组织靶向复合物的制作方法
本发明涉及组织靶向复合物及其用途,特别是在眼科学中的用途。
背景技术:
在分析和诊断领域,给组织例如膜染色的染料是重要的工具。适用于染色的染料应当尽可能选择性地结合至特定类型的组织或膜,使得该染料的结合是其存在或位置的标记。适用于该目的的染料通常必须是耐受的并且应当具有从进行染色的周围区域突显的颜色。
在眼科学中,染料用于给随后将通过手术去除的膜染色。用于该目的的染料应当是水溶性的,因为在像眼睛一样敏感的器官中应当避免溶剂,应当具有允许容易与周围区域区分开的颜色,应当在使用后容易去除,并且当然应当不刺激或损伤眼睛。虽然已经存在一些用于给眼睛中的膜染色的染料,但是迄今为止所有这些染料都具有本发明旨在克服的缺点。
对于比如黄斑变性、视网膜病变、视网膜改变等的眼部疾病,通常需要进行玻璃体切除术,即,其中玻璃体或其部分被去除以便然后允许去除视网膜的内界膜或任何可能在眼睛内部形成的视网膜前膜以便诱导愈合过程和/或防止对视网膜的进一步损伤的手术。在这种手术中,使用标准手术器械将所涉及的膜从视网膜剥离,并且外科医生能够区分待剥离的膜和下面的视网膜是非常重要的,以便在手术期间不损伤视网膜。
视网膜由具有独立功能的多个层组成。脉络膜含有大部分的光感受器并且另外地负责充足的血液供应。它构成了视网膜的外部界限;视网膜的内侧与玻璃体相邻。内界膜是苗勒氏(müller)细胞的基膜,即,将玻璃体与视网膜分离并且从而构成视网膜的内部界限的细胞外膜。它不仅附连至苗勒氏细胞突,而且经由原纤维附连至玻璃体。这种附连特征在于玻璃体中的胶原纤维粘合至ilm。ilm由不同类型的糖蛋白和胶原组成。该层的厚度高达2.5μm。该边界层经常受到引起视力损伤并且可能/必须通过外科手术治疗的病症的影响。
例如,所谓的视网膜前膜可以在视网膜上形成;由于早期的病症或手术,这些膜可以在没有任何诱因的情况下发展或形成。这些膜可以收缩,导致视网膜变形和随后的视力损伤。因此需要去除拉动视网膜或黄斑的这种变形。
ilm去除也是治疗视网膜脱离和黄斑变性的一种选择。为了能够通过“ilm剥离”去除该薄膜,用可以选择性地给待去除的组织染色的染料来识别它是有帮助的。
已知用于该目的的染料的实例是亮蓝g、亮蓝r、专利蓝v、和台盼蓝以及吲哚菁绿。这些染料显示出毒性作用或用于外科手术的染色能力不足。
过去已经努力寻找用于给膜染色的染料,其尽可能选择性地粘附至待染色的膜。所以很难找到显示出与生理可接受性、水溶性和合适的色度相关的足够水平的选择性的用于单个膜的特定染色的合适的染料。
技术实现要素:
因此本发明的目的在于提供一种充分地给膜染色以使其可见的染色剂。本发明的目的还在于提供具有通过简单取代可调节的色度的试剂。本发明的进一步的目的在于提供一种染色剂,其不能渗透到更深的细胞层,但能够在目标区域选择性地相互作用,使得染色后染料以足够的量和在足够长的时间内保持在预期位置。
所有这些目的都通过权利要求书中限定的根据本发明的组织靶向复合物实现。
本发明涉及组织靶向复合物,其包括至少一个靶向基元[a-l-q+-alkb-],其中a是锚定基团,l是连接体,q+是季铵基团,b-是眼科学上可接受的抗衡离子,并且alk是具有4至12个碳原子的链长的烷基链;至少一个发色团,和任选地载体分子。
现已发现,令人惊讶地,适合或适于分别结合至眼组织或用于给眼组织染色的复合物可以通过使用包括至少两种基元——即至少一个靶向基元和至少一个发色团和/或至少一个载体分子的复合物形成。例如,根据本发明的组织靶向复合物可以包括发色团和另外地作为第三组分的载体分子。利用根据本发明的该组合,可以选择性地靶向膜并且用赋予期望的颜色的染料给它们染色。
通过提供具有与所期望的环境相互作用的功能和用于染色的单独单元实现有利的性质。这使得可以提供具有期望色度但对待染色组织没有选择性的发色团,其为允许相应组织被选择性地染色的形式。
定义
如本文所使用的术语“复合物”是指执行(包括彼此独立地执行)不同功能的分子组分的组合。
本发明的组织靶向复合物是包括靶向单元和染色或官能单元的复合物,该染色或官能单元在与目标组织接触时,在那里相互作用并且给目标区域染色以使其直接对眼睛可见,或者在用合适的波长照射下对眼睛可见。
术语“发色团”描述了可以直接或者通过用合适波长的光照射产生颜色的分子、元素、单元或基团。所产生的颜色是对眼睛可见的颜色并且因而是在380至780nm的范围内。术语“生理上可接受的”和“眼科学上可接受的”是指不损伤施用含有所述化合物、材料或物质的组合物的生物体并且符合其中普遍存在的条件的化合物、物质、材料等。此外,术语“眼科学上可接受的”意思是,所考虑的物质不刺激或损伤特别敏感的眼睛。
术语“载体分子”描述了能够充当用于根据本发明的基元比如靶向基元或发色团的载体的任何类型的分子。“携带”,其中其在该上下文中是指基元,意思是对载体分子的任何类型的固定,其可以通过共价键合或通过相互作用,例如通过吸附。基元通常共价键合至载体分子。载体分子可以是适合其功能的任何形式。因而,载体分子可以采取链、球、纳米颗粒或微粒或用于惰性载体分子的其他常见形状的形式。
如本文所使用,术语“连接体单元”是指连接两个基团的分子,其中连接体单元基本上是惰性的或中性的,这意味着其连接的基元的功能不会受到显著影响。连接体单元可以是连接两个单元的任何分子。连接体分子具有适用于特定目的的长度;为了使待连接的基元不要相距太远,单元不应当太长。连接体单元通常不贡献电荷或官能性。
“季铵离子”是带正电荷的基团,并且由氮和附连至其的四个基团组成。
与聚合物并且与根据本发明的组织靶向复合物相关联时,术语“水溶性的”和“水相容性的”互换地使用。由于对于含有聚合物的大分子,比如根据本发明的组织靶向复合物,通常无法精确确定分散液和溶液之间的梯度,因此水溶性/水相容性的组织靶向复合物应理解为可以与水混合,并且不会沉淀下来。
术语“目标区域”是指其中待完成结合或染色的区域。对于本发明,这通常是ilm和/或视网膜前膜。
当与靶向基元的选择性关联使用时,术语“相互作用”描述了靶向基元和膜之间的任何类型的相互作用,其导致靶向基元并因而根据本发明的复合物保留在其中发生相互作用的位置。该术语涵盖了任何类型的物理相互作用,前提是复合物保持在其发挥其作用的位置。
术语“眼膜”用于描述眼睛中的膜,特别是视网膜前膜和ilm。
根据本发明的复合物包括至少一个靶向基元,其将复合物引导至待染色的目标区域并且其中染料和/或载体分子将在期望的时间段内经历吸附、结合或其他这种相互作用。因此,靶向基元是与目标区域特异性地相互作用的基元。
现已令人惊讶地发现,结构基元与眼膜特异性地相互作用。该结构基元[a-l-q+-alkb-]由锚定基团、连接体、季铵基团和具有4至12个碳原子长度的烷基链组成。锚定基团由杂原子或官能团形成并且具有附连至发色团或至载体分子的功能,该杂原子可以是n、o、s、p或适于附连的另外的杂原子。杂原子可以特别地是n、s或o。对于官能团,考虑可以容易地形成键的基团,比如醇基、羰基、羧基等。
锚定基团经由连接体连接至季铵基团。连接体分子是将锚定基团连接至季铵基团、使两个基团保持预定距离并且还有助于柔韧性的分子。具有在1至10个原子,优选地1至8个并且尤其是1至6个原子范围内的链长并且实现了所述功能的任何分子在此都是合适的。已发现特别合适的烷基链是具有1至6个,优选地2至4个碳原子的烷基链。
季铵基团q+——其一端经由连接体附连至锚定基团并且另一端也带有烷基链,具有两个另外的取代基。这两个取代基是非关键性的,前提是它们在将分子引导至目标区域方面是中性的,即,不影响期望的功能。合适的实例是c1-c4烷基。这两个取代基优选独立地是h或甲基。
此外,对于季铵基团,存在抗衡离子b-。抗衡离子本身是非关键性的。因此可以使用任何分别在生理上或眼科学上合适的抗衡离子。优选使用卤素离子作为抗衡离子,比如氯离子或溴离子,或磷酸氢根离子或硫酸氢根离子。
根据本发明的靶向基元的第三部分是具有4至12个碳原子,优选地6至10个碳原子链长的烷基链。发现附连至季铵基团的烷基链有助于与ilm的相互作用。如果烷基链具有少于4个碳原子,则不再实现选择性结合。如果链长超过12个碳原子,则根据本发明的复合物的水溶性存在问题。另外,已发现,如果烷基链含有不饱和键将是不利的,因为这些限制了链的柔韧性。支化烷基链也是合适的,前提是支化不损害柔韧性。如果使用支化烷基,则关键因素是最长直链的长度,其在此描述为链长。链中存在的支链通常是甲基。
靶向基元可以经由锚定基团附连至发色团或至载体分子,或附连至二者,并且作为经由连接体又附连的特异性结合的基元,具有带有柔性烷基链的季铵离子,这对于与眼膜的相互作用是必要的。
令人惊讶地是,发现正是季铵离子和柔性烷基链的组合导致复合物与眼膜的特异性相互作用。不受理论的束缚,认为,季铵离子的正电荷与疏水烷基链一起导致分子被吸引到眼膜并且由于电荷分布和疏水性而保持在那里。因此,靶向基元应当以这样的方式键合至发色团和/或载体分子使得季铵基团和疏水组分易于与目标区域相互作用。这种相互作用的其他优势是染色剂或载体分子去到期望染色或结合的地方-眼膜处-并且停留在那里,这意味着与非特异性粘合的染料相比可以减少染色剂的量,由于非特异性附连染色剂目标区域外存在较少的视觉模糊,并且当与眼膜一起剥离膜时去除了大部分的染色剂。
根据本发明的组织靶向复合物的第二必要组分分别是至少一个发色团或至少一个载体分子。由于染色复合物旨在用于给眼膜染色,染料或发色团必须分别满足上述要求,即,它必须不具有刺激性或毒性并且它必须达到充分染色并形成水溶性的或水相容性的染色复合物。
所使用的发色团本身可以是任何已知的染料,前提是它满足了上述要求。蓝色至绿色色度范围内的染料特别适用于给眼睛中的膜染色,以便提供针对红色背景的最大对比度。此外,染料的染色能力必须尽可能地高,因为染色强度越高,染料的浓度就可以越低。期望的是,浓度尽可能地低,以便需要使用非常低的剂量,这也可以达到更好的耐受性。
根据本发明的复合物还有助于保持剂量低于其他已知染料的情况。期望的膜的选择性染色和靶向基元引起的附连意味着可以减少染料的量。特别地,染料被选择地带到所期望的位置并且保持在那里。
已经发现蒽醌基团非常适合用于根据本发明的用途并且作为根据本发明的复合物的一部分。蒽醌染料本身是众所周知的。可以通过衍生化调节颜色,这意味着所期望的色度可以通过适当的衍生化达到所期望的色度。此外,可以引入/附连进一步的基元比如靶向基元、桥接基元或载体分子。
在整个可见光范围内具有吸收的染料可以通过改变取代基的性质及其在蒽醌基础骨架中的位置来合成。特别地,在1、4、5和8位,即在蒽醌的α-位的取代,对染料的吸收具有强烈影响。未取代的蒽醌在405nm处具有最大吸收。电子供体引起红移,即朝向更长波长位移。因而,例如,1-氨基蒽醌具有λmax=475nm的最大吸收;1,4-二氨基蒽醌的最大吸收朝向更长波长位移115nm。这伴随着消光系数的增加,例如在1,4-二氨基蒽醌的情况下其高三倍。在2、3、6和7位即在所谓的β-位的取代基对所产生的颜色和反应性具有不太明显的影响。这些位置可以任选地用于附连其他基元。由于通过引入官能团已经改变了色度,还可以使用其他α-位的一个来附连进一步的基元而不限制颜色选择。
蒽醌染料的进一步的优势是它们对热效应、化学品和光的高稳定性。
因而蒽醌的α-位可以被选择性地衍生化以使其适合所期望的色度。此外,蒽醌骨架提供了引入靶向基元的选择,因为存在足够的可以用于衍生化的位置。如实施例中所述,衍生化本身可以通过标准方法相对容易地完成。
如以上所述,有利的是,提供具有在蓝色至绿色色度范围内的颜色的染料。已经发现,在两个苯环的一个的两个α-位上用氨基取代的蒽醌给出蓝色,而其中氨基和卤素基团附连在苯环的α-位的蒽醌相反具有在红色区域中的颜色。如果附连不同的杂原子,例如o和s,颜色同样改变。在这种情况下,第二苯环可以是未取代的。因此本发明的一个实施方式使用在两个α-位用取代的氨基取代的蒽醌作为发色团以赋予蓝色。
所使用的发色团特别地是式i的化合物:
其中基团r10、r11、r12和r13的每个独立地是氢、c1-c4烷基、如权利要求1中所限定的锚定基团或连接体分子,
基团r14、r15、r16和r17的每个独立地是氢、c1-c4烷基或连接体单元。
在一个实施方式中,提供蒽醌,其不仅具有优选的蓝色色度,而且带有靶向基元,该靶向基元通过使用引入到蒽醌的两个氨基实现了蓝色并且同时用作靶向基元的锚定基团而具有选择性地结合至眼膜、ilm和erm的性质。靶向基元不会对染色产生不利影响并且蒽醌不会对特异性结合产生不利影响。因此,在优选的实施方式中,蒽醌在两个α-位带有用c1-c6烷基取代的氨基,而未附连至蒽醌氨基氮的烷基的另一端带有又附连至烷基链的季铵离子。蒽醌染料和靶向基元的这种组合既赋予了蓝色,又有助于眼膜处的相互作用。因而提供了具有许多有利性质的组织靶向复合物。
蒽醌可以在一个至四个α-位用杂原子基团,特别地用氨基取代。如以上所述,在此取代基的数量可以用作适当地影响色度的手段。如果使用两个取代基,那么它们可以都附连在一个苯基的α-位或者一个氨基可以附连至两个苯基的每个上。
在一个优选的实施方式中,氨基被取代使得它同时还是复合物的靶向基元。
在另一个实施方式中,通过在仍然空着的位置的一个上引入靶向基元可以进一步衍生化通过适当的取代被赋予期望的色度的蒽醌。因此,在另一个实施方式中,蒽醌的1个、2个、3个或4个α-位带有影响色度的取代基,例如电子供体比如烷基氨基,并且其余位置的一个——其还可以是例如β-位——带有经由锚定基团附连的靶向基元。
这些根据本发明优选的蒽醌是水溶性的或水相容性的并且因此可以以这种形式用于给眼膜染色。
现在已经发现,取决于取代基,蒽醌能够渗透到细胞的更深区域,例如在眼膜附近。通常不期望的是,用于染色或处理的化合物能够更深地渗透到细胞,例如渗透到细胞核中,并且能够发挥细胞毒性作用。此外,不期望的是未去除并且因此保留在体内的细胞的染色。
在旨在防止根据本发明的组织靶向复合物能够渗透到细胞的一个实施方式中,除了至少一个靶向基元和至少一个发色团之外组织靶向复合物还含有载体分子。选择载体分子以便防止更深地渗透到细胞中。同时,载体分子必须是水相容性的,必须不会不利地影响靶向基元的特异性结合能力,并且必须不会刺激细胞或对细胞有害。换句话说,它是生理上和眼科学上可接受的聚合物。这种聚合物本身是已知的。
合适的载体分子是带有官能团的聚合物,通过该官能团可以附连发色团和任选地靶向基元。
任何载体分子都是合适的,前提是它在所形成的染色复合物中是水相容性的,不会对靶向功能或染色产生不利影响,并且能够附连至发色团和/或靶向基元。适合于此的是水相容性的聚合物,比如,例如,衍生自乙烯基化合物的聚合物和共聚物、衍生自多元醇的聚合物和共聚物、丙烯酸类聚合物和共聚物、以及天然和衍生的聚合物;也可以考虑肽或蛋白质。衍生自乙烯基化合物的聚合物和共聚物的实例是聚乙烯胺、聚乙烯醇、聚乙烯吡咯烷酮和相应的共聚物。衍生自多元醇的聚合物和共聚物的实例是聚甘油,多糖,聚糊精,环糊精,聚乙二醇,纤维素,和纤维素衍生物比如羟丙基甲基纤维素、羟乙基纤维素,透明质酸,和透明质酸衍生物。丙烯酸类聚合物可以是聚-n-(2-羟丙基)甲基丙烯酰胺和n-(2-羟丙基)甲基丙烯酰胺。其他合适的聚合物是聚谷氨酸,基于丙交酯和/或乙交酯的聚合物,例如聚乳酸,包括共聚物形式的聚谷氨酸,聚酰胺胺,和满足上述要求的其他聚合物。也可以是所提及的聚合物的混合物。
载体分子的尺寸可以在很宽的范围内变化并且取决于在个别情况下选择的聚合物、电荷等。可以考虑具有在900至1000万范围内的分子量的聚合物。适合于此的是具有5000至500000或1百万,例如优选地20000至200000,特别地30000至100000范围内的分子量的载体分子。
在一个实施方式中,发色团和/或靶向基元可以附连至具有反应性基团的聚合物。适合于此的是,例如,衍生自乙烯基胺的均聚物或共聚物,其中胺基可以被适当取代的蒽醌攻击并且与其共价键合。用于载体分子的聚合物还可以是肽或蛋白质。特别适用于该实施方式的是卤素取代的蒽醌,即在指定用于附连的位置带有卤素基团的蒽醌。因此卤素基团应当处于期望附连聚合物的位置。
使用蒽醌染料的优势是可获得反应性和较低反应性的位置用于附连在两个苯环上并且可获得影响颜色的位置(α)和对所得分子的颜色没有明显影响的位置(β)。
因此,一个实施方式使用蒽醌衍生物,其在至少一个α-位具有靶向基元并且任选地在β-位具有用于载体分子的附连位置,例如卤素比如氟或溴。
因而,在一个实施方式中,根据本发明的组织靶向复合物可以在至少一个α-位带有生色和靶向基团并且在β-位经由连接基团附连至载体分子。在优选的实施方式中,蒽醌衍生物在一个苯环上带有一个或两个靶向基元并且在另一个苯环的β-位附连至载体分子。
载体分子的功能之一是防止染色复合物渗透到细胞中。这意味着载体基元应当具有足够的尺寸以防止复合物吸收到细胞中。另一方面,聚合物不能太大而变得不溶和/或不再是溶解形式或在水中分散的形式。载体分子可以是任何合适的形状。它可以是发色团和靶向基元以任何顺序附连至其的聚合物链。它还可以是其中在分支点附连发色团和靶向基元的支链聚合物。在进一步的实施方式中,聚合物可以采取珠的形式,在其表面上附连发色团和/或靶向基元。
由于靶向基元的结构意味着它还可以附连至发色团,在另一个实施方式中,复合物具有这样的结构,其中载体分子具有与其附连的发色团单元,该发色团单元又具有附连至它们的靶向基元。
附连至载体分子的发色团和/或靶向基元的数量取决于载体分子的尺寸、单体单元的类型、靶向基元的结合能力、发色团(一种或多种)的染色能力。本领域技术人员能够使用简单的常规测试来确定个别情况下载体分子/发色团/靶向基元的最佳比例。发色团单元与靶向基元的比例同样是非关键的,并且,例如,通过发色团的颜色强度、靶向基元的亲和力、所期望的色度来引导。已经发现,相对于发色团过量的靶向基元通常是有利的。发色团单元与靶向基元的摩尔比在1∶20至5∶1,特别地在1∶10至2∶1的范围内被认为是合适的。
对于根据本发明的组织靶向复合物,通常仅使用一种类型的发色团。然而,还可以将不同的发色团附连至载体分子或者使用每个带有不同的发色团的载体分子的混合物。在此最重要的是达到所期望的色度以便然后给眼膜染色。
根据本发明的组织靶向复合物的结构还使得可以提供根据所使用的光源看起来具有不同颜色的染色剂。例如,可以使用具有不同发色团的根据本发明的组织靶向复合物的组合,其中一个发色团在一个波长处染色并且另一个发色团在不同波长的入射光下变得可见。
还可以使用具有荧光性质的化合物作为发色团。
根据本发明的组织靶向复合物具有与眼膜选择性相互作用的靶向基元。因而可以选择性地给眼膜染色为目标区域。因此本发明进一步提供了根据本发明的组织靶向复合物用于染色眼膜的用途。
附图说明
附图用于说明本发明,其中
图1显示了在0.005至0.08mg/ml的浓度下bbg对照物(蓝色)和bbgms溶液(红色)的所获得的uv/vis光谱数据的曲线图。
图2显示了在0.005至0.16mg/ml的浓度下7a和7b对照物(蓝色和红色)以及7a和7bms溶液(紫色和绿色)的所获得的uv/vis光谱数据的曲线图。
图3显示了在0.005至0.16mg/ml的浓度下7c和7d对照物(蓝色和红色)以及7c和7dms溶液(紫色和绿色)的所获得的uv/vis光谱数据的曲线图。
图4显示了在0.005至0.04mg/ml的浓度下icg对照物(蓝色)和icgms溶液(红色)的所获得的uv/vis光谱数据的曲线图。
实施例
通过以下实施例进一步说明了本发明,但不受它们的限制。实施例中给出的所有浓度均是指每ml的总溶液的底物的mg数。除非另有说明,否则所有反应均在室温下进行,即近似25℃。
以下步骤和条件用于分析:
1h核磁共振波谱(1hnmr波谱)
使用brukeravanceiiift-nmr波谱仪在室温下记录300mhz和600mhznmr。参考作为内标的氘代溶剂的信号报告化学位移。自旋多重度已缩写为s(单峰),d(双峰),dd(双二重峰),t(三重峰)或m(多重峰)。
13c核磁共振波谱(13cnmr波谱)
同样在brukeravanceiiift-nmr波谱仪上在75mhz和150mhz并且在室温下进行测量。参考作为内标的氘代溶剂的信号报告化学位移。
maldi-tof-ms
使用brukerultraflextof(时间偏光)质谱仪记录测量值。使用337nm氮气激光器操作仪器,该337nm氮气激光器为线性模式和反射器模式二者。将样品溶解在合适的溶剂中并且使用2-(4-羟基苯基偶氮)苯甲酸(haba)转移。
esi-ms
使用finniganlcqdeca离子阱api质谱仪记录测量值。将待测定的物质以1mg/ml的浓度溶解在合适的溶剂中。电离是通过电喷雾电离进行。
lc-ms
使用与agilent四极杆质谱仪组合的带有吸收检测器(214nm)的agilenttechnologies6120仪器进行测量。使用1.8μm(2.1x50mm)agilentsb-c18柱,其中在室温下h2o∶mecn+0.1%甲酸作为溶剂混合物。流速为0.4ml/min,其中溶剂混合物为95∶5h2o∶mecn和5∶95h2o∶mecn。
gc-ms
使用thermofinnigantracedsq(双级四极杆)记录测量值。通过离子碰撞电离(ei)使样品电离。
ftir光谱
使用带有lotatr附件的nicolet6700ftir波谱仪在室温下记录ftir光谱。使用hene激光校准波数刻度。在4000-300cm-1范围内记录测量值。
uv/vis光谱
使用analytikjenaag
薄层色谱
使用merck板(在铝箔上的硅胶60f254)进行薄层色谱。
hplc
使用具有吸收和荧光检测器的agilent1200记录rp-hplc(反相高效液相色谱)测量值。在60℃下以1ml/min的流速使用agilentzorbaxeclipsexdb-c18柱(4.6x100mm)。采用含有0.1%三氟乙酸的h2o和mecn的二元洗脱液混合物。通过在10min的时间段以乙腈/水5∶95至95∶5的梯度洗脱进行测量。
实施例1
1,4-二氟蒽醌(3)的合成
将13.4g(0.9mol)的邻苯二甲酸酐(1)、48.8g(0.37mol)的alcl3和108ml(2mol)的1,4-二氟苯(2)加热回流48h。然后通过蒸馏回收过量的1,4-二氟苯。将400ml的1n盐酸添加至棕色残余物并且用三份600ml氯仿萃取残余物。将合并的有机相在减压下浓缩至大约50ml。然后将中间产物用至少100ml的低沸点石油醚沉淀。滤出褐色固体并且然后干燥。(产率:理论值的66.7%)将固体放置在200g的多磷酸中并且在140℃下加热2h。在冷却至室温后,将粘性反应混合物添加至1l的冰水中并且用三份400ml二氯甲烷萃取。将有机相在减压下浓缩,并且然后可以通过硅胶柱色谱纯化(己烷∶乙酸乙酯[7∶1])分离产物(3)。
产率:13.2g(54.1mmol)理论值的60.1%。(文献值=50%)
ftir(diamant):v=3084(s,υc=c),1678(s,υc=o),1587(m,υc=c),1249(vs,υc-f),719(s,δar-h)cm-1。
1hnmr(300mhz,cdcl3,rt):δ=8.20-8.29(m,2h,ar-h3,6),7.76-7.85(m,2h,ar-h1,2),7.43-7.53(m,2h,ar-h11,12)ppm。
13cnmr(300mhz,dmso-d6,rt):δ=180.23(c9,17),(158.55,155.10)(c10,13),134.40(c1,2),133.04(c4,5),126.23(c3,6),124.6-125.25(c7,8),121.43-121.60(c11,12)ppm。
ei-msm/z:244[m]
实施例2
1,4-双[[2-(二甲基氨基)丙基]氨基]蒽醌(5)的合成
在氮气氛围下,25ml的dmso中,将3.0g(12.3mmol)的1,4-二氟蒽醌(3)与3.1ml(24.6mmol)的n,n-二甲基氨基丙胺(4)在大约100℃下加热。在搅拌24h之后,将反应混合物冷却至室温并且用水稀释。将蓝色粗产物用chcl3萃取,在减压下浓缩,并且通过硅胶柱色谱纯化(ch2cl2∶meoh∶net3[3∶4∶0.1])。
产率:2.1g(5.2mmol)理论值的42.3%。
ftir(diamant):ν=3425(w,υnh),3068(w,υar-h),2948(m,υch2),2859(m,υch2),2787(m,υ(n-ch3)c-h),2766(m,υ(n-ch3)n-c),1642(w,υc=o),1591(m,υc=c),1567(m,υc=c),1556(s,υc=c),1457(m,δch2,ch3),731(s,υar-h)cm-1。
1hnmr(300mhz,cdcl3,rt):δ=10.83(t,3jh,h=5.35hz,2h,nh15,16),8.34(dd,3j=5.92,4j=3.27hz,2h,ar-h3,6),7.69(dd,3j=5.91,4j=3.31hz,2h,ar-h1,2),7.30(s,2h,ar-h11,12),3.48(m,4h,ch219,20),2.45(t,3j=6.94hz,4h,ch222,27),2.27(s,12h,nme224,24,29,30),1.92(t,3j=7.1hz,4h,ch221,26)ppm。
13cnmr(75mhz,dmso-d6,rt):δ=182.41(c9,17),146.42(c10,13),134.70(c1,2),132.07(c4,5),126.12(c3,6),123.80(c7,8),109.85(c11,12),57.78(c22,27),45.67(c24,25,29,30),41.03(c19,20),28.05(c21,26)ppm。
esi-msm/z:205.3[m+2h]2+
元素分析:理论值:c:70.56;h:7.90;n:13.71
分析结果:c:70.65;h:7.70;n:13.58
实施例3
n,n′-(((9,10-二氧-9,10-二氢蒽-1,4-二基)双(氮烷二基))双(丙烷-3,1-二基))双(n,n-二甲基丁烷-1-铵)溴化物(7a)的合成
将0.5g(1.23mmol)的1,4-双[[2-(二甲基氨基)丙基]氨基]蒽醌(5)与1.68g(1.3ml,12.25mmol)的1-溴丁烷(6a)在大约5ml的丙酮中加热回流24h。在将溶液冷却至室温后,将产物在己烷中完全沉淀,过滤,并且用己烷洗涤。将蓝色固体在高真空下干燥。
产率:0.81g(1.19mmol)理论值的97%。
ft-ir(diamant):ν=3404(br,υnh),3006(w,υar-h),2958(m,υch2),2871(m,υch2),1641(w,υc=o),1607(w,υc=c),1594(m,υc=c),1572(s,υc=c),1519(m,υc=c),1467(m,δch2,ch3),724(s,υar-h)cm-1。
1hnmr(300mhz,dmso-d6,rt):δ[ppm]=10.79(t,3j=5.82hz,2h,nh15,16),8.24(dd,3j=5.85hz,4j=3.4hz,2h,ar-h3,6),7.83(m,2h,ar-h1,2),7.57(s,2h,ar-h11,12),3.55(m,4h,ch219,20),3.44(m,4h,ch222,26),3.31(m,4h,ch224,28),3.07(s,12h,nme235-38),2.09(m,4h,ch221,25),1.64(m,4h,ch229,32),1.29(m,4h,ch230,33),0.91(t,3j=7.3hz6h,ch331,34)ppm。
13cnmr(75mhz,dmso-d6,rt):δ=181.12(c9,17),145.63(c10,13),133.72(c1,2),132.55(c4,5),125.67(c3,6),124.52(c7,8),108.84(c11,12),62.89(c24,28),60.62(c22,26),50.24(c35,36,37,38),39.5(c19,20在dmso-d6信号下),23.67(c29,32),22.73(c21,25),19.13(c30,33),13.40(c31,34)ppm。
lc-msm/z:261.2[m-2br]2+
hplc:97.07%6.03min
实施例4
n,n′-(((9,10-二氧-9,10-二氢蒽-1,4-二基)双(氮烷二基))双(丙烷-3,1-二基))双(n,n-二甲基辛烷-1-铵)溴化物(7b)的合成
将0.65g(1.33mmol)的1,4-双[[2-(二甲基氨基)丙基]氨基]蒽醌(5)与2.6g(2.3ml,13.3mmol)的1-溴辛烷(6b)在大约5ml的丙酮中加热回流24h。在将溶液冷却至室温后,将产物在己烷中完全沉淀,过滤,并且用己烷洗涤。
将蓝色固体在高真空下干燥。
产率:0.99g(1.25mmol)理论值的94%。
ft-ir(diamant):ν=3419(br,υnh),3004(w,υar-h),2953(m,υch2),2923(m,υch2),2854(m,υch2),1645(w,υc=o),1595(w,υc=c),1576(m,υc=c),1588(s,υc=c),1467(m,δch2,ch3),717(s,υar-h)cm-1。
1hnmr(300mhz,dmso-d6,rt):δ[ppm]=10.79(t,3j=5.84hz,2h,nh15,16),8.25(dd,3j=5.90hz,4j=3.30hz,2h,ar-h3,6),7.82(m,2h,ar-h1,2),7.57(s,2h,ar-h11,12),3.55(m,4h,ch219,20),3.43(m,4h,ch222,26),3.29(m,4h,ch224,28),3.06(s,12h,nme235-38),2.09(m,4h,ch221,25),1.64(m,4h,ch229,32),1.12-1.31(m,20h,ch230,31,33,34,39-41,43-45),0.82(m,6h,ch342,46)ppm。
13cnmr(75mhz,dmso-d6,rt):δ=181.10(c9,17),145.63(c10,13),133.74(c1,2),132.56(c4,5),125.71(c3,6),124.56(c7,8),108.84(c11,12),62.85(c24,28),60.38(c22,26),50.32(c35-38),39.5(c19,20dmso-d6信号下),31.16(c40,44),28.50(c31,34,39,43),25.80(c30,33),22.73(c29,32),22.02(c21,25),21.71(c41,45),13.94(c42,46)ppm。
lc-msm/z:317.3[m-2br]2+
hplc:96.98%7.75min
实施例5
n,n′-(((9,10-二氧-9,10-二氢蒽-1,4-二基)双(氮烷二基))双(丙烷-3,1-二基))双(n,n-二甲基癸烷-1-铵)溴化物(7c)的合成
将0.33g(0.81mmol)的1,4-双[[2-(二甲基氨基)丙基]氨基]蒽醌(5)与1.69ml(8.1mmol)的1-溴癸烷(6c)在5ml的丙酮中加热回流大约48h。在将溶液冷却至室温后,将产物在己烷中完全沉淀,过滤,并且用己烷洗涤。将蓝色固体在高真空下干燥。
产率:0.68g(0.8mmol)理论值的99%。
ft-ir(diamant):ν=3419(br,υnh),3007(w,υar-h),2948(m,υch2),2920(m,υch2),2852(m,υch2),1639(w,υc=o),1595(m,υc=c),1579(m,υc=c),1562(s,υc=c),1467(m,δch2,ch3),731(s,υar-h)cm-1。
1hnmr(300mhz,dmso-d6,rt):δ=10.79(t,3j=5.73hz,2h,nh15,16),8.24(dd,3j=5.88hz,4j=3.31hz,2h,ar-h3,6),7.82(m,2h,ar-h1,2),7.57(s,2h,ar-h11,12),3.55(m,4h,ch219,20),3.43(m,4h,ch222,26),3.29(m,4h,ch224,28),3.06(s,12h,nme235-38),2.08(m,4h,ch221,25),1.63(m,4h,ch229,32),1.12-1.32(m,28h,ch230,31,33,34,39-43,45-49),0.83(m,6h,ch344,50)ppm。
13cnmr(75mhz,dmso-d6,rt):δ=181.17(c9,17),145.70(c10,13),133.79(c1,2),132.62(c4,5),125.77(c3,6),124.62(c7,8),108.90(c11,12),62.87(c24,28),60.41(c22,26),50.39(c35-38),39.5(c19,20在dmso-d6信号下),31.31(c42,48),28.57-28.94(c31,34,39-41,45-47),25.82(c30,33),22.75(c29,32),22.11(c21,25),21.74(c43,49),13.98(c44,50)ppm。
lc-msm/z:345.4[m-2br]2+
hplc:96.92%8.57min
元素分析:理论值:c:62.11;h:8.77;n:6.58
分析值:c:61.87;h:8.72;n:6.42
实施例6
n,n′-(((9,10-二氧-9,10-二氢蒽-1,4-二基)双(氮烷二基))双(丙烷-3,1-二基))双(n,n-二甲基十二烷-1-铵)溴化物(7d)的合成
将0.35g(0.86mmol)的1,4-双[[2-(二甲基氨基)丙基]氨基]蒽醌(5)与0.85g(0.89ml,3.43mmol)的1-溴十二烷(6d)在大约2ml的丙酮中加热回流24h。在将溶液冷却至室温后,将产物在己烷中完全沉淀,过滤,并且用己烷洗涤。将蓝色固体在高真空下干燥。
产率:0.71g(0.79mmol)理论值的91%。
ft-ir(diamant):ν=3425(br,υnh),3006(w,υar-h),2948(m,υch2),2919(s,υch2),2851(m,υch2),1639(w,υc=o),1595(m,υc=c),1580(m,υc=c),1562(s,υc=c),1468(m,δch2,ch3),731(s,υar-h)cm-1。
1hnmr(300mhz,dmso-d6,rt):δ=10.78(m,2h,nh15,16),8.25(m,2h,ar-h3,6),7.82(m,2h,ar-h1,2),7.56(s,2h,ar-h11,12),3.55(m,4h,ch219,20),3.21-3.46(m,ch222,26,24,28被h2o信号遮盖),3.05(s,12h,nme235-38),2.08(m,4h,ch221,25),1.63(m,4h,ch229,32),1.11-1.32(m,36h,ch230,31,33,34,39-51,53),0.84(m,6h,ch352,54)ppm。
13cnmr(75mhz,dmso-d6,rt):δ=181.17(c9,17),145.65(c10,13),133.77(c1,2),132.61(c4,5),125.74(c3,6),124.54(c7,8),108.89(c11,12),62.80(c24,28),60.38(c22,26),50.38(c35-38),39.5(c19,20在dmso-d6信号下),31.29(c44,50),28.53-29.1(c31,34,39-43,45-49),25.78(c30,33),22.70(c29,32),22.09(c21,25),21.69(c51,53),13.96(c52,54)ppm。
lc-msm/z:373.4[m-2br]2+
hplc:96.44%9.79min
实施例7
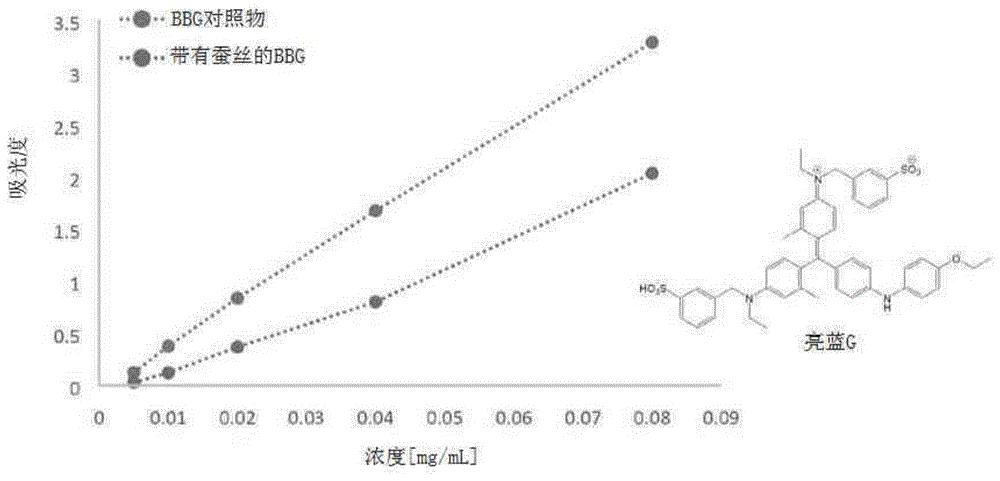
使用类似于ilm的ilm模型底物测试已知染料和根据本发明的染色复合物的结合能力。已经发现丝颗粒(seidengranulat)显示出作为ilm模型底物的良好适应性。为了检查各种染料与ilm模型底物的结合,在用模型底物处理之前和之后,在各种浓度下进行染料亮蓝g(bbg)、化合物7a-d和吲哚菁绿(icg)的uv/vis测量。这些测量的目的是为了比较各个染料的结合强度。结果在以下显示并且每个在图1至4中以图表形式描绘。
使用在生理ph7.4下的磷酸盐缓冲溶液(pbs)制备具有5mg/ml浓度的染料的原液。由于icg在盐溶液中较差的溶解度,使用双蒸馏水。所制备的浓度在表中显示。
表1:结合至ilm模型底物的用于uv/vis-监测研究的染料溶液的制备
为了确保测量尽可能精确,染料模型底物溶液和对照物不是分开制备的,而是同样用作原液。这是通过制备表1所显示的所需量的两倍半来完成的。
随后,将6mg的ilm模型底物加6ml的染料溶液和仅包含6ml染料溶液的对照溶液搅拌整整10分钟,并且然后在6000rpm下一起离心8分钟。将溶液拍照并通过uv/vis光谱分析小心倒出的上清液。计算出模型底物的最大吸收量λmax与对照物的最大吸收量λmax的比率。这可以比较各个染料的结合强度。结果的曲线图在图1中显示。所得数据的列表汇总如下所示。
a)活性染料亮蓝g(bbg)的uv/vis测量(比较)
如前所述,亮蓝g是在染色玻璃体切除术中常用的染料,其选择性给内界膜染色。这意味着,在与模型底物的结合研究中,也可以预期良好的染色,并因此在用模型底物处理后其最大吸收明显降低。
如由图1可证明,证实了与模型底物的预期良好结合。在此仅考虑了最强的最大吸收,其负责赋予蓝色。在此对照物的最大值是585nm。用模型底物处理的样品显示出最大值的轻微偏移,随着浓度的增加,该最大值接近对照物的最大值。对照物的吸收比用模型底物处理的样品的吸收强得多。这表明染料和模型底物之间的良好的相互作用。表2显示了每种染料溶液的最大吸收。通过结合强度的曲线图确认模型底物的λmax与相关对照物的λmax的比率。在0.005mg/ml的浓度下,用模型底物处理的样品仅显示了对照物最大值的24%。这等于76%的染料结合。随着溶液浓度增加,结合降低。在0.08mg/ml的浓度下,可以证明仅48%的染料结合。
表2:在0.005至0.08mg/ml的浓度下bbg对照物和bbg模型底物(ms)溶液的所获得的uv/vis光谱数据
b)(根据本发明的)蒽醌染料7a-d的uv/vis测量
同样测试了根据本发明的蒽醌染料7a-d与模型底物的结合强度。由于与已知的bbg相比,最大吸收较低,因此在这种情况下测量了0.005mg/ml至0.16mg/ml的6种浓度。所获得的uv/vis光谱数据的曲线图在图2和3显示出。
图2和3清楚地证明了蒽醌染料7b-d与模型底物的非常良好的结合。由于蒽醌染料具有两个最大吸收,该图显示了两条对照线和两条样品线。最大吸收是在588nm和632-635nm。在此,对照最大吸收远高于用模型底物处理的那些值。无法记录更高的浓度,因为对照物的吸收太高了。表3显示了ms样品相对于相应对照物的的λmax值的比率。在表4至7中显示了uv/vis光谱研究所获得的精确测量的数据。随着烷基长度增加,染料与模型底物的相互作用增强。尽管带有丁基的化合物7a具有仅在大约50至80%之间的λmax比率,但染料7b-d可实现改进值。然而清楚的是,随着烷基长度增加,染料的消光系数也降低。这是化合物的分子量和增加的疏水性二者的结果。
表3:在0.005至0.16mg/ml的浓度下7对照物和7ms溶液的所获得的uv/vis光谱数据
表4:在0.005至0.16mg/ml的浓度下7a对照物和7ams溶液的所获得的uv/vis光谱数据
表5:在0.005至0.16mg/ml的浓度下7b对照物和7bms溶液的所获得的uv/vis光谱数据
表6:在0.005至0.16mg/ml的浓度下7c对照物和7cms溶液的所获得的uv/vis光谱数据
表7:在0.005至0.16mg/ml的浓度下7d对照物和7dms溶液的所获得的uv/vis光谱数据
c)活性染料吲哚菁绿(icg)的uv/vis测量
吲哚菁绿是染色玻璃体切除术中最重要的染料之一,特别是在ilm剥离领域。然而,该染料的确引起了一些问题并且还带来了处理困难。虽然它很早就用于给内界膜和视网膜前膜染色,但是研究已显示该染料具有许多缺点。除了手术后视力下降之外,在使用该染料时,在光存在下的低稳定性也是严重的问题。此外,在磷酸盐缓冲溶液中无法测量或使用icg,因为这导致化合物沉淀。这意味着,随后的测量也必须在双蒸馏水中进行,因为在缓冲溶液中的测量显示出仅在离心后沉淀出了染料。图4中显示了所获得的uv/vis数据的曲线图。
由所呈现的数据清楚的是,icg在当前的测量中也显示了一些问题。尽管染料显示了在高达0.02mg/ml的浓度下几乎没有吸收,但是0.04mg/ml的浓度足以使吲哚菁绿获得3.5822的吸收,并且因而获得最大可能测量值。由于过高的吸收以及随后的不精确的测量,不可能使用更高的浓度。由表8可见,虽然对照物的λmax值与模型底物-染料溶液的λmax值的比率显示出染料与模型底物的正结合,但是由于较低的浓度,结果在误差范围内会发生很大变化。这可能是由于在低浓度下的弱吸收和光解分解的结果。在进行测量方法期间,不可能确保完全排除光。
表8:在0.005至0.04mg/ml的浓度下icg对照物和icgms溶液的所得的uv/vis光谱数据
实施例8
以下显示了一些用于制备染料的实例合成方法和方案。使用1,4-二氟蒽醌的6-甲基-取代的衍生物,其由4-甲基邻苯二甲酸酐和1,4-二氟苯合成并且然后必须进行氧化。该方法允许修饰侧基而没有由于酸的潜在副作用。它还提供了简化纯化的可能性。在6-位的甲基的随后氧化应产生期望的酸基。方案1中显示了1,4-二氟-6-甲基蒽醌(14)的合成。
方案1:1,4-二氟-6-甲基蒽醌(14)的合成
该反应以类似于合成1,4-二氟蒽醌(1)的方式进行。在通过柱色谱法纯化之后,通过1hnmr、13cnmr和ir光谱,以及通过元素分析验证以约20%的较低产率的化合物14的成功合成。接着,用n,n-二甲基氨基丙胺(4)修饰以得到双官能化的产物1,4-双((3-(二甲基氨基)丙基)氨基)-6-甲基蒽醌(15)(方案2)。在通过柱色谱法纯化产物混合物之后,通过1hnmr明确验证了化合物15。也在这种情况下,在成功转化为染料15之后,邻近氟的质子产生单重态。
方案2:1,4-二氟-6-甲基蒽醌(14)与n,n-二甲基氨基丙胺(4)生成染料10的反应
因为此时已经知道具有c10-取代基的季蒽醌染料的更高的选择性,因此仅用1-溴癸烷进行双改性的1,4-二氟-6-甲基蒽醌15的四元化。方案3显示了所进行的反应。
方案3:双官能化1,4-二氟-6-甲基蒽醌(15)与1-溴癸烷(6c)的反应
蒽醌染料16的成功合成已通过1hnmr明确验证。通过胺结合的甲基的信号从2.21ppm到3.04ppm的位移指示了合成的成功。随后通过具有碳酸钠的高锰酸钾和相转移催化剂“aliquat336”在二氯甲烷/水(1;1)中氧化化合物16中的甲基是不成功的。因此,直接由偏苯三酸酐(17)和1,4-二氟苯(1)做出进一步的尝试以获得羧基改性的蒽醌化合物。所进行的反应如方案4所显示。通过1hnmr光谱以及通过质谱验证5,8-二氟-9,10-二氧代-9,10-二氢蒽-2-羧酸(18)。
方案4:在6-位用酸基改性的染料5,8-二氟-9,10-二氧-9,10-二氢蒽-2-羧酸的合成(18)
通过与n,n-二甲基氨基丙胺(4)反应,以类似于先前方法的方式进一步转化为19,并且在方案5中显示。
方案5:羧基修饰的1,4-二氟蒽醌(18)与n,n-二甲基氨基丙胺(4)以得到5,8-双((3-(二甲基氨基)丙基)氨基)-9,10-二氧-9,10-二氢蒽-2-羧酸的反应
通过1hnmr光谱和质谱分析证实了蒽醌染料5,8-双((3-(二甲基氨基)丙基)氨基)-9,10-二氧-9,10-二氢蒽-2-羧酸(19)的成功合成。通过与1-溴癸烷反应并入正电荷的最终步骤是不成功的。许多副反应会干扰该反应,从而阻止产物形成。正电荷也使产物的随后纯化和分离变得更加困难。这意味着无法成功验证目标结构。
实施例9聚合蒽醌染料
尝试了基于化合物n,n′-(((蒽醌-1,4-二基)双(氮烷二基))双(丙烷-3,1-二基))双(n,n-二甲基癸烷-1-铵)溴化物(7c)的另外的聚合结构的合成。当用于人体时,聚合物防止扩散到更深的细胞层,从而防止毒性作用。由于二氟蒽醌朝向伯胺的高反应性,聚乙烯胺(pva)被用作聚合基础结构。为了将染料附连至该聚合物,因此使用了单官能化的衍生物,其在化合物5的合成中作为副产物形成并且可以通过柱色谱法纯化以纯净的形式分离。如方案6中所显示等摩尔量的两种起始原料的使用允许化合物20以良好的产率分离。
方案6:单官能化染料1-((3-(二甲基氨基)丙基)氨基)-4-氟蒽醌(20)的合成
通过1hnmr和13cnmr光谱验证了化合物20的成功合成。通过与双官能化衍生物比较,邻近氟的芳族质子可以清楚地显示出产生两个信号,因为它们不是化学上等同的。此外,信号显示出更高的多重性,因为同样可以观察到与氟原子的耦合。在1hnmr光谱中,邻近氟原子的氢原子显示出两个3j耦合,而第二个氢原子产生出3j耦合和4j耦合二者。这允许信号的明确分配。
进一步的反应是类似于化合物7a-d的方式。这是通过单官能化的染料20与1-溴癸烷(6c)在丙酮中反应完成的。在此,产物也在反应中沉淀并且可以通过过滤分离出来。通过1hnmr光谱验证了成功合成。反应在方案7中显示。
方案7:单官能化染料21的合成
通过化合物21中的游离氟原子经由聚乙烯胺(pva)中的伯胺实现了附连至聚合结构。该反应在含有痕量三乙胺的thf/h2o中进行,以防止氨基的可能质子化。经由胺的与21的类似聚合物附连意味着随后形成的聚合物是蓝色的。反应方案如下所示。
方案8:单官能化化合物21与pva(22)的反应
将蒽醌染料以按重量计10%和20%的含量并入聚合物中。可以通过由取代带来的颜色变化用肉眼,和通过薄层色谱法监测反应的过程。不完全转化意味着,聚合物仍然含有游离氨基,这既干扰了与要染色的组织的结合,又干扰了分子量的gpc监测。因此,没有采用进一步的分析方法。结合强度的评估在3.1.2节中显示。
- 还没有人留言评论。精彩留言会获得点赞!