用于制备类视黄醇X受体-特异性类视黄醇的化合物和合成方法与流程
关于联邦政府赞助的研究或开发的声明
本发明是在美国国立卫生研究院授予的授权号2r44ai112512-02a1的政府支持下完成的。政府拥有本发明的某些权利。
相关申请
本申请要求于2018年5月14日提交的美国临时专利申请号62/671,137和2017年11月17日提交的美国临时专利申请号62/588,163的优先权。这些申请的全部内容通过引用并入本发明。
背景技术:
已经描述了具有类视黄醇生物活性的化合物。类视黄醇是类视黄醇x受体(rxr)的激动剂,其临床前研究表明,选择性激活rxr可以调节与分化相关的功能,抑制细胞生长、凋亡和转移,可用于治疗与rxr调节的生化功能相关的多种疾病。
技术实现要素:
本发明提供了可用于制备具有类视黄醇生物活性的化合物的化合物。在一个实施方案中,描述了(2e,4e)-3-甲基-5-((1s,2s)-2-甲基-2-(5,5,8,8-四甲基-5,6,7,8-四氢萘-2-基)环丙基)戊-2,4-二烯酸(化合物a)。
本发明还提供了制备化合物a的合成方法。
一方面,化合物38或化合物a通过图2所示的包括一个或多个合成步骤的方法制备。
另一方面,化合物38或化合物a通过图3所示的包括一个或多个合成步骤的方法制备。
另一方面,化合物38或化合物a通过图4所示的包括一个或多个合成步骤的方法制备。
另一方面,化合物38或化合物a通过图5所示的包括一个或多个合成步骤的方法制备。
另一方面,化合物38或化合物a通过图6所示的包括一个或多个合成步骤的方法制备。
附图说明
图1示出了用于制备化合物a的合成路线。
图2示出了用于制备化合物a的合成路线。
图3示出了用于制备化合物a的合成路线。
图4示出了用于制备化合物a的合成路线。
图5示出了用于制备化合物a的合成路线。
图6示出了用于制备化合物a的合成路线。
具体实施方式
定义
如本发明所用,“施用”是指根据需要施用以达到所需效果的化合物。
术语“烷基”是指具有指定碳原子数的直链或支链饱和烃。烷基取代基中的碳原子数可以由前缀“cx-y”表示,其中x是取代基中的最小碳原子数,y是取代基中的最大碳原子数。同样,cx链是指含有x个碳原子的烷基链。
术语“烯基”是指具有指定碳原子数的直链或支链不饱和(例如,至少一个、至少两个、至少三个或至少四个不饱和基,即碳-碳双键)烃基。烯基取代基中的碳原子数可以由前缀“cx-y”表示,其中x是取代基中的最小碳原子数,y是取代基中的最大碳原子数。同样,cx链是指含有x个碳原子的烯基链。
术语“芳基”是指包含一个或多个具有指定碳原子数的芳族环系统的单环或多环碳环系统。芳基取代基中的碳原子数可以由前缀“cx-y”表示,其中x是取代基中的最小碳原子数,y是取代基中的最大碳原子数。同样,cx链是指含有x个碳原子的芳基链。
如本发明所用,“赋形剂”包括可用于制备药物组合物的生理相容性添加剂。药学上可接受的载体和赋形剂的实例可以例如在remingtonpharmaceuticalscience,第16版中找到。
“卤素”或“卤代”是指氟、氯、溴或碘基团。优选地,卤素是氟、氯或溴。
“药学上可接受的载体”是指可用于制备药物组合物的载体:与组合物的其他成分相容,对接受者无害,并且在生物学上或其他方面都不有害。“药学上可接受的载体”包括一种和一种以上的载体。实施方案包括用于局部、眼、肠胃外、静脉内、腹膜内肌内、舌下、经鼻和口服给药的载体。“药学上可接受的载体”还包括用于制备水分散体的试剂和用于注射或分散体的无菌粉末。
如本发明所用,“治疗有效量”是指有效影响、减少或抑制本发明所述的受体的活性或防止其活化的化合物或组合物的剂量。如本发明中所使用的,该术语还可以指在不充分激活rar的情况下有效地在动物,优选人中产生期望的体内效应的量。
化合物
(2e,4e)-3-甲基-5-((1s,2s)-2-甲基-2-(5,5,8,8-四甲基-5,6,7,8-四氢萘-2-基)环丙基)戊-2,4-二烯酸是rxr特异性类视黄醇(本发明称为化合物a)。化合物a具有两个手性中心,并且具有s,s的绝对立体化学。先前已经描述了用于制备化合物a的合成方法以及化合物a的用途。
尽管文献提供了制备化合物a的方法,但仍需要制备化合物a的化合物和合成方法,这些化合物和合成方法至少可改善合成中的总收率、对映体过量、成本、合成期间的安全性、合成期间的便利性或化合物的分离。
本发明提供了可用于制备(2e,4e)-3-甲基-5-((1s,2s)-2-甲基-2-(5,5,8,8-四甲基-5,6,7,8-四氢萘-2-基)环丙基)戊-2,4-二烯酸(化合物a)的化合物。
因此,在一个方面,本发明提供了一种化合物,其中所述化合物为:
其盐、水合物或溶剂化物。
在另一方面,本发明提供式i的化合物:
或其盐、水合物或溶剂化物,
其中r1为c1-20-烷基;被独立选自-nh2、-nh(c1-10-烷基)、-n(c1-10-烷基)(c1-10-烷基)、-oh、卤素、-c1-10烷基、-c1-10卤代烷基、-o-(c1-10烷基)或-o-(c1-10卤代烷基)的一个或多个取代基取代的c1-20-烷基;c1-20-烯基;被独立选自-nh2、-nh(c1-10-烷基)、-n(c1-10-烷基)(c1-10-烷基)、-oh、卤素、-c1-10烷基、-c1-10卤代烷基、-o-(c1-10烷基)或-o-(c1-10卤代烷基)的一个或多个取代基取代的c1-20烯基;c6-14-芳基;或被独立选自-nh2、-nh(c1-10-烷基)、-n(c1-10-烷基)(c1-10-烷基)、-oh、卤素、-c1-10烷基、-c1-10卤代烷基、-o-(c1-10烷基)或-o-(c1-10卤代烷基)的一个或多个取代基取代的c6-14-芳基。
在另一方面,本发明提供式i的化合物:
或其盐、水合物或溶剂化物,
其中r1为c4-20-烷基、c1-20-烯基或c6-16-芳基。
在另一方面,本发明提供式ii的化合物:
或其盐、水合物或溶剂化物,
其中r1为c4-20-烷基、c1-20-烯基或c6-16-芳基。
在另一方面,本发明提供式iii的化合物:
或其盐、水合物或溶剂化物,
其中r1为c1-20-烷基;被独立选自-oh、卤素、-c1-10-烷基、-c1-10-卤代烷基、-o-(c1-10-烷基)或-o-(c1-10-卤代烷基)的一个或多个取代基取代的c4-20烷基;c1-20-烯基;被独立选自-oh、卤素、-c1-10-烷基、-c1-10-卤代烷基、-o-(c1-10-烷基)或-o-(c1-10-卤代烷基)的一个或多个取代基取代的c1-20烯基;c6-14-芳基;或被独立选自-oh、卤素、-c1-10-烷基、-c1-10-卤代烷基、-o-(c1-10-烷基)或-o-(c1-10-卤代烷基)的一个或多个取代基取代的c6-14-芳基。
在另一方面,本发明提供式iii的化合物:
或其盐、水合物或溶剂化物,
其中r1为c4-20-烷基、c1-20-烯基或c6-16-芳基。
在本发明提供的通式的一些实施方案中,r1为c4-10-烷基、c1-20-烯基或c6-14-芳基。在一些实施方案中,r1为c4-10-烷基、c1-10-烯基或c6-14-芳基。在一些实施方案中,r1为c1-20-烯基或苯基。在一些实施方案中,r1为c1-10-烯基或苯基。在一些实施方案中,r1为c4-20-烷基。在一些实施方案中,r1为c4-10-烷基。在一些实施方案中,r1为c4-8-烷基。在一些实施方案中,r1为甲基。在一些实施方案中,r1为乙基。在一些实施方案中,r1为丙基。在一些实施方案中,r1为c1-20-烯基。在一些实施方案中,r1为c1-10-烯基。在一些实施方案中,r1为c1-6-烯基。在一些实施方案中,r1为c6-16-芳基。在一些实施方案中,r1为c6-14-芳基。在一些实施方案中,r1为c6-10-芳基。在一些实施方案中,r1为苯基。
在另一方面,本发明提供式iv的化合物:
或其盐、水合物或溶剂化物,
其中r2是c1-20-烷基、c1-20-烯基或c6-16-芳基。
在另一方面,本发明提供式v的化合物:
或其盐、水合物或溶剂化物,
其中r2为c1-20-烷基、c1-20-烯基或c6-16-芳基。
在另一方面,本发明提供式vi的化合物:
或其盐、水合物或溶剂化物,
其中r2为c1-20-烷基、c1-20-烯基或c6-16-芳基。
在本发明提供的通式的一些实施方案中,r2为c1-10-烷基、c1-10-烯基或c6-14-芳基。在一些实施方案中,r2为c1-20-烯基或c6-14-芳基。在一些实施方案中,r2为c1-10-烯基或c6-14-芳基。在一些实施方案中,r2为c1-20-烷基。在一些实施方案中r2为c1-10-烷基。在一些实施方案中,r2为c1-8-烷基。在一些实施方案中,r2为c1-4-烷基。在一些实施方案中,r2为c4-10-烷基。在一些实施方案中,r2为乙基。在一些实施方案中,r2为c1-20-烯基。在一些实施方案中,r2为c1-10-烯基。在一些实施方案中,r2为c1-6-烯基。在一些实施方案中,r2为c6-14-芳基。在一些实施方案中,r2为c6-10-芳基。在一些实施方案中,r2为苯基。
在另一方面,本发明提供式vii的化合物:
或其盐、水合物或溶剂化物,
其中r3为芳基。
在另一方面,本发明提供式viii的化合物:
或其盐、水合物或溶剂化物,
其中r3为芳基,或被独立选自-oh、卤素、-c1-10-烷基、-c1-10-卤代烷基或-o-(c1-10-烷基)的一个或多个取代基取代的芳基。
在另一方面,本发明提供式viii的化合物:
或其盐、水合物或溶剂化物,
其中r3为芳基。
在另一方面,本发明提供式ix的化合物:
或其盐、水合物或溶剂化物,
其中r3为芳基,或被独立选自-oh、卤素、-c1-10-烷基、-c1-10-卤代烷基或-o-(c1-10-烷基)的一个或多个取代基取代的芳基。
在另一方面,本发明提供式ix的化合物:
或其盐、水合物或溶剂化物,
其中r3为芳基。
在另一方面,本发明提供式x的化合物:
或其盐、水合物或溶剂化物,
其中r3为芳基,
其中r3为芳基,或被独立选自-oh、卤素、-c1-10-烷基、-c1-10-卤代烷基或-o-(c1-10-烷基)的一个或多个取代基取代的芳基。
在另一方面,本发明提供式x的化合物:
或其盐、水合物或溶剂化物,
其中r3为芳基。
在一些实施方案中,r3为c6-14-芳基。在一些实施方案中,r3为c6-10-芳基。在一些实施方案中,r3为c14-芳基。在一些实施方案中,r3为c10-芳基。在一些实施方案中,r3为苯基。
在一些实施方案中,所述化合物为:
或其盐、水合物或溶剂化物。
在一些实施方案中,所述化合物为:
或其盐、水合物或溶剂化物。
在一些实施方案中,所述化合物为:
或其盐、水合物或溶剂化物。
在一些实施方案中,所述化合物为:
或其盐、水合物或溶剂化物。
在一些实施方案中,所述化合物为:
或其盐、水合物或溶剂化物。
在一些实施方案中,所述化合物为:
或其盐、水合物或溶剂化物。
在一些实施方案中,所述化合物为式(xia),或其水合物或溶剂化物,
其中所述化合物具有至少约80.0%的对映体过量的式(xi)化合物,或其水合物或溶剂化物,
在一些实施方案中,所述化合物为化合物38,或其药学上可接受的盐,
其中:
化合物38具有至少约98.0%的对映体过量的化合物a,
;并且
所述化合物是通过合成方法制备的,所述合成方法包括中间体化合物的制备方法,其中所述中间体化合物为式(xia),或其水合物或溶剂化物,
其中所述中间体化合物具有至少约80.0%的对映体过量的式(xi)化合物,或其水合物或溶剂化物,
在一些实施方案中,所述对映体过量为至少约98.0%。
在一些实施方案中,所述中间体化合物(例如,式(xia))的制备方法包括:
使式(xii)化合物或其溶剂化物与ch2i2、et2zn、zni2和
从而形成中间体化合物。
在一些实施方案中,所述
在一些实施方案中,所述中间体化合物(例如,式(xia))的制备方法包括:
(i)在溶液中在ch2i2和et2zn的存在下,使式(xii)化合物或其溶剂化物与式(xiii)化合物或其溶剂化物接触,
以形成式(xii)化合物或其溶剂化物的反应产物;和
(ii)随后,使步骤(i)的式(xii)化合物或其溶剂化物的反应产物与h2o2接触,从而制备中间体化合物。
在一些实施方案中,所述中间体化合物(例如,式(xia))的制备方法包括:
在ch2i2和二烷基锌的存在下,使式(xii)化合物或其溶剂化物与式(xiv)化合物或其对映体或其溶剂化物接触,
从而制备中间体化合物。
在一些实施方案中,所述中间体化合物(例如,式(xia))的制备方法包括:
在约0℃下在ch2i2和et2zn存在下,使式(xii)化合物或其溶剂化物与式(xv)化合物或其对映体或其溶剂化物接触,
从而制备中间体化合物。
在一些实施方案中,本发明提供的式或化合物(例如化合物5、6、9、10、11、13、19、20、21、22、28、29、30、31、32、33、34、37、38、39或40,或式ii、iii、v、vi、viii、ix或x;例如化合物5、6、9、10、11、13、20、22、28、31、34、39,或式iii或vi)的对映体过量为至少90%。在一些实施方案中,本发明提供的式或化合物的对映体过量为至少95%。在一些实施方案中,本发明提供的式或化合物的对映体过量为至少98%。在一些实施方案中,本发明提供的式或化合物的对映体过量为至少99%。在一些实施方案中,本发明提供的式或化合物的对映体过量为至少99.5%量。在一些实施方案中,本发明提供的式或化合物的对映体过量为至少99.9%。在一些实施方案中,本发明提供的式或化合物的对映体过量为至少99.95%。在一些实施方案中,本发明提供的式或化合物的对映体过量为至少99.99%。在一些实施方案中,本发明提供的式或化合物的对映体过量为至少约90%、91%、92%、93%、94%、95%、96%、97%、98%、99%、99.2%、99.4%、99.5%、99.6%、99.8%、99.9%、99.95%、99.99%或以这些值中的任何两个为边界的范围。
在一些实施方案中,化合物38(即化合物a)具有对映体过量的化合物a,其基本上消除了化合物a的对映体(例如化合物b)或将其降低至不可检测的水平。
在一些实施方案中,本发明提供的化合物以其悬浮液或溶剂的形式提供。
在一些实施方案中,本发明提供了包含本发明提供的化合物的组合物。
在一些实施方案中,本发明提供了药物组合物,其包含本发明提供的化合物和药学上可接受的赋形剂或载体。
流程
本发明提供了(2e,4e)-3-甲基-5-((1s,2s)-2-甲基-2-(5,5,8,8-四甲基-5,6,7,8-四氢萘-2-基)环丙基)戊-2,4-二烯酸(化合物a)的制备方法。表1中显示了可用于本发明提供的方法的某些化合物。本发明提供了可用于本发明提供的方法的某些化合物,如式i、式ii、式iii、式iv、式v、式vi、式vii、式viii、式ix或式x。
表1
已发现,根据以下方案,催化量的对映选择性诱导剂(本发明提供的化合物是适于对映选择性诱导的催化剂,例如化合物39、化合物40或化合物5),约1.0至约1.2当量的ch2i2,约1.0至约1.2当量的et2zn和约1.0当量的zni2与化合物8反应以形成化合物9。
因此,本发明提供了化合物9,或其盐、水合物或溶剂化物的制备方法:
包括:
使化合物8或其盐、水合物或溶剂化物与ch2i2、et2zn、zni2和催化量的化合物40或其盐、水合物或溶剂化物接触,
从而形成化合物9或其盐、水合物或溶剂化物。
在一些实施方案中,化合物8与化合物40的摩尔比为约1.0:0.1。在一些实施方案中,化合物8与ch2i2的摩尔比为约1.0:(1.0-1.2)。在一些实施方案中,化合物8与et2zn的摩尔比为约1.0:(1.0-1.2)。在一些实施方案中,化合物8与zni2的摩尔比为约1.0:1.0。
因此,本发明还提供了化合物9,或其盐、水合物或溶剂化物的制备方法:
包括:
使化合物8或其盐、水合物或溶剂化物与ch2i2、et2zn、zni2和催化量的化合物5或其盐、水合物或溶剂化物接触,
从而形成化合物9或其盐、水合物或溶剂化物。
在一些实施方案中,化合物8与化合物5的摩尔比为约1.0:0.1。在一些实施方案中,化合物8与ch2i2的摩尔比为约1.0:(1.0-1.2)。在一些实施方案中,化合物8与et2zn的摩尔比为约1.0:(1.0-1.2)。在一些实施方案中,化合物8与zni2的摩尔比为约1.0:1.0。
已发现,根据以下方案,催化量的对映选择性诱导剂(本发明提供的化合物是适于对映选择性诱导的催化剂,例如化合物3),约1.0至约1.2当量的ch2i2,约1.0至约1.2当量的et2zn与化合物8反应以形成化合物9。
因此,本发明提供了化合物9,或其盐、水合物或溶剂化物的制备方法:
包括:
使化合物8或其盐、水合物或溶剂化物与ch2i2、et2zn和催化量的化合物39或其盐、水合物或溶剂化物接触,
从而形成化合物9或其盐、水合物或溶剂化物。
在一些实施方案中,化合物9的对映体过量为至少约80.0%(例如,至少约85%、90%、91%、92%、93%、94%、95%、96%、97%、98%或99%)。在一些实施方案中,化合物9的对映体过量为至少约98.0%(例如,至少约98.5%、99.0%、99.5%、99.6%、99.7%、99.8%、99.9%、99.99%或99.999%)。
在一些实施方案中,化合物8与化合物39的摩尔比为约1.0:0.1。在一些实施方案中,化合物8与ch2i2的摩尔比为约1.0:(1.0-1.2)。在一些实施方案中,化合物8与et2zn的摩尔比为约1.0:(1.0-1.2)。
已经发现,kobut与替代试剂n-buli相比更便宜并且更易于操作,其根据以下方案与化合物12协同反应以将化合物11转化为化合物13。
因此,本发明提供了化合物13,或其盐、水合物或溶剂化物的制备方法:
包括:
使化合物11或其盐、水合物或溶剂化物与叔丁醇钾和化合物12或其盐、水合物或溶剂化物接触,
从而形成化合物13或其盐、水合物或溶剂化物。
在一些实施方案中,化合物13的对映体过量为至少约80.0%(例如,至少约85%、90%、91%、92%、93%、94%、95%、96%、97%、98%或99%)。在一些实施方案中,化合物13的对映体过量为至少约98.0%(例如,至少约98.5%、99.0%、99.5%、99.6%、99.7%、99.8%、99.9%、99.99%或99.999%)。
化合物13与koh接触后可进行水解以形成化合物a。
类似地,kobut根据以下方案与式i的化合物协同反应以将化合物11转化为式iii的化合物。
因此,本发明提供了式iii化合物,或其盐、水合物或溶剂化物的制备方法:
包括:
使化合物11或其盐、水合物或溶剂化物与叔丁醇钾和式i化合物或其盐、水合物或溶剂化物接触,
从而形成式iii化合物或其盐、水合物或溶剂化物,
其中r1为c1-20-烷基;被独立选自-nh2、-nh(c1-10-烷基)、-n(c1-10-烷基)(c1-10-烷基)、-oh、卤素、-c1-10烷基、-c1-10卤代烷基、-o-(c1-10烷基)或-o-(c1-10卤代烷基)的一个或多个取代基取代的c1-20-烷基;c1-20-烯基;被独立选自-nh2、-nh(c1-10-烷基)、-n(c1-10-烷基)(c1-10-烷基)、-oh、卤素、-c1-10烷基、-c1-10卤代烷基、-o-(c1-10烷基)或-o-(c1-10卤代烷基)的一个或多个取代基取代的c1-20烯基;c6-14-芳基;或被独立选自-nh2、-nh(c1-10-烷基)、-n(c1-10-烷基)(c1-10-烷基)、-oh、卤素、-c1-10烷基、-c1-10卤代烷基、-o-(c1-10烷基)或-o-(c1-10卤代烷基)的一个或多个取代基取代的c6-14-芳基。
在一些实施方案中,r1为c4-20-烷基;被独立选自-nh2、-nh(c1-10-烷基)、-n(c1-10-烷基)(c1-10-烷基)、-oh、卤素、-c1-10烷基、-c1-10卤代烷基、-o-(c1-10烷基)或-o-(c1-10卤代烷基)的一个或多个取代基取代的c4-20-烷基;c1-20-烯基;被独立选自-nh2、-nh(c1-10-烷基)、-n(c1-10-烷基)(c1-10-烷基)、-oh、卤素、-c1-10烷基、-c1-10卤代烷基、-o-(c1-10烷基)或-o-(c1-10卤代烷基)的一个或多个取代基取代的c1-20烯基;c6-14-芳基;或被独立选自-nh2、-nh(c1-10-烷基)、-n(c1-10-烷基)(c1-10-烷基)、-oh、卤素、-c1-10烷基、-c1-10卤代烷基、-o-(c1-10烷基)或-o-(c1-10卤代烷基)的一个或多个取代基取代的c6-14-芳基。
在一些实施方案中,r1为c4-20-烷基;被独立选自-oh、卤素、-c1-10烷基、-c1-10卤代烷基、-o-(c1-10烷基)或-o-(c1-10卤代烷基)的一个或多个取代基取代的c4-20-烷基;c1-20-烯基;被独立选自-oh、卤素、-c1-10烷基、-c1-10卤代烷基、-o-(c1-10烷基)或-o-(c1-10卤代烷基)的一个或多个取代基取代的c1-20烯基;c6-14-芳基;或被独立选自-oh、卤素、-c1-10烷基、-c1-10卤代烷基、-o-(c1-10烷基)或-o-(c1-10卤代烷基)的一个或多个取代基取代的c6-14-芳基。
在一些实施方案中,r1为c4-10-烷基;被独立选自-nh2、-nh(c1-6-烷基)、-n(c1-6-烷基)(c1-6-烷基)、-oh、卤素、-c1-3烷基、-c1-3卤代烷基、-o-(c1-3烷基)或-o-(c1-3卤代烷基)的一个或多个取代基取代的c4-10-烷基;c1-10-烯基;被一个或多个独立选自-nh2、-nh(c1-6-烷基)、-n(c1-6-烷基)(c1-6-烷基)、-oh、卤素、-c1-3烷基、-c1-3卤代烷基、-o-(c1-3烷基)或-o-(c1-3卤代烷基)的取代基取代的c1-10烯基;c6-10-芳基;或被独立选自-nh2、-nh(c1-6-烷基)、-n(c1-6-烷基)(c1-6-烷基)、-oh、卤素、-c1-3烷基、-c1-3卤代烷基、-o-(c1-3烷基)或-o-(c1-3卤代烷基)的一个或多个取代基取代的c6-10-芳基。
在一些实施方案中,r1为c4-10-烷基;被独立选自-nh2、-nh(c1-3-烷基)、-n(c1-3-烷基)(c1-3-烷基)、-oh、卤素、-c1-3烷基、-c1-3卤代烷基、-o-(c1-3烷基)或-o-(c1-3卤代烷基)的一个或多个取代基取代的c4-10-烷基;c1-10-烯基;被独立选自-nh2、-nh(c1-3-烷基)、-n(c1-3-烷基)(c1-3-烷基)、-oh、卤素、-c1-3烷基、-c1-3卤代烷基、-o-(c1-3烷基)或-o-(c1-3卤代烷基)的一个或多个取代基取代的c1-10烯基;c6-10-芳基;或被独立选自-nh2、-nh(c1-3-烷基)、-n(c1-3-烷基)(c1-3-烷基)、-oh、卤素、-c1-3烷基、-c1-3卤代烷基、-o-(c1-3烷基)或-o-(c1-3卤代烷基)的一个或多个取代基取代的c6-10-芳基。
在一些实施方案中,r1为c4-10-烷基;被独立选自-oh、卤素、-c1-3烷基、-c1-3卤代烷基、-o-(c1-3烷基)或-o-(c1-3卤代烷基)的一个或多个取代基取代的c4-10-烷基;c1-10-烯基;被独立选自-oh、卤素、-c1-3烷基、-c1-3卤代烷基、-o-(c1-3烷基)或-o-(c1-3卤代烷基)的一个或多个取代基取代的c1-10烯基;c6-10-芳基;或被独立选自-oh、卤素、-c1-3烷基、-c1-3卤代烷基、-o-(c1-3烷基)或-o-(c1-3卤代烷基)的取一个或多个代基取代的c6-10-芳基。
在一些实施方案中,r1为c4-10-烷基;被独立选自f、cl、br、甲基、-ch2f、-chf2、-cf3、-ch2cl、-chcl2、-ccl3、-o-甲基、-o-ch2f、-o-chf2、-o-cf3、-o-ch2cl、-o-chcl2或-o-ccl3的一个或多个取代基取代的c4-10-烷基;c1-10-烯基;被独立选自f、cl、br、甲基、-ch2f、-chf2、-cf3、-ch2cl、-chcl2、-ccl3、-o-甲基、-o-ch2f、-o-chf2、-o-cf3、-o-ch2cl、-o-chcl2或-o-ccl3的一个或多个取代基取代的c1-10-烯基;c6-10-芳基;或被独立地选自f、cl、br、甲基、-ch2f、-chf2、-cf3、-ch2cl、-chcl2、-ccl3、-o-甲基、-o-ch2f、-o-chf2、-o-cf3、-o-ch2cl、-o-chcl2或-o-ccl3的一个或多个取代基取代的c6-10-芳基。
在一些实施方案中,r1为c4-6-烷基;被独立选自-nh2、-nh(c1-3-烷基)、-n(c1-3-烷基)(c1-3-烷基)、f、cl、br、-ch2f、-chf2、-cf3、-ch2cl、-chcl2、-ccl3、-o-甲基、-o-ch2f、-o-chf2、-o-cf3、-o-ch2cl、-o-chcl2或-o-ccl3的一个或多个取代基取代的c4-6-烷基;c1-4-烯基;被独立选自-nh2、-nh(c1-3-烷基)、-n(c1-3-烷基)(c1-3-烷基)、f、cl、br、甲基、-ch2f、-chf2、-cf3、-ch2cl、-chcl2、-ccl3、-o-甲基、-o-ch2f、-o-chf2、-o-cf3、-o-ch2cl、-o-chcl2或-o-ccl3的一个或多个取代基取代的c1-4-烯基;苯基;或被独立地选自-nh2、-nh(c1-3-烷基)、-n(c1-3-烷基)(c1-3-烷基)、f、cl、br、甲基、-ch2f、-chf2、-cf3、-ch2cl、-chcl2、-ccl3、-o-甲基、-o-ch2f、-o-chf2、-o-cf3、-o-ch2cl、-o-chcl2或-o-ccl3的一个或多个取代基取代的苯基。
在一些实施方案中,r1为c1-4-烷基;被独立选自-nh2、-nh(c1-3-烷基)、-n(c1-3-烷基)(c1-3-烷基)、f、cl、br、-ch2f、-chf2、-cf3、-ch2cl、-chcl2、-ccl3、-o-甲基、-o-ch2f、-o-chf2、-o-cf3、-o-ch2cl、-o-chcl2或-o-ccl3的一个或多个取代基取代的c1-4-烷基。
在一些实施方案中,r1为c1-4-烯基;被独立选自-nh2、-nh(c1-3-烷基)、-n(c1-3-烷基)(c1-3-烷基)、f、cl、br、甲基、-ch2f、-chf2、-cf3、-ch2cl、-chcl2、-ccl3、-o-甲基、-o-ch2f、-o-chf2、-o-cf3、-o-ch2cl、-o-chcl2或-o-ccl3的一个或多个取代基取代的c1-4-烯基。
在一些实施方案中,r1为苯基;或被独立地选自-nh2、-nh(c1-3-烷基)、-n(c1-3-烷基)(c1-3-烷基)、f、cl、br、甲基、-ch2f、-chf2、-cf3、-ch2cl、-chcl2、-ccl3、-o-甲基、-o-ch2f、-o-chf2、-o-cf3、-o-ch2cl、-o-chcl2或-o-ccl3的一个或多个取代基取代的苯基。
在一些实施方案中,r1为c4-6-烷基;被独立选自f、cl、br、-ch2f、-chf2、-cf3、-ch2cl、-chcl2、-ccl3、-o-甲基、-o-ch2f、-o-chf2、-o-cf3、-o-ch2cl、-o-chcl2或-o-ccl3的一个或多个取代基取代的c4-6-烷基;c1-4-烯基;被独立选自f、cl、br、甲基、-ch2f、-chf2、-cf3、-ch2cl、-chcl2、-ccl3、-o-甲基、-o-ch2f、-o-chf2、-o-cf3、-o-ch2cl、-o-chcl2或-o-ccl3的一个或多个取代基取代的c1-4-烯基;苯基;或被独立地选自f、cl、br、甲基、-ch2f、-chf2、-cf3、-ch2cl、-chcl2、-ccl3、-o-甲基、-o-ch2f、-o-chf2、-o-cf3、-o-ch2cl、-o-chcl2或-o-ccl3的一个或多个取代基取代的苯基。
在一些实施方案中,r1为c1-4-烷基;被独立选自f、cl、br、-ch2f、-chf2、-cf3、-ch2cl、-chcl2、-ccl3、-o-甲基、-o-ch2f、-o-chf2、-o-cf3、-o-ch2cl、-o-chcl2或-o-ccl3的一个或多个取代基取代的c1-4-烷基。
在一些实施方案中,r1为c1-4-烯基;被独立选自f、cl、br、甲基、-ch2f、-chf2、-cf3、-ch2cl、-chcl2、-ccl3、-o-甲基、-o-ch2f、-o-chf2、-o-cf3、-o-ch2cl、-o-chcl2或-o-ccl3的一个或多个取代基取代的c1-4-烯基。
在一些实施方案中,r1为苯基;或被独立地选自f、cl、br、甲基、-ch2f、-chf2、-cf3、-ch2cl、-chcl2、-ccl3、-o-甲基、-o-ch2f、-o-chf2、-o-cf3、-o-ch2cl、-o-chcl2或-o-ccl3的一个或多个取代基取代的苯基。
在一些实施方案中,r1为c4-20-烷基、c1-20-烯基或c6-14-芳基。
在一些实施方案中,r1为c1-20-烯基或c6-14-芳基。
在一些实施方案中,r1为c1-10-烯基或c6-10-芳基。
在一些实施方案中,r1为c1-10-烯基或苯基。
在一些实施方案中,r1为c1-10-烷基。
在一些实施方案中,r1为c1-6-烷基。
在一些实施方案中,r1为甲基、乙基、丙基、异丙基、丁基、异丁基或叔丁基。
在一些实施方案中,式(iii)化合物的对映体过量为至少约80.0%(例如,至少约85%、90%、91%、92%、93%、94%、95%、96%、97%、98%或99%)。在一些实施方案中,式(iii)化合物的对映体过量为至少约98.0%(例如,至少约98.5%、99.0%、99.5%、99.6%、99.7%、99.8%、99.9%、99.99%、或99.999%)。
式iii化合物在与koh接触后可以进行水解以形成化合物a。
在一些实施方案中,化合物a的对映体过量为至少约80.0%(例如,至少约85%、90%、91%、92%、93%、94%、95%、96%、97%、98%或99%)。在一些实施方案中,化合物a的对映体过量为至少约98.0%(例如,至少约98.5%、99.0%、99.5%、99.6%、99.7%、99.8%、99.9%、99.99%或99.999%)。
已发现使化合物16与化合物17和催化剂接触产生化合物33,如以下方案所示。
因此,本发明提供了化合物33,或其盐、水合物或溶剂化物的制备方法:
包括:
使化合物16或其盐、水合物或溶剂化物与催化剂和化合物17或其盐、水合物或溶剂化物接触,
从而形成化合物33或其盐、水合物或溶剂化物。
在一些实施方案中,化合物33的对映体过量为至少约80.0%(例如,至少约85%、90%、91%、92%、93%、94%、95%、96%、97%、98%或99%)。在一些实施方案中,化合物33的对映体过量为至少约98.0%(例如,至少约98.5%、99.0%、99.5%、99.6%、99.7%、99.8%、99.9%、99.99%或99.999%)。
已发现使化合物19与n-buli和b(ome)3接触产生化合物21,如以下方案所示。
因此,本发明提供了化合物21,或其盐、水合物或溶剂化物的制备方法:
包括:
使化合物19或其盐、水合物或溶剂化物与n-buli或t-buli,和b(ome)3接触,
从而形成化合物21或其盐、水合物或溶剂化物。
在一些实施方案中,化合物21的对映体过量为至少约80.0%(例如,至少约85%、90%、91%、92%、93%、94%、95%、96%、97%、98%或99%)。在一些实施方案中,化合物21的对映体过量为至少约98.0%(例如,至少约98.5%、99.0%、99.5%、99.6%、99.7%、99.8%、99.9%、99.99%或99.999%)。
类似地,使式viii化合物与n-buli和b(ome)3接触产生式ix化合物,如以下方案所示。
因此,本发明提供了式ix化合物,或其盐、水合物或溶剂化物的制备方法:
包括:
使式viii化合物或其盐、水合物或溶剂化物与n-buli或t-buli,和b(ome)3接触,
从而形成式ix化合物或其盐、水合物或溶剂化物,
其中r3为芳基;或被独立选自-oh、卤素、-c1-10烷基、-c1-10卤代烷基或-o-(c1-10烷基)的一个或多个取代基取代的芳基。
在一些实施方案中,r3为c6-14-芳基;或被独立选自-oh、卤素、-c1-10烷基、-c1-10卤代烷基、-o-(c1-10烷基)或-o-(c1-10-卤代烷基)的一个或多个取代基取代的c6-14-芳基。
在一些实施方案中,r3为苯基;或被独立选自-oh、卤素、-c1-10烷基、-c1-10卤代烷基、-o-(c1-10烷基)或-o-(c1-10-卤代烷基)的一个或多个取代基取代的苯基。
在一些实施方案中,r3为苯基;或被独立选自-oh、卤素、-c1-3烷基、-c1-3卤代烷基、-o-(c1-3烷基)或-o-(c1-3-卤代烷基)的一个或多个取代基取代的苯基。
在一些实施方案中,r3为苯基;或被独立选自f、cl、br、甲基、-ch2f、-chf2、-cf3、-ch2cl、-chcl2、-ccl3、-o-甲基、-o-ch2f、-o-chf2、-o-cf3、-o-ch2cl、-o-chcl2或-o-ccl3的一个或多个取代基取代的苯基。
在一些实施方案中,式(ix)化合物的对映体过量为至少约80.0%(例如,至少约85%、90%、91%、92%、93%、94%、95%、96%、97%、98%或99%)。在一些实施方案中,式(ix)化合物的对映体过量为至少约98.0%(例如,至少约98.5%、99.0%、99.5%、99.6%、99.7%、99.8%、99.9%、99.99%、或99.999%)。
已发现使化合物21与化合物3和pd(0)接触产生化合物30,如以下方案所示。
因此,本发明提供了化合物30,或其盐、水合物或溶剂化物的制备方法:
包括:
使化合物21或其盐、水合物或溶剂化物与pd(0)和化合物3或其盐、水合物或溶剂化物接触,
从而形成化合物30或其盐、水合物或溶剂化物。
在一些实施方案中,化合物30的对映体过量为至少约80.0%(例如,至少约85%、90%、91%、92%、93%、94%、95%、96%、97%、98%或99%)。在一些实施方案中,化合物30的对映体过量为至少约98.0%(例如,至少约98.5%、99.0%、99.5%、99.6%、99.7%、99.8%、99.9%、99.99%或99.999%)。
类似地,使式ix化合物与化合物3和pd(0)接触产生式x化合物,如以下方案所示。
因此,本发明提供了式x化合物,或其盐、水合物或溶剂化物的制备方法:
包括:
使式ix化合物或其盐、水合物或溶剂化物与pd(0)和化合物3或其盐、水合物或溶剂化物接触,
从而形成式x化合物或其盐、水合物或溶剂化物;
其中r3为芳基;或被独立选自-oh、卤素、-c1-10烷基、-c1-10卤代烷基或-o-(c1-10烷基)的一个或多个取代基取代的芳基。
在一些实施方案中,r3为c6-14-芳基;或被独立选自-oh、卤素、-c1-10烷基、-c1-10卤代烷基、-o-(c1-10烷基)或-o-(c1-10-卤代烷基)的一个或多个取代基取代的c6-14-芳基。
在一些实施方案中,r3为苯基;或被独立选自-oh、卤素、-c1-10烷基、-c1-10卤代烷基、-o--(c1-10烷基)或-o-(c1-10-卤代烷基)的一个或多个取代基取代的苯基。
在一些实施方案中,r3为苯基;或被一个或多个独立选自-oh、卤素、-c1-3烷基、-c1-3卤代烷基、-o-(c1-3烷基)或-o-(c1-3-卤代烷基)的取代基取代的苯基。
在一些实施方案中,r3为苯基;或被独立选自f、cl、br、甲基、-ch2f、-chf2、-cf3、-ch2cl、-chcl2、-ccl3、-o-甲基、-o-ch2f、-o-chf2、-o-cf3、-o-ch2cl、-o-chcl2或-o-ccl3的一个或多个取代基取代的苯基。
在一些实施方案中,式(x)化合物的对映体过量为至少约80.0%(例如,至少约85%、90%、91%、92%、93%、94%、95%、96%、97%、98%或99%)。在一些实施方案中,式(x)的化合物的对映体过量为至少约98.0%(例如,至少约98.5%、99.0%、99.5%、99.6%、99.7%、99.8%、99.9%、99.99%、或99.999%)。
已发现使化合物8与化合物6接触产生化合物9,如以下方案所示。
因此,本发明提供了式(xi)化合物,或其溶剂化物的制备方法:
包括:
(i)在溶液中在ch2i2和et2zn的存在下,使式(xii)化合物或其溶剂化物与式(xiii)的化合物或其溶剂化物接触;和
(ii)随后,使步骤(i)的溶液与h2o2接触,从而制备式(xi)化合物或其溶剂化物,其中式(xi)化合物的对映体过量至少为80.0%。
已发现使化合物8与催化量的化合物41(或其对映体)接触产生化合物9,如以下方案所示。
因此,本发明提供了式(xi)化合物,或其溶剂化物的制备方法:
包括:
在ch2i2和二烷基锌的存在下,使式(xii)化合物或其溶剂化物与式(xiv)化合物或其对映体或其溶剂化物接触,
从而制备式(xi)化合物或其溶剂化物。
在一些实施方案中,所述二烷基锌为znet2。
在一些实施方案中,式(xi)化合物的对映体过量为至少约80.0%(例如,至少约85%、90%、91%、92%、93%、94%、95%、96%、97%、98%或99%)。在一些实施方案中,式(xi)化合物的对映体过量为至少约98.0%(例如,至少约98.5%、99.0%、99.5%、99.6%、99.7%、99.8%、99.9%、99.99%、或99.999%)。
已发现,使化合物8与催化量的化合物40(或其对映异构体)接触产生化合物9,如以下方案所示。
因此,本发明提供了式(xi)化合物,或其溶剂化物的制备方法:
包括:
在约0℃下在ch2i2存在下,使式(xii)化合物或其溶剂化物与式(xv)化合物或其对映体或其溶剂化物接触,
从而制备式(xi)化合物或其溶剂化物。
在一些实施方案中,式(xii)与式(xv)的摩尔比为约1.0:0.05至约1.0:0.3。在一些实施方案中,式(xii)与式(xv)的摩尔比为约1.0:0.1。
已发现使化合物4与化合物7接触产生化合物8,如以下方案所示。
因此,本发明提供了式(xii)化合物,或其溶剂化物的制备方法:
包括:
在pd/c和碱的存在下,使式(xvi)化合物或其溶剂化物与式(xvii)化合物或其溶剂化物接触,
从而制备式(xii)化合物或其溶剂化物。
在一些实施方案中,所述碱为k2co3。
已发现使化合物16与化合物10接触产生化合物33,如以下方案所示。
因此,本发明提供了式(xvii)化合物,或其溶剂化物的制备方法:
包括:
在n2ch2co2et的存在下,使式(xviii)化合物或其溶剂化物与式(xix)化合物或其对映体或其溶剂化物接触,
从而制备式(xvii)化合物或其溶剂化物。
在一些实施方案中,式(xvii)化合物的对映体过量为至少约80.0%(例如,至少约85%、90%、91%、92%、93%、94%、95%、96%、97%、98%或99%)。在一些实施方案中,式(xvii)化合物的对映体过量为至少约98.0%(例如,至少约98.5%、99.0%、99.5%、99.6%、99.7%、99.8%、99.9%、99.99%、或99.999%)。
在一些实施方案中,本发明提供的方法在溶剂或溶剂组合(例如至少一种溶剂,例如一种溶剂、两种溶剂、三种溶剂或四种或更多种溶剂)的存在下进行。
在一些实施方案中,所述溶剂为非极性溶剂或极性非水溶剂、水性溶剂或其组合。
在一些实施方案中,所述溶剂为乙腈、氯仿、二氯甲烷、1,3-二甲基-3,4,5,6-四氢-2(1h)-嘧啶酮、乙醇、乙醚、h+(aq)(例如,hcl(aq)、hbr(aq)、hooac(aq)等)、庚烷、己烷、异丙醇、甲醇、甲基叔丁基醚、2-甲基-四氢呋喃、(oh)-(aq)(例如naoh(aq)、koh(aq)、nh3·h2o(aq)等)、四氢呋喃、甲苯、水。
在一些实施方案中,本发明提供的方法在作为脱水剂的筛(例如
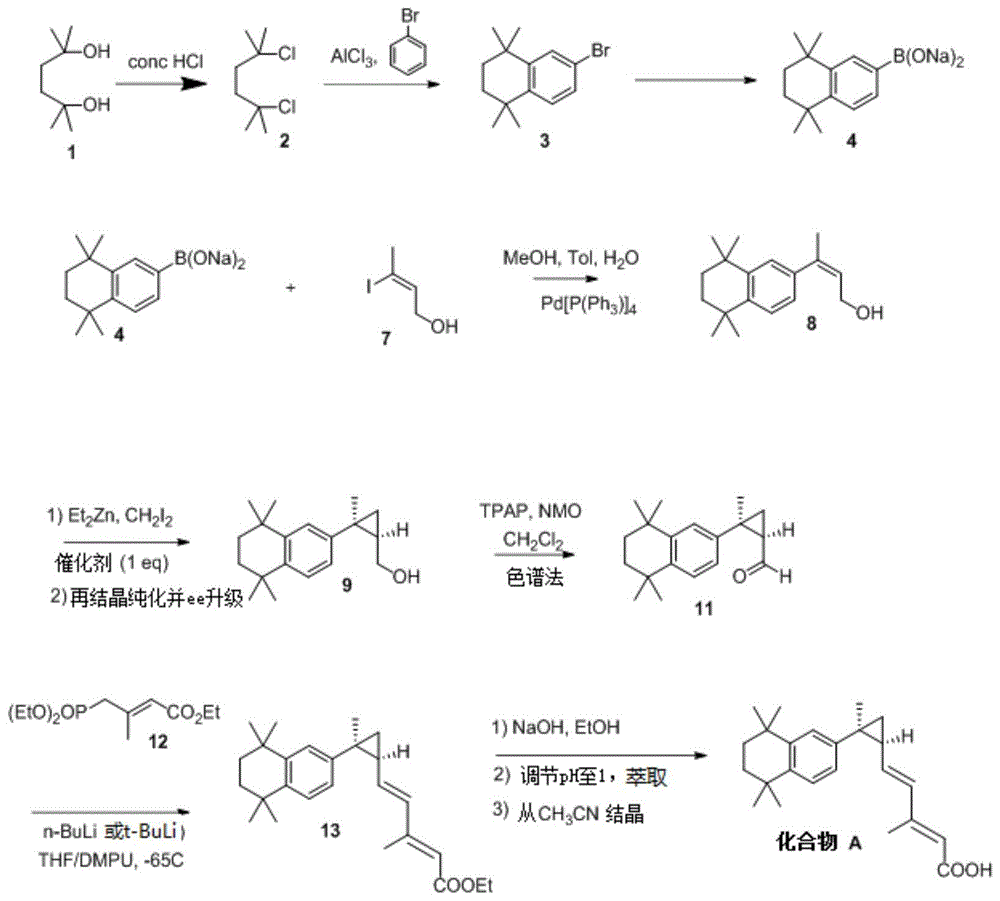
在一些实施方案中,如图1所示制备化合物a。正如对本领域技术人员显而易见的,本发明提供的某些式可以代替图1所示的方案的某些中间化合物,以达到制备化合物a的替代方法。
在一些实施方案中,如图2所示制备化合物a。正如对本领域技术人员显而易见的,本发明提供的某些式可以代替图2所示的方案的某些中间化合物,以达到制备化合物a的替代方法。
在一些实施方案中,如图3所示制备化合物a。正如对本领域技术人员显而易见的,本发明提供的某些式可以代替图3所示的方案的某些中间化合物,以达到制备化合物a的替代方法。
在一些实施方案中,如图4所示制备化合物a。正如对本领域技术人员显而易见的,本发明提供的某些式可以代替图2所示的方案的某些中间化合物,以达到制备化合物a的替代方法。
在一些实施方案中,如图5所示制备化合物a。正如对本领域技术人员显而易见的,本发明提供的某些式可以代替图5所示的方案的某些中间体化合物,以达到制备化合物a的替代方法。
在一些实施方案中,如图6所示制备化合物a。正如对本领域技术人员显而易见的,本发明提供的某些式可以代替图6所示的方案的某些中间化合物,以达到制备化合物a的替代方法。
方法
rxr激动剂在调节细胞生长和分化中具有多种活性。化合物a是一种rxr选择性激动剂,已发现可用于治疗多种疾病或病症,包括癌症、神经系统疾病、肌肉疾病、脱髓鞘疾病和自身免疫疾病。通过还施用甲状腺激素已经获得了改善的协同作用。
作为选择性rxr激动剂,化合物a以比其活化视黄酸受体(rar)低得多的浓度活化rxr同二聚体和异二聚体受体。这很重要,因为rar的激活会带来不需要和有害的副作用,并有可能抵消rxr激动剂活性产生的有益作用。如表2和表3所示,化合物a在0.1到1nm的浓度下将rxr激活至最大(ec90)的90%,具体取决于受体亚型,但在实际上更高浓度为200-300nm下,仅诱导最小(10%)rar激活(ec10),具体取决于受体亚型。化合物a是对rxr具有纳摩尔级亲和力的强效和特异性rxr激动剂,而化合物b是具有显著rar活性的显著低效的rxt激动剂(请参见表4)。rar激活已显示出对多巴胺能神经元(帕金森病中垂死的神经元)的存活有害。另外,rar激活可能抵消rxr激活在治疗癌症中的有益作用。
表2.化合物a的rxrec90和rarec10值。
表3.化合物a的rxrec90与rarec10之比。
表4.化合物a和化合物b的rxr和rar活性。
20mg/m2的口服剂量不超过200nm的全身性浓度,约0.014mg/m2的口服剂量可产生约0.1nm的瞬时全身性浓度。因此,一些实施方案需要0.014至20mg/m2的剂量。鼻剂量可能低10倍之多。
rxr激动剂调节基因表达,从而抑制某些癌症的生长或消退。化合物a在体外或动物模型中已显示出对多种癌症的活性,包括血液系统癌症,如白血病和皮肤t细胞淋巴瘤,肺癌(小细胞和非小细胞),乳腺癌(雌激素受体阳性和阴性),子宫颈癌,胰腺癌和前列腺癌。化合物a在癌症治疗中的用途的进一步描述可见于uspn8,101,662,用特定rxr激动剂治疗癌症,和wo2017/075612,使用rxr激动剂和甲状腺激素的组合治疗癌症,其关于该用途的所有教导均通过引用并入本发明。
还发现化合物a具有免疫调节作用,特别是对th17/treg比的调节有利于treg细胞,导致免疫反应和炎症的减弱。进一步,已经发现化合物a具有神经保护作用并且促进少突胶质细胞分化和髓鞘再生。因此,化合物a可用于治疗具有神经变性和/或自身免疫成分的多种疾病,包括肌肉和神经系统疾病。可以用化合物a治疗的示例性疾病包括帕金森病、阿尔茨海默病、多发性硬化症、精神分裂症、肌萎缩性侧索硬化症、缺血性损伤、外伤性损伤、抑郁症或与年龄相关的神经变性。在一些实施方案中,合适的剂量在0.001至100mg/kg/天的范围内。在其他实施方案中,剂量可以是0.001至0.2mg/kg/天或0.1至3mg/kg/天。化合物a在治疗神经变性和自身免疫疾病中的用途的进一步描述可见于:uspn10,034,854,使用rxr激动剂的自身免疫疾病治疗;和usppn2015-0038585,使用选择性rxr激动剂同时缓解髓鞘再生和免疫调节作用来治疗疾病;uspn9,877,941,使用rxr激动剂和甲状腺激素的组合治疗神经系统疾病;wo2017/155577,使用rxr激动剂和甲状腺激素的组合治疗自身免疫疾病;wo2017/155578,使用rxr激动剂和甲状腺激素的组合治疗肌肉疾病;其关于此类用途的所有教导,均通过引用并入本发明。
因此,在一些实施方案中,本发明提供了治疗癌症的方法,其包括向有此需要的受试者施用有效量的化合物38或其药学上可接受的盐,其中:化合物38具有对映体过量的化合物a,至少约98.0%;所述化合物是通过合成方法制备的,并且所述合成方法包括本发明所述的中间体化合物的制备方法。在一些实施方案中,所述中间化合物为式(xia)。在一些实施方案中,式(xia)通过本发明提供的方法制备。
在一些实施方案中,本发明提供了治疗癌症的方法,其包括向有此需要的受试者施用化合物(即rxr激动剂,例如:化合物38具有对映体过量至少约98%(例如,至少约99.0%,例如至少约99.5%)的化合物a,具有对映体过量至少约98%(例如,至少约99.0%,例如至少约99.5%)的化合物a,其组合物或其药物组合物),治疗有效量为约0.1至约20mg/m2/天。
在一些实施方案中,rxr激动剂的治疗有效量为低于视黄酸受体(rar)激活阈值且等于或高于rxr有效剂量的剂量。
在一些实施方案中,所述癌症为血液恶性肿瘤、肺癌、前列腺癌、乳腺癌、胰腺癌、结肠癌或宫颈癌。
在一些实施方案中,所述治疗还包括向受试者施用甲状腺激素。
本发明还提供了在有此需要的受试者中治疗神经系统疾病、肌肉疾病、脱髓鞘疾病或自身免疫疾病的方法,包括向所述受试者施用治疗有效量的化合物(即rxr激动剂,例如:化合物38具有对映体过量至少约98%(例如,至少约99.0%,例如至少约99.5%)的化合物a,具有对映体过量至少约98%(例如,至少约99.0%,例如至少约99.5%)的化合物a,其组合物或其药物组合物),其中治疗有效量为0.001mg/kg/天至约100mg/kg/天。
在一些实施方案中,所述神经系统疾病为帕金森病、阿尔茨海默病、多发性硬化症、精神分裂症、肌萎缩性侧索硬化症、缺血性损伤、外伤性损伤、抑郁症或与年龄相关的神经变性。
在一些实施方案中,所述治疗还包括施用甲状腺激素。
在一些实施方案中,所述治疗有效量为约0.001mg/kg/天至约0.2mg/kg/天。
在一些实施方案中,所述治疗有效量为约0.1mg/kg/天至约3.0mg/kg/天。
在一些实施方案中,以治疗有效量施用rxr激动剂,其中该剂量低于视黄酸受体(rar)激活阈值且等于或高于rxr有效剂量。
在一些实施方案中,rxr激动剂具有对映体过量的化合物a,其基本上消除了化合物a的对映体(例如,化合物b)的rar活化或其降低至不可检测的水平。
试剂盒和制品
本发明还提供了试剂盒,其包含本发明提供的化合物。
本发明还提供了制品,其包含本发明提供的化合物。
在一些实施方案中,所述试剂盒或制品还包括其使用说明书。
实施例
实施例1:化合物29的合成
在二碘甲烷(1-3当量)存在下,用zn-cu对(1-3当量)或类似的试剂处理,在无水thf或无水乙醚(10ml)中的化合物18(1当量),并在适当的温度搅拌数小时,并用水淬灭反应。滤出固体zn-cu混合物,分离有机层并除去溶剂。将粗产物纯化,得到环丙基产物19。
在适当的温度下,在无水thf(10ml)中的环丙基产物19(1当量)与正丁基锂或仲丁基锂或叔丁基锂反应,并搅拌适当的时间,然后用硼酸三甲酯或类似的硼酸酯试剂处理,然后用稀酸淬灭反应,然后与naoh(aq)反应,得到硼酸酯产物21。
在溴四氢萘化合物3(1当量),在pd(pph3)4(催化量)和1当量溴甲烷存在下,在dme或thf(10ml)和一些meoh中的硼酸酯化合物21(1当量)加热适当的时间,并在适当的温度下适当的时间长度,得到粗制的环丙基四氢萘化合物30。将粗反应物用乙酸乙酯萃取,干燥并纯化,得到化合物30。
在适当压力下,在氢气气氛下,化合物30(1当量)与乙酸乙酯或meoh(10ml)中的pd-c(催化量)反应,得到环丙醇29。除去溶剂后滤出pd-c固体,然后纯化,得到环丙醇29。
正如对本领域技术人员显而易见的是,本发明提供的某些式可以代替实施例1中所示方案的某些中间化合物,以获得制备化合物29的替代方法。
实施例2:化合物37的合成
在惰性气体(氮气或氩气)下,在适当温度下用kot-bu(1.1当量)在thf中处理化合物12(1.1当量)适当的时间。然后在适当的温度下向该混合物中加入在thf或乙醚溶剂中的化合物32(1当量),并搅拌适当的时间长度,然后用水淬灭反应,并用乙酸乙酯萃取。乙酸乙酯层经干燥和纯化得到产物37。
实施例3:化合物34的合成
向三氟甲磺酸铜(cuotf,6.8mg,0.027mmol)的悬浮液中,加入手性催化剂(8.1mg,0.028mmol)的氯仿溶液(9ml),形成手性催化剂化合物10。溶液通过插管,插管由针头和装有玻璃棉的针座组成。向该混合物中缓慢地(历时1.5小时)添加在无水氯仿(10ml)中的化合物16(3.19g,14mmol)和n2chco2et(309mg,2.71mmol)。14小时后,将混合物真空浓缩。混合物通过硅胶柱色谱法纯化。通过手性色谱法或mplc分离所需化合物34(masamuneetaltet.letts.1990,31,6005;evansetalj.a.c.s.1991,113,726;masamuneetaltet.letts.1991,32,7373)。
实施例4:化合物11的合成
向化合物34(312mg,1mmol)的无水二氯甲烷的冷(-78℃)溶液(6ml)中,缓慢地(超过5min)加入二异丁基氢化铝的二氯甲烷溶液(1m溶液,1.1ml)。将冷的反应搅拌4小时或直至反应完成。通过加入水(1ml)将反应淬灭,用二氯甲烷(10ml)稀释,用稀(5%)aq.hcl或nahco3溶液(5ml)洗涤,干燥并蒸馏出溶剂。分离出化合物11,并将其用于下一步。
实施例5:化合物13的合成
向化合物12(290mg,1.1mmol)的冷(-78℃)无水四氢呋喃溶液(6ml)中加入正丁基锂(1.6m溶液,0.7ml)。将冷的反应搅拌20分钟,然后加入化合物11(264mg,1mmol)的无水四氢呋喃溶液(3ml),并将混合物搅拌3小时或直至反应完成。通过加入水(1ml)淬灭反应,用乙酸乙酯(30ml)稀释,用盐水(5ml)洗涤,干燥并蒸馏出溶剂。柱色谱纯化后分离化合物13。
实施例6:化合物a的合成
向化合物13(290mg,1.1mmol)的甲醇溶液(2ml)中加入koh(aq)(1m溶液,3ml),加热至80℃持续2小时或直至反应完成。将反应混合物冷却至环境温度,并用稀hcl的水溶液(10%,3ml)酸化。用乙酸乙酯(3×10ml)萃取反应物,干燥并蒸馏出溶剂。通过重结晶或色谱纯化分离化合物a。
实施例7:化合物33的合成
在环境温度下,将n2chco2et(228mg,2mmol)的无水二氯甲烷溶液(3ml)缓慢地(历时5小时)添加到化合物16(2.28g,10mmol)、铑催化剂(4.4mg,0.01mmol)和二氯甲烷的溶液中。将混合物搅拌两个小时或直至反应完成,然后使其通过短的氧化铝柱,其中二氯甲烷为洗脱剂,以除去rh(oac)2催化剂。然后,通过蒸馏除去溶剂和过量的化合物16。通过色谱隔离并分离顺式异构体33和反式异构体40的混合物。化合物33用于下一步(callot,h.j.;metz,f.;tetrahedron1985,41,4495;maxwell,j.l.etalorganometallics92,11,645;callot,h.j.;piechoeki,c.;tetrahedronlett.1980,21,3489)。
实施例8:化合物32的合成
向化合物33(312mg,1mmol)的冷(-78℃)无水二氯甲烷溶液(6ml)中,缓慢地(超过5min)加入二异丁基氢化铝的二氯甲烷溶液(1m溶液,1.1ml)。将冷的反应搅拌4小时或直至反应完成。通过加入水(1ml)将反应淬灭,用二氯甲烷(10ml)稀释,用稀(5%)aq.hcl或nahco3溶液(5ml)洗涤,干燥并蒸馏出溶剂。分离出化合物32,并将其用于下一步。
实施例9:化合物37的合成
向化合物12(290mg,1.1mmol)的冷(-78℃)无水四氢呋喃溶液(6ml)中添加正丁基锂(1.6m溶液,0.7ml)。将冷的反应搅拌20分钟,然后加入化合物32(264mg,1mmol)的无水四氢呋喃溶液(3ml),并将混合物搅拌3小时或直至反应完成。通过加入水(1ml)淬灭反应,用乙酸乙酯(30ml)稀释,用盐水(5ml)洗涤,干燥并蒸馏出溶剂。柱色谱纯化后分离化合物37。
实施例10:化合物38的合成
向化合物37(290mg,1.1mmol),甲醇(2ml)的溶液中添加koh(aq)(1m溶液,3ml),并加热至80℃持续2小时或直到反应完成。将反应混合物冷却至环境温度,并用稀hcl的水溶液(10%,3ml)酸化。用乙酸乙酯(3×10ml)萃取反应物,干燥并蒸馏出溶剂。通过重结晶或色谱纯化分离化合物38。
在本发明提供的一些实施方案中,化合物38具有至少约80.0%(例如,至少约85%、90%、91%、92%、93%、94%、95%、96%、97%、98%或99%)对映体过量的化合物a,
。在一些实施方案中,化合物38具有至少约98.0%(例如,至少约98.5%、99.0%、99.5%、99.6%、99.7%、99.8%、99.9%、99.99%或99.999%)的对映体过量。
实施例11:化合物3的合成
在惰性气氛下,向装有化合物8(256mg,1mmol)和磺酰胺催化剂(化合物5或其对映体,27mg,0.1mmol)的冷瓶(烧瓶a)(0℃)中加入ch2cl2(3ml)。向该溶液中添加et2zn(113μl,1.1mmol),并搅拌10分钟。向另一个烧瓶(烧瓶b)中加入碘(508mg,2mmol)和ch2cl2(10ml),将该悬浮液冷却至0℃,并加入et2zn(103μl,1mmol)搅拌10分钟。在另一个烧瓶(烧瓶c)中,加入ch2i2(161μl,2mmol)和ch2cl2(24ml),将溶液冷却至0℃,加入et2zn(103μl,1mmol)并搅拌5分钟。将烧瓶a的内容物在0℃下插管到烧瓶b中并搅拌2分钟,然后将这些内容物插管到烧瓶c中,在0℃下搅拌烧瓶c中的反应内容物直至反应完成。通过加入naoh(aq)(2m溶液,13ml)淬灭反应。分离水溶液层,并用ch2cl2(2×15ml)萃取,干燥合并的有机层并除去溶剂,并通过硅胶柱色谱法纯化,并且分离出产物(化合物9)(denmark,s.e.;christenson,b.l.;oconnor,s.p.;tetrahedronletts.1995,36,2219;denmark,s.e.;christenson,b.l.;oconnor,s.p.;murase,n.;pureappl.chem.,1996,68,23;denmark,s.e.;oconnor,s.p.;j.org.chem.1997,62,584)。
实施例12:化合物11的合成
向化合物9(270mg,1mmol)的ch2cl2溶液(6ml)中加入分子筛(1g)、tpap(3mg)、nmo(141mg,1.2mmol),并在环境下搅拌直至反应完成。使反应通过短硅胶柱,并用乙酸乙酯-己烷混合物洗脱。除去溶剂后,分离出产物(化合物11)。
实施例13:化合物12的合成
化合物36:提供的方法使用在己烷和乙醚的混合物中的pbr3以79%的收率将醇35转化为溴化物36。后处理表明,该产物被大量源自2-methf(nmr)的开环材料所污染。
结果:化合物36运行1。在-5至-10℃下,向26g(0.18mol)35的400ml庚烷和150mlmethf溶液中添加pbr3(放热,51.3g,0.189mol)。将混合物冷搅拌1小时,然后在室温下过夜。离子对色谱法(ipc)显示峰到峰转化。通过添加225ml盐水(放热)将混合物淬灭(冷)。汽提有机层,得到51g(138%)油,其显示出(nmr)与methf反应衍生的大量杂质。将该油重新溶解在400ml庚烷中,并用8%nahco3(2×100ml)洗涤。将溶液干燥(mgso4),过滤并汽提,得到41g(110%)油。nmr较纯,但不干净。该物质经色谱分离或蒸馏纯化。
可替代地,二氯甲烷被用作唯一的溶剂。由于1当量的pbr3可以完全转化为36(以上)并与methf反应,因此不止一种br与醇反应。因此,以0.1倍以上的规模,在<-5℃下用0.5当量的pbr3处理2.6g35的40mldcm溶液。离子对色谱法(ipc)显示,中间体形成并随着时间的流逝逐渐减少。但是,原料和极性中间体没有被完全消耗掉。加入另外0.25当量(总计0.75当量),并使反应进行过夜。后处理得到3.7g(定量的)产物。但是,更长的保留时间杂质(z异构体)比上一次运行的杂质含量高得多(7%)。查看液相色谱痕迹,可以看出它会随着时间增长。这些数据表明,原始方法中的醚起弱路易斯碱的作用,可调节hbr副产物的作用。
结果:化合物36运行2。在-5至-10℃下,向2.6g(0.018mol)35的40mldcm溶液中添加pbr3(放热,2.3g,0.009mol,0.5当量)。将混合物在室温下搅拌5小时,但没有完成。将混合物冷却,再添加0.25当量,并将反应在室温搅拌过夜。将反应冷却,并用10ml水(放热)淬灭,干燥(mgso4)并汽提,得到3.7g(定量)的澄清油。hplc显示不期望的z异构体(nmr,gcms)为约7%。
实施例14:化合物9的合成
化合物9(simmons-smith反应):所有添加均在至少约20分钟和约-5℃下进行。步骤:用氮气吹扫反应器。向10l反应器中装入8(200g)、dcm(4l)和硼酸酯催化剂(250g)。打开搅拌器。冷却至-5至10℃。在20-60min内,第一次添加et2zn溶液(800ml,1.1当量)保持-5至10℃(放热)。冷搅拌约20-30分钟。在20-60min内,添加第一批ch2i2(228克)保持-5至10℃(轻微放热)(当开始加入ch2i2时罐变暗)。在20-60min内,添加第二批et2zn溶液(800ml,1.1当量)保持-5至-10℃(温和放热)。冷搅拌约20-30分钟。在20至60分钟内,添加第二批ch2i2(228g)保持-5至10℃(轻微放热)。第二次添加ch2i2后,胶状固体凝结成可搅拌的团块。随着温度升高至0℃,胶状固体似乎稀薄。过夜后,肿块完全破裂;罐内是充分搅拌的不溶物浆液。在0℃下搅拌过夜。拉ipc。加热至20℃。拉ipc。在20至60分钟内,添加第三批et2zn溶液(800ml,1.1当量),保持-4至-7℃(不放热)。冷搅拌约20-30分钟。在20至60分钟内,添加第三批ch2i2(228克)保持-5至10℃(略有放热)。冷搅拌约20-30分钟。加热至20℃。搅拌30分钟。添加另外的12%的1.1当量的加料,并且在第二个过夜时间之后反应完成。完成后,冷却至0-5℃,并添加3nhcl(2l)。前200ml放热(至20℃),并带有一些泡沫和废气(易于控制)。再次冷却至0℃后,剩余的酸仅在9℃的放热条件下加料。在15-25℃下搅拌30分钟,然后切割层。下层为橙色,无混浊。有一个黑色碎层迁移到界面并大部分溶解。在可能的情况下,黑色碎层应与水一起用。蒸馏除去(真空)大部分dcm。出于机械原因,这次运行是在旋转蒸发仪中完成的。作为一般的安全预防措施,使用真空保持温度下降。进行dcm的去除至少是为了:a)减少工作体积,更重要的是,b)控制有机溶液的密度并防止随后的层反转。最初的4.0升中总共删除了3.2升。确切的数量并不重要。此时去除dcm也使最终的汽提塔(甲苯)更容易(几乎没有沸腾器)。用1nhcl(1l)洗涤。切割各层并装入naoh溶液(250克50%+890克水的总量:1l)。冷却至5℃。加入35%h2o2(200克)。添加的前80ml是放热的:温度保持在<12℃。其余的添加不会明显放热。加热至20℃。切割层。加入20%na2so3(625g),搅拌5-15分钟,切割层。该添加不是放热的。随后,将这种洗涤液与h2o2废物级分合并在一个单独的烧瓶中,在25℃至40℃的温度下缓慢放热(5-10min)。较小的受控放热表明可以将na2so3添加到反应器中,而无需事先分离h2o2。洗涤250ml盐水。通过solkafloc40过滤有机层以除去浊度。真空除去溶剂(浴液50℃,7torr)。
后处理得到238g(113%)的橙色油,其中含有20mol%(8wt%)的甲苯。手性hplc显示错误的对映体为11%,而在类似的20g运行中为4%。
通过上述在环丙烷化步骤中依次添加试剂,提高了对映选择性。另外,使试剂的当量最小化,从而节省了成本并简化了后处理条件。
实施例15:丁基硼酸(化合物25)的合成
在10l反应器中以中等规模制备丁基硼酸,方法是在-42至-50℃的温度下,将buli添加到硼酸三异丙酯中,得到197g(76%)。vmax为6.5l(32.5l/kg)。
实施例16:化合物27的合成
二乙醇胺络合物(27)由上述丁基硼酸通过在mtbe和dcm的混合溶剂体系中与1.1当量的二乙醇胺(dea)以
在一些实施方案中,化合物6的形成(例如原位)是通过在合适的溶剂中使化合物27与化合物28接触以形成化合物6。
产率:258g,79%(纯度96%,nmr)。还收获了第二批产物(49g,15%),纯度仅为75%(nmr)。化合物27用作保持潜在不稳定的丁基硼酸的稳定固体。
实施例17:化合物27的合成
步骤:用n2吹扫反应器。向10l反应器中加入bub(oh)2(196g)、dea(222g)、dcm(2.1l)和mtbe(2.1l)。加料分子筛(345g)。二乙醇胺(dea)是粘性液体(mp=27-31℃),难以完全倾倒和转移。保留一些溶剂,以便在过筛后冲洗装料漏斗。搅拌2-24小时。拉ipc。滤出固体,并用dcm(0.5l)洗涤。去除溶剂。将残余物溶于dcm(1.1l)。残余物可溶于dcm。加料mtbe(2.5l)。加料mtbe时,混合物变得混浊。让其在冷藏室中过夜。静置(不搅拌)过夜后,晶体分离良好。分块。滤出固体,并用最少的mtbe洗涤。真空干燥(30℃,n2)至恒重:258g(79%)。浓缩滤液以引发进一步的结晶并再次收获:49g(15%)。
实施例18:化合物9的重结晶
结晶包括以下步骤:将粗产物溶解在meoh中并汽提以从粗产物中除去可能影响结晶的残留溶剂。将粗产物溶于3体积的meoh(过滤以除去浊度)。用7.6体积的乙二醇(eg,晶种)稀释。缓慢或快速添加eg仅会产生浑浊(有或没有晶种无结晶)。用2体积的水(晶种)慢慢稀释。逐滴添加水会产生不溶性(油)物质,因为每滴液滴都会撞击液体(有或没有晶种无结晶)。逐渐地,在添加过程中,胶粘材料会分离,并可能粘在墙壁和搅拌器上。最终,形成了充分搅拌的浆液(胶粘材料仍然粘附在墙上;即不会重新溶解或重悬)。过夜。滤出固体。用5:1eg:meoh洗涤,然后用水洗涤浆液。
该过程不是结晶,而是胶凝,其大部分转变成晶体。确实提高了对映体纯度,尽管没有达到期望的程度。
来自结晶的水湿滤饼不能直接进行第二结晶,而是必须首先干燥(空气或重新溶解)。溶剂的比例似乎很重要。
第二次结晶提高了对映体纯度,但不充分(需要99:1)。
最终浆液的等分试样(过滤之前)加热(喷枪)以得到完整溶液。静置时,会形成一些非晶质和一些晶体。
化合物9(重结晶):将实施例14的汽提的粗产物(238g,113%)溶解于0.9l的meoh中,汽提至干(116g),以除去可能干扰结晶的残余甲苯。目标溶剂比为meoh-3,乙二醇-7.6,水-2(相对于理论重量9的体积)。因此,在5l机械搅拌的烧瓶中,将刚汽提的粗产物溶于meoh(600ml),并迅速添加乙二醇,直到混合物变浑浊(1520ml的约90%)。剩余物与晶种缓慢加入(1-2小时);没有晶体形成。400ml水中的约50ml与晶种缓慢加入(1-2小时),然后搅拌过夜;没有晶体形成。剩余物与晶种缓慢加入(2-3小时)。胶粘的棉状晶体分离并搅拌均匀(不凝结)。搅拌过夜后,将固体滤出(2l粗玻璃料),并用5:1eg:meoh(2×250ml,置换),水(3×700ml,浆液)洗涤。手性hplc纯度不够,因此需要第二次结晶。通过溶解在己烷中(分离水)并在第二次结晶后汽提以得到183g(参见表1)来回收化合物9。
实施例19:化合物4的合成
步骤:用n2吹扫反应器。在2l反应器中装入溴化物3(106.7g)和thf(350ml)。冷却至-55℃。放热。以添加速度保持温度(约50分钟:澄清溶液)。加入保持在<-50℃(50分钟)的buli溶液(275ml)。搅拌20-45分钟。在老化过程中不溶物形成稀浆。在搅拌悬浮液的同时,加入保持<-50℃(50分钟)的硼酸异丙酯(116ml)。吸热。以添加速率保持温度(约50分钟)。搅拌30分钟。在1-2小时内升温至15℃(可接受过夜)。加料3nhcl(440ml)。加料时放热10℃。不溶物溶解为两个透明层。搅拌10分钟。将水(约750ml)排放到分液漏斗中。用methf(350ml)萃取,合并有机层。搅拌至平衡。排出残留的水。将有机层转移至旋转蒸发仪并剥离至干燥(浴液45℃,终真空30torr);145g残留物。加入庚烷(100ml),然后再次干燥。125g残留物,静置后固化。重新溶于450ml庚烷(在旋转烧瓶中,浴液45℃),然后转移回2l反应器(混浊溶液)。在大约20分钟内,在45-55℃下加入naoh溶液(由55ml的50%naoh和268ml水制备);固体沉淀。冷却至40℃并过滤掉固体。800ml介质玻璃料过滤器与介质一起使用,以减慢过滤速度。在暴露表面之前用庚烷洗涤滤饼,以避免滤饼从壁上收缩。滤饼用硅胶片压制。湿滤饼:200克。用庚烷(3x100ml)洗涤滤饼。干重:105g(95%)。真空干燥40-45℃。将庚烷层汽提至干,得到17g所示油(gcms),为起始原料的87:13混合物,其中br被丁基和h代替(参见gc/ms)。
实施例20:化合物8的合成
步骤:用n2吹扫反应器。向2l反应器中加入硼酸酯4(110g)、甲苯(400ml)、meoh(260ml)和水(140ml)。打开搅拌器。加料k2co3(115克)。喷射(n2)10分钟。加料碘化物7(83g)。喷射(n2)15分钟。加料催化剂(2.5克)(混合物为两个液相加固体)。加热至70-72℃。搅拌1小时并提取样品进行ipc。再搅拌2小时。冷却至20-25℃。加料200ml水,搅拌5分钟。停止搅拌器并切割水层。用300ml甲苯萃取水层。用水(3x200ml)洗涤合并的有机层,然后用盐水洗涤。剥离至干燥(无浑浊的黄色油,静置过夜开始结晶)。加料150ml庚烷并汽提至干以除去残留的甲苯。加热(60℃)庚烷并热过滤;产品极易溶于热。滤出1.4克淡橙色粉末。种子要等到31℃才能保持。产品在25℃时像棉一样呈纤维状出现。在约20℃下持续搅拌6-8小时,可将纤维状外观改变为更细的浆状(粉状悬浮液)。冷却至40℃,然后每2℃缓慢冷却并添加种子。缓慢冷却至室温并搅拌过夜。冷却至-10℃。过滤掉固体。用<-10℃的庚烷洗涤滤饼。真空干燥40-45℃。最终干产率:49g(48%)。
实施例21:化合物9的合成
向ch2i2(160ml,2mmol)的ch2cl2溶液(5ml)中滴加et2zn(100ml,1mmol)。将所得溶液在0℃下搅拌15分钟,并形成白色沉淀。将该溶液冷却至-40℃,并加入taddol-ti催化剂的溶液(化合物39;通过混合(4r,5r)-2,2-二甲基-a,a',a'-四苯基-1,3-二氧戊环-4,5-二甲醇(taddol)(140mg,0.29mmol)(或其对应的对映体)和加入ti(oi-pr)4(74ml,0.25mmol)的
实施例22:化合物8的合成
向氮气吹扫的反应器中装入化合物4(1.651kg)、3-碘丁-2-烯-(z)-醇(化合物7)(1.408kg,7.11摩尔)和etoh(14l)。在搅拌该混合物的同时,将碳酸钾(1.962kg,14.22摩尔)、pd-c(0.115kg)和水(0.75kg)添加至反应器,并向反应混合物中鼓入氮气5分钟。将反应加热至65-70℃2小时。继续反应直至硼酸酯4小于反应混合物的1%。将反应混合物冷却至室温,并通过solkafloc40(0.8kg)过滤。滤饼用etoh(8l)洗涤,将合并的有机层溶剂在旋转蒸发仪上减压除去。向该粗产物中加料水(3.4l)、甲基叔丁基醚(mtbe)(0.8l)和庚烷(7.8l)。将混合物加热至45-55℃,分离各层,有机层用水(2x2l)洗涤。通过solkafloc40过滤有机层。从滤液中蒸出溶剂至干。将该粗产物在60℃的温度下吸收于庚烷(4.4l)中,并将溶液缓慢冷却至40℃,每1℃添加种子。将浆液在30℃下搅拌过夜。然后将混合物在-10至-12℃之间冷却30分钟。然后使用solkafloc40(粗玻璃料)滤出固体。固体滤饼用冷(0℃)庚烷洗涤,并在40-45℃之间真空干燥。分离出白色固体的产物(化合物8),为1.62kg(84%收率)。
实施例23:化合物9的合成
向装有加料漏斗、热电偶和氮气入口的50l反应器中加料ch2cl2(12.8l),dme(2.76l),并在搅拌下将所得溶液冷却至-25至-15℃。使用加料漏斗在保持<-15c的稳定气流中加入znet215%的甲苯溶液(17.8l,19.4mol,3.6当量),然后加入甲苯(用70ml冲洗加料漏斗)。缓慢加入ch2i2(3.25l,40.37mol,7.46当量),并在加入过程中保持温度在-15±5℃(延迟放热)。在-15±5℃下搅拌20-30分钟后,加料硼酸酯催化剂(1.755kg,6.5mol,1.2当量)的ch2cl2(200ml)溶液,然后在35分钟内立即滴加在ch2cl2(2.1l)中的乙醇8(1.4kg,5.418mol,1.0当量)。在-15±5℃下搅拌后,将混合物加热至-5℃,搅拌过夜。
小心地加入3mhcl溶液(6l),并将混合物剧烈搅拌直至所有固体溶解(15-30分钟)。分离各相并汽提dcm。有机层用水洗涤,与naoh(aq)(7.6l的50%溶液;18.75mol在水中)一起搅拌。此时,进行氧化处理,添加0.6l30%h2o2(注:放热反应非常明显。冷却并缓慢添加)。搅拌15-30分钟后,切割出水层,有机相用2.4l的10%na2so3和半盐卤水(3l)洗涤。然后将混合物汽提成油,溶于8l庚烷中,汽提(2×),得到化合物9的浆液(1.98kg,130%)。将其从甲醇和乙二醇中重结晶。
1)将粗产物溶解在3体积的meoh中(过滤除去浊度)。
2)用7.6体积的乙二醇稀释(eg,晶种)。
3)用2倍体积的水缓慢稀释(晶种)。
4)过夜。
5)通过过滤收集固体。
重结晶产率为87.5%,ee为98.3%。
实施例24:化合物9的合成
向ch2i2(160μl,2mmol)的ch2cl2溶液(5ml)中滴加et2zn(100ml,1mmol)。将所得溶液在0℃下搅拌15分钟,并形成白色沉淀。将该溶液冷却至-40℃,并加入taddol-ti催化剂的溶液(化合物39;通过混合(4r,5r)-2,2-二甲基-a,a',a'-四苯基-1,3-二氧戊环-4,5-二甲醇(taddol)(140mg,0.29mmol)(或其对应的对映体)和加入ti(oi-pr)4(74ml,0.25mmol)的
实施例25:化合物9的合成
在250mlrbf中加料ch2cl2(50ml)、dme(10ml),并在冷却浴中冷却至-20±5℃。加入znet2溶液(70ml,1.0m在庚烷中,70mmol,3.5当量),然后加入ch2i2(38.0g,142mmol,7.1eq)至纯净的烧瓶中,用2x2mlch2cl2冲洗,大约30分钟。将所得溶液再搅拌20分钟,并将手性酒石酰胺催化剂8c-d溶于ch2cl2中,并加至烧瓶中,保持温度在-15±3℃。然后在15分钟内加入烯丙醇8(5.16g,20mmol,1.0当量)的ch2cl2溶液(20ml)。添加结束后,移去冷却浴,并使反应混合物缓慢温热至环境温度,搅拌过夜。第二天早晨,通过hplc分析混合物,在混合物中发现约2%的8,因此开始后处理。加入hcl(aq)溶液(3m,40ml,6当量),并将混合物搅拌直至所有固体溶解。停止搅拌并分离各层。用ch2cl2反萃取水层(在水相中检测到一些产物),合并的有机萃取液用naoh(aq)(20%,30ml),na2so3(aq)(30ml,饱和),然后盐水(2x15ml)洗涤。将得到的油状物吸收在温暖的庚烷/2-methf(80ml/15ml)中,将固体滤出,并将溶液浓缩以提供6.0g的浅黄色油状物(产率:112%;hplc纯度:97.9%;手性纯度:88%,等于76%ee(对映体过量)。
实施例26:化合物a分析
通过lc/ms(esi+)、1h-nmr(300mhz/cdcl3)和hplc(面积%)分析本发明制备的灰白色固体化合物38(例如化合物a)。化合物a的对映体纯度为99.9%(按hplc面积),没有可检测到的化合物b(按hplc面积)。还确定了熔点(dsc),其起始温度为142.7℃,峰值为144.7℃。
除非另有说明,否则在说明书和权利要求书中使用的所有表示成分数量,性质例如分子量,反应条件等的数字均应理解为在所有情况下均由术语“约”修饰。如本发明所用,术语“约”和“大约”是指在10%至15%之内,优选在5%至10%之内。因此,除非有相反的指示,否则说明书和所附权利要求书中列出的数字参数是近似值,其可以根据本发明试图获得的所需特性而变化。至少,并且不试图将等同原则的应用限制于权利要求的范围,每个数字参数应至少根据所报告的有效数字的数量并通过应用普通的舍入技术来解释。尽管阐述本发明的广泛范围的数值范围和参数是近似值,但是在具体实施例中阐述的数值被尽可能精确地报道。但是,任何数值都固有地包含某些误差,这些误差必定是由它们各自的测试测量中的标准偏差引起的。
在描述本发明的上下文中使用的术语“一”、“一个”、“该”和类似指示物(尤其是在以下权利要求的上下文中)应解释为涵盖单数和复数,除非本发明另有说明或与上下文明显矛盾。本发明中数值范围的列举仅旨在用作分别指代该范围内的每个单独数值的简写方法。除非本发明另外指出,否则每个单独的值都被并入说明书中,就好像它在本发明中被单独引用一样。除非本发明另外指出或与上下文明显矛盾,否则本发明描述的所有方法可以以任何合适的顺序执行。本发明提供的任何和所有实施方案或示例性语言(例如“如”)的使用仅旨在更好地阐明本发明提供的主题。说明书中的任何语言都不应解释为表示对实施本发明必不可少的任何未要求保护的要素。
本发明公开的本发明的替代元件或实施例的分组不应解释为限制。每个组成员可以单独引用或要求保护,也可以与该组的其他成员或此处找到的其他元素组合使用。预期出于方便和/或可专利性的原因,组中的一个或多个成员可以包括在组中或从中删除。当发生任何这样的包含或删除时,说明书被认为包含经修改的基团,从而满足所附权利要求中使用的所有马库什基团的书面描述。
本发明描述了本发明的某些实施方案,包括发明人已知的用于实施本发明的最佳方式。当然,在阅读了前面的描述后,这些描述的实施方案的变化对本领域的普通技术人员来说是显而易见的。发明人期望熟练的技术人员适当地采用这样的变型,并且发明人希望以不同于本发明具体描述的方式来实践本发明。因此,本发明包括适用法律所允许的所附权利要求书中所述主题的所有修改和等同物。而且,除非本发明另外指出或与上下文明显矛盾,否则本发明涵盖上述元件在其所有可能的变化中的任何组合。
本发明公开的特定实施方案可以使用由……组成或基本上由...组成的语言构成的权利要求中可能进一步受到限制。当在权利要求书中使用时,无论是按申请案提出还是按修正案添加,过渡术语“由……组成”不包括权利要求书中未指定的任何要素、步骤或成分。过渡术语“基本上由...组成”将权利要求的范围限制为指定的材料或步骤以及那些不会实质性影响基本和新颖特征的材料或步骤。如此要求保护的本发明的实施方案在本发明中被固有地或明确地描述和启用。
此外,在整个说明书中,已经对专利和印刷出版物进行了许多参考。以上引用的参考文献和印刷出版物中的每一个均通过引用以其整体并入本发明。
最后,应理解,本发明公开的本发明的实施方案是对本发明原理的说明。可以采用的其他修改在本发明的范围内。因此,作为实施方案而非限制,可以根据本发明的教导来利用本发明的替代构造。因此,本发明不限于精确地如所示出和描述的那样。
实施方案
实施方案1.一种化合物,其中所述化合物为:
或其水合物或溶剂化物。
实施方案2.一种式ix化合物或其溶剂化物的制备方法:
包括:
使式viii化合物或其溶剂化物与n-buli或t-buli,和b(ome)3接触,
从而形成式ix化合物或其溶剂化物,
其中r3为芳基;或被独立选自-oh、卤素、-c1-10烷基、-c1-10卤代烷基或-o-(c1-10烷基)的一个或多个取代基取代的芳基。
实施方案3.根据实施方案2所述的方法,其中r3为c6-14-芳基;或被独立选自-oh、卤素、-c1-10烷基、-c1-10卤代烷基、-o-(c1-10烷基)或-o-(c1-10卤代烷基)的取一个或多个代基取代的c6-14-芳基。
实施方案4.根据实施方案2所述的方法,其中:
所述式ix化合物为
所述式viii化合物为
实施方案5.一种式x化合物或其溶剂化物的制备方法:
包括:
使式ix化合物
或其溶剂化物与pd(0)和
从而形成式x化合物或其溶剂化物,
其中r3为芳基;或被独立选自-oh、卤素、-c1-10烷基、-c1-10卤代烷基或-o-(c1-10烷基)的一个或多个取代基取代的芳基。
实施方案6.根据实施方案5所述的方法,其中r3为c6-14-芳基;或被独立选自-oh、卤素、-c1-10烷基、-c1-10卤代烷基、-o-(c1-10烷基)或-o-(c1-10卤代烷基)的一个或多个取代基取代的c6-14-芳基。
实施方案7.根据实施方案5所述的方法,其中:
所述式ix化合物为
所述式x化合物为
实施方案8.根据实施方案5所述的方法,其中:
所述式ix化合物为
所述式x化合物为
实施方案9.一种2-甲基-2-(5,5,8,8-四甲基-5,6,7,8-四氢萘-2-基)环丙烷甲酸乙酯
包括:
使1,1,4,4-四甲基-6-(丙-1-烯-2-基)-1,2,3,4-四氢萘
实施方案10.根据实施方案9所述的方法,其中所述2-甲基-2-(5,5,8,8-四甲基-5,6,7,8-四氢萘-2-基)环丙烷甲酸乙酯或其溶剂化物为
实施方案11.一种(2-甲基-2-(5,5,8,8-四甲基-5,6,7,8-四氢萘-2-基)环丙基)甲醇
包括:
使(z)-3-(5,5,8,8-四甲基-5,6,7,8-四氢萘-2-基)丁-2-烯-1-醇
实施方案12.根据实施方案11所述的方法,其中所述(2-甲基-2-(5,5,8,8-四甲基-5,6,7,8-四氢萘-2-基)环丙基)甲醇或其溶剂化物为
所述n,n'-(环己烷-1,2-二基)二甲磺酰胺或其溶剂化物为
实施方案13.一种式iii化合物或其溶剂化物的制备方法:
包括:
使(1r,2s)-2-甲基-2-(5,5,8,8-四甲基-5,6,7,8-四氢萘-2-基)环丙烷甲醛
从而形成式iii化合物或其溶剂化物,
其中r1为c1-20-烷基;被独立选自-nh2、-nh(c1-10-烷基)、-n(c1-10-烷基)(c1-10-烷基)、-oh、卤素、-c1-10烷基、-c1-10卤代烷基、-o-(c1-10烷基)或-o-(c1-10卤代烷基)的一个或多个取代基取代的c1-20-烷基;c1-20-烯基;被独立选自-nh2、-nh(c1-10-烷基)、-n(c1-10-烷基)(c1-10-烷基)、-oh、卤素、-c1-10烷基、-c1-10卤代烷基、-o-(c1-10烷基)或-o-(c1-10卤代烷基)的一个或多个取代基取代的c1-20烯基;c6-14-芳基;或被独立选自-nh2、-nh(c1-10-烷基)、-n(c1-10-烷基)(c1-10-烷基)、-oh、卤素、-c1-10烷基、-c1-10卤代烷基、-o-(c1-10烷基)或-o-(c1-10卤代烷基)的一个或多个取代基取代的c6-14-芳基。
实施方案14.根据实施方案13所述的方法,其中r1为c1-20-烯基或c6-14-芳基。
实施方案15.根据实施方案2-14中任意一项所述的方法,其中所述方法在溶剂或溶剂组合中进行。
实施方案16.根据实施方案15所述的方法,其中所述溶剂为非极性溶剂或极性非水溶剂、水性溶剂或其组合。
实施方案17.一种式(xi)化合物或其溶剂化物的制备方法:
包括:
(i)在溶液中在ch2i2和et2zn的存在下,使式(xii)化合物或其溶剂化物与式(xiii)的化合物或其溶剂化物接触,和
(ii)随后,使步骤(i)的溶液与h2o2接触,从而制备式(xi)化合物或其溶剂化物,其中式(xi)化合物的对映体过量至少为98%。
实施方案18.一种式(xi)化合物或其溶剂化物的制备方法:
包括:
在ch2i2和二烷基锌的存在下,使式(xii)化合物或其溶剂化物与式(xiv)化合物(或其对映体)或其溶剂化物接触,
从而制备式(xi)化合物或其溶剂化物。
实施方案19.根据实施方案18所述的方法,其中所述二烷基锌为znet2。
实施方案20.一种式(xi)化合物或其溶剂化物的制备方法:
包括:
在约0℃下在ch2i2和et2zn存在下,使式(xii)化合物或其溶剂化物与式(xv)化合物或其对映体或其溶剂化物接触,
从而制备式(xi)化合物或其溶剂化物。
实施方案21.根据实施方案20所述的方法,其中式(xii)与式(xv)的摩尔比为约1.0:0.05至约1.0:0.3。
实施方案22.根据实施方案20所述的方法,其中式(xii)与式(xv)的摩尔比为约1.0:0.1。
实施方案23.一种式(xii)化合物或其溶剂化物的制备方法:
包括:
在pd/c和碱存在下,使式(xvi)化合物或其溶剂化物与式(xvii)化合物或其溶剂化物接触,
从而制备式(xii)化合物或其溶剂化物。
实施方案24.根据实施方案23所述的方法,所述碱为k2co3。
实施方案25.一种式(xvii)化合物或其溶剂化物的制备方法:
包括:
在n2ch2co2et的存在下,使式(xviii)化合物或其溶剂化物与式(xix)化合物或其溶剂化物接触,
从而制备式(xvii)化合物或其溶剂化物。
实施方案26.根据实施方案18或20所述的方法,其中式(xi)化合物的对映体过量为至少约98%。
实施方案27.根据实施方案25所述的方法,其中式(xvii)的化合物的对映体过量为至少约98%。
实施方案28.一种式(xi)化合物或其水合物或溶剂化物,
其中式(xi)化合物的对映体过量为至少约80.0%。
实施方案29.根据实施方案28所述的化合物,其中式(xi)的化合物的对映体过量为至少约98%。
实施方案30.根据实施方案28所述的化合物,其中所述化合物通过实施方案11、12、17、18或20所述的方法制备。
实施方案31.一种组合物,其包含实施方案28所述的化合物。
实施方案32.一种式(xvii)化合物或其水合物或溶剂化物,
其中式(xvii)化合物的对映体过量为至少约80.0%。
实施方案33.根据实施方案32所述的化合物,其中式(xvii)化合物的对映体过量为至少约98%。
实施方案34.根据实施方案32所述的化合物,其中所述化合物通过实施方案9、10或25所述的方法制备。
实施方案35.一种组合物,其包含实施方案32所述的化合物。
- 还没有人留言评论。精彩留言会获得点赞!