基于分子动力学测试电场作用下离子扩散系数的方法与流程
本发明属于微观纳米材料技术领域,尤其涉及基于分子动力学测试电场作用下离子扩散系数的方法。
背景技术:
近年来,我国一些混凝土结构耐久性设计规范中,推荐使用非稳态氯离子快速迁移试验方法——rcm法,测定混凝土的氯离子扩散系数。氯离子是造成混凝土中钢筋锈蚀的主要原因之一,其渗透扩散性是反映混凝土抵抗氯离子侵入和钢筋抗腐蚀能力的一个重要参数,一般用氯离子扩散系数dt表示,目前其主要确定方法有自然扩散法和电迁移试验法,但两种方法各有不足。
自然扩散法原理简单,容易令人接受,且表观扩散系数比较接近实际情况,可信度高,经修正后可用于实际寿命预测,其缺点为:在于试验周期漫长,要进行切片、研粉、浸取、电化学滴定、数学拟合等多个步骤,试验过程繁琐。电迁移试验,主要有astmc1202法、csiro改进法、acmt法、rcm法和nel法,rcm法可以得到氯离子扩散系数,可以用于混凝土结构耐久寿命预测,但缺点为:需人工读取氯离子渗透深度,试验结果受人为因素影响较大;此外,其阴极溶液nacl浓度较高,最高达10%,这不仅增加试验材料和费用,试验后的溶液也会造成污染。
此外,电量法可以得到6h电量值,电流由仪器测出,准确、快速,能定性评价混凝土的抗氯离子性能;但不能得到氯离子扩散系数,不能用于混凝土寿命预测,常规的浓度加速试验由于试验周期长,速度慢,工作比较繁琐,一般情况下要在3个月才能得到一个测试结果。
技术实现要素:
根据以上现有技术的不足,本发明提供了基于分子动力学测试电场作用下离子扩散系数的方法,其能有效解决现有技术中的在氯离子扩散系数预测中耗时时间久、操作繁琐、准确性不高或易污染环境等问题。
本发明解决的技术问题采用的技术方案为:
基于分子动力学测试电场作用下离子扩散系数的方法,包括:
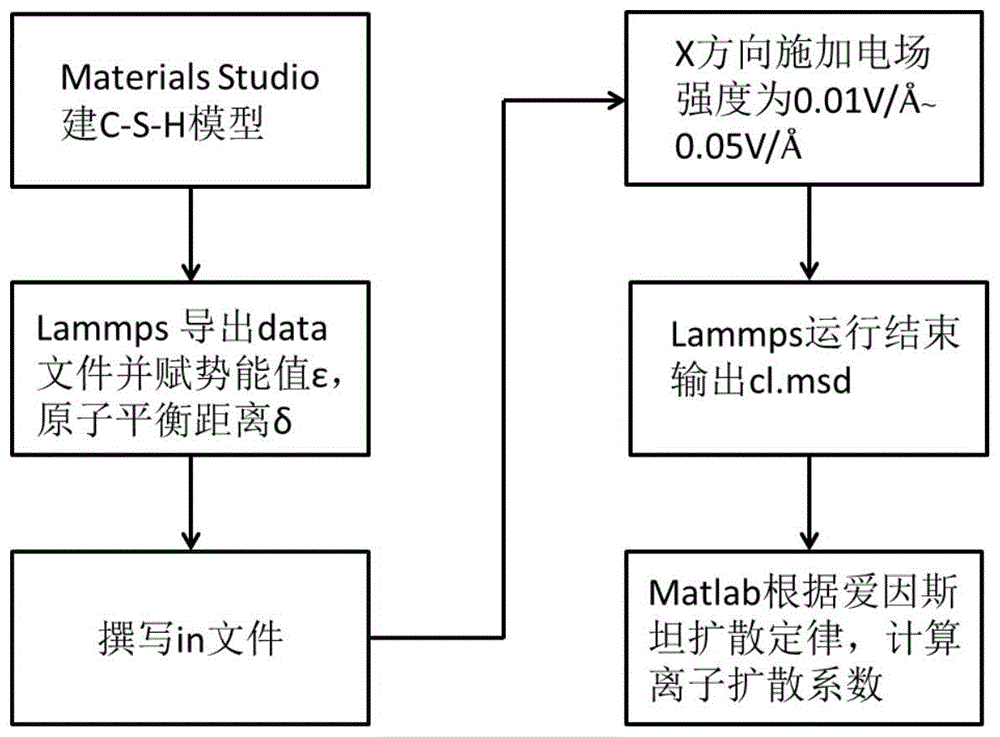
步骤1、用分子动力学仿真软件materialsstudio构建c-s-h模型,也就是水化硅酸钙模型;分别给氯离子、钠离子、钙离子、硅氧四面体中的硅原子以及氧原子赋电荷,保持电荷平衡,电荷平衡时为0,采用cvff力场能量最小化保证模型模拟时能量最低,materialsstudio最后导出car,cor文件,把导出的car,cor文件复制到lammps中。
基于对水化系统中金属-氧原子交互作用中离子键或共价键类型成分的描述,适用于水化晶体复合物及其固/液界面处的分子模拟,已成功应用在氧化物、氢氧化物的结构模拟和层状结构材料的层间水和离子行为的模拟中。近年来,clayff力场被引入了水泥基体系中,对氢氧化钙、单硫型水化硫铝酸钙(afm)和托贝莫来石(tobermorite)的成功模拟表明其同样适用于钙硅体系,因此本专利将主要使用clayff力场模拟c-s-h凝胶的结构、传输等特性。
clayff力场中使用spc(simplepointcharge)模型来表示水分子,利用harmonic势场函数来分别表示o-h共价键的拉伸、h-o-h角度的弯曲作用:
ebondstretchij=k1(rij-r0)2(1),
eanglebendijk=k2(θijk-θ0)2(2),
k1,k2分别表示拉伸和扭转刚度,r0代表o-h键的平衡长度,θ0表示h-o-h的平衡角度,rij是为o和h原子间的分离距离,θijk是金属氧和氢的键角;
步骤2、基于大规模原子分子并行模拟器lammps导出data文件,期间采用clayff力场中规定的原子参数设置模型各原子键角,键长强度值;通过clayff力场中明确的参数分别赋给data文件中钠离子、氯离子、钙离子、硅氧四面体中的硅原子和氧原子不同的势能值ε,以及钠离子、氯离子、钙离子、硅氧四面体中的硅原子以及氧原子之间的平衡距离δ;
lj势函数用于估计模型中所有原子的大小及非键结作用势能的最低值,clayff力场使用lj势函数来表示钠离子、氯离子、钙离子、硅氧四面体中的硅原子和氧原子间的范德瓦尔斯力,模拟真实状态的模型中所有原子间作用,表达式为:
其中rij为i,j两个原子间距离,i,j为涉及模拟体系中发生阴阳离子之间的相互作用的钠离子、氯离子、钙离子或硅氧四面体中的氧原子,d0,ij、r0,ij是c-s-h模型与观测到的结构和物理特性数据拟合得到的经验参数,分别表示i,j两个原子之间的能量与距离常数;i,j为涉及模拟体系中发生阴阳离子之间相互作用的钠离子、氯离子、钙离子或硅氧四面体中的氧原子。
模拟过程中,离子之间存在库伦作用力,其静电库伦作用表达式为:
式中qi、qj表示模拟体系中相互作用的两种原子i,j各自的部分电荷;e是电子的电荷;rij为i,j原子之间的分离距离;i,j为涉及模拟体系中发生阴阳离子之间相互作用的钠离子、氯离子、钙离子或硅氧四面体中的氧原子;ε0为真空的介电常数,值为8.8542*10-12f/m;计算值ε和δ查阅参考文献clayff力场获得;
步骤3、撰写in文件,运行in文件包括四部分:
(1)c-s-h模型沿三维坐标x轴、y轴、z轴施加周期性边界;
(2)固定c-s-h模型最底层原子,让溶液离子跑1000ps达到均匀分布;
(3)解除c-s-h基底,让溶液和基底充分驰豫,给c-s-h模型中x轴,y轴,z轴方向分别赋初始力值大小为0,0,0;
(4)设定lammps模拟参数:在in文件中,采用nvt系综,时间步长1fs,温度设定300k让整个盒子驰豫达到平衡,保证氯化钠溶液在c-s-h模型中扩散均匀,设定电场大小,以及电场方向;
步骤4、给c-s-h模型在x轴方向或者y轴方向施加电场,继续跑2000ps,让离子在电场作用下运动,最后输出2000ps的原子轨迹进行matlab编程分析,即可计算不同电场大小下的均方位移大小,即msd的大小;
步骤5、lammps运行结束,输出均方位移文件cl.msd文件,均方位移文件为原子在电场作用下充分驰豫,平衡后输出的氯离子模拟数据;
步骤6、根据爱因斯坦的扩散定律通过matlab分析以及origin做图,得到离子的扩散系数dt。
进一步地,在步骤4中,施加的电场大小为
进一步地,在步骤6中,离子的扩散系数d(t)的具体计算为:
式中,n为模型的不同维数,此处选n=3为三维模型,d(t)为离子的扩散系数,ri(t)为时间t时c-s-h模型中原子的位置,ri(0)为初始时间原子的位置,<>表示平均值,t表示时间。
本发明具有以下有益效果:(1)本发明采用分子动力学模拟计算,通过materialsstudio建立模型,通过大规模原子分子并行模拟器lammps系统输出msd文件,达到快速计算离子在电场作用下的传输速度,通过图像计算出离子的msd,继而根据爱因斯坦方程计算得到离子在电场作用下的扩散系数,时间短、计算方便、效率高,避免了繁琐的计算过程,且不会产生因人为因素造成的误差,结果准确;(2)能够弥补试验当中无法观察到离子微观层面的离子之间的反应,能够更加形象的表观离子在电场作用下的趋势,节约成本,可操作性强,风险成本低;(3)计算机可以连续半年以上工作,56核cpu运算器,大大提高计算效率,能快速预测离子在不同电场大小作用下的扩散系数,为离子在微观实验中提供理论基础和依据。
附图说明
图1是本发明所述方法的流程框架图;
图2是本发明实施例中用materialsstudio建的c-s-h纳米孔道模型示意图;
图3是本发明实施例中用matlab以及origin分析3.4nmc-s-h纳米孔道中不同电场大小氯离子的均方位移图;
图4是本发明实施例中计算出电场作用下离子在3.4nm纳米孔道和5.6nm孔道中的离子扩散系数;
图中:1、硅氧四面体2、钙离子3、氯离子4、钠离子5、水分子。
具体实施方式
下面结合附图对本发明做进一步描述。
实施例一:
如图1~图4所示,本发明所述的基于分子动力学测试电场作用下离子扩散系数的方法,包括构建c-s-h纳米孔道模型,lammps模拟计算2000帧的原子轨迹,对其进行分析得到电场作用下离子的均方位移,利用matlab编程提取离子的均方位移,进一步根据爱因斯坦扩散定律通过matlab代码编程计算离子的扩散系数,以此实现电场作用下对c-s-h纳米孔道扩散系数的计算,本发明的实施可以按以下步骤进行:
第一步:首先构建c-s-h纳米孔道模型。用分子动力学仿真软件materialsstudio构建水化硅酸钙(c-s-h)模型,首先采用托贝莫来石(tobermorite
第二步:基于大规模原子分子并行模拟器(lammps)导出data文件,期间采用clayff力场中规定的原子参数设置模型各原子键角,键长强度值;通过clayff力场中明确的参数分别赋给原子势能值ε,原子之间的平衡距离δ;lennardjones(lj)势函数用于估计分子的大小及非键结作用势能的最低值,此外,clayff力场使用lennard-jones作用来表示原子间的范德瓦尔斯力,模拟真实状态的原子间作用。
第三步:给模型在x方向施加电场,计算不同电场大小下的均方位移大小;lammps运行结束,输出cl.msd文件,均方位移文件为原子在电场作用下充分驰豫,平衡后输出的2000帧氯离子模拟数据;模型结束之后,取模型最后一帧在电场作用下原子轨迹图,用materialsstudio还原该模型轨迹的最后一帧,得到相应的图像,然后根据钠离子、氯离子的分布状态解释电场作用下氯离子扩散较快,钠离子扩散较慢的传输机理并且通过还原的模型能够发现c-s-h界面
本发明采用分子动力学模拟计算,通过materialsstudio建立模型,通过大规模原子分子并行模拟器lammps系统输出msd文件,达到快速计算水和离子在电场作用下的传输速度,通过图像计算出离子的msd,继而根据爱因斯坦方程计算得到离子在电场作用下的扩散系数,时间短、计算方便、效率高,避免了繁琐的计算过程,且不会产生因人为因素造成的误差结果准确。
以上所述为本发明的实施例,并非因此限制本发明的专利范围,凡是利用本发明说明书以及附图内容所作的等效结构或等效流程变换,或直接或间接运用在其他相关的技术领域,均同理包括在本发明的专利保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!