一种双药配位聚合物抗结核纳米药物的制备方法与流程
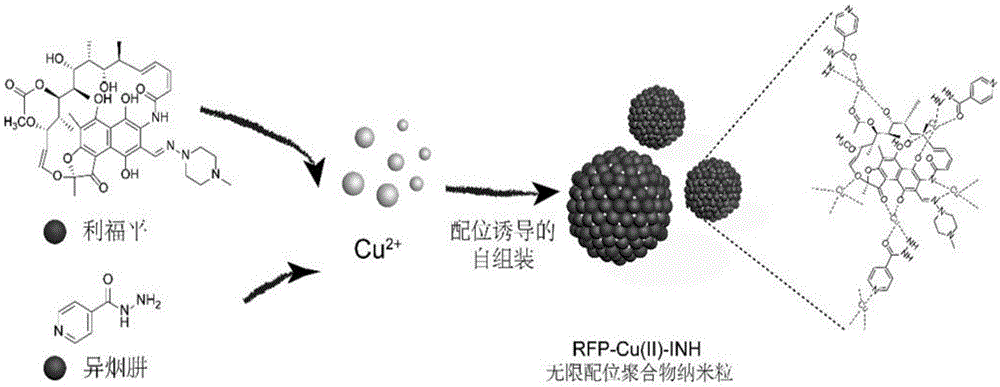
本发明涉及纳米药物范畴,特别涉及一种双药配位聚合物抗结核纳米药物的制备方法。
背景技术:
利福平,异烟肼,吡嗪酰胺和乙胺丁醇联合使用是目前国际上通用的标准化抗结核治疗方案。利福平为暗红色结晶粉末,在丙酮,二氯甲烷,乙醇中可溶解,几乎不溶于水,其具有广谱抗菌性能,抗菌机制为与细菌中依赖dna的rna多聚酶的β亚基结合,抑制结核分枝杆菌rna的合成,防止该酶与dna连接,从而阻断rna转录过程,使dna和蛋白的合成停止,进而阻碍细菌增殖。异烟肼为白色粉末,易溶于水,可溶解于乙醇(溶解度大于3mg/ml),是具有高选择性的抗结核药物,主要作用机制为抑制结核分枝杆菌细胞壁合成分枝菌酸,或同结核菌的菌体辅酶相结合,发挥干扰脱氧核糖核酸或核糖核酸合成的作用,进而抑制或阻止细胞的代谢繁殖。乙胺丁醇易溶于水,可溶于乙醇,单独使用时对结核杆菌的杀菌效果较弱,但在杀灭耐药型结核杆菌方面具有优势。吡嗪酰胺略溶于水,微溶于甲醇和乙醇,是一种酸依耐性抗结核药物,单独使用时疗效较差。
研究表明:单独使用一种抗结核药物时易产生耐药性,疗效较差。因此,在临床治疗中必须选用两种或两种以上的抗结核药物来防止耐药性发生,增强抗菌效果,缩短疗程,实现理想的抗结核治疗效果。很多研究者针对简化联合给药方式作了相应研究。这类发明主要致力于提高药物稳定性和实现药物复方给药。其中,钱令等人制备了口服复方利福平异烟肼胶囊(公开号:cn204798407u);王曦等人制备了含利福平异烟肼的药物组合物(公开号:cn1989966a);k·阿密斯等人制备了包含利福平、异烟肼、乙胺丁醇和吡嗪酰胺的抗结核病的组合物(公开号:cn105407874a)。
但是多种药物联合治疗时,受限于药物溶解性差异,难以实现理想的时空分布;并且到达病灶部位时的药物比例也难于控制。构建多药协同纳米药物是解决这一问题的有效途径。然而,目前有关一线抗结核药物的纳米药物递送系统的发明报道十分有限。目前已有的发明有:g·k库勒等人制备了一种包封有抗结核药物的聚dl-丙交酯-共-乙交酯纳米微粒(公开号:cn101160119a);欧阳五庆等人制备了一种利福平纳米乳剂抗生素药物(公开号:cn1875941a);聂丽娜等人制备了一种利福平纳米混悬剂(公开号:cn102370616a)。现有的方法大多利用表面活性剂对疏水的利福平进行包裹,或者使用多孔纳米载体对药物分子进行担载,但此类包裹方式通常包裹率较低,不能长时间稳定存在,并且大量引入表面活性剂及载体材料易引起系统毒性进而不利于在生物体内的治疗应用。有关多药协同抗结核纳米药物的发明更鲜有报道。戈朝晖等人制备了一种载有异烟肼、利福平的白蛋白纳米粒制剂(公开号:cn103271911a),但是由于利福平和异烟肼亲疏水性能差异很大,这种吸附-解吸附型药物搭载和释放方式在体循环过程中无法实现药物的时空同步释放。更重要的是受载体性质的影响,现有方法无法精确设计和担载确定比例的药物分子,进而无法实现最优的协同作用效果。已有的发明无法解决上述难题。
无限配位聚合物是由金属盐离子和双(多)齿配体通过自组装途径构成的配位聚合物纳米粒。用于制备无限配位聚合物的金属离子和配体分子容易得到且高度可选择,几乎所有具有配位能力的小分子功能剂及链状分子都能进行无限配位聚合物纳米粒的制备。更为优势的是:无限配位聚合物纳米粒通常具有可调节的尺寸、光学特征和其他形态依耐性特征,并且易于进行进一步的表面改性。因而,这种由重复配体通过金属节点连接的结构形式是非常多功能的。
目前,基于无限配位聚合物的发明主要集中在催化、气体储存、分离、光电及生物传感系统等领域。典型的发明有:吴仁安等人制备了一种糖-金属配位聚合物材料(公开号:cn108070090a);刘东等人制备了一种基于配位聚合物的荧光光控开关(公开号:cn107880276a);黄启谷等人制备了烯烃配位聚合催化剂(公开号:cn103880990a);张所江等人制备了一种离子型稀土金属有机配位聚合物(公开号:cn101863910a)。将这种技术用于药物递送系统的发明鲜有报道。可查询到的相关发明有r·莫泽等人制备的用于诊断成像和放射治疗的叶酸轭合物和相应的金属螯合物络合物(公开号:cn101646672a);郭丽华等人制备了一种具有抗癌活性的半三明治结构铱配合物(公开号:cn107652330a)。但是需要指出的是,这些发明所提金属有机配合物药物大多是非纳米结构,且配体均不是临床药物,他们在抗肿瘤时的毒副作用不可预测。poon等人首次以药物分子为配体制备了抗肿瘤配位聚合物纳米粒(非专利文献1:journalofcontrolledreleasevolume201,10march2015,pages90-99)。我们课题组也制备了药物-金属抗肿瘤纳米药物(非专利文献2:biomaterialsvolume154,february2018,pages197-212)。但应用于多药协同抗结核治疗的无限配位聚合物纳米药物当前尚无报道。
技术实现要素:
为了解决结核病化疗时,因联合化疗的两种药物溶解性差异及体内代谢路径不同,而引起时空分布不一致的问题;以及疗效差,剂量需求大,毒副作用大的问题;本发明的目的在于提供一种双药配位聚合物抗结核纳米药物的制备方法,首先将亲水及亲脂性抗结核药物按特定比例溶解在乙醇等溶剂中制成药物混合原液,然后将过渡金属(或镧系元素)氯化盐乙醇溶液快速加入药物混合原液,再向其中加入含表面活性剂的碱性溶液调节ph到中性,避光室温反应12-48小时得到产物;利用磷酸缓冲盐溶液对所得纳米药物进行透析,除去未包裹的药物;本发明制备方法简单高效,药物包裹率达100%,载药量水平高达96.27%,该纳米体系无需额外载体材料,在临床静脉注射给药时,能避免大量载体材料带来的额外毒副作用,减轻病人消化、代谢和排出载体材料时的额外负担。
为了达到上述目的,本发明的技术方案为:
一种双药配位聚合物抗结核纳米药物的制备方法,包括以下步骤:
步骤一:制备药物混合原液:
把1.0-5.0mg亲水性抗结核药物与1.0-5.0mg亲脂性抗结核药物共溶解于1ml溶剂中,在60-80khz超声下进行分散,分散时间为8-20分钟;
所述亲水性抗结核药物包括:异烟肼、吡嗪酰胺或乙胺丁醇;
所述亲脂性抗结核药物包括利福平;
所述溶剂包括:无水乙醇、二甲基亚砜、水或两种或以上任意比例混合;
步骤二:制备药物-金属离子预反应混合液:
取10mg/ml的过渡金属氯化盐和/或镧系元素氯化盐乙醇溶液10-300μl,以每分钟120-160滴的速度滴加到1ml步骤一制备的药物混合原液中,并按500-800转/分钟边滴加边进行磁力搅拌,滴加完毕后,继续搅拌5-10分钟,制备得药物-金属离子预反应混合液;
所述过渡金属氯化盐包括:二水合氯化铜、四水合二氯化铁或六水合三氯化铁;
所述镧系元素氯化盐为六水合氯化钆;
步骤三:制备双药配位聚合物纳米粒:
向步骤二所制混合液中快速加入碱性溶液和表面活性剂的混合溶液,表面活性剂在碱性溶液中的质量浓度为0.2wt%-1wt%,调节ph至中性,在此操作过程中保持1600转/分钟磁力搅拌;制备完成后,将溶液避光搅拌12-48小时,制备得双药配位聚合物纳米粒;
所述碱性溶液为1.21mg/ml的三羟甲基氨基甲烷,0.04mg/ml的氢氧化钠,体积浓度0.1%的三乙胺水溶液、磷酸二氢钾-氢氧化钠缓冲溶液、硼酸-硼砂缓冲液、甘氨酸-氢氧化钠缓冲液或硼砂-氢氧化钠缓冲溶液,缓冲溶液ph值范围8-10,选用其中一种即可;
所述表面活性剂包括:普罗尼克系列、十六烷基三甲基氯化铵、十六烷基三甲基溴化铵、吐温系列或非离子表面活性剂曲拉通x-100;
步骤四:透析浓缩:
将步骤三所得产物装入截留分子量为1500道尔顿的透析袋中,300转/分钟磁力搅拌条件下,在磷酸缓冲盐溶液中透析72-96小时,随后使用截留量30000道尔顿的离心透析管进行浓缩得到最终产物。
本发明首先利用溶剂制备药物混合原液,随后加入过渡金属(和、或镧系元素)氯化盐乙醇溶液,再加入碱性溶液调节ph值。利用配位诱导的自组装原理形成药物分子-金属配位聚合物纳米粒。其自聚机制是,药物分子作为多齿配体能够与过度金属离子形成三维空间上延续的配位键。在该过程中,药物原液中的各药物分子作为配体与中心金属原子共配位。在金属离子足量的情况下该纳米粒的包裹率可达100%。由于在该方法下反应条件温和故而无法生成金属有机框架,所得配位聚合物纳米粒为无定型状态,又称无限配位聚合物纳米粒。
本发明提出了一种基于配位诱导的自组装的一线抗结核药物递送系统。该系统克服了常规给药模式下药物亲疏水性质不同带来的双药体内时空分布不可控的缺点。特别地,得益于100%包裹率的特点,该系统实现了的双药比例的精确设计和控制,为获得最佳协同作用、实现最小毒副作用提供了平台。本发明的优势是:该体系中参与纳米粒形成的双药比例精确可控,为获得最佳协同作用和实现最小毒副作用提供了可能;本发明适用于利福平、异烟肼、乙胺丁醇、吡嗪酰胺等一线抗结核药物及具有配位功能的其他药物分子,具有良好的生物利用前景。
附图说明
图1为配位诱导的自组装法制备双药配位聚合物抗结核纳米药物的制备流程,以实例一为例。
图2为是实施例一、实施例二、实施例三、实施例四、实施例五、实施例六所得纳米药物的马尔文粒度仪测量的粒径照片。
图3为实施例所得纳米药物投射电镜照片,其中3a为实施例一、3b实施例二、3c实施例三,标尺=50nm。
图4a为实施例一x射线衍射图谱;4b为实施例一化学结构推测。
图5为实施例一x射线电子能谱表征,其中图5a为实施例一的xps全谱分析;图5b为cucl2的高分辨cu2p的xps谱分析;图5c为实施例一所得纳米药物的高分辨cu2p的xps谱分析;图5d为实施例一所得纳米药物与游离利福平、异烟肼的傅里叶变换红外吸光光谱比较。
图6为实例一所得利福平/异烟肼双载药纳米药物中主要元素的外层电子结合能变化。
具体实施方式
下面结合附图以及具体实施例对本发明进行详细阐述。
实施例一
参照流程图1,本实施例包括以下步骤:
步骤一:将3.0mg异烟肼和3.0mg利福平共溶解于1ml无水乙醇中,在60khz超声下进行分散,分散时间为8分钟,制成药物混合原液。
步骤二:取10mg/ml的二水合氯化铜乙醇溶液200μl,以每分钟120滴的速度迅速滴加到1ml步骤一所述利福平-异烟肼药物混合原液中,在此过程中500转/分钟避光磁力搅拌,滴加完毕后保持搅拌5分钟。
步骤三:向步骤二所述混合液中快速加入含质量浓度0.2wt%普罗尼克f127的1.21mg/ml的三羟甲基氨基甲烷,调节ph至7.4,此过程中保持1600转/分钟磁力搅拌;之后避光搅拌48小时。
步骤四:将所得产物装入截留分子量为1500道尔顿的透析袋中,300转/分钟磁力搅拌,在磷酸缓冲盐溶液中透析72小时;后用截留量30000道尔顿的离心透析管进行浓缩得到最终产物。
本实例中所得利福平/异烟肼(1:1质量比)双搭载纳米药物的粒径经马尔文粒度仪测量(详见图2中的a线条),其粒径为8.2nm。计算得药物包裹率为100%,总载药量为88.95%,其中利福平载药量为44.48%,异烟肼载药量为44.48%。
图3a为实施例一的透射电镜照片,展示了本实施例所得产物的形貌,标尺为50nm。
图4a为实施例一的x射线衍射结果,展示了该实施例所得纳米粒为无定型纳米粒,无明显晶体结构。图4b为根据药物分子化学结构及x射线电子能谱结论推断所得纳米药物的化学结构。
图5为实施例一x射线电子能谱表征。其中图5a为实施例一的xps全谱分析。图5b为cucl2的高分辨cu2p的xps谱分析。图5c为实施例一所得纳米药物的高分辨cu·2p的xps谱分析。图5d为实施例一所得纳米药物与游离利福平、异烟肼的傅里叶变换红外吸光光谱比较。上述表征结果证明药物分子与金属离子形成纳米粒的过程为配位诱导的自组装。
图6为实例一所得利福平/异烟肼双载药纳米药物中主要元素的外层电子结合能变化,说明所得双载药抗结核纳米药物中利福平和异烟肼分子均参与配位。
实施例二
本实施例包括以下步骤:
步骤一:将3.0mg利福平和3.0mg吡嗪酰胺共溶解于1ml乙醇、水体积比1:1混合溶液中,在60khz超声下进行分散,分散时间为10分钟,制成药物混合原液(此时为悬浮液,不影响后续制备)。
步骤二:取10mg/ml的二水合氯化铜乙醇溶液300μl,以每分钟120滴的速度迅速滴加到1ml步骤一所述吡嗪酰胺-利福平药物混合原液中,在此过程中600转/分钟避光磁力搅拌,滴加完毕后保持搅拌8分钟。
步骤三:向步骤二所述混合液中快速加入0.5wt%的十六烷基三甲基氯化铵的0.1%(v/v)三乙胺水溶液,调节ph至7.4,此过程中保持1600转/分钟磁力搅拌;之后避光搅拌24小时。
步骤四:将所得产物装入截留分子量为1500道尔顿的透析袋中,300转/分钟磁力搅拌,在磷酸缓冲盐溶液中透析72小时,截留量30000道尔顿的离心透析管进行浓缩。
本实施例中所得利福平/吡嗪酰胺(1:1质量比)双搭载纳米药物的粒径经马尔文粒度仪测量(详见图2中的b线条),其粒径为9.9nm;计算得药物包裹率为100%,总载药量为84.27%,其中利福平载药量为42.13%,吡嗪酰胺为42.13%。
图3b为实施例二的透射电镜照片,展示了本实例所得纳米药物的形貌。
实施例三
本实施例包括以下步骤:
步骤一:将3.0mg利福平和3.0mg乙胺丁醇共溶解于1ml乙醇中,在80khz超声下进行分散,分散时间为16分钟,制成药物混合原液。
步骤二:取10mg/ml的六水合三氯化铁乙醇溶液100μl,以每分钟120滴的速度迅速滴加到1ml步骤一所述利福平-异烟肼-乙胺丁醇药物混合原液中,在此过程中500转/分钟避光磁力搅拌,滴加完毕后保持搅拌5分钟。
步骤三:向步骤二所述混合液中快速加入1wt%的x-100的0.04mg/ml氢氧化钠溶液,调节ph至7.4,此过程中保持1600转/分钟磁力搅拌,之后避光搅拌12小时。
步骤四:将所得产物装入截留分子量为1500道尔顿的透析袋中,300转/分钟磁力搅拌,在磷酸缓冲盐溶液中透析84小时,截留量30000道尔顿的离心透析管进行浓缩。
本实例中所得利福平/乙胺丁醇(1:1质量比)多搭载纳米药物的粒径经马尔文粒度仪测量(详见图2中的c线条),其粒径为10.4nm;计算得药物包裹率为100%,总载药量为96.27%,其中利福平、乙胺丁醇的载药量分别为48.33%。
图3c为实施例三的透射电镜照片,展示了本实例所得纳米药物的形貌。
实施例四
本实施例包括以下步骤:
步骤一:将3.0mg利福平和3.0mg乙胺丁醇共溶解于1ml二甲基亚砜、水体积比为3:1混合溶液中,在80khz超声冰浴下进行分散,分散时间为10分钟,制成药物混合原液(此时为悬浮液,不影响后续制备流程)。
步骤二:取10mg/ml的二水合氯化铜乙醇溶液150μl,以每分钟150滴的速度滴加到1ml步骤一所述利福平-异烟肼-乙胺丁醇-吡嗪酰胺药物混合原液中,在此过程中600转/分钟避光磁力搅拌,滴加完毕后保持搅拌10分钟。
步骤三:向步骤二所述混合液中快速加入0.5wt%的吐温(t-20)的0.1%(v/v)三乙胺水溶液,调节ph至7.4,此过程中保持1600转/分钟磁力搅拌,之后避光搅拌16小时。
步骤四:将所得产物装入截留分子量为1500道尔顿的透析袋中,300转/分钟磁力搅拌,在磷酸缓冲盐溶液中透析96小时,截留量30000道尔顿的离心透析管进行浓缩。
本实施例中所得利福平/乙胺丁醇(1:1质量比)多搭载纳米药物的粒径经马尔文粒度仪测量(详见图2中的d线条),其粒径为10.4nm;计算得药物包裹率为100%,总载药量为91.48%,其中利福平、乙胺丁醇的载药量相同,分别为45.74%。
实施例五
本实施例包括以下步骤:
步骤一:将3.0mg利福平和3.0mg乙胺丁醇共溶解于1ml乙醇中,在60khz超声下进行分散,分散时间为16分钟,制成药物混合原液(此时为悬浮液,不影响后续制备流程)。
步骤二:取10mg/ml的二水合氯化铜乙醇溶液60μl与10mg/ml的六水合氯化钆乙醇溶液10μl混合,以每分钟120滴的速度迅速滴加到1ml步骤一所述利福平-异烟肼-乙胺丁醇药物混合原液中,在此过程中600转/分钟避光磁力搅拌,滴加完毕后保持搅拌6分钟。
步骤三:向步骤二所述混合液中快速加入0.2wt%的普罗尼克f127的1.21mg/ml的三羟甲基氨基甲烷,调节ph至7.4,此过程中保持1600转/分钟磁力搅拌,之后避光搅拌48小时。
步骤四:将所得产物装入截留分子量为1500道尔顿的透析袋中,300转/分钟磁力搅拌,在磷酸缓冲盐溶液中透析84小时,截留量30000道尔顿的离心透析管进行浓缩。
本实例中所得的利福平/乙胺丁醇(1:1质量比)双搭载纳米药物的粒径经马尔文粒度仪测量(详见图2中的e线条),其粒径为12.4nm;计算得药物包裹率为100%,总载药量为95.76%,其中三种药物载药量相同,分别为47.88%。
实施例六
本实施例包括以下步骤:
步骤一:将3.0mg利福平和1.0mg异烟肼共溶解于1ml乙醇中,在60khz超声下进行分散,分散时间为8分钟,制成药物混合原液。
步骤二:取10mg/ml的二水合氯化铜乙醇溶液160μl,以每分钟120滴的速度滴加到1ml步骤一所述利福平-异烟肼药物混合原液中,在此过程中800转/分钟避光磁力搅拌,滴加完毕后保持搅拌9分钟。
步骤三:向步骤二所述混合液中快速加入含0.2wt%普罗尼克f127的10mm的1.21mg/ml的三羟甲基氨基甲烷,调节ph至7.4,此过程中保持1600转/分钟磁力搅拌,之后避光搅拌48小时。
步骤四:将所得产物装入截留分子量为1500道尔顿的透析袋中,300转/分钟磁力搅拌,在磷酸缓冲盐溶液中透析72小时,截留量30000道尔顿的离心透析管进行浓缩。
本实例中所得利福平/异烟肼(3:1质量比)双搭载纳米药物的粒径经马尔文粒度仪测量(详见图2中的f线条),其粒径为13.2nm;计算得药物包裹率为100%,总载药量为87.05%,其中利福平的载药量为65.29%,异烟肼的载药量为21.76%。
实施例七
本实施例包括以下步骤:
步骤一:将1.0mg利福平和3.0mg异烟肼共溶解于1ml乙醇中,在60khz超声下进行分散,分散时间为12分钟,制成药物混合原液。
步骤二:取10mg/ml的二水合氯化铜乙醇溶液220μl,以每分钟160滴的速度滴加到1ml步骤一所述利福平-异烟肼药物混合原液中,在此过程中800转/分钟避光磁力搅拌,滴加完毕后保持搅拌10分钟。
步骤三:向步骤二所述混合液中快速加入含0.2wt%f127的1.21mg/ml的三羟甲基氨基甲烷,调节ph至7.4,此过程中保持1600转/分钟磁力搅拌,之后避光搅拌36小时。
步骤四:将所得产物装入截留分子量为1500道尔顿的透析袋中,300转/分钟磁力搅拌,在磷酸缓冲盐溶液中透析72小时,截留量30000道尔顿的离心透析管进行浓缩。
本实例中所得利福平/异烟肼双搭载纳米药物中双药比例为利福平:异烟肼=1:3(质量比);计算得药物包裹率为100%,总载药量为83.0%,其中利福平的载药量为20.74%,异烟肼的载药量为62.25%。
实施例八
本实施例包括以下步骤:
步骤一:将3.0mg利福平和1.0mg吡嗪酰胺共溶解于1ml二甲基亚砜、水体积比为1:1混合溶液中,在80khz超声下进行分散(冰浴),分散时间为20分钟,制成药物混合原液。
步骤二:取10mg/ml的二水合氯化铜乙醇溶液120μl,以每分钟160滴的速度滴加到1ml步骤一所述利福平-异烟肼药物混合原液中,在此过程中500转/分钟避光磁力搅拌,滴加完毕后保持搅拌10分钟。
步骤三:向步骤二所述混合液中快速加入含0.5wt%吐温(t-40)的磷酸二氢钾-氢氧化钠缓冲溶液,调节ph至7.4,此过程中保持1600转/分钟磁力搅拌,之后避光搅拌24小时。
步骤四:将所得产物装入截留分子量为1500道尔顿的透析袋中,300转/分钟磁力搅拌,在磷酸缓冲盐溶液中透析72小时,截留量30000道尔顿的离心透析管进行浓缩。
本实施例中所得利福平/吡嗪酰胺多搭载纳米药物中各药比例为利福平:吡嗪酰胺=3:1(质量比);计算得药物包裹率为100%,总载药量为89.95%,其中利福平的载药量为67.46%,吡嗪酰胺的载药量为22.49%。
实施例九
本实施例包括以下步骤:
步骤一:将1.0mg利福平和3.0mg吡嗪酰胺共溶解于1ml乙醇、水体积比为1:1混合溶液中,在65khz超声下进行分散(冰浴),分散时间为10分钟,制成药物混合原液(此时为悬浮液,不影响后续制备流程)。
步骤二:取10mg/ml的四水合二氯化铁乙醇溶液100μl,以每分钟120滴的速度滴加到1ml步骤一所述利福平-异烟肼-吡嗪酰胺药物混合原液中,在此过程中500转/分钟避光磁力搅拌,滴加完毕后保持搅拌10分钟。
步骤三:向步骤二所述混合液中快速加入含1wt%十六烷基三甲基溴化铵的甘氨酸-氢氧化钠缓冲液缓冲溶液,调节ph至7.4,此过程中保持1600转/分钟磁力搅拌,之后避光搅拌36小时。
步骤四:将所得产物装入截留分子量为1500道尔顿的透析袋中,300转/分钟磁力搅拌,在磷酸缓冲盐溶液中透析96小时,截留量30000道尔顿的离心透析管进行浓缩。
本实施例中所得利福平/吡嗪酰胺多搭载纳米药物中各药比例为利福平:吡嗪酰胺=1:3(质量比);计算得药物包裹率为100%,总载药量为90.06%,其中利福平的载药量为22.51%,吡嗪酰胺的载药量为67.54%。
实施例十
本实施例包括以下步骤:
步骤一:将3.0mg利福平和1.0mg乙胺丁醇共溶解于1ml乙醇中,在70khz超声下进行分散(冰浴),分散时间为12分钟,制成药物混合原液(此时为悬浮液,不影响后续制备流程)。
步骤二:取10mg/ml的二水合氯化铜乙醇溶液40μl与10mg/ml的六水合氯化钆乙醇溶液10μl混合,以每分钟140滴的速度滴加到1ml步骤一所述利福平-异烟肼-乙胺丁醇药物混合原液中,在此过程中600转/分钟避光磁力搅拌,滴加完毕后保持搅拌10分钟。
步骤三:向步骤二所述混合液中快速加入含0.5wt%吐温(t-60)的硼砂-氢氧化钠缓冲溶液,调节ph至7.4,此过程中保持1600转/分钟磁力搅拌,之后避光搅拌26小时。
步骤四:将所得产物装入截留分子量为1500道尔顿的透析袋中,300转/分钟磁力搅拌,在磷酸缓冲盐溶液中透析96小时,截留量30000道尔顿的离心透析管进行浓缩。
本实施例中所得利福平/乙胺丁醇多搭载纳米药物中各药比例为利福平:乙胺丁醇=3:1(质量比);计算得药物包裹率为100%,总载药量为95.90%,其中利福平载药量为71.94%,乙胺丁醇载药量为23.98%。
实施例十一
本实施例包括以下步骤:
步骤一:将5.0mg利福平和1.0mg异烟肼共溶解于1ml乙醇中,在70khz超声下进行分散(冰浴),分散时间为12分钟,制成药物混合原液(此时为悬浊液不影响后续制备)。
步骤二:取10mg/ml的六水合三氯化铁乙醇溶液150μl与10mg/ml的六水合氯化钆乙醇溶液10μl混合,以每分钟130滴的速度迅速滴加到1ml步骤一所述利福平-异烟肼-乙胺丁醇药物混合原液中,在此过程中680转/分钟避光磁力搅拌,滴加完毕后保持搅拌10分钟。
步骤三:向步骤二所述混合液中快速加入含0.5wt%十六烷基三甲基氯化铵的1.21mg/ml的三羟甲基氨基甲烷缓冲溶液,调节ph至7.4,此过程中保持1600转/分钟磁力搅拌,之后避光搅拌36小时。
步骤四:将所得产物装入截留分子量为1500道尔顿的透析袋中,300转/分钟磁力搅拌,在磷酸缓冲盐溶液中透析96小时,截留量30000道尔顿的离心透析管进行浓缩。
本实例中所得利福平/异烟肼双搭载纳米药物中各药比例为利福平:异烟肼=5:1(质量比);计算得药物包裹率为100%,总载药量为90.90%,其中利福平的载药量为75.75%,异烟肼的载药量为15.15%。
实施例十二
本实施例包括以下步骤:
步骤一:将1.0mg利福平和5.0mg异烟肼1ml二甲基亚砜、水体积比为1:1混合溶液中,在60khz超声下进行分散(冰浴),分散时间为12分钟,制成药物混合原液(此时为悬浮液,不影响后续制备流程)。
步骤二:取10mg/ml的二水合氯化铜乙醇溶液200μl与10mg/ml的六水合氯化钆乙醇溶液10μl混合,以每分钟150滴的速度滴加到1ml步骤一所述利福平-异烟肼-吡嗪酰胺药物混合原液中,在此过程中800转/分钟避光磁力搅拌,滴加完毕后保持搅拌10分钟。
步骤三:向步骤二所述混合液中快速加入含0.5wt%吐温(t-60)的0.1%(v/v)三乙胺水溶液,调节ph至7.4,此过程中保持1600转/分钟磁力搅拌,之后避光搅拌48小时。
步骤四:将所得产物装入截留分子量为1500道尔顿的透析袋中,300转/分钟磁力搅拌,在磷酸缓冲盐溶液中透析96小时,截留量30000道尔顿的离心透析管进行浓缩。
本实施例中所得利福平/异烟肼双搭载纳米药物中各药比例为利福平:异烟肼=1:5(质量比);计算得药物包裹率为100%,总载药量为88.40%,其中利福平的载药量为14.73%,异烟肼的载药量为73.67%。
实施例十三
本实施例包括以下步骤:
步骤一:将3.0mg异烟肼和3.0mg利福平共溶解于1ml无水乙醇中,在60khz超声下进行分散,分散时间为8分钟,制成药物混合原液。
步骤二:取10mg/ml的六水合氯化钆乙醇溶液110μl,以每分钟120滴的速度滴加到1ml步骤一所述利福平-异烟肼药物混合原液中,在此过程中700转/分钟避光磁力搅拌,滴加完毕后保持搅拌10分钟。
步骤三:向步骤二所述混合液中快速加入0.5wt%吐温(t-60)的0.1%(v/v)三乙胺水溶液,调节ph至7.4,此过程中保持1600转/分钟磁力搅拌;之后避光搅拌56小时。
步骤四:将所得产物装入截留分子量为1500道尔顿的透析袋中,300转/分钟磁力搅拌,在磷酸缓冲盐溶液中透析72小时,后用截留量30000道尔顿的离心透析管进行浓缩得到最终产物。
本实施例中所得利福平/异烟肼双搭载纳米药物中各药比例为利福平:异烟肼=1:1(质量比);计算得药物包裹率为100%,总载药量为93.41%,其中利福平和异烟肼的载药量均为46.71%。
- 还没有人留言评论。精彩留言会获得点赞!