核酸结构中核苷酸与核苷酸相互作用预测的方法与流程
本发明属于生物分子结构分析研发领域,具体是指一种核酸结构中核苷酸与核苷酸相互作用预测的方法。
背景技术:
rna是细胞中最基本生物大分子之一,除传递遗传信息与蛋白质合成的生物学功能外,rna还参与基因调控等其它生物学功能,与肿瘤和神经系统等多种人类疾病有密切的关联。rna的生物学功能依赖于它的三维空间结构,由于rna易于降解,实验上测定rna的三维空间结构需耗费大量的资源,极大限制了rna相关的生物学功能研究。近几年,rna三维空间结构理论预测的出现改变了rna结构-功能研究领域的研究现状。但是,现有的rna三级结构预测方法大多数不能很好预测rna的长程空间结构相互作用,无法精确预测复杂拓扑结构的rna分子。rna长程空间结构相互作用可以稳定rna的拓扑结构,对rna折叠与三级结构十分重要。因此,亟需发展一种核酸结构中核苷酸与核苷酸相互作用预测的方法。
针对现有技术的缺陷或改进需求,本发明提供了一种核酸结构中核苷酸与核苷酸相互作用预测的方法。其目的在于利用受限玻尔兹曼机模型分析核酸的结构特征,利用直接耦合分析模型分析核酸序列的共进化特征。结合和两者优势预测核苷酸与核苷酸相互作用,解决现有技术对核苷酸与核苷酸相互作用预测精度较低,无法搭建复杂核酸三级结构的技术问题。
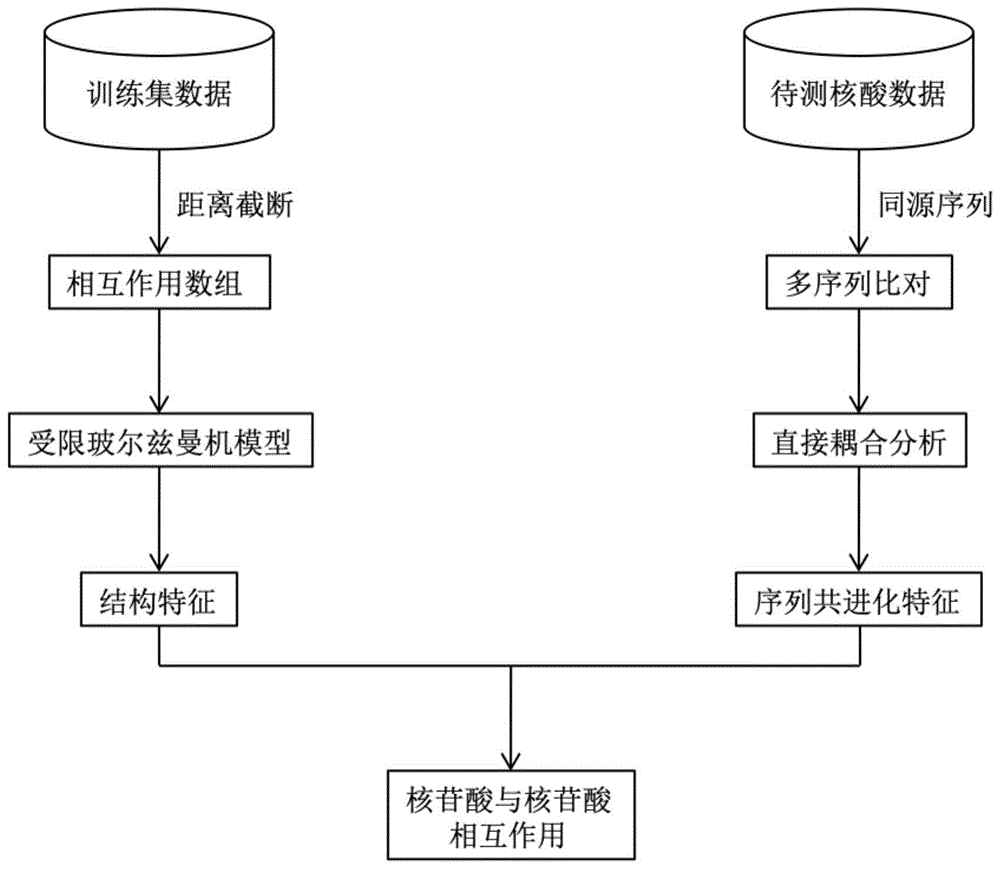
为解决上述技术问题,本发明提供了一种核酸结构中核苷酸与核苷酸相互作用预测的方法,其特征在于:包括如下步骤:
(1)核酸序列和结构数据的选取:
该步骤中的核酸序列提取自核酸信息分类数据库rfam,结构数据提取自核酸三级结构数据库rna3dhub,version3.21(数据统计截止至2018年5月11日,具体信息见http://rna.bgsu.edu/rna3dhub/nrlist/release/3.21);
(2)受限玻尔兹曼机学习核酸结构特征:
首先将结构数据中的核酸结构以
受限玻尔兹曼机的能量表达式为
e(v,h|w,b,c)=-btv-cth-htwv(1)
其中w是可见层单位v与隐藏层单位h之间的连接权重矩阵,h、b、c是作为补偿的偏置单元。v、h的概率给定如下:
z(w,b,c)=∑v,he-e(v,h|w,b,c)(3)
其中z(w,b,c)是对所有可能的v和h求和的配分函数;通过随机梯度下降(sgd)对经验数据的负对数似然进行受限玻尔兹曼机训练;l(w,c,b,t)定义为损失函数,希望它在sgd期间最小化
p(v|w,b,c)定义如下
p(v|w,b,c)=∑hp(v,h|w,b,c)(5)
t为样本数据;通过最小化损失函数,根据以下公式更新参数:w,b,c
上述利用线性插值的方法将结构数据中的核苷酸与核苷酸相互作用数组统一调整到100x100的大小,并将数组的下三角转换成一维数组;
上述利用
上述利用随机梯度下降法sgd和对比散度cd算法对受限玻尔兹曼机模型进行训练,更新权重;
上述受限玻尔兹曼机模型学习率参数的设置为0.1;
(3)直接耦合分析模型预测序列共同进化:
在进行多序列比对即msa时去除序列插空大于50%的序列,然后计算单个核苷酸和一对核苷酸的核苷酸频率;
其中两个位点之间相互作用强度的直接耦合定义为
借助一个孤立双位点模型
(4)受限玻尔兹曼机与直接耦合分析模型相结合:
核苷酸与核苷酸相互作用预测结果综合了受限玻尔兹曼机学习的结构特征和直接耦合分析学习的序列共进化特征
direct=di×w2(11)
其中,direct为核苷酸与核苷酸预测结果,di为直接耦合分析得到的序列共进化信息,w为受限玻尔兹曼机学习得到的结构特征;通过计算公式direct=di×w2综合受限玻尔兹曼机学习的结构特征和直接耦合分析学习序列的共进化特征。
总体而言,通过本发明所构思的以上技术方案与现有技术相比,能够取得下列有益效果:
(1)本发明开创性地提出了在基于受限玻尔兹曼机的核酸结构特征提取方法。
(2)本发明提供的核酸结构中核苷酸与核苷酸相互作用预测的方法优于现有的传统方法(mfdca和plmdca),在测试例中分别提高了15%和4%。
附图说明
图1是本发明方法的流程示意图。
图2是本发明的预测精度与现有传统方法的比较图。
具体实施方式
以下结合附图及实验结果对本发明作进一步地详细说明:
本发明核酸结构中核苷酸与核苷酸相互作用预测的方法。具体包括以下步骤:
(1)核酸序列和结构数据的选取
该步骤中的核酸序列提取自核酸信息分类数据库(rfam),结构数据提取自核酸三级结构数据库(rna3dhub,version3.21,数据统计截止至2018年5月11日,具体信息见http://rna.bgsu.edu/rna3dhub/nrlist/release/3.21)。
本实例中的训练集为rna3dhub(version3.21)中长度为50nt到120nt,分辨率
(2)受限玻尔兹曼机学习核酸结构特征
该步骤首先将训练集结构数据中的核酸结构以
受限玻尔兹曼机的能量表达式为
e(v,h|w,b,c)=-btv-cth-htwv(1)
其中w是可见层单位v与隐藏层单位h之间的连接权重矩阵,h、b、c是作为补偿的偏置单元。v、h的概率给定如下:
z(w,b,c)=∑v,he-e(v,h|w,b,c)(3)
其中z(w,b,c)是对所有可能的v和h求和的配分函数。通过随机梯度下降(sgd)对经验数据的负对数似然进行受限玻尔兹曼机训练。l(w,c,b,t)定义为损失函数,希望它在sgd期间最小化
p(v|w,b,c)定义如下
p(v|w,b,c)=∑hp(v,h|w,b,c)(5)
t为样本数据。通过最小化损失函数,我们可以根据以下公式更新参数:w,b,c
(3)直接耦合分析模型预测序列共同进化:
本实例中的测试集为6个核糖开关分子,pdbid分别为1y26、2gdi、2gis、3irw、3owi和3vrs。首先通过核酸信息分类数据库(rfam)提取测试集中6个待测核糖开关分子相应的多序列比对信息。然后,删除多序列比对中插空大于50%的序列,并计算单个核苷酸和一对核苷酸的核苷酸频率。
其中两个位点之间相互作用强度的直接耦合定义为
借助一个孤立双位点模型
(4)受限玻尔兹曼机与直接耦合分析模型相结合:
核苷酸与核苷酸相互作用预测结果综合了步骤2中受限玻尔兹曼机学习的结构特征和步骤3中直接耦合分析学习的序列共进化特征
direct=di×w2(11)
其中,direct为核苷酸与核苷酸相互作用的预测结果,di为步骤3中通过直接耦合分析得到的序列共进化信息,w为步骤2中通过受限玻尔兹曼机学习得到的结构特征。
为了评估核酸结构中核苷酸与核苷酸相互作用预测的精度,本发明采用标准的预测精度计算方法对测试集进行了统计分析,并与传统的基于平均场模型直接耦合分析(mfdca)和最大似然估计模型直接耦合分析(plmdca)方法进行了比较。具体的预测精度(ppv)的计算方法为
其中tp为真阳性的预测相互作用数目,表示预测正确的核苷酸与核苷酸相互作用;fp为假阳性的预测相互作用数目,表示预测错误的核苷酸与核苷酸相互作用。
表2为本发明方法与传统的基于平均场模型直接耦合分析(mfdca)在测试集中的预测精度比较分析结果。在排名前100的相互作用中,本发明方法对核糖开关1y26预测正确的相互作用数为34,预测精度(ppv)为0.34,传统的基于平均场模型直接耦合分析(mfdca)方法预测正确的相互作用数为30,预测精度(ppv)为0.3,本发明方法的预测精度提高了13%。对于另外5个核糖开关(2gdi,2gis,3irw,3owi和3vrs),在前100相互作用中,本发明方法预测正确的相互作用数分别为45,38,33,24,17;传统的基于平均场模型直接耦合分析(mfdca)方法预测正确的相互作用数分别为34,33,33,20,16,本发明方法的预测精度分别提高了32%,15%,0,20%,6%。因此,本发明方法与传统的基于平均场模型直接耦合分析(mfdca)相比,平均准确率提高了15%。
表3为本发明方法与最大似然估计模型直接耦合分析(plmdca)在测试集中的预测精度比较分析结果。在排名前100的相互作用中,本发明方法对测试集中的核糖开关(1y26,2gdi,2gis,3irw,3owi和3vrs)预测正确的相互作用数分别为29,46,32,37,27,21;最大似然估计模型直接耦合分析(plmdca)方法预测正确的相互作用数分别为29,46,33,35,20,24。本发明方法与最大似然估计模型直接耦合分析(plmdca)相比,平均准确率提高了4%。
本发明提供的核酸结构中核苷酸与核苷酸相互作用预测的方法可以有效预测核酸结构中的长程空间结构相互作用,帮助精确预测和搭建核酸分子的空间三级结构,对理解核酸分子的折叠机制与生物学功能有重要的帮助。
表格1.实例中的训练集rna结构信息
表2.测试集中6个核糖开关在本发明方法和传统mfdca中的预测精度
表3.测试集中6个核糖开关在本发明方法和传统plmdca中的预测精度
- 还没有人留言评论。精彩留言会获得点赞!