SGC-003药物组合物及其制备方法与用途与流程
本发明属于药物制剂领域,涉及一种含4,6-二氨基-2-[1-(3-氟噻吩-2-基)甲基-1h-吡唑并[3,4-b]吡啶-3-基]-5-嘧啶基-n-甲基氨基甲酸甲酯(简称为sgc-003)的药物组合物。本发明还涉及该药物组合的制备方法及用途。
背景技术:
肺动脉高压(pulmonaryarterialhypertension,pah)是临床上一种较少见的慢性疾病,主要是由于血管的异常生长、增生、不可逆重构、收缩,导致血栓形成,引起外周血管阻力的增加和肺动脉压力的上升,使右心室功能发生障碍,最终导致右心室衰竭甚至死亡。pah的临床症状不典型,是非特异性的。虽然发病率低,但误诊率高达90%,通常易被误诊为一般心脏病或者哮喘。由于误诊或者诊断不明,导致许多患者在确诊时已是该病的中晚期,不利于预后。pah的平均发病年龄为36岁,75%患者集中于20-40岁年龄段,15%患者年龄在20岁以下,且女性发病率高于男性。
临床上常用于治疗pah的药物有前列环素及其类似物、内皮素受体拮抗剂、磷酸二酯酶-5抑制剂(phosphodiesterase-5,pde-5)和可溶性鸟苷酸环化酶(solubleguanylatecyclase,sgc)激动剂。前列环素具有扩血管作用,并可促进环磷酸腺苷(cyclicadenosinemonophosphate,amp)形成从而抑制血管平滑肌细胞增殖和血小板的聚集。目前在我国批准上市的只有吸入式伊洛前列素。由内皮细胞分泌的内皮素-1(endothelin-1,et-1)是目前已知的作用最强的收缩血管的物质。et-1受体包括eta和etb,et-1与不同的受体结合,对血管产生不同的作用。血管收缩主要由eta介导,血管平滑肌细胞的增殖由eta和etb共同介导。内皮素受体拮抗剂可以阻滞et-1受体,从而导致血管的扩张和肺血管增生抑制。pde-5过度表达时会导致环磷酸鸟苷(cyclicguanosinephosphate,cgmp)大量水解成gmp而失活,导致一氧化氮(no)的血管扩张作用和细胞增殖抑制作用无法发挥。pde-5抑制剂抑制pde-5活性,减少了cgmp的降解,使得no能够发挥作用,从而加强cgmp介导的肺血管扩张和血管平滑肌细的抗增殖能力。sgc激动剂的作用机制是直接刺激sgc,并增强sgc对低水平no的敏感度,导致cgmp生成增加与随后血管扩张,从而降低血压。bayer公司开发的riociguat(品牌名:adempas;中文名:利奥西呱)即属于sgc激动剂。
本发明人以利奥西呱为先导化合物进行结构改造得到了一系列化合物,从中筛选得到sgc-003(相对分子质量424.8)作为候选化合物,结构式为4,6-二氨基-2-[1-(3-氨噻吩-2-基)甲基-1h-吡唑并[3,4-b]吡啶-3-基]-5-嘧啶基-n-甲基氨基甲酸甲酯(简称为sgc-003)为可溶性鸟苷酸环化酶激动剂,其化学结构式如式i所示。
药效学评价研究表明,预防给予sgc-003能明显降低大鼠肺动脉压和右心指数,改善右心室肥大,相比利奥西呱具有更强的药理作用。检测sgc-003对肥大的心肌细胞表面积、总蛋白质含量和心房利钠肽基因mrna表达的影响,发现sgc-003对心肌细胞的保护作用更为明显,且不良反应较少,临床上安全性和有效性评价良好,绝对生物利用度高于利奥西呱,具有十分广阔的应用前景。
本发明根据药物特点和临床用药的需求研发了sgc-003口服制剂。
技术实现要素:
本发明人经过深入的研究和创造性劳动,制得了一种含sgc-003的药物组合物。本发明人惊奇地发现,该药物组合物特别是口服片剂具有服用方便、减少胃内崩解所需时间、溶出速率快、起效快等优点;动物药代试验结果显示该组合物具有较好的生物利用度和药代动力学性质。由此提供了下述发明:
本发明的第一方面涉及一种药物组合物,其包含式i所示的化合物或其可药用盐,以及一种或多种药学上可接受的辅料。
其中,按照药物组合物的重量百分比计算,式i化合物或其可药用盐的含量为0.01%-1%,优选为0.05%-0.5%,更优选为0.06%-0.3%,例如0.06%、1.2%或3%。
在本发明的一些实施方案中,所述的药物组合物,其为固体制剂,优选为片剂。
在本发明的一些实施方案中,所述的药物组合物,其中,所述药物组合物为用于治疗和/或预防肺动脉高压的药物组合物;优选地,所述肺动脉高压为慢性血栓栓塞性肺高压。
在本发明的一些实施方案中,所述的药物组合物,其中,式i化合物或其可药用盐为唯一活性成分。
在本发明的一些实施方案中,所述的药物组合物,其中,所述式i化合物或其可药用盐为第一活性成分,并且所述药物组合物还包含具有抗肺动脉高压的不同于第一活性成分的第二活性成分;优选地,所述第二活性成分为选自前列环素及其类似物(例如伊洛前列素)、内皮素受体拮抗剂(例如内皮素-1)、磷酸二酯酶-5抑制剂和除式i化合物或其可药用盐之外的可溶性鸟苷酸环化酶激动剂中的一种或多种。
在本发明的一些实施方案中,所述的药物组合物,其中,所述辅料为选自填充剂、崩解剂、粘合剂、润滑剂和包衣剂中的一种或多种;优选地,其还含有润湿剂。
在本发明的一些实施方案中,所述的药物组合物,其特征在于如下的(1)至(6)项中的任意一项或者多项:
(1)所述填充剂为选自乳糖、微晶纤维素、玉米淀粉、糊精和预胶化淀粉中的一种或多种,优选为乳糖和/或微晶纤维素;
(2)所述崩解剂为选自微晶纤维素、交联聚维酮(例如交联聚维酮xl-10)、淀粉、羧甲基淀粉钠、交联羧甲基纤维素钠和二氧化硅中的一种或多种,优选为交联聚维酮和/或微晶纤维素;
(3)所述粘合剂为选自羟丙基甲基纤维素、羟丙基纤维素、羧甲基纤维素钠、甲基纤维素和乙基纤维素中的一种或多种,优选为羟丙基甲基纤维素;
(4)所述润滑剂为选自硬脂酸镁、滑石粉、硬脂酸,富马酸钠、peg6000、十二烷基硫酸钠和微粉硅胶中的一种或多种,优选为硬脂酸镁;
(5)所述包衣剂为欧巴代包衣预混剂和/或羟丙甲纤维素,优选为欧巴代包衣预混剂;
(6)所述润湿剂选自水、乙醇和乙醇水溶液,优选为水。
在本发明的一些实施方案中,所述的药物组合物,其中,按照药物组合物的重量百分比计算,
所述填充剂的含量为70%-90%,
所述崩解剂的含量为2%-5%,
所述粘合剂的含量为2%-10%,
所述润滑剂的含量为0.1%-1%,
所述包衣剂的含量为2%-3%;
所述润湿剂的含量为24%-25%。
本发明的第二方面涉及一种制备本发明的第一方面中任一项所述的药物组合物的方法,包括下述步骤:
(1)将填充剂、崩解剂混合均匀,得到第一混合物;
(2)粘合剂加入到润湿剂中,得到第二混合物;
(3)将第二混合物加入到第一混合物中,通过湿法制粒,然后整粒、烘干,得到颗粒;
(4)将式i化合物或其可药用盐的原料药进行微粉化处理,得到微粉;
(5)将步骤(4)中的微粉均匀喷向步骤(3)的颗粒上,然后干燥,得到干燥颗粒;
(6)向步骤(5)的干燥颗粒中加入润滑剂,混合均匀,然后压片,得到裸片;
(7)将步骤(6)中的裸片进行包衣,制得片剂。
本发明的第三方面涉及第一方面中任一项所述的药物组合物在制备治疗和/或预防肺动脉高压的药物中的用途;优选地,所述肺动脉高压为慢性血栓栓塞性肺高压。
本发明中,如果没有特别说明,所述“第一”(例如第一活性成分等)或“第二”(例如第二活性成分等),仅仅是为了指代上的区分或者表述上的清楚,并不具有严格的次序或者顺序的含义。
附图说明
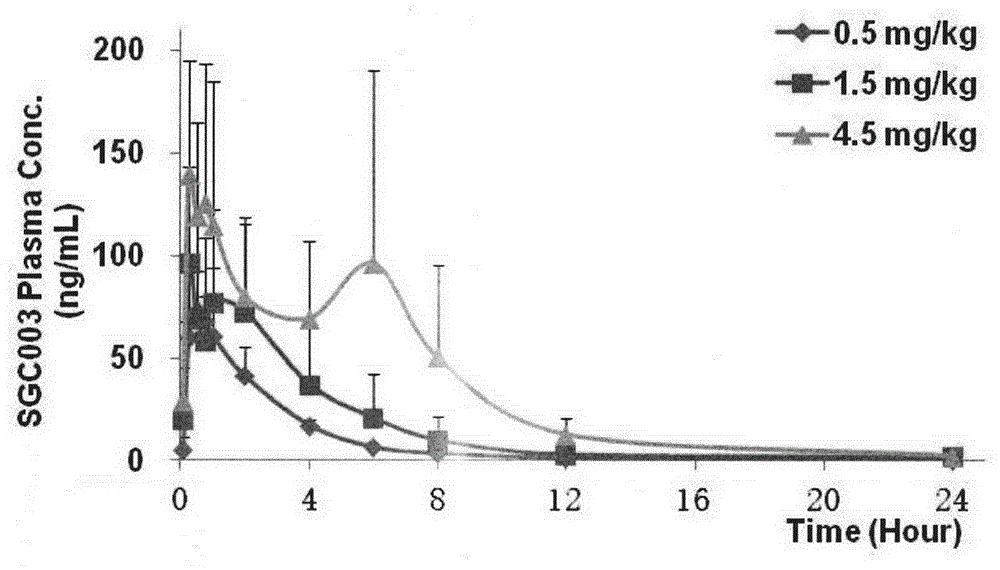
图1:雄性比格犬灌胃不同剂量sgc-003后的平均药-时曲线(n=5,mean±sd)。
图2:雄性比格犬静脉或灌胃sgc-003(0.5mg/kg)后的平均药-时曲线(n=5,mean±sd)。
具体实施方式
下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
根据临床用药需求,确定本品的剂型为口服片剂,设置三个规格0.5mg/片、1.0mg/片和2.5mg/片。三个规格的片重及大小均一致,通过不同颜色的薄膜包衣区分,除去薄膜衣后均显白色或者类白色,通过调节乳糖的处方量来控制片重。依据《化学药物制剂研究指导原则》,对本品处方组成、生产工艺、包装材料、稳定性等进行考察,确定本晶的处方和制备工艺。
实施例1:sgc-003口服片剂(规格0.5mg)
(1)将填充剂乳糖和微晶纤维素70%-90%、崩解剂微晶纤维素和交联聚维酮xl-102%-5%混合均匀,得到第一混合物;
(2)粘合剂羟丙基甲基纤维素加入到纯水溶液中,配制3%羟丙基甲基纤维素水溶液,得到第二混合物;
(3)将第二混合物加入到第一混合物中,通过湿法制粒,然后整粒、烘干,得到颗粒;
(4)将sgc-003原料药进行微粉化处理,得到微粉;
(5)将步骤(4)中的微粉按照5g/1万片的规格均匀喷向步骤(3)的颗粒上,然后干燥,得到干燥颗粒;
(6)向步骤(5)的干燥颗粒中加入润滑剂硬脂酸镁0.1%-1%,混合均匀,然后压片,得到裸片;
(7)将步骤(6)中的裸片加入欧巴代预混剂进行包衣,增重2%-3%制得片剂。
实施例2:sgc-003口服片剂(规格1.0mg)
(1)将填充剂乳糖和微晶纤维素70%-90%、崩解剂微晶纤维素和交联聚维酮xl-102%-5%混合均匀,得到第一混合物;
(2)粘合剂羟丙基甲基纤维素加入到纯水溶液中,配制3%羟丙基甲基纤维素水溶液,得到第二混合物;
(3)将第二混合物加入到第一混合物中,通过湿法制粒,然后整粒、烘干,得到颗粒;
(4)将sgc-003原料药进行微粉化处理,得到微粉;
(5)将步骤(4)中的微粉按照10g/1万片的规格均匀喷向步骤(3)的颗粒上,然后干燥,得到干燥颗粒;
(6)向步骤(5)的干燥颗粒中加入润滑剂硬脂酸镁0.1%-1%,混合均匀,然后压片,得到裸片;
(7)将步骤(6)中的裸片加入欧巴代预混剂进行包衣,增重2%-3%制得片剂。
实施例3:sgc-003口服片剂(规格2.5mg)
(1)将填充剂乳糖和微晶纤维素70%-90%、崩解剂微晶纤维素和交联聚维酮xl-102%-5%混合均匀,得到第一混合物;
(2)粘合剂羟丙基甲基纤维素加入到纯水溶液中,配制3%羟丙基甲基纤维素水溶液,得到第二混合物;
(3)将第二混合物加入到第一混合物中,通过湿法制粒,然后整粒、烘干,得到颗粒;
(4)将sgc-003原料药进行微粉化处理,得到微粉;
(5)将步骤(4)中的微粉按照25g/1万片的规格均匀喷向步骤(3)的颗粒上,然后干燥,得到干燥颗粒;
(6)向步骤(5)的干燥颗粒中加入润滑剂硬脂酸镁0.1%-1%,混合均匀,然后压片,得到裸片;
(7)将步骤(6)中的裸片加入欧巴代预混剂进行包衣,增重2%-3%制得片剂。
上述制备例制得的样品用于药代动力学测试,见下面实施例。
实施例4:sgc-003不同片剂比格犬生物利用度比较
1)实验动物:
比格犬,体重10±2kg。购于北京玛斯生物技术有限公司,实验动物许可证号:scxk-(京)2011-0003。在军事医学研究院动物中心一级动物房饲养。
2)实验试剂:
ll-5,粉末,纯度99%;由军事医学研究院毒物药物研究所药物合成室提供。
乙腈,色谱纯,fisherscientific公司产品。
甲酸,色谱纯,dikma公司产品。
dmso,色谱纯,sigma-aldrich公司产品。
sgc-003的peg溶液,按以下方法制备:
①准确量取peg-400,在注射用生理盐水中配置成peg-400含量为5%(v/v)的溶液。
②准确称取sgc-003原料药,加入上述5%peg-400溶液中溶解,置于超声水浴中超声5分钟使溶解完全。sgc-003的浓度为0.25mg/ml或0.5mg/ml(m/v)。
sgc-003的cmc混悬液,按以下方法制备:
①准确称取羧甲基纤维素钠(carboxymethylcellulosesodium,cmc),在注射用生理盐水中配置成cmc含量为2%(m/v)的溶液,在搅拌器上搅拌4-8小时使完全溶解。
②准确称取sgc-003原料药,加入上述2%cmc溶液中,充分搅拌,并置于超声水浴中超声5分钟使混悬液均匀。sgc-003的浓度为0.25mg/ml(m/v)。
3)样品处理:
取大鼠血浆或比格犬血浆0.050ml,加入0.005ml空白生理盐水后混匀,再加入0.15ml内标乙腈溶液(ll-5,200ng/ml),充分振荡后,14000rpm离心10分钟,取上清10μl进样于lc-ms/ms分析测定。
4)实验方法:
比较不同片剂类型及溶剂类型对sgc-003生物利用度的影响。实验共有peg溶液静脉给药组,peg溶液口服给药组,cmc混悬液口服给药组,200目筛片口服给药组,微粉化片口服给药组5个不同处理组。200目筛片,微粉化片均含有sgc-0032.5mg每片,每次给药每只犬一片,剂量约为0.25mg/kg。sgc-003的peg溶液与cmc混悬液均配制为0.25mg/ml,每只犬每次给药10ml。
4只体重约为10kg的比格犬进行5周期自身对照交叉给药,每两次给药期间清洗期为一周。每次给药前动物禁食12小时,自由饮水,给药后立即灌水约10ml。口服给药组于给药前及给药后5、15、30、45min及1、2、4、6、8、12、24h由四肢静脉分别交替采血2ml,静脉给药组于给药前及给药后2、5、15、30、45min及1、2、4、6、8、12、24h由四肢静脉分别交替采血2ml。采血样品2000g离心15min,分离血浆置-20℃冰箱存放待处理。
采用
5)实验结果:
给药后各时间点比格犬sgc-003的血药浓度值见下表1。由winnonlin软件,可计算得到各主要药代动力学参数,如下表2所示。
表1:sgc-003给药后各时间点比格犬体内的血药浓度(n=4)
表2:比格犬sgc-003给药后的主要药代动力学参数(n=4)
以200目筛片剂口服组为参照组,其它各组与其比较,进行针对cmax、auclast与aucinf_obs三个参数的类等效性分析,可以得到表1参数。由表中参数可得到,相较于口服200目筛片剂,sgc-003微粉化后片剂的cmax或auc均有所提高(比值大于100%),且具有显著性(在90%置信区间下,比值未能落在80%-120%的范围内)。同时,口服peg溶液或cmc混悬液也可获得相对200目筛片较高的cmax或auc,且这种差异是具有显著性的。这说明sgc-003在比格犬中的吸收可能与其溶解状态相关,当溶解状态较好时药物在体内的暴露会增加。微粉化片剂较200目筛片剂更有利于药物的溶解,因此也能获得较好的药物暴露。
实施例5:比格犬口服sgc-003的生物利用度研究
1)实验方法
参考大鼠给药剂量,每组为5只雄性比格犬。
上述5只比格犬的0.5mg/kg剂量组,在灌胃给药至少一周后(超过清洗期),再次等剂量静脉注射给药,计算绝对生物利用度。
采用双周期交叉,自身对照实验设计,比格犬分别静注给药(0.5mg/kg;n=5)与灌胃给药(0.5mg/kg;n=5)。期间间隔一周洗净期。给药前禁食12小时,自由饮水。给药前及给药后2、5、15、30、45分钟和1、2、4、6、8、12、24小时由未给药的三肢静脉分别交替采血2ml,2000g离心15分钟,分离出血浆置于-20℃冰箱存放待处理。
2)实验结果
比格犬分别口服和静注sgc-003(0.5mg/kg)后的平均血药浓度见表3,血药浓度-时间曲线见图2,主要药代动力学参数见表4。
表3:静注与灌胃sgc-003(0.5mg/kg)后比格犬体内平均血药浓度(n=5)
表8:比格犬静注与灌胃sgc-003(0.5mg/kg)后的主要药代动力学参数(n=5)
结果表明,用auc(0-t)和auc(0-∞)分别计算绝对生物利用度为78%和80%,显示比格犬口服sgc-003吸收较好。
尽管本发明的具体实施方式已经得到详细的描述,本领域技术人员将会理解。根据已经公开的所有教导,可以对那些细节进行各种修改和替换,这些改变均在本发明的保护范围之内。本发明的全部范围由所附权利要求及其任何等同物给出。
- 还没有人留言评论。精彩留言会获得点赞!