一种微量溶质元素晶界偏聚的筛选方法与流程
本发明涉及一种微量溶质元素晶界偏聚的筛选方法,利用该方法能够迅速有效地获取溶质元素在晶界偏聚的各种信息,为改善材料的力学性能提供科学依据,属于材料设计技术领域。
背景技术:
实际使用的固体材料绝大部分是多晶体,而不是按单一定向的晶格排列的单晶体。多晶体中相邻两个不同取向晶粒之间的界面叫做晶界。在多晶材料中往往也不可避免地存在着各种溶质元素,如杂质和合金添加剂等。这些外来元素由于原子沿着晶界的扩散比在体相中扩散所需要的激活能小,因此更易于渗入晶界处分布,这些必然使晶界具有非常复杂的性质,进而影响材料的整体力学性能。因此,无论是金属或是非金属,晶界性质都尤为重要。上世纪八十年代由日本科学家渡边忠雄最先提出晶界工程,同时也可称为晶界设计与控制。通过晶界设计与控制提高材料性能已经被试图广泛用于设计和发展高性能的结构和功能材料。
随着超级计算机的计算能力和访问技术的迅速提高,材料模拟仿真在材料设计领域得到越来越广泛的应用。它能够直接从原子尺度描述微观组织对材料性能的影响,为新合金的实验开发和设计提供理论指导。尤其量子力学方法随着计算能力的增强更适用于实际材料体系。量子力学第一原理的优势在于几乎不会采用任何带有经验的参数,而是直接从给定的原子构型开始运用量子力学的基本计算,具有很高的准确度和预言性。大量理论计算已经表明杂质或合金添加元素对材料力学性能的影响与它引起的结合能的改变(原子从晶界迁移至自由表面所需的能量)是紧密联系的,此项规则可用于判断偏聚元素是否起脆化作用。
技术实现要素:
本发明的目的在于提供一种微量溶质元素晶界偏聚的筛选方法,采用该筛选方法可以简单快捷地获取微量溶质元素在基体晶界处偏聚的各种信息,与以往的单一的实验方法相比,大幅度降低了实验操作的复杂性以及研究成本的投入,进而从晶界设计与控制角度出发改善了材料的力学性能。
为实现上述目的,本发明采用以下技术方案:
一种微量溶质元素晶界偏聚的筛选方法,包括以下步骤:
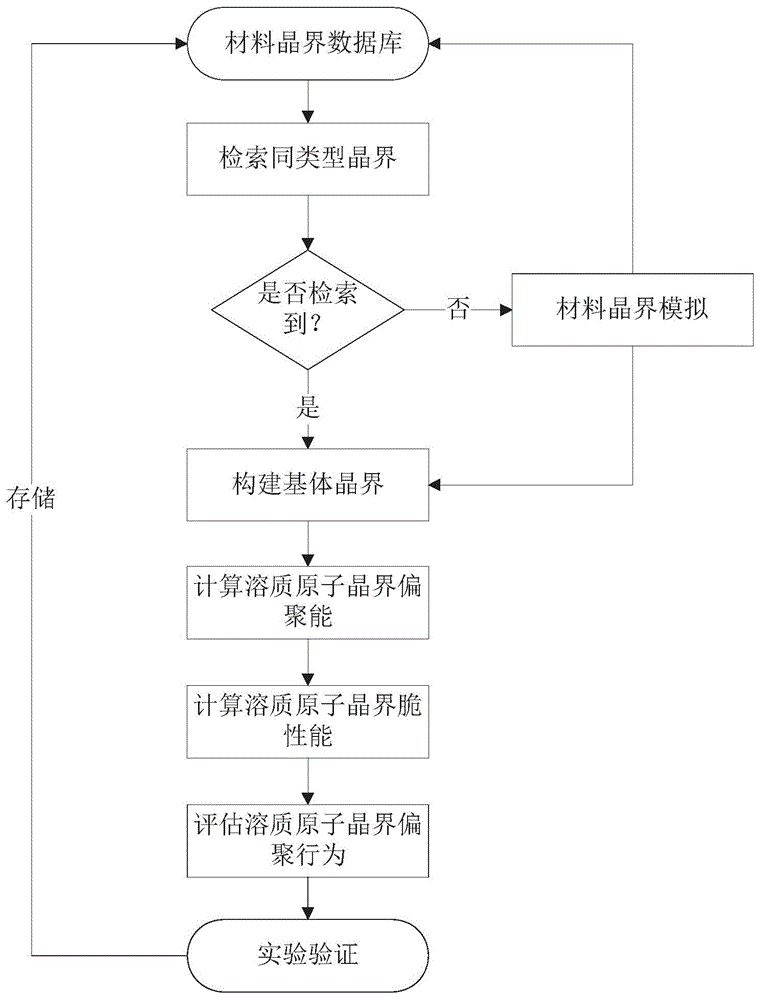
(1)获取基体晶界特征:从材料晶界数据库中检索同类型晶界,若存在,依据数据库信息构建晶界,若不存在,通过材料模拟软件建立对应的晶界,材料模拟软件包括量子力学第一原理软件vasp以及分子动力学计算软件lammps等。材料晶界数据库主要包括实验和计算模拟获取的材料的晶格常数、晶界原子点阵分布和晶界形成能等信息;
(2)获取微量溶质元素晶界偏聚能:利用材料模拟软件设置相关参数获取微量溶质元素晶界偏聚能曲线,并判断溶质元素在晶界是否偏聚以及其最可能的晶界占据位置;
(3)获取微量溶质元素晶界脆化能:利用材料模拟软件设置相关参数获取微量溶质元素在基体表面的偏聚能,从而计算出微量溶质元素从晶界迁移至自由表面所需的能量,即晶界脆化能,最终根据以上结果判断出微量溶质元素对晶界力学性能的影响程度,完成溶质元素晶界偏聚的评估,并根据脆化能的数值筛选出需要的溶质元素。
在本发明的筛选方法中,所述材料晶界数据库材料晶界数据库主要包括实验和计算模拟获取的材料的晶格常数、晶界原子点阵分布和晶界形成能等信息。将所述步骤(3)获取的有关的晶界晶格常数、晶界原子点阵分布和晶界形成能等信息存储到材料晶界数据库。所述步骤(2)中通过量子力学第一原理或其它计算方法设置相关参数绘制偏聚能曲线,用于确定微量溶质元素在晶界的偏聚位置。
基体晶界特征的获取方式有两种:一是直接从材料晶界数据库中检索同类型晶界,通常选取晶界形成能低且实际中存在的晶界;二是基于材料模拟软件直接模拟构建同类型晶界,且优先构建含有自由表面的晶界模型,使得计算模型包括晶界、体相与表面三个部分。同时保证拓展原胞每层至少含有4个原子,才可适用于模拟微量元素偏聚体系。
本发明的优点在于:
本发明对微量溶质元素晶界偏聚的筛选主要通过构建晶界模型,并利用计算软件获取微量溶质元素晶界偏聚能曲线及脆化能而最终评估溶质元素在晶界的偏聚行为。以往的科学研究已经表明溶质元素晶界偏聚行为与原子从晶界迁移至自由表面所需的能量紧密相关,但没有提出适当的建立模型方法以及完整流程的设计方法。本发明设计的一种微量溶质元素晶界偏聚的筛选方法,依据材料晶界数据库中检索同类型晶界或者材料模拟软件建立对应的晶界,并通过模拟获取微量溶质元素晶界偏聚能和脆化能,很大程度减少了传统实验方法人力物力的支出,明显地提高材料设计的效率。用本发明的方法筛选晶界偏聚的微量溶质元素具有很强的实用价值。
附图说明
图1为本发明实施的流程图。
图2为图1al∑5(210)[001]晶界的侧视图和俯视图。大球和小球分别代表(001)平面和(002)平面的原子。晶界附近不同的原子层从晶界面开始依次用数字标注。
图3为晶界偏聚能曲线。
具体实施方式
下面结合附图和实施例对本发明作详细说明,但并不意味着对本发明保护范围的限制。
如图1所示,为本发明实施的流程图,包括如下步骤:从材料晶界数据库中检索所需的基体材料同类型晶界,依据数据库信息构建晶界或基于材料模拟软件直接模拟并构建同类型晶界,接着通过模拟手段获取微量溶质元素在晶界的偏聚能以及脆性能等,并与实验结果进行校对,最终根据以上结果筛选出在晶界上偏聚的溶质元素种类以及判断它对晶界力学性能的影响。
实施例
以al晶界为例,微量溶质元素晶界偏聚的筛选包括如下步骤:
1、获取al基体晶界特征:从材料晶界数据库中检索面心立方结构的金属al晶界的特征,确定存在al∑5(210)[001]晶界。基于量子力学第一原理模拟计算得到晶格常数为
2、获取微量溶质元素晶界偏聚能:偏聚能的定义为偏聚元素从体相位置移至晶界附近时所获得的能量。因此,对于处于晶界gbn位置的元素偏聚能表达式可以写成:
根据公式1,我们获取了溶质元素zn和mg的偏聚能曲线,如图3所示。计算结果表明zn容易偏聚到晶界处gb2位置,偏聚能为-0.18ev;而mg容易偏聚到晶界处gb3位置,偏聚能为-0.17ev。
3、获取微量溶质元素晶界脆化能:将单个zn和mg原子分别置于模型的表面,通过量子力学第一原理方法计算zn和mg在表面的偏聚能分别为-0.25ev和-0.27ev。通过两种偏聚能的数值比较,我们获得溶质元素zn和mg脆性能分别为-0.07ev和-0.10ev,这表明它们更易于富集到自由表面。此模拟结果证实zn和mg都是脆性元素。
此模拟结果与实验观测到的现象一致。利用本发明,能够方便快捷地评估出微量溶质元素zn和mg原子偏聚到al晶界而引起的材料力学性能变化,具有很强的实用价值。
- 还没有人留言评论。精彩留言会获得点赞!