并且还额外包括cdb差异计算模块,所述模块能够执行下列步骤:
当样本的染色质相互作用结果具有至少2次重复(例如2次、3次、4次、5次、6次、7次、8次、9次或10次)时,
i.对于计算模块所得到的每个条件下的数据,合并位于一个区间(bin)内的cdb;随后针对不同重复进行库深度的归一化;
ii.并且计算每个基因组区间(bin)内不同重复的平均ri;优选的,每次重复使用kr标准化来校正样本内偏差;
iii.)每个重复乘以一个用于校正文库大小差异的库深度调整因子(sizefactor),所述sizefactor为每个hi-c重复矩阵总和的平均值除以所有重复的矩阵总和;
iv)应用ma归一化校正相同条件的重复之间的系统偏差;
v)如果两个条件之间的平均ri值的差异高于所有cdb平均ri差异的90%分位数,或者其平均ri值在不同条件之间显著不同(p<0.05,t检验)同时所述差异高于所有cdb的50%分位数,则认为cdb存在差异;相反,则认为不存在差异;
当样本的染色质相互作用结果不存在重复时,进行如下操作:
i’)对于计算模块所获得的每个条件下的数据,合并位于一个区间(bin)内的cdb;随后进行库深度的归一化;
ii’)通过交集确定差异cdb。
在本发明的第五个方面中,提供了一种鉴定调控染色质拓扑结构域或cdb的试剂的方法,其包括将使样本与一种或多种试剂接触,利用第一个或第二个方面所述的方法分析cdb或者cdb差异,以及
鉴定相比于不添加试剂的对照组能够改变cdb的试剂。
在本发明的第六个方面中,提供了一种分析细胞分化、发育或病变过程中遗传物质高级结构改变的方法,其包括本发明第二个方面中所述的步骤。
在本发明的第七个方面中,提供了一种鉴定染色质结构变异的方法,其包括本发明第二个方面中所述的步骤。
在本发明的第八个方面中,提供了一种鉴定能够调控遗传物质高级结构或引起染色质结构变异的调控试剂的方法,其包括将使样本与一种或多种试剂接触,利用本发明第二个方面所述的方法分析cdb差异,以及
鉴定相比于不添加试剂的对照组能够改变cdb的试剂。
在本发明的第九个方面中,提供了一种与染色质结构改变相关的疾病的诊断方法,其包括进行本发明第二个方面的方法所述的步骤,其中样本为来自受试者的样品,并根据cdb差异分析的结果判断是否可能患有疾病;所述疾病优选是遗传疾病或癌症。
在本发明的第十个方面中,提供了一种潜在的与cdb相关的染色质结构蛋白的分析或鉴定方法,其中包括利用第一个方面所述的方法识别cdb位置,并鉴定出在多个cdb中富集的蛋白,即为所述潜在的与cdb相关的染色质结构蛋白。
本发明的方法与现有技术已有的其他方法相比,不但能够检测到数目更多cdb,而且与其他方法的一致性很高,在多次重复样本中结果也具有极佳的重复性。并且在使用计算机实施所述分析方法时,10kb下的全基因组数据只需10分钟即可完成,大大节约了时间并且降低了成本。此外,hicdb在各个阈值下的ctcf富集程度都是最高的,并且将在10kb分辨率下多于两种方法检测到的染色质边界作为金标准,也可发现hicdb具有最佳的灵敏性和特异性。
附图说明
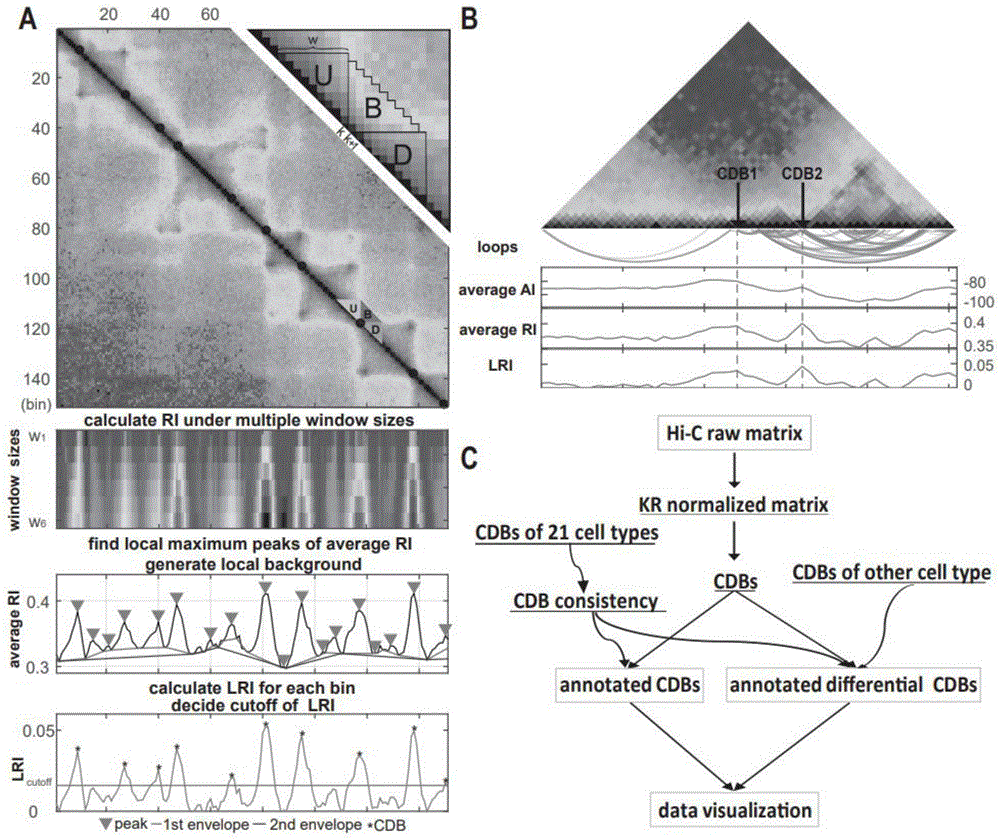
图1(a)显示了hicdb流程。图示区域是从10-kbgm12878hi-c矩阵中提取的。检测到的cdb在热图中显示为蓝点。其中右上角的小图详细展示了如何计算相对绝缘性。hi-c图谱下面的图显示了如何计算ri和lri的过程。(b)ai,ri和lri之间差异的示意图。cdb1代表具有高ai和高lri的cdb。cdb2代表在高度连接区域中具有低ai但高lri的cdb。(c)hicdb分析的总结。该流程图介绍了其主要功能和顺序操作,该流程可以以软件方式呈现。黄色方块是输入数据,绿色方块表示可选输出。带箭头的曲线表示可选步骤。
图2显示了hicdb与cdb的现有检测方法之间的比较。(a-b)不同方法之间的一致性。紫色条表示每种方法检测到的cdb总数。蓝色条表示通过任何其他方法确认的cdb数量以及比例。在10kb数据中计算一致性时允许出现一个bin的错误。(c-d)在40kb和10kb数据集中通过不同方法鉴定的cdb处每40kb/10kb的峰数量的聚集。(e-f)通过不同方法各自唯一预测的cdb处每40kb/10kb的峰数汇总。(g)不同方法的可重复性。如果方法具有排序的cdb输出,则在不同的截止值下计算cdb可重复性,否则,方法显示为单个点。通过将重叠的cdb数除以在两个hi-c重复上检测到的平均cdb数来计算可重复性。在10kb数据中计算可重复性时允许出现一个bin错误。(h)不同染色体的平均运行时间(计算机配置:cpu242.6ghz)。
图3显示了与代表性蛋白质结合位点重叠的cdb的百分比例。在10kb分辨率gm12878数据集中计算的百分比允许一个bin的错误。ctcf、cohesion亚基rad21和polr2a均对hicdb鉴定出的cdb表现出更高的偏好。
图4显示了不同截止值下cdb的ctcf结合百分比。不同方法的总检测到的cdb数量不同,这使得ctcf百分比比较困难。为了公平地比较这些方法,当方法具有排序的cdb输出时,计算了不同截止值下的ctcf结合百分比,而没有排序输出的方法在图中显示为单个点。在不同的截止值下,hicdb鉴定的cdb具有最高的ctcf结合百分比,这证明了hicdb的特异性。该图还表明ctcf结合百分比与lri相关。此外,类似gsea的截止选项不会偏向ctcf富集在hicdb检测到的cdb上。在10kb分辨率gm12878数据集中计算的ctcf结合百分比允许一个bin错误。
图5显示了hicdb对较小规模的cdb的识别。(a)不同方法得到的cdb距离分布。(b)使用在深度测序imr90样品上的至少两种方法检测的cdb作为金标准,在40-kbimr90hi-c图谱中,不同方法检测cdb的性能。(c)对深度测序的gm12878样品代表性区域(chr21:32.30-34.30mb)使用不同方法检测的比较。
图6显示了hi-c环锚和cdb的比较。(a)维恩图显示56%的cdb与利用hiccups调出的hi-c染色质环锚点(anchor)相重叠。此交集允许一个bin错误。(b)bin是基于与ctcf介导的染色质环锚点和polr2a介导的chia-pet环的锚点是否重叠而进行分类。特异性的hi-c染色质环锚点倾向于由ctcf介导的染色质环控制。
图7显示了cdb的表观遗传特征。(a)gm12878中chr21:42,50-46,50m的整体结构,其中显示了40kb和10kbgm12878数据集中由hicdb检测到的cdb。与hi-c环锚不重叠的10kbcdb标记为红色。(b)在两种分辨率下检测到cdb上的tf富集。x轴显示与某些tf结合位点重叠的cdb百分比;y轴显示与随机区域相比,tf结合位点在cdb处的富集情况。
图8gm12878和imr90的cdb预测结果比较。(a)以差异cdb为中心进行hi-c图的聚合。hi-c图显示了细胞类型特异性cdb处的绝缘性变强。(b)细胞特异性活性调节信号中富集的细胞类型特异性cdb。(c)在差异cdb附近的差异表达基因的倍数变化分布。使用wilcoxon秩和检验计算p值。(d)由hicdb检测的gm12878和imr90之间存在的差异区域(chr9:36.50-37.50mb)。该区域具有b细胞重要调节因子pax5,其在imr90中不表达。e1-e3标记了hicdb检测到的pax5的三种潜在增强子。
图9显示了由great分析的差异cdb附近的goterm。great的关联区域是基础区域+扩展区域:上游5kb,下游1kb,最大扩展500kb。
具体实施方式
还可进一步通过实施例来理解本发明,然而,要理解的是,这些实施例不限制本发明。现在已知的或进一步开发的本发明的变化被认为落入本文中描述的和以下要求保护的本发明范围之内。
样品
术语“样品”可以是或者可以源自一种或多种细胞、一种或多种细胞核、或一种或多种组织样品。实体可以是或者可为可源自存在核酸(如染色质)的任何实体。样品可以是或者可以源自一种或多种分离的细胞或一种或多种分离的组织样品,或者一种或多种分离的细胞核。
样品可以是或者可以源自活细胞和/或死细胞和/或核裂解物和/或分离的染色质。
样品可以是或者可以源自患病和/或非患病受试者的细胞。
样品可以是或者可以源自怀疑患有疾病的受试者。
样品可以是或者可以源自要测试他们将来会患有疾病的可能性的受试者。
样品可以是或者可以源自存活或非存活患者材料。
术语“条件”可以指某一种样本所处的外部特定环境,所述环境的变化会导致样本内部状态的改变;或者所述条件也可以指代两种或多种不同种类,并且为了实验目的进行比较的样本,每个样本即视为一个条件。
染色质
染色质中最丰富的蛋白质是组蛋白。染色质的结构取决于几个因素。总体结构取决于细胞周期的阶段:在分裂间期期间,染色质是结构上松散的,从而容许接近转录和复制dna的rna和dna聚合酶。分裂间期期间的染色质的局部结构取决于dna上存在的基因:活跃转录的dna编码基因是最松散包装的,并且发现它们与rna聚合酶联合(称为常染色质),而发现编码无活性基因的dna与结构蛋白联合,并且是更为紧密包装的(异染色质)。染色质中的结构蛋白的表遗传化学修饰也改变局部染色质结构,特别是通过甲基化和乙酰化对组蛋白蛋白质的化学修饰。由于细胞准备分裂,即进入有丝分裂或减数分裂,染色质更紧密包装以促进后期期间的染色体分离。在真核细胞的细胞核中,分裂间期染色体占据独特的染色体区域。
染色质相互作用
染色质相互作用是指一个核苷酸区段通过直接与另外一个核苷酸区段通过折叠成环等高级结构直接接触或结合,或者是一个核苷酸区段结合一个特定的中介分子(如蛋白质),该中介分子同时还与另外的一个或更多个核苷酸区段直接接触或结合,或者是一个核苷酸区段结合第一中介分子(如蛋白质),该中介分子又与与另外的一个或更多个核苷酸区段所结合的第二中介分子(如蛋白质)直接接触或结合,从而实现核苷酸区段之间的相互作用。在本发明中,染色质相互作用也可以被称为染色质环,
hi-c是检测染色质空间构象的关键技术,基于hi-c技术又演变出了多种染色质相互作用的分析技术,例如singlecellhi-c、dilutionhi-c、insituhi-c、dnasehi-c、capture-c和bl-hi-c,通过hi-c可以产生全基因组范围存在的大规模染色质相互作用的数据,现有技术中所有可用于染色质空间构象、染色质相互作用分析的方法均可用于本发明的方法中,以产生染色质相互作用的数据。在分析中,部分染色质区域或区段之间由于空间折叠相互相靠近,从而在染色质相互作用的分析,显示出相互相互作用的信号。这些信号经过分析转换为频率值后,被称为“接触强度”、“染色质接触强度”、“接触频率”、“染色质邻接频率”。
基于hi-c数据的染色质相互作用的检测方法,主要是基于互作矩阵(在本发明中,其也被称为染色质相互作用矩阵、染色质相互作用图谱、hi-cmap、hi-c互作图谱和hi-c矩阵)的基础上建模并计算的。
hi-c互作矩阵记录了hi-c实验检测到的基因组不同区域之间相互作用的配对读段的个数,可用来衡量基因组间相互作用的频率。通常,可将基因组各个染色体先划分为大小相同的区间(例如1mb,10kb等,区间的大小用来表示hi-c互作矩阵分辨率的高低),然后可统计不同区间之间检测到的配对读段的个数。例如1号染色体1mb分辨率的hi-c互作矩阵m的第i行第j列(下标从1开始)的数值mij表示该染色体上[i-1,i]mb区域和[j-1,j]mb区域之间的相互作用的读段总数。
染色质相互作用在互作矩阵中常常表示为局部峰值。例如,若基因组中相隔较远的两端区域a和b之间形成染色质环,a和b由于蛋白复合体等连接在一起,在三维空间中非常靠近。所以通过hi-c实验,捕获到的a和b区域之间的相互作用的读段数目就会较多,即表现为hi-c互作矩阵中的局部峰值。
染色质拓扑结构域(tad)
在染色质中存在的的兆碱基大小的局部染色质相互作用域,称作“拓扑相关结构域(tad)”,是由连续的大片段的染色质折叠缠绕形成的三维结构。同一个拓扑结构域内部的染色质相互作用相对富集,不同的拓扑结构域之间的相互作用则非常少。这些域与约束异染色质扩散的基因组区域相关联。所述域在不同细胞类型间稳定并且在物种间高度保守,并且彼此间具有相互作用,也为基因组形成高级结构提供了基础。
不同的拓扑结构域之间存在一个边界,这些边界被称为contactdomainboundaries(cdbs),边界上往往有ctcf蛋白和cohesin蛋白复合体的结合。
方法
标准hicdb方法
本发明提供了一种染色质拓扑结构域边界的识别方法,也被称为hicdb法,其理论依据在于cdb是具有高绝缘强度的局部峰。为了测量上述绝缘强度,本发明方法构建了一种的被称为局部相对绝缘(localrelativeinsulation,lri)的度量,将二维的hi-c图谱转换为一维向量。hicdb法具体包括以下步骤(图1a):
首先,设立一个新的统计量“相对绝缘性”ri来表示染色质结构域的相对绝缘性,使用相对绝缘性而非绝对绝缘性有利于找到tad内部的sub-tad结构,因为这些sub-tad之间的绝缘性不高,但是sub-tad内部的相互作用非常频繁,因此相对绝缘性较高。给定窗长w,介于k和k+1个区间(bin)之间的每个基因组位置s,定义相对绝缘性ri(w,s)如下所示:
其中,u、d和b分别表示位点s的上游、下游以及中间区域的平均染色质相互作用频率(图1a)。
随后在多个窗长下计算相对绝缘谱,加和平均得到每个基因组位置的平均相对绝缘性(ri),从而使得域边界更加明显(图1a)。根据相对绝缘性的定义,相对绝缘性受当前位置的绝对绝缘数值和两侧区域染色质相互作用的聚集程度影响。平均ri值越高则越有可能是染色质结构域的边界,因而检测结构域边界变成检测平均ri的局部峰值,利用matlab中的内置函数findpeaks来检测峰值,这些峰值位置即是候选染色质结构域结构边界:
随后,利用平均ri减去平滑背景后得到局部相对绝缘性(lri),以进一步增强cdb信号:
其中,lower_envelope被定义为下包络,指平均ri的局部极小值包络,通过线性插值拟合ri的局部极小峰值即可得到。lri的截止值可以由用户决定,也可以由hicdb参照ctcf结合位点富集程度输出。
lri度量结合了接触域的自关联和绝缘属性来检测cdb,这相比与现有技术此前的方法仅使用单一属性来讲具有显著的优势。例如,insulationscore和topdom测量了域边界的绝对绝缘(ai),而没有参考局部背景,这倾向于低估活跃区域中cdb的绝缘强度(crane,e.,bian,q.,mccord,r.p.,lajoie,b.r.,wheeler,b.s.,ralston,e.j.,uzawa,s.,dekker,j.andmeyer,b.j.(2015)condensin-drivenremodellingofxchromosometopologyduringdosagecompensation.nature,523,240;shin,h.,shi,y.,dai,c.,tjong,h.,gong,k.,alber,f.andzhou,x.j.(2015)topdom:anefficientanddeterministicmethodforidentifyingtopologicaldomainsingenomes.nucleicacidsres.,44,e70-e70.)。在这些模型中,对于每个基因组位点s,通过仅平均相互作用(b)计算平均ai:
ai和lri之间差异的示意图如图1b所示。cdb1代表具有高ai和高lri的cdb,而cdb2代表低ai但在密集的self-associated接触结构域中具有高lri的cdb。在使得绝缘强度度量在整个基因组中具有可比性并且将更多cdb(包括在整体具有更高染色质接触频率的区域内的cdb)与噪声区分开来方面,lri度量要明显优于ai。
总之,hicdb通过考虑接触域的self-association和绝缘特性以及通过应用多尺度聚合和背景去除的特异性来提高其在cdb检测中的灵敏度。
截止值(cutoff)的选择
已知ctcf是染色质结构域边界上的主要结构蛋白,为选择具有生物意义的相对绝缘性阈值来确定染色质结构域边界。可以将候选边界按照相对绝缘性从高到低排列,根据其是否在ctcf的motif附近计算统计量富集分数,取富集分数最大值前的候选边界为最终染色质结构域边界。在本发明中,hicdb法进一步提供了具有生物学意义的cdb截断值选项,该选项基于适应基因集富集分析(gsea)的方法(clark,n.r.andma’ayan,a.(2011)introductiontostatisticalmethodsforanalyzinglargedatasets:gene-setenrichmentanalysis.sci.signal.,4,tr4-tr4;subramanian,a.,tamayo,p.,mootha,v.k.,mukherjee,s.,ebert,b.l.,gillette,m.a.,paulovich,a.,pomeroy,s.l.,golub,t.r.andlander,e.s.(2005)genesetenrichmentanalysis:aknowledge-basedapproachforinterpretinggenome-wideexpressionprofiles.proc.nat.acad.sci.usa,102,15545-15550.)考虑ctcf基序的富集(29,30)。当表明hicdb截止选项时,hicdb将首先根据其lri对候选cdb进行排名。然后,通过浏览列表计算富集分数es,该列表反映候选cdb列表顶部的ctcf基序富集。es(i)的定义如下:
es是一个运行总和统计量,当它遇到具有ctcf基序的峰值时会增加,否则会减少。s表示具有ctcf基序的候选cdb集合。li表示第i候选cdb。lrii表示第i候选cdb的局部相对绝缘。nhit是s中候选cdb的数量,而n表示候选cdb的总数。选择在最大es处的lri作为cdb检测截止值。没有ctcf基序但具有比截止值更高的lri的候选cdb也保留在输出中,因为这实际上也具有生物学意义。该截止值选项在cdb检测数和ctcf富集之间保持平衡,但它不会偏向ctcf富集在hicdb检测的cdb上(例如参见图4)。通过使用来自jaspar数据库(heinz,s.,benner,c.,spann,n.,bertolino,e.,lin,y.c.,laslo,p.,cheng,j.x.,murre,c.,singh,h.andglass,c.k.(2010)simplecombinationsoflineage-determiningtranscriptionfactorsprimecis-regulatoryelementsrequiredformacrophageandbcellidentities.mol.cell,38,576-589;bryne,j.c.,valen,e.,tang,m.-h.e.,marstrand,t.,winther,o.,dapiedade,i.,krogh,a.,lenhard,b.andsandelin,a.(2007)jaspar,theopenaccessdatabaseoftranscriptionfactor-bindingprofiles:newcontentandtoolsinthe2008update.nucleicacidsres.,36,d102-d106)的ctcfpwm矩阵的homer基序分析获得全基因组ctcf基序位点。
差异cdb检测
为了识别多次重复的hi-c数据中具有差异的边界,本发明中,进一步计算每个条件下直接叠加原始hi-c矩阵上的cdb,并首先将得到的cdb汇集在一起,合并距离在1个bin内的cdb。然后,在对hi-c矩阵进行样本内、库深度、样本间的归一化后,对每个bin的不同重复计算平均ri。每次重复使用kr标准化来校正样本内偏差(例如酶切等的影响)(kalhor,r.,tjong,h.,jayathilaka,n.,alber,f.andchen,l.(2012)genomearchitecturesrevealedbytetheredchromosomeconformationcaptureandpopulation-basedmodeling.nat.biotechnol.,30,90)。接着对每个重复乘以一个用于校正文库大小差异的库深度调整因子(sizefactor),其被定义为每个hi-c重复矩阵总和的平均值除以所有重复的矩阵总和。
进一步应用ma归一化(djekidel,m.n.,chen,y.andzhang,m.q.(2018)find:differentialchromatininteractionsdetectionusingaspatialpoissonprocess.genomeres.,28,412-422.)校正相同条件的重复之间的系统偏差。为了控制假阳性,仅针对在一种条件下检测到的cdb检验其在不同样品间的平均ri值是否有显著性差异。如果两个条件之间的平均ri值的差异高于所有cdb平均ri差异的90%分位数,或者其平均ri值在不同条件之间显著不同(p<0.05,t检验)同时所述差异高于所有cdb的50%分位数,则认为cdb存在差异。对于没有重复的hi-c数据集,检测每个条件下经过库深度归一化处理后矩阵中的cdb,并通过交集确定差异cdb。
cdb分析
在一些实施方式中,还公开了cdb的分析方法,包括下列步骤:(1)以密集(denseformat)或稀疏格式(sparseformat)对原始hi-c矩阵执行kr归一化。(2)利用前述的标准hicdb方法,在步骤(1)获得的经过kr标准化的hi-c图中进行cdb检测。优选的,schmitt等人(schmitt,a.d.,hu,m.,jung,i.,xu,z.,qiu,y.,tan,c.l.,li,y.,lin,s.,lin,y.andbarr,c.l.(2016)acompendiumofchromatincontactmapsrevealsspatiallyactiveregionsinthehumangenome.cellrep.,17,2042-2059)生成针对21种细胞类型的hi-c数据被预先计算了cdb的存在和分布,连同所述cdb在不同细胞类型中的一致性,可作为在新样品中注释检测到的cdb的参考。(3)优选的,还可以针对步骤(2)中的hi-c数据(不论是否具有重复数据),进行差异cdb检测。(4)优选的,对单一hi-c图进行可视化和/或对已标注cdb的两个hi-c图之间进行比较。
数据源
用于比较cdb检测方法的中等分辨率(40-kb)hi-c数据的原始矩阵来自http://chromosome.sdsc.edu/mouse/hi-c/download.html(dixon,j.r.,selvaraj,s.,yue,f.,kim,a.,li,y.,shen,y.,hu,m.,liu,j.s.andren,b.(2012)topologicaldomainsinmammaliangenomesidentifiedbyanalysisofchromatininteractions.nature,485,376)。
hiccups检测到的高分辨率(10-kb)hi-c数据集和hi-cloops来自ncbi,登录号为gse63525(rao,s.s.,huntley,m.h.,durand,n.c.,stamenova,e.k.,bochkov,i.d.,robinson,j.t.,sanborn,a.l.,machol,i.,omer,a.d.andlander,e.s.(2014)a3dmapofthehumangenomeatkilobaseresolutionrevealsprinciplesofchromatinlooping.cell,159,1665-1680)。
对于imr90,两个重复的hi-c矩阵不可用,因此使用了juicer的计算结果(durand,n.c.,shamim,m.s.,machol,i.,rao,s.s.,huntley,m.h.,lander,e.s.andaiden,e.l.(2016)juicerprovidesaone-clicksystemforanalyzingloop-resolutionhi-cexperiments.cellsys.,3,95-98)。
从ncbi获得21个人类细胞系和原代组织的hi-c矩阵,登录号为gse87112(schmitt,a.d.,hu,m.,jung,i.,xu,z.,qiu,y.,tan,c.l.,li,y.,lin,s.,lin,y.andbarr,c.l.(2016)acompendiumofchromatincontactmapsrevealsspatiallyactiveregionsinthehumangenome.cellrep.,17,2042-2059)。
gm12878细胞系的ctnc和rna聚合酶ii(polr2a)的chia-pet数据从ncbi下载,登录号为gse72816(tang,z.,luo,o.j.,li,x.,zheng,m.,zhu,j.j.,szalaj,p.,trzaskoma,p.,magalska,a.,wlodarczyk,j.andruszczycki,b.(2015)ctcf-mediatedhuman3dgenomearchitecturerevealschromatintopologyfortranscription.cell,163,1611-1627)。
从encode数据库(neph,s.,vierstra,j.,stergachis,a.b.,reynolds,a.p.,haugen,e.,vernot,b.,thurman,r.e.,john,s.,sandstrom,r.andjohnson,a.k.(2012)anexpansivehumanregulatorylexiconencodedintranscriptionfactorfootprints.nature,489,83)下载所有chip-seq和rna-seq数据。差异基因利用deseq2进行识别(调整p值<0.01,log2-倍变化>1),(love,m.i.,huber,w.andanders,s.(2014)moderatedestimationoffoldchangeanddispersionforrna-seqdatawithdeseq2.genomebiol.,15,550)。
实施例1hicdb与已有的cdb检测方法的比较。
本实施例使用中等分辨率(40kb)和更高分辨率(10kb)的原始hi-c数据比较了不同cdb的检测方法的性能,并测量了几个定量标准,包括cdb数量、一致性、蛋白质结合富集、鲁棒性和时间复杂性。基于40kb数据集,将hicdb与armatus、di、hicseg、ic-finder、insulation和topdom进行了比较。在10-kb数据集中,di和hicseg由于其过高的计算时间复杂度而被排除在比较之外。同时由于arrowhead被设计用于染色质环水平的分辨率的hi-c实验,并且相比用于40-kb数据集的其他方法调用更少数量的域边界而被包括在比较中。
首先,分析了各方法一致性用以反映cdb检测的准确性(图2a和b)。hicdb检测到5768个cdb,在40kbimr90数据集的实际计数中最高的,其中有76%被其它方法检测到。在10kb的数据集中,arrowhead识别的cdb具有最高的一致性比率(86%),而hicdb则具有相近的一致性比率85%。尽管armatus和ic-finder分别在40kb和10kb数据集上确定了最多的cdb,但它们的一致性比率和数量都低于hicdb。
ctcf和cohesin富集指标进一步用于比较不同的方法,因为它们是广泛接受的域边界特征,常用于域检测方法的比较中。同时还考虑了cdb上的polr2a结合,因为已知活跃的转录与cdb形成相关。在两个数据集的方法中,由hicdb所检测到的与ctcf,cohesin和polr2a结合位点重叠的比例在各个方法中都是最高的(图3)。值得注意的是,对于不同的截止值,hicdb检测的cdb与ctcf的结合百分比总是最高的(图4)。
进一步通过通过聚集图(aggregationplot)检查了cdb上的结构蛋白或组蛋白修饰信号的分布(图2c和d)。结构蛋白和活性转录信号都集中在hicdb检测的cdb的中心,特别是在10kb的数据集上,而其他方法具有更宽的富集区域,表明hicdb可能检测到确切的功能位点。
除了hicdb检测的cdb与其他方法具有的高度一致性之外,在两个数据集中,hicdb所检测到的独有cdb富集了最多的结构和调节信号(图2e和f)。在40-kbimr90数据集中,topdom、ic-finder和di方法检测出较低的结构和调节信号的富集,这与上述方法所预测的独特cdb的侧翼区域仅具有模糊的绝缘边界的情况是一致的,这表明这些cdb位置的预测并不准确。此外,只有hicdb所独特预测的cdb在10-kbgm12878数据集上显示出清晰的绝缘和高度丰富的结构和调节信号。
同时,hicdb具有很高的鲁棒并且具有很快的速度。针对重复数据集的再现性对于评估鲁棒性非常重要。所有方法都应用于40-kbhesc数据集以及10-kbgm12878hi-c数据集重复以获得它们的再现性比率。在两种分辨率数据集的不同截止值下,hicdb在再现性方面优于其他方法(图2g)。另外,hicdb的时间复杂度是o(n),其中n是hi-c邻接矩阵的行/列数。在上机时,hicdb花了大约2分钟来计算全基因组cdb,这比分析40kb数据时第二快方法的绝缘分数(insulationscore)快2.5倍。hicdb分析10kb数据花了大约10分钟,使其比arrowhead和绝缘分数快两倍(图2h)。
实施例2hicdb可以准确识别较小规模的cdb。
本实施例比较了不同方法所鉴定的cdb距离分布(图5a)。armatus倾向于检测两个数据集上聚集在一起的许多小区域(图5c)。在40kb数据集中,hicdb检测的cdb之间平均距离为505kb,除了armatus之外,上述距离在所有方法中是最短的。利用10-kb数据,hicdb、arrowhead、topdom和ic-finder鉴定出的cdb之间距离约为200kb。值得注意的是,arrowhead和topdom的cdb距离分布具有两个峰值,这意味着这两种方法检测到的cdb的一小部分彼此紧密定位(图5c)。
由于深度测序数据的信噪比较高,基于10kb染色质邻接矩阵检测到的cdb比基于40kb矩阵的cdb更准确和完整。接下来将在10kbimr90hi-c矩阵中多于两种方法检测到的cdb作为“金标准“(即被认为是真实的cdb),用于定量评估评估在40kb分辨率的数据下各方法的特异性和灵敏度(图5b)。
结果表明:相比于其它方法,hicdb的灵敏度(34.1%)和特异性(69.0%)最高。这也表明hicdb可以在40kb数据集上比其他方法更准确的检测到较小规模的cdb。其次是topdom,灵敏度为26.7%,特异性为67.5%;再次是ic-finder;至于di、绝缘分数和hicseg,由于其最初被设计用于低分辨率hi-c数据中的tad边界检测,这导致它们的灵敏度相对较低。
由于缺乏评估10kb数据集性能的合适参照,于是根据其它的表观遗传注释评估不同方法检测到的cdb。图5c显示了gm12878基因组中一段代表性的两兆碱基的区域(chr21:32.30-34.30mb),其含有由hicdb、arrowhead、armatus、ic-finder、topdom和insulationscore法分别检测得到的15个、13个、9个、7个、7个和6个cdb。由于hicdb法对该区域主要结构进行了准确识别,因而还检测到另外五个cdb,即b1-b5。这些只被hicdb识别的位置处于内部相互作用密集的相互作用域下,并且具有较高的相对绝缘性。其中,b1、b2和b3位于ctcf介导的染色质环的锚点(anchor)附近,而b4和b5是由活性组蛋白标记覆盖的polr2a介导的染色质环簇的边界。此外,在该区域中检测到的hi-c环往往是强ctcf介导的环,并且未能在强烈的自相关结构域中预测具有诸如b1-b7的锚的环。由上述结果可知,本发明hicdb可以在不同分辨率下从hi-c数据中准确地检测到较小规模的cdb,而所检测到的cdb与已知的结构蛋白(例如ctcf和cohesin)结合位点以及活性转录调节信号具有准确的重合。此外,hicdb的再现性比率也优于其他测试方法,从而有效的用于一致性和差异性cdb的检测。
实施例3hicdb检测的cdb富集结构蛋白与细胞特异性转录因子
本实施例验证了hicdb所预测的cdb与hi-c环、chia-pet环以及转录因子的结合位点。所有分析均在gm12878细胞系上进行。
首先,将hicdb所预测的cdb与chromhmm注释进行重叠,以显示这些cdb与染色质状态的关系。在40-kb数据集中,cdb显著富集了绝缘体(2.11倍)和启动子(1.75倍)。同时,在10-kb数据集中检测到的cdb富集了活性启动子(5.86倍),绝缘子(3.36倍)和增强子(3.23倍)。
随后将cdb与使用hiccuphi-c数据上提取的另一特征hi-c染色质环进行了比较。结果发现56%的cdb与hi-c染色质环锚定一致(图6)。在只被识别为cdb而未被识别为hi-c染色质环锚点的基因组位置中,有25%与仅有polr2a介导的染色质环重合。另外整体而言,cdb相对hi-c染色质环更加富集细胞特异的转录因子,而hi-c染色质环更加富集结构性的蛋白,如ctcf,yy1,cohesin等(参见图6)。可见相对于染色质环,cdb更偏功能性。进一步的观察见图7a,其中显示了在gm12878的4m碱基区域(chr21:42,50-46,50m)上检测到的cdb。结果表明,40-kbcdb和hi-c环主要与ctcfchia-pet锚点重叠,而10-kbcdb也反映了polr2a相互作用簇的锚点。而其中有11个cdb与polr2a染色质环的锚点重叠,但是未被该区域的hi-c环捕获。
接下来分析了除ctcf和polr2a之外,其他蛋白质是否与cdb相关。于是将cdb与来自encode数据库的转录因子(tf)和组蛋白修饰的229个chip-seq数据集进行比较(neph,s.,vierstra,j.,stergachis,a.b.,reynolds,a.p.,haugen,e.,vernot,b.,thurman,r.e.,john,s.,sandstrom,r.andjohnson,a.k.(2012)anexpansivehumanregulatorylexiconencodedintranscriptionfactorfootprints.nature,489,83)。除了结构蛋白ctcf和cohesin,znf143、yy1、trim22和转录因子如ikzf1,runx3,bhlhe40出现在半数左右的cdb上,另外细胞特异的转录因子rxra、irf3、myc和brca1等虽然只在一部分cdb上出现,但是相对基因组随机区域,富集程度在40kb和10kb时分别达到2倍和6倍以上(图7b)。这些转录因子的富集程度比在hi-c上检测到的染色质环上的富集程度更高,表明了cdb相比于目前hi-c图谱上检测到的染色质环与细胞特异性更为相关。另外有趣的是,trim22蛋白在cdb上的富集程度与经典的结构蛋白cohesin类似,经检验,这种现象不仅出现在gm12878细胞系中,也出现在mcf7细胞系中(数据未显示),因此trim22很可能是一种未被报道的结构蛋白。
以上结果表明了,hicdb检测的cdb会富集结构蛋白与细胞特异性转录因子,因此hicdb在检测功能性cdb方面更接近生物体的真实状况,具有独特的优势。
实施例4细胞类型特异性cdb与细胞类型特异性组蛋白修饰和细胞特异性基因的激活相关
首先对10-kbgm12878和imr90数据集使用hicdb法进行了差异cdb检测,并分别预测了gm12878和imr90特异性cdb。hi-c聚合热图证实了在差异cdb处绝缘性具有变化(图8a)。
随后利用10kb分辨率hi-c数据分析了gm12878和imr90中特异性cdb的异同,揭示了差异cdb与细胞特异基因激活的相关性。比较hicdb在gm12878和imr90两种细胞系上的检测到的cdb,发现细胞特异的cdb上富集着细胞特异的组蛋白信号。具体的,将在gm12878和imr90中cdb处的polr2a、h3k4me3、h3k27ac和h3k27me3的信号进行汇集,以研究调节元件在差异cdb中的富集情况(图8b)。在gm12878中,发现活性调节信号,特别是增强子标记h3k27ac,更富集于gm12878特异性的cdb上而非imr90特异性cdb。相比之下,作为抑制性组蛋白标记的h3k27me3则不存在于gm12878特异性cdb处,而是富基于imr90特异性cdb处。这表明细胞类型特异性cdb与细胞类型特异性组蛋白修饰相关。
然后,利用great分析两种细胞系差异cdb周边基因的功能(标注goterm),发现gm12878特异的cdb周边基因主要富集在b细胞激活和干扰素伽玛信号通路上,imr90特异的cdb周边基因主要富集在肺发育通路上(图9)。imr90中有235个基因在两种细胞中表达水平相差50倍并且启动子区域与imr90特异的cdb重合,其中230个基因是相对于gm12878上调的,只有5个基因是相对于cdb下调的(图8c),这个结果显示细胞特异cdb的出现与邻近细胞特异基因的激活相关。
pax5是b细胞分化的重要调控因子,pax5的变异和重组会诱发肿瘤,该蛋白在gm12878中有表达,而在imr90中没有表达。hicdb检测到pax5邻域(chr9:36.50-37.50mb)在gm12878和imr90中具有截然不同的cdb(图8d),除了由hi-c染色质环发现的启动子(p)和pax5的远程增强子之外,三个其它的增强子(e1-e3)与gm12878中检测到的cdb重叠。然而,在imr90中则并未检测到p、e1和e3。也就是说,hicdb检测到的cdb与多个pax5增强子相重合,且具有活跃的组蛋白修饰信号。
以上结果表明通过hicdb检测到的细胞类型特异性的cdb与细胞类型特异性活性的组蛋白修饰、以及细胞特异性基因的上调具有密切关联。这更进一步说明了hicdb能够更加准确的检测得到真实的cdb。
- 还没有人留言评论。精彩留言会获得点赞!