一种基因型校正的装置和方法与流程
本发明属于生物信息分析领域,涉及一种基因型校正的装置和方法。
背景技术:
由于人类基因组是二倍体,在基因组同一位点的来源于两条同源染色体的碱基均可能不同于参考基因组上的碱基,若来源于两条同源染色体的碱基均不同于参考基因组上的碱基,则为纯合突变型;若来源于两条同源染色体的碱基只有一个不同于参考基因组上的碱基,则为杂合突变型,若来源于两条同源染色体的碱基与参考基因组上的碱基相同,则为野生型。
基于高通量测序测定和分析基因型是确定个体基因是否存在变异的主要手段之一。目前,对于每个位点的基因型判定是依据待检测位点测序数据中的突变型碱基深度与测序深度的比值,即突变频率(allelefrequency,af)来判别。然而,片段扩增、序列比对、测序错误等原因会造成测序偏倚,从而导致突变频率的测量值与真实值会有一定偏差,例如,采用常规变异检测软件(如tvc)检出的变异基因型会偶有出现异常的情况,即野生型的被检测成突变型,杂合突变被检测成野生型或纯合突变。
因此,迫切需要一种对变异检出的基因型进行校正的装置和方法,提高高通量测序分析基因型的敏感性和特异性。
技术实现要素:
针对现有技术的不足及实际的需求,本发明提供一种基因型校正的装置和方法,所述装置通过贝叶斯模型对检测到的数据建立统计模型,构建先验分布,确定总体分布信息,经过核密度估计计算出条件概率值p(af|gt),并最终获得后验概率值p(gt|af),通过判定后验概率值与其对应的阈值关系来对该位点的基因型进行判别,敏感性和特异性好,值得推广应用。
为达此目的,本发明采用以下技术方案:
第一方面,本发明提供一种基因型校正的装置,所述装置包括如下单元:
(1)样本收集单元:收集不同基因型的初始样本,确定初始样本各位点的基因型;
(2)数据采集单元:对初始样本进行测序和变异分析,采集突变频率;
(3)模型构建单元:采用贝叶斯模型结合核密度估计的方法,根据单元(2)采集的突变频率和单元(1)确定的已知基因型构建贝叶斯模型,制定阈值;
(4)检测校正单元:对待测样本进行测序,采集突变频率,将根据贝叶斯模型结合核密度估计的方法计算得到的后验概率值p(gt|af),通过比较后验概率值p(gt|af)与单元(3)制定的阈值,判别校正该位点的基因型。
本发明中,本发明利用核密度估计结合贝叶斯模型,将无法覆盖的af值范围通过核密度估计构建连续型数据,计算出该位点的条件概率值p(af|gt)和后验概率值p(gt|af),通过判定后验概率值与其对应的阈值关系来对该位点的基因型进行判别和校正;在从而达到变异检测软件所检出该位点af值状态下,对其检出的基因型发生概率进行校正的目的,通过定制野生型、杂合型和纯合型的临界范围(阈值),检测准确度达到99.9%。
本发明通过贝叶斯模型对检测到的数据建立统计模型,构建先验分布,确定总体分布信息,并最终计算出在已知基因型分类的情况下发生该af频率的条件概率值p(af|gt)和已知af值的情况下判定某种基因型的后验概率值p(gt|af),通过判定后验概率值与其对应的阈值关系来对该位点的基因型进行判别。
贝叶斯预测模型是一种统计学方法,用来估计统计量的某种性质。目前,贝叶斯理论已经广泛应用于数据挖掘、医疗诊断、工业控制、投资风险、预测、人工智能等领域,具有非常广泛的研究前景,可以用来解决医学、市场预测、风险评估、信号估计、概率推理等一系列不确定的问题。在高通量测序领域,常用的变异(snp/indel)检测软件如gatk,tvc等也都是以贝叶斯预测模型作为基因型判别分型的重要依据。
优选地,单元(1)所述基因型包括野生型、纯合突变型或杂合突变型中的任意一种或至少两种的组合,优选为野生型、纯合突变型和杂合突变型的组合。
优选地,单元(1)所述确定的方法为一代测序法。
优选地,单元(2)所述测序的方法为半导体测序法。
优选地,单元(2)所述突变频率为突变型碱基深度与测序深度的比值。
优选地,单元(3)所述贝叶斯模型的公式为:
其中,所述gti表示为某种基因型事件,af表示突变频率事件;
所述p(gti)为先验概率,即在不附加任何条件下发现某种基因型的概率值。
优选地,所述条件概率p(gti|af)为后验概率,即在观察到af频率后对应的基因型进行判定的概率值;
所述条件概率p(af|gti)为计算过程中的条件概率,即在事件gti已知的条件下事件af发生的概率值。
本发明贝叶斯公式如下:
公式描述:
公式中,gti表示为某种基因型事件,af表示突变频率事件。条件概率p(gti|af)为后验概率,即在观察到af频率后对应的基因型进行判定的概率值;p(gti)为先验概率,即在不附加任何条件下发现某种基因型的概率值,可以通过已构建的数据集合中基因型分布比例计算;在事件gti已知的条件下事件af发生的条件概率为p(af|gti),公式当中,对于p(af|gti)的计算是最关键的。事件af发生的条件下事件gti发生的条件概率为p(gti|af),即后验概率。
优选地,所述条件概率p(af|gti)通过核密度估计的方法计算;
所述核密度估计的公式如下:
其中,p(xaf)为概率密度函数,对其积分能够求得p(af|gti);
概率n(xi,z)为高斯分布,z为设定参数,称为带宽,n为数据点总数,带宽影响密度估计,反映kde曲线整体的平坦程度。
优选地,所述带宽的选取公式如下:
其中,z为带宽,n为数据点总数,σ为标准差。
对于p(af|gti)的计算,本申请采用核密度估计的方法计算,核密度估计kerneldensityestimation(kde)是在概率论中用来估计未知的密度函数,属于非参数检验方法之一,已经广泛为人们所用。相比较于参数估计,非参数估计的优势在于不需要证明数据服从特定的分布或进行任何假设,适合于任何数据集合的概率密度估计。
核密度函数的原理比较简单,在我们知道某一事物的概率分布的情况下,如果某一个数在观察中出现了,我们可以认为这个数的概率密度很大,和这个数比较近的数的概率密度也会比较大,而那些离这个数远的数的概率密度会比较小。密度估计其实就是通过核函数(如高斯)将每个数据点的数据+带宽当作核函数的参数,得到n个核函数,再线性叠加就形成了核密度的估计函数,归一化后就是核密度概率密度函数了。简而言之,核密度估计,就是采用平滑的峰值函数(“核”)来拟合观察到的数据点,从而对真实的概率分布曲线进行模拟。
本发明采用的核函数核密度估计公式如下:
公式描述:
p(xaf)概率密度函数,对其积分可以求得p(af|gti),概率n(xi,z)为高斯分布,z为设定参数,也称为带宽(bandwidth)或平滑参数,n为数据点总数。带宽影响密度估计,反映kde曲线整体的平坦程度。
带宽可依据如下公式进行选取:
其中,z为带宽,n为数据点总数,σ为标准差。
优选地,单元(4)所述测序为半导体测序。
优选地,单元(4)所述校正的判定标准为:满足p(gt|af)≥阈值时,则判定为该基因型。
作为优选技术方案,一种基因型校正的装置,具体包括如下单元:
(1)样本收集单元:收集野生型、纯合突变型和杂合突变型的初始样本,一代测序法确定初始样本各位点的基因型;
(2)数据采集单元:采用半导体测序法对初始样本进行测序和变异分析,采集突变频率,即突变型碱基数与测序深度的比值;
(3)模型构建单元:采用贝叶斯模型结合核密度估计的方法,根据单元(2)采集的突变频率和单元(1)确定的已知基因型制定阈值;
贝叶斯模型的公式为:
其中,所述gti表示为某种基因型事件,af表示突变频率事件;
所述p(gti)为先验概率,即在不附加任何条件下发现某种基因型的概率值;
所述条件概率p(gti|af)为后验概率,即在观察到af频率后对应的基因型进行判定的概率值;
所述条件概率p(af|gti)为计算过程中的条件概率,即在事件gti已知的条件下事件af发生的概率值,通过核密度估计的方法计算;
所述核密度估计的公式如下:
其中,p(xaf)为概率密度函数,对其积分能够求得p(af|gti);
概率n(xi,z)为高斯分布,z为设定参数,称为带宽,n为数据点总数,带宽影响密度估计,反映kde曲线整体的平坦程度;
带宽的选取公式如下:
其中,z为带宽,n为数据点总数,σ为标准差;
(4)检测校正单元:采用半导体测序方法对待测样本进行测序,采集突变频率,将根据贝叶斯模型结合核密度估计的方法计算得到的条件概率值与单元(3)制定的阈值比较,判别校正该位点的基因型;
校正的判定标准为:满足p(gt|af)≥阈值时则判定为该基因型。
第二方面,本发明提供一种基因型校正的方法,采用如第一方面所述的装置,
优选地,所述方法包括如下步骤:
(1)收集样本:收集野生型、纯合突变型和杂合突变型的样本,一代测序法确定样本各位点的基因型;
(2)采集数据:采用半导体测序法对样本进行测序和变异分析,采集突变频率,即突变型碱基数与测序深度的比值;
(3)制定阈值:采用贝叶斯模型结合核密度估计的方法,根据步骤(2)采集的突变频率和步骤(1)确定的已知基因型制定阈值;
贝叶斯模型的公式为:
其中,所述gti表示为某种基因型事件,af表示突变频率事件;
所述p(gti)为先验概率,即在不附加任何条件下发现某种基因型的概率值;
所述条件概率p(gti|af)为后验概率,即在观察到af频率后对应的基因型进行判定的概率值;
所述条件概率p(af|gti)为计算过程中的条件概率,即在事件gti已知的条件下事件af发生的概率值,通过核密度估计的方法计算;
所述核密度估计的公式如下:
其中,p(xaf)为概率密度函数,对其积分能够求得p(af|gti);
概率n(xi,z)为高斯分布,z为设定参数,称为带宽,n为数据点总数,带宽影响密度估计,反映kde曲线整体的平坦程度;
带宽的选取公式如下:
其中,z为带宽,n为数据点总数,σ为标准差;
(4)检测校正:采用半导体测序方法对待测样本进行测序,采集突变频率,将根据贝叶斯模型结合核密度估计的方法计算得到的条件概率值与步骤(3)制定的阈值比较,判别校正该位点的基因型;
校正的判定标准为:满足p(gt|af)≥阈值时,则判定为该基因型。
与现有技术相比,本发明具有如下有益效果:
本发明利用核密度估计结合贝叶斯模型,将无法覆盖的af值范围通过核密度估计构建连续型数据,计算出该位点的条件概率值p(af|gt)和后验概率值p(gt|af),通过判定后验概率值与其对应的阈值关系来对该位点的基因型进行判别和校正,在从而达到变异检测软件所检出该位点af值状态下,对其检出的基因型发生概率进行校正的目的,通过定制野生型、杂合型和纯合型的临界范围(阈值),检测准确度达到99.9%。
附图说明
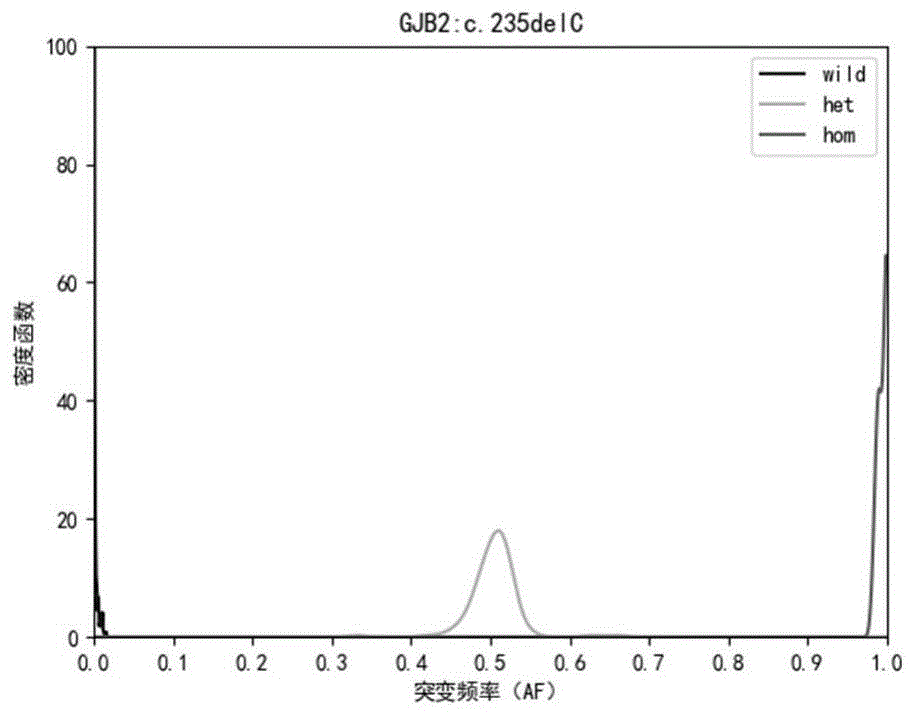
图1为本发明的gjb2:c.235delc的核密度函数曲线图,“wild”表示野生型,“het”表示杂合突变型,“hom”表示纯合突变型;
图2为本发明的gjb2:c.299-300delat的核密度函数曲线图,“wild”表示野生型,“het”表示杂合突变型,“hom”表示纯合突变型;
图3为本发明的gjb2:c.176-191del16的核密度函数曲线图,“wild”表示野生型,“het”表示杂合突变型,“hom”表示纯合突变型;
图4为本发明的gjb2:c.257c>g的核密度函数曲线图,“wild”表示野生型,“het”表示杂合突变型,“hom”表示纯合突变型;
图5为本发明的gjb2:c.512insaacg的核密度函数曲线图,“wild”表示野生型,“het”表示杂合突变型,“hom”表示纯合突变型;
图6为本发明的gjb2:c.427c>t的核密度函数曲线图,“wild”表示野生型,“het”表示杂合突变型,“hom”表示纯合突变型;
图7为本发明的gjb2:c.35delg的核密度函数曲线图,“wild”表示野生型,“het”表示杂合突变型,“hom”表示纯合突变型;
图8为本发明的gjb2:c.109g>a的核密度函数曲线图,“wild”表示野生型,“het”表示杂合突变型,“hom”表示纯合突变型;
图9为本发明的gjb2:c.35insg的核密度函数曲线图,“wild”表示野生型,“het”表示杂合突变型,“hom”表示纯合突变型;
图10为本发明的gjb3:c.538c>t的核密度函数曲线图,“wild”表示野生型,“het”表示杂合突变型,“hom”表示纯合突变型;
图11为本发明的slc26a4:ivs7-2a>g的核密度函数曲线图,“wild”表示野生型,“het”表示杂合突变型,“hom”表示纯合突变型;
图12为本发明的slc26a4:c.2168a>g的核密度函数曲线图,“wild”表示野生型,“het”表示杂合突变型,“hom”表示纯合突变型;
图13为本发明的slc26a4:c.1174a>t的核密度函数曲线图,“wild”表示野生型,“het”表示杂合突变型,“hom”表示纯合突变型;
图14为本发明的slc26a4:c.1975g>c的核密度函数曲线图,“wild”表示野生型,“het”表示杂合突变型,“hom”表示纯合突变型;
图15为本发明的slc26a4:c.1226g>a的核密度函数曲线图,“wild”表示野生型,“het”表示杂合突变型,“hom”表示纯合突变型;
图16为本发明的slc26a4:c.1229c>t的核密度函数曲线图,“wild”表示野生型,“het”表示杂合突变型,“hom”表示纯合突变型;
图17为本发明的slc26a4:ivs15+5g>a的核密度函数曲线图,“wild”表示野生型,“het”表示杂合突变型,“hom”表示纯合突变型;
图18为本发明的slc26a4:c.2027t>a的核密度函数曲线图,“wild”表示野生型,“het”表示杂合突变型,“hom”表示纯合突变型;
图19为本发明的slc26a4:c.589g>a的核密度函数曲线图,“wild”表示野生型,“het”表示杂合突变型,“hom”表示纯合突变型;
图20为本发明的slc26a4:c.1079c>t的核密度函数曲线图,“wild”表示野生型,“het”表示杂合突变型,“hom”表示纯合突变型;
图21为本发明的slc26a4:c.281c>t的核密度函数曲线图,“wild”表示野生型,“het”表示杂合突变型,“hom”表示纯合突变型;
图22为本发明的slc26a4:c.754t>c的核密度函数曲线图,“wild”表示野生型,“het”表示杂合突变型,“hom”表示纯合突变型;
图23为本发明的slc26a4:ivs14+1g>a的核密度函数曲线图,“wild”表示野生型,“het”表示杂合突变型,“hom”表示纯合突变型;
图24为本发明的slc26a4:c.1336c>t的核密度函数曲线图,“wild”表示野生型,“het”表示杂合突变型,“hom”表示纯合突变型;
图25为本发明的slc26a4:c.1343c>t的核密度函数曲线图,“wild”表示野生型,“het”表示杂合突变型,“hom”表示纯合突变型;
图26为本发明的slc26a4:c.1693insa的核密度函数曲线图,“wild”表示野生型,“het”表示杂合突变型,“hom”表示纯合突变型;
图27为本发明的slc26a4:c.2086c>t的核密度函数曲线图,“wild”表示野生型,“het”表示杂合突变型,“hom”表示纯合突变型;
图28为本发明的slc26a4:c.387delc的核密度函数曲线图,“wild”表示野生型,“het”表示杂合突变型,“hom”表示纯合突变型;
图29为本发明的slc26a4:c.917insg的核密度函数曲线图,“wild”表示野生型,“het”表示杂合突变型,“hom”表示纯合突变型;
图30为本发明的slc26a4:ivs13+9c>t的核密度函数曲线图,“wild”表示野生型,“het”表示杂合突变型,“hom”表示纯合突变型;
图31为本发明的12srrna:m.1555a>g的核密度函数曲线图,“wild”表示野生型,“mutant”表示突变型;
图32为本发明的12srrna:m.1494c>t的核密度函数曲线图,“wild”表示野生型,“mutant”表示突变型;
图33为本发明的mt-tl1:m.3243a>g的核密度函数曲线图,“wild”表示野生型,“mutant”表示突变型。
具体实施方式
为更进一步阐述本发明所采取的技术手段及其效果,以下结合附图并通过具体实施方式来进一步说明本发明的技术方案,但本发明并非局限在实施例范围内。
实施例1
遗传性耳聋相关基因检测遗传性耳聋5个常见基因的33个突变位点,对于每个位点均设计了pcr引物,通过多重pcr技术扩增目标区域片段,对于每个位点的基因型判定是依据待检测位点测序数据中的突变型碱基数与测序深度的比值,即突变频率(allelefrequency,简写为af)来判别的,对于高通量测序的检测结果,由于片段扩增、序列比对、测序错误等原因会造成的测序偏倚,检测结果突变频率的测量值与真实值会有一定偏差。
本申请依据贝叶斯模型(bayesianmodels),构建每个位点的基因型频率分布模型,通过核密度估计的方法计算该位点的条件概率值p(af|gt)和后验概率值p(gt|af),通过判定后验概率值与其对应的阈值关系来对该位点的基因型进行判别,具体方案如下:
首先,用已知基因型的样品,制定各位点的贝叶斯模型阈值;
a.样本收集:收集各位点各种基因型(包括野生型和突变型)临床病人的样本,采用一代测序法确定样品各位点的基因型,见表1;
表1用于阈值制定的样品基因型统计
*:未列出的位点基因型为野生型。
b.基因检测及数据采集:采用遗传性耳聋相关基因检测试剂盒(半导体测序法)对收集的临床样品进行检测,并使用半导体测序仪进行测序,对下机数据进行变异分析,采集突变测序数据中的突变型碱基深度与测序深度的比值,即突变频率(allelefrequency,af)用于构建贝叶斯模型并制定阈值阈值。
各位点基因型数据统计如表2所示;
表2
表2制定阈值各基因型数据统计
c.阈值的计算:采用贝叶斯模型结合核密度估计的方法,根据各检测位点的af数据和已知基因型计算阈值;
本发明通过贝叶斯模型对检测到的数据建立统计模型,构建先验分布,确定总体分布信息,并最终计算出在已知基因型分类的情况下发生该af频率的条件概率值p(af|gt)和已知af值的情况下判定某种基因型的后验概率值p(gt|af),通过判定后验概率值与其对应的阈值关系来对该位点的基因型进行判别。
贝叶斯预测模型是一种统计学方法,用来估计统计量的某种性质。目前,贝叶斯理论已经广泛应用于数据挖掘、医疗诊断、工业控制、投资风险、预测、人工智能等领域,具有非常广泛的研究前景,可以用来解决医学、市场预测、风险评估、信号估计、概率推理等一系列不确定的问题。在高通量测序领域,常用的变异(snp/indel)检测软件如gatk,tvc等也都是以贝叶斯预测模型作为基因型判别分型的重要依据。
本发明贝叶斯公式如下:
公式描述:
公式中,gti表示为某种基因型事件,af表示突变频率事件。条件概率p(gti|af)为后验概率,即在观察到af频率后对应的基因型进行判定的概率值;p(gti)为先验概率,即在不附加任何条件下发现某种基因型的概率值,可以通过已构建的数据集合中基因型分布比例计算;在事件gti已知的条件下事件af发生的的条件概率为p(af|gti);因此公式当中,对于p(af|gti)的计算是最关键的,事件af发生的条件下事件gti发生的条件概率p(gti|af),即后验概率。
对于p(af|gti)的计算,本申请采用核密度估计的方法计算,核密度估计kerneldensityestimation(kde)是在概率论中用来估计未知的密度函数,属于非参数检验方法之一,已经广泛为人们所用,相比较于参数估计,非参数估计的优势在于不需要证明数据服从特定的分布或进行任何假设,适合于任何数据集合的概率密度估计。
核密度函数的原理比较简单,在我们知道某一事物的概率分布的情况下,如果某一个数在观察中出现了,我们可以认为这个数的概率密度很大,和这个数比较近的数的概率密度也会比较大,而那些离这个数远的数的概率密度会比较小。密度估计其实就是通过核函数(如高斯)将每个数据点的数据+带宽当作核函数的参数,得到n个核函数,再线性叠加就形成了核密度的估计函数,归一化后就是核密度概率密度函数了;简而言之,核密度估计,就是采用平滑的峰值函数(“核”)来拟合观察到的数据点,从而对真实的概率分布曲线进行模拟。
本发明采用的核函数核密度估计公式如下:
公式描述:
p(xaf)概率密度函数,对其积分可以求得p(af|gti),概率n(xi,z)为高斯分布,z为设定参数,也称为带宽(bandwidth)或平滑参数,n为数据点总数。带宽影响密度估计,反映kde曲线整体的平坦程度。
带宽可依据如下公式进行选取:
其中,z为带宽,n为数据点总数,σ为标准差。
各位点阈值制定及敏感性特异性见表3,满足p(gt|af)≥0.999时则判定为该基因型;如p(wild|af)≥0.999,则判定为野生型。
表3各位点阈值制定
各位点数据的核密度函数曲线见图1-图33,各位点通过贝叶斯预测模型的方法计算后,对于常染色体基因野生型,纯合突变型,杂合突变型的检测敏感性和特异性均为1.00;对于线粒体基因突变型和野生型的检测,敏感性和特异性也均为1.00;可以看出,各位点在选定的阈值对于阴性数据和阳性数据的区分较好,表明选定的阳性判断值可以很好的区分出阴性数据和阳性数据。从图1-图33各位点的核密度估计函数曲线图可以看出,各位点的阳性判断值对于位点的基因型区分能力较强。
实施例2结果验证
本发明通过对730份样品进行阈值的制定,各样品各位点的灵敏度和特异度均为1;同时采用了645份样品对制定的模型阈值进行了验证,验证结果见表4:
表4模型阈值的验证结果
由表4可知,所有阴性数据的位点灵敏度和特异度均为1,这表明计算出的各检测位点的灵敏度和特异性均很好,能满足临床样品的检测需求,检测结果可靠。
实施例3
与未进行贝叶斯结合核密度估计校正的变异检出结果对比,结果见表5
表5
由表5可知,未进行贝叶斯结合核密度估计校正的变异检出结果存在检出的变异基因型会偶有出现异常的情况,即野生型的被检测成突变型,杂合突变被检测成野生型或纯合突变,而进行校正后能提高高通量测序分析基因型的敏感性和特异性。
综上所述,本发明提供一种基因型校正的装置和方法,所述装置通过贝叶斯模型对检测到的数据建立统计模型,构建先验分布,确定总体分布信息,并最终计算出后验概率值p(gt|af),通过判定后验概率值与其对应的阈值关系来对该位点的基因型进行判别,敏感性和特异性好,值得推广应用。
申请人声明,本发明通过上述实施例来说明本发明的详细方法,但本发明并不局限于上述详细方法,即不意味着本发明必须依赖上述详细方法才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明产品各原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
- 还没有人留言评论。精彩留言会获得点赞!