一种基于异构生物网络融合的关键蛋白质识别方法与流程
本发明涉及系统生物学领域,具体涉及一种基于异构生物网络融合的关键蛋白质识别方法。
背景技术:
被剔除后造成有关蛋白质复合物功能丧失,并导致生物体无法生存或发育的蛋白质称为关键蛋白质,识别关键蛋白质有助于理解细胞生存和发展的最低需求,对合成生物学起到至关重要的作用。研究关键蛋白质为医学等相关学科提供有价值的信息,特别是在疾病诊疗、药物设计上有重要的应用前景。在生物学中,关键蛋白质主要是通过生物医学实验识别,这些方法代价高、效率低、适用的物种有限,因此提出高效的识别关键蛋白质的计算方法成为人们关注的热点问题之一。
目前,关键蛋白质识别的计算方法大致可以分为基于机器学习的方法、基于网络拓扑结构的方法和基于多元生物信息融合的方法三种。
(1)基于机器学习的方法
chen等结合蛋白质进化率、蛋白质尺寸、度中心性等,采用神经网络和svm成功地预测了酵母的关键蛋白质;saha等做了类似的研究工作,他们的分类器结合k-最近邻居和svm算法;gustafson等选择大量关键性相关属性,包括上游尺寸、旁系同源等序列特性和网络拓扑特性;hwang等结合orf长度、链和phy等序列特征,以及共同功能度等拓扑特征建立svm分类器;acencio等通过整合酵母相互作用网络、调控网络和代谢网络构造整合网络,然后用个体特征和整合特征训练基于决策树的元分类器。
这类方法面临的挑战是如何根据已知的关键蛋白质训练分类器并应用于其他未知物种。许多研究者仅依靠已知关键蛋白质的同源映射,同源映射基于已知关键蛋白质的同源蛋白质也很有可能是关键蛋白质的概念。但是这种方法有一些限制:首先,同源映射限制在保守的直系同源物种之间进行,通常占目标基因组的小部分;其次,关键蛋白质倾向于保守性,但是存在大量保守的关键蛋白质和非关键蛋白质在参考物种中没有直系同源。
(2)基于网络拓扑结构的方法
h.jeong等提出中心性-致死性法则,指出蛋白质的关键性与网络拓扑结构紧密相关;zotenko等发现,虽然高度连通节点倾向表现出关键性,但是网络中仍然存在很大一部分节点具有很高的度,却不是关键蛋白质;li等提出了基于局部连通性的关键蛋白质识别方法lac。
ning等提出一种基于蛋白质网络反向最近邻居的中心性测度;lin等着眼于对邻居节点属性的研究,提出最大邻居分量和最大邻居分量密度的概念;tew等结合功能相似性,提出邻居节点功能中心性方法;hart等指出关键性是复合物的一种属性;song等考虑复合物和生物过程内外的物理相互作用,深入理解了蛋白质的关键性;wang等提出基于边聚集系数的中心性方法预测关键蛋白质;estrada采用蛋白质的二分性预测关键蛋白质,发现越不具备二分性的蛋白质越有可能是关键蛋白质;yu等发现,网络中的瓶颈节点往往是关键蛋白质。此外,基于节点删除的策略也是一种衡量节点重要性的方法;chua等提出结合现有中心性测度方法识别关键蛋白质;rio等分析了18个不同的重构代谢网络上的16种不同的中心性测度,发现任意结合两种都能提高预测性能;qi等基于局部相互作用稠密度,提出拓扑中心性方法lid。
上述这类方法同样存在一些限制:首先,相互作用数据包含大量的假阳性和假阴性,这将影响关键蛋白质识别的准确性;其次,大多数的方法很少分析其他已知关键蛋白质的内在属性,而只是使用网络的拓扑属性。
(3)基于多元生物信息融合的方法
为克服基于网络拓扑结构方法的局限,研究者们结合网络拓扑特性和其他的生物信息识别关键蛋白质。ren等结合网络拓扑特性和复合物信息提出复合物中心性;li等结合相互作用数据和基因表达数据提出名为pec的关键蛋白质预测方法;zhang等通过改进pec,提出名为coewc的关键蛋白质挖掘方法,该方法结合网络拓扑特征和共表达的特性;zhao等结合基因表达数据和网络拓扑属性,提出名为poem的关键蛋白质识别方法;peng等结合同源信息和相互作用网络,提出迭代的关键蛋白质预测方法ion。
但,基于多元生物信息融合的方法来识别关键蛋白质也存在一定局限性。因此,有必要改进异构生物网络融合的方式,设计一种全新的关键蛋白质识别方法。
技术实现要素:
本发明需要解决的技术问题是提供一种基于异构生物网络融合的关键蛋白质识别方法,以解决现有技术中存在的关键蛋白质预测性能差的技术缺陷。
为解决上述技术问题,本发明所采取的技术方案如下。
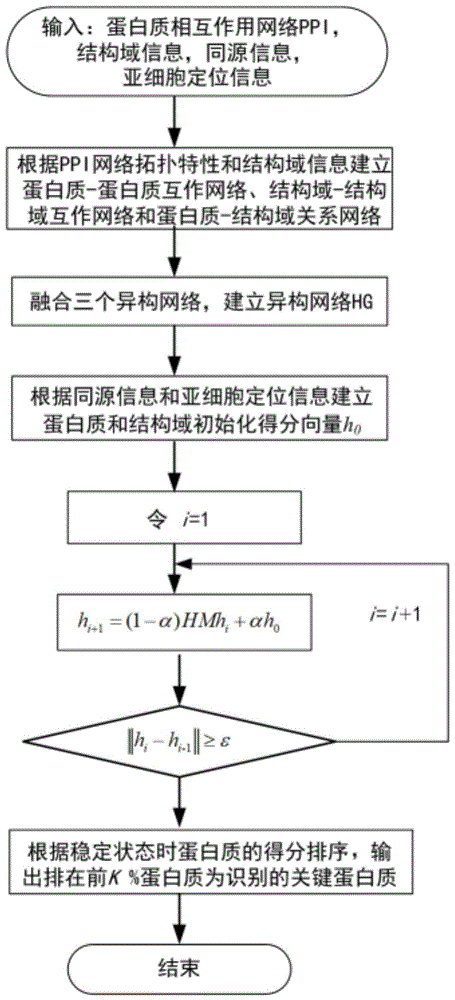
一种基于异构生物网络融合的关键蛋白质识别方法,包括以下步骤:
s1:获取酵母蛋白质相互作用网络拓扑结构、蛋白质结构域信息、蛋白质同源信息以及蛋白质亚细胞定位信息;
s2:根据蛋白质相互作用网络拓扑结构和蛋白质结构域信息,分别建立蛋白质-蛋白质互作网络p_g、结构域-结构域互作网络d_g和蛋白质-结构域关系网络pd_g;
s3:融合蛋白质-蛋白质互作网络p_g、结构域-结构域互作网络d_g和蛋白质-结构域关系网络pd_g三个网络,建立异构网络hg;
s4:根据蛋白质同源信息和亚细胞定位信息,建立蛋白质和结构域的初始化得分向量h0;
s5:基于随机游走模型,迭代地计算蛋白质和结构域的得分向量ht,直到稳定状态;
s6:根据稳定状态时蛋白质的得分进行排序,输出排在前k%的蛋白质为识别的关键蛋白质。
优选地,通过聚集系数对蛋白质相互作用网络进行加权获得,蛋白质-蛋白质互作网络加权的计算公式如下:
ni和nj分别表示蛋白质pi和蛋白质pj的邻居节点的集合,ni∩nj表示蛋白质pi和蛋白质pj的共同邻居节点集合。
优选地,步骤s2中所述结构域-结构域互作网络d_g根据蛋白质-蛋白质互作网络p_g及其蛋白质与结构域之间的关系建立,结构域与结构域之间相互作用的权值计算公式如下:
其中,p(di)和p(dj)分别表示包含结构域di和结构域dj的蛋白质集合,s(py,p(dj))表示蛋白质py和蛋白质集p(dj)之间的语义相似性。
优选地,步骤s2中所述蛋白质-结构域关系网络pd_g根据结构域信息直接建立,若蛋白质pi包含结构域dj,则mpd(i,j)=1,否则,mpd(i,j)=0。
优选地,步骤s3中所述的异构网络hg通过邻接矩阵hm表示,然后通过归一化操作,建立分块转移矩阵。
上述邻接矩阵hm表示为:
其中mp表示蛋白质-蛋白质互作网络p_g对应的邻接矩阵,md表示结构域-结构域互作网络d_g对应的邻接矩阵,mpd表示蛋白质-结构域关系网络对应的邻接矩阵。
优选地,建立分块转移矩阵的计算公式如下:
从蛋白质pi到蛋白质pj的转移概率为
从蛋白质di到蛋白质dj的转移概率为
从蛋白质pi到结构域dj的转移概率为
从蛋白质pi到蛋白质pj的转移概率为
其中,β为参数,表示从蛋白质-蛋白质互作网络p_g移动到结构域-结构域互作网络d_g的移动概率。
优选地,步骤s4中所述蛋白质和结构域的初始化得分向量h0的构建步骤如下:
s401:计算亚细胞位置的重要性得分:
其中,|pi|表示与第i个亚细胞位置相互作用的蛋白质数量,n表示亚细胞位置的数量;
s402:计算蛋白质的位置得分:
其中,s(pi)表示与蛋白质pi相互作用的亚细胞位置列表;
s403:根据同源信息计算蛋白质的保守性得分:
s404:蛋白质pi的初始化得分计算公式如下:
h0(pi)=s_score(pi)+i_score(pi)
s405:结构域dj的初始化得分计算公式如下:
其中,s_p(dj)表示包含结构域dj的蛋白质列表。
优选地,步骤s5中采用随机游走算法迭代计算蛋白质和结构域的得分向量ht的方法主要包括以下步骤:
s501:得分向量h的得分如下:
hi+1=(1-α)hmhi+αh0
其中,参数α用来来调节初始得分与上次迭代得分的比重,h0为跳转概率。
s502:如果||hi-hi-1||≤ε,使i=i+1使返回s501继续进行迭代,否则,迭代终止。
由于采用了以上技术方案,本发明所取得技术进步如下。
本发明改进了关键蛋白质识别方法研究中多源生物数据的融合方式,结合蛋白质相互作用网络和蛋白质结构域信息建立加权的蛋白质-蛋白质互作网络、结构域-结构域互作网络以及蛋白质-结构域关系网络;融合三个异构网络,基于随机游走模型,设计了新的关键蛋白质识别方法。本发明在建立蛋白质初始化得分向量时,考虑关键蛋白质的保守特性和结构特性,结合同源信息和亚细胞定位信息,来识别关键蛋白质,大大提高了关键蛋白质的识别准确率。
附图说明
图1是本发明的流程图;
图2是本发明融合异构生物网络的示意图;
图3是本发明提出的方法与其他十种关键蛋白质预测方法dc、ic、bc、cc、sc、nc、coewc、pec、poem、ion分别预测前1%,5%,10%,15%,20%,25%个关键蛋白质的准确度比较图。
具体实施方式
下面将结合附图和具体实施例对本发明进行进一步详细说明。
一种基于异构生物网络融合的关键蛋白质识别方法,该方法的流程如图1所示,主要是结合蛋白质相互作用网络和蛋白质结构域信息建立加权的蛋白质-蛋白质互作网络、结构域-结构域互作网络以及蛋白质-结构域关系网络;融合三个异构网络,结合同源信息和亚细胞定位信息,建立蛋白质和结构域的初始化得分向量,基于随机游走模型,来识别关键蛋白质。
本实施例以酵母蛋白质为例,在酵母蛋白质相互作用网络运行本方法,来验证本发明的有效性。具体方法包括以下详细步骤。
s1:获取酵母蛋白质相互作用网络拓扑结构、蛋白质结构域信息、蛋白质同源信息以及蛋白质亚细胞定位信息。
蛋白质相互作用网络来源于酿酒酵母(面包酵母)dip数据库,由5,023个蛋白质和22,570条边组成;已经通过基因敲除实验被很好地特征化,并被广泛应用于关键蛋白质的评估。
上述三种数据均源于internet网上的公共数据库。其中,蛋白质结构域数据从pfam数据库下载得到,包含1107个不同的结构域,涉及ppi网络中的3,056个蛋白质;蛋白质亚细胞定位数据从compartments数据库中获取,版本为2014-4-20;蛋白质的直系同源信息来自于inparanoid数据库,版本号是7;inparanoid数据库包含了100个物种(99个真核生物和1个原核生物)间的直系同源蛋白质数据。
s2:根据蛋白质相互作用网络拓扑结构和蛋白质结构域信息,分别建立蛋白质-蛋白质互作网络p_g、结构域-结构域互作网络d_g和蛋白质-结构域关系网络pd_g。
(1)建立蛋白质-蛋白质互作网络p_g
聚集系数用于刻画网络中某个节点与其邻居之间的亲疏程度,也是复杂网络中最重要的拓扑特征之一。本发明采用聚集系数对蛋白质相互作用网络进行加权获得,蛋白质-蛋白质互作网络加权的计算公式如下:
ni和nj分别表示蛋白质pi和蛋白质pj的邻居节点的集合,ni∩nj表示蛋白质pi和蛋白质pj的共同邻居节点集合。
(2)建立结构域-结构域互作网络d_g
d_g网络根据蛋白质-蛋白质互作网络p_g及其蛋白质与结构域之间的关系建立,结构域与结构域之间的权重计算公式如下:
其中,p(di)和p(dj)分别表示包含结构域di和结构域dj的蛋白质集合,s(py,p(dj))表示蛋白质py和蛋白质集p(dj)之间的语义相似性。
(3)建立蛋白质-结构域关系网络pd_g
蛋白质-结构域关系网络pd_g根据结构域信息直接建立。若蛋白质pi包含结构域dj,则mpd(i,j)=1,否则,mpd(i,j)=0。
s3:融合p_g、d_g和pd_g三个网络,建立异构网络hg,异构网络hg的示意图如图2所示。
异构网络hg可以通过邻接矩阵hm表示为:
其中mp表示蛋白质-蛋白质互作网络p_g对应的邻接矩阵,md表示结构域-结构域互作网络d_g对应的邻接矩阵,mpd表示蛋白质-结构域关系网络对应的邻接矩阵。
然后通过归一化操作,建立分块转移矩阵,分块转移矩阵的计算公式如下:
从蛋白质pi到蛋白质pj的转移概率为
从蛋白质di到蛋白质dj的转移概率为
从蛋白质pi到结构域dj的转移概率为
从蛋白质pi到蛋白质pj的转移概率为
其中,β为参数,表示从蛋白质-蛋白质互作网络p_g移动到结构域-结构域互作网络d_g的移动概率。
s4:根据蛋白质同源信息和亚细胞定位信息,建立蛋白质和结构域的初始化得分向量h0。具体方法如下。
(1)计算亚细胞位置的重要性得分:
其中,|pi|表示与第i个亚细胞位置相互作用的蛋白质数量,n表示亚细胞位置的数量。
(2)计算蛋白质的位置得分:
其中,s(pi)表示与蛋白质pi相互作用的亚细胞位置列表。
(3)根据同源信息计算蛋白质的保守性得分:
(4)蛋白质pi的初始化得分计算公式如下:
h0(pi)=s_score(pi)+i_score(pi)
(5)结构域dj的初始化得分计算公式如下:
其中,s_p(dj)表示包含结构域dj的蛋白质列表。
s5:基于随机游走模型,迭代地计算蛋白质和结构域的得分向量ht,直到稳定状态。迭代步骤如下:
(1)计算蛋白质和结构域的得分向量h的得分如下:
hi+1=(1-α)hmhi+αh0
其中,参数α用来来调节初始得分与上次迭代得分的比重,h0为跳转概率。
(2)如果||hi-hi-1||≥ε,使i=i+1使返回上一步继续进行迭代,否则,迭代终止。
其中ε是一个控制迭代终止的参数,本发明中,默认地设置为10-5。
s6:根据稳定状态时蛋白质的得分进行排序,输出排在前k%的蛋白质为识别的关键蛋白质。
上述为本发明应用于酵母蛋白质的关键蛋白质识别过程中,为验证本发明的准确性,同时还采用了其他十种关键蛋白质预测方法dc、ic、bc、cc、sc、nc、coewc、pec、poem、ion进行酵母蛋白质的关键蛋白质识别,通过分别预测前1%、5%、10%、15%、20%、25%个关键蛋白质,可对是一种关键蛋白质识别方法的准确度进行比较,具体如图3所示。
从图3中可以看出,本发明用于识别关键蛋白质的方法相对于其他方法具有较高的准确度。
- 还没有人留言评论。精彩留言会获得点赞!