一种姜黄素固体分散体的制备方法与流程
本发明涉及药物化学技术领域,特别涉及一种姜黄素固体分散体的制备方法。
背景技术:
固体分散技术是提高水不溶性或难溶性药物分散度、溶解度和生物利用度的有效途径之一。固体分散体制备时,应根据药物和载体性质,制剂目的等选择载体材料。固体分散体材料有水溶性、难溶性和肠溶性3大类。选择合适的载体材料及制备方法进行固体分散体的制备,是提高药物生物利用度最有效的途径。
姜黄素固体分散体能够增加姜黄素的水溶性,因为姜黄素在固体分散体中以无定型形态存在,能够增加溶解时的比表面积;同时载体材料能与姜黄素分子的酚羟基形成氢键,通过氢键使姜黄素分子分散于大分子中,由于载体材料的亲水性,利于增加其水中分散性或溶解性。采用有机溶剂法制备姜黄素固体分散体,能够提高姜黄素溶出度和溶解度,但存在溶剂残留的问题。
药物与载体相容是形成稳定固体分散体的前提。水为果胶的良溶剂,果胶除具备传统载体的良好水溶性和相容性外,自身还具有很多生理活性,通过果胶分子中的亲水基团对姜黄素起到增溶效应,以改善药物水中分散度或溶解度。目前固体分散体的制备方法多以单一方法为主。
技术实现要素:
本发明的目的在于提供一种姜黄素固体分散体的制备方法,本发明选择无机溶剂,将溶剂法和沉淀法相结合,利用分子自组装,在酸性条件下团簇、聚集,形成亮黄色沉淀,进行固体分散体的制备,避免了有机溶剂残留带来的用药毒副作用,解决了现有技术中采用有机溶剂法制备姜黄素固体分散体存在溶剂残留的问题存在溶剂残留的问题。
为实现上述目的,本发明提供如下技术方案:
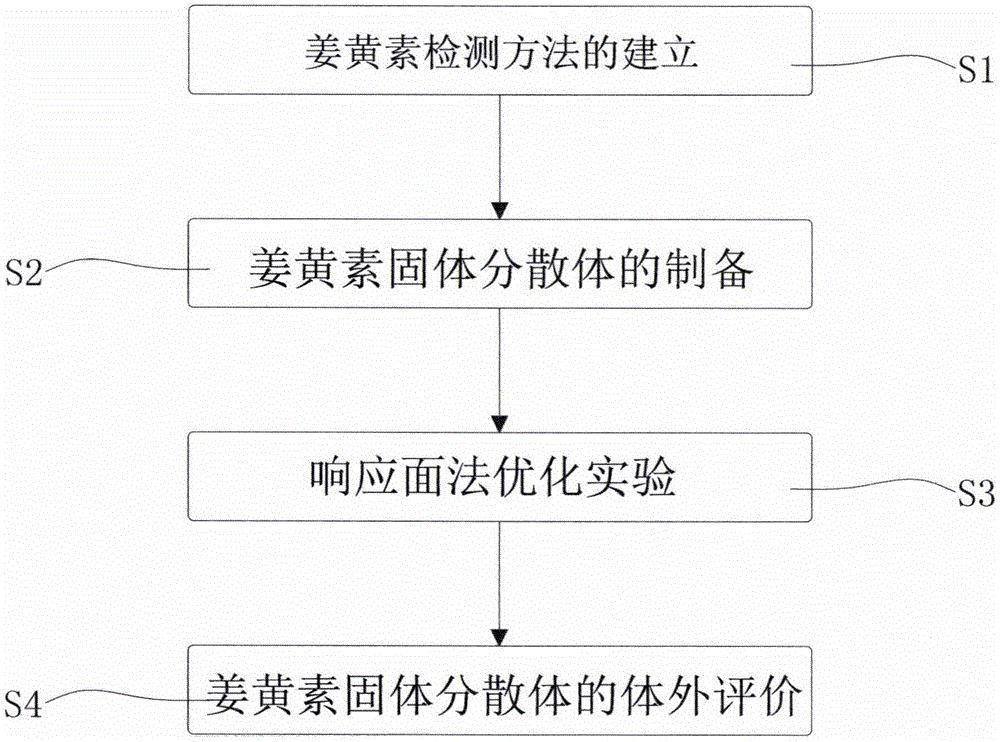
一种姜黄素固体分散体的制备方法,包括如下步骤:
s1:姜黄素检测方法的建立
s2:姜黄素固体分散体的制备
实验将溶剂法和沉淀法相结合,利用分子自组装,进行固体分散体的制备;
s3:响应面法优化实验
在单因素实验基础上,根据box-behnken实验设计原理,采用design-expert8.0软件,设计4因素3水平共29组实验进行响应面实验;
s4:姜黄素固体分散体的体外评价。
进一步地,s1还包括如下步骤:
s101:载药量和包封率的测定;
s102:稳定性实验
准确称取定量的姜黄素,分别溶于浓度为0.1,0.5,1.0和5.0%的naoh溶液中,于相同时间点取样测其吸光度;
s103:标准曲线的绘制;
s104:精密度实验
分别配制低、中、高三种浓度的姜黄素碱溶液,在3小时内不同时间点取样测其吸光度,每个样平行测定三次。
进一步地,s4还包括如下步骤:
s401:热特性分析(dsc)
精确称取在干燥器中平衡24h的固体分散体样品3~5mg,封于带孔铝制锅内;未装样品的铝制锅作为参比;
s402:x-射线衍射特征分析(xrd)
将固体分散体直接压实在样品槽内,采用cu-kα射线(λ=0.154nm)下进行xrd测试;
s403:扫描电镜分析(sem)
先将导电胶粘贴于铜制样品台上,再将干燥固体分散体用吸耳球吹散分布于导电胶上,喷金台上喷金5min后,置于样品室进行×1000倍微观形态的观察;
s404:体外释药实验。
进一步地,s103还包括如下步骤:
s1031:检测波长的确定;
s1032:标准曲线的建立;
进一步地,s404还包括如下步骤:
s4041:模拟体液的配制:模拟体液使用不同ph的磷酸盐缓冲液(pbs)进行配制;
s4042:检测波长的确定:以空白固体分散体的pbs缓冲液和乙醇为参比,在300~700nm波长范围内扫描,确定最大吸收波长λmax均为427nm;
s4043:标准曲线的建立:采用步骤s1032的建立步骤,溶剂为75%乙醇,得到标准曲线方程;
s4044:姜黄素累积释放度的测定。
与现有技术相比,本发明的有益效果是:本发明提出的姜黄素固体分散体的制备方法,选择无机溶剂,将溶剂法和沉淀法相结合,利用分子自组装,在酸性条件下团簇、聚集,形成亮黄色沉淀,进行固体分散体的制备,避免了有机溶剂残留带来的用药毒副作用,解决了现有技术中采用有机溶剂法制备姜黄素固体分散体存在溶剂残留的问题存在溶剂残留的问题。
附图说明
图1为本发明的制备流程图;
图2为本发明以不含见黄素的固体分散体作为空白对照波长范围内扫描曲线图;
图3为本发明以空白固体分散体的pbs缓冲液和乙醇为参比波长范围内扫描图;
图4为本发明果胶和姜黄素配比对包封率和载药率影响示意图;
图5为本发明cacl2浓度对固体分散体的影响为显著性影响示意图;
图6为本发明反应时间对固体分散体的影响示意图;
图7为本发明包封率和载药量随溶剂浓度的变化情况示意图;
图8为本发明的反应温度对固体分散体的影响示意图;
图9为本发明姜黄素固体分散体的热性能研究差示扫描量热法示意图;
图10为本发明x射线粉末衍射示意图;
图11为本发明不同加速电压下对姜黄素固体分散体颗粒的形貌扫描电镜示意图;
图12为本发明的em元素分析结果示意图;
图13为本发明药物累积释放度、绘制其与时间之间的关系曲线图;
图14为本发明姜黄素分散体在模拟胃肠液中的释放行为图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
参阅图1,一种姜黄素固体分散体的制备方法,包括如下步骤:
步骤3:姜黄素检测方法的建立
3.1载药量和包封率的测定
re=maq/mtq×100%(1)
l=maq/mwp×100%(2)
式中:re为包封率(%);l为载药量(%);maq为载入果胶的药物质量(mg);mtq为投入药物的质量(mg);mwp为固体分散体总质量(mg);
3.2稳定性实验
准确称取定量的姜黄素,分别溶于浓度为0.1,0.5,1.0和5.0%的naoh溶液中,于相同时间点取样测其吸光度,根据吸光度变化曲线,最终确定变化幅度最小、稳定性较高的0.5%naoh溶液进行分散体中姜黄素含量的测定;
3.3标准曲线的绘制;
(1)检测波长的确定:以不含见黄素的固体分散体作为空白对照,在300~700nm波长范围内扫描(如图2),确定最大吸收波长λmax为469nm。
(2)标准曲线的建立:精密称取5.0mg姜黄素标准品,置于100ml棕色容量瓶中,加0.5%naoh溶液溶解并定容至刻度线。精密移取0.5,1.0,1.5,2.0,2.5ml标准品溶液置于25ml棕色容量瓶中,加碱液定容至刻度线,摇匀,于469nm处测其吸光度。以浓度c(μg/ml)为横坐标,吸光度a为纵坐标,绘制标准曲线,并进行线性回归。得到标准曲线方程为:a=0.1424c+0.0016,其中r=0.9999。结果表明,姜黄素在1.0~5.0μg/ml浓度范围内存在良好的线性关系。
3.4精密度实验
由于姜黄素在碱液中以不稳定的烯醇式存在,为了确保测定值的准确性,分别配制低、中、高三种浓度的姜黄素碱溶液(分别为2.0μg/ml,5.0μg/ml和20.0μg/ml),在3小时内不同时间点(间隔30min)取样测其吸光度,每个样平行测定三次,在测定时间内,精密度的rsd分别为3.41%,3.39%和3.31%。由于姜黄素在碱液中的吸光度会随时间降低,为了尽可能的减小测定误差,实验操作尽可能在半小时内完成吸光度的测定。
步骤2:姜黄素固体分散体的制备
实验将溶剂法和沉淀法相结合,利用分子自组装,在酸性条件下团簇、聚集,形成亮黄色沉淀,进行固体分散体的制备,实验以姜黄素和果胶配比、cacl2浓度、温度、时间和溶剂浓度为考察因素,以包封率和载药量为指标;
步骤3:响应面法优化实验
在单因素实验基础上,根据box-behnken实验设计原理,实验选取cacl2浓度(x1)、反应时间(x2)、溶剂浓度(x3)和反应温度(x4)为独立自变量,以载药量(y1)和包封率(y2)为响应值,采用design-expert8.0软件,设计4因素3水平共29组实验进行响应面实验。
步骤4:姜黄素固体分散体的体外评价;
4.1热特性分析(dsc)
参照chuaham等的方法测定。精确称取在干燥器中平衡24h的固体分散体样品3~5mg,封于带孔铝制锅内;未装样品的铝制锅作为参比。设置n2流速为150μml/min,温度范围0~300℃,加热速率为10℃/min。
4.2x-射线衍射特征分析(xrd)
将固体分散体直接压实在样品槽内,采用cu-kα射线(λ=0.154nm)下进行xrd测试。扫扫描速度2θ/min,衍射角范围5~80°,采用连续扫描方式。
4.3扫描电镜分析(sem)
先将导电胶粘贴于铜制样品台上,再将干燥固体分散体用吸耳球吹散分布于导电胶上,喷金台上喷金5min后,置于样品室进行×1000倍微观形态的观察。
4.4体外释药实验
(1)模拟体液的配制:模拟体液使用不同ph的磷酸盐缓冲液(phosphatebuffersaline,pbs)进行配制。
(2)检测波长的确定:以空白固体分散体的pbs缓冲液和乙醇为参比,在300~700nm波长范围内扫描(如图3),确定最大吸收波长λmax均为427nm。
(3)标准曲线的建立:方法同3.3(2),溶剂为75%乙醇。得到标准曲线方程为:a=0.1424c-0.0018,其中r=0.9999。结果表明,姜黄素在1.0~5.0μg/ml浓度范围内存在良好的线性关系。
(4)姜黄素累积释放度的测定
称取固体分散体10.0mg,分别置于装有30ml释放介质的棕色释药瓶中,置于温度为37℃、转速为100r/min的恒温水浴振荡器中,按照一定的释药时间间隔,取样5ml,同时补加等温、等体积相应ph的模拟体液,以维持介质体积的恒定,通过分光光度法,以对应空白固体分散体为参比,427nm处测定释放介质中姜黄素含量,按(3)式计算累积释放度,绘制不同释放介质中药物累积释放度和时间之间的关系曲线图:
式中:re%是累积释放度;cn为第n次取样后释放介质中药物浓度;v0为释放介质的体积;vi为每次取样的体积;ci为第i次取样置换时释放介质中药物浓度;m为试样中药物含量。
步骤5:进行结果与分析
5.1果胶和姜黄素配比对固体分散体的影响
果胶和姜黄素配比对包封率(0.01<p<0.05)和载药率(p>0.05)影响的显著性不同。如图4所示,随料药配比的增大,包封率和载药量均呈现先增加后减小的变化趋势;由于果胶分子自身结构的复杂性,且具有较高分子量,实验设定的加量范围内,包封率和载药量的变化幅度较小。由于果胶为聚阴酸性多糖,其载药机制包括物理包覆和静电吸附两种。在碱液中与naoh发生化学反应,甚至分子内部发生断链,反应结束经酸化处理后,低浓度果胶分子中的羧酸根离子浓度降低,致使与姜黄素分子间的吸附作用降低;且形成的果胶-钙有效交联网络结构区较少,降低了果胶对姜黄素的包覆作用。随果胶浓度的增加,有效的果胶-钙三维交联网络结构增多,药物与载体之间的吸附作用增强,促使载药量和包封率提高;当料药配比为3.0∶1时,与ca2+之间形成稳定的网络交联结构,且果胶分子中羧基的质子化效应最强,形成强的分子间氢键作用力,表现为包封率和载药量分别达到最大值42.70%和6.70%。随着果胶浓度的进一步增加,分子间斥力作用增强,未与钙离子交联的果胶分子增多,引起载药量和包封率的下降。
5.2cacl2浓度对固体分散体的影响
cacl2浓度对固体分散体的影响为显著性影响(0.01<p<0.05)。如图5所示,随cacl2浓度增加,包封率和载药量均呈现先增加后减小的变化趋势。当ca2+浓度较低时,形成的果胶-钙交联网络结构较少;当ca2+浓度达到一定浓度时,能够形成稳定的果胶-钙桥联“蛋-盒”结构区,致使包封率和载药量达到最大值;当ca2+浓度进一步增加,稳定的“蛋-盒”网络结构被破坏,形成更为致密的不稳定三维网络交联结构,包封率和载药量随之下降。
5.3反应时间对固体分散体的影响
反应时间对固体分散体的影响具有极显著性(p<0.01)。如图6所示,前两个小时内,二者几乎保持不变,包封率和载药量分别达到65%和10%以上;随时间继续增加,包封率和载药量均呈现下降趋势,且下降趋势明显。由于搅拌时间较长,可能造成果胶分子大幅度断链,形成有效的果胶-钙交联网络结构区减少;且果胶分子与碱液发生化学反应,导致羧基的质子化作用降低,分子链之间的静电斥力增强,表现为包封率和载药量的下降。
5.4溶剂浓度对固体分散体的影响
溶剂浓度对固体分散体的影响具有极显著性(p<0.01)。包封率和载药量随溶剂浓度的变化情况如图7所示,随溶剂浓度增大,二者均呈现先增大后减小的变化趋势;当溶剂浓度为0.60%时,包封率和载药量分别达到最大值66.48%和10.32%。naoh浓度能够对果胶分子的结构和链段大小造成很大影响[179],碱液浓度较低时,果胶分子链多以聚集状态存在,形成“蛋-盒”桥联结构较少,造成包封率和载药量较低。当碱液浓度增加至大于0.60%时,可能造成果胶分子链发生断裂,长链变成短链,甚至影响到分子结构的变化,致使形成较为松散的三维网络结构;同时,较高的碱液浓度会使更多na+与果胶分子中的-cooh-通过静电吸引力而聚集,从而降低了果胶分子对药物的吸附作用,以上因素共同作用,导致包封率和载药量降低。
5.5反应温度对固体分散体的影响
反应温度对固体分散体的影响具有极显著性(p<0.01)。如图8,当温度为50℃时,包封率和载药量均为最大值;温度小于50℃时,随温度升高,包封率和载药量均呈增加趋势,包封率增幅较载药量小;温度大于50℃时,随温度升高,二者均呈现降低趋势,包封率降幅明显。果胶分子链的分散状态主要受温度影响,当温度较低时,果胶分子以聚集、蜷缩状态存在,形成的有效“蛋-盒”桥联区较少,造成包封率和载药量不高;当温度较高时,果胶分子链以舒展态存在,由于搅拌和溶剂的共同作用,致使分子链断链几率增加,分子量降低,果胶的紧密结合区域进一步破坏,固体分散体的包封率和载药量下降。
5.5参数优化
实验设定的cacl2浓度、反应时间、溶剂浓度和反应温度四个因素对载药量和包封率2个响应指标有不同的影响,需综合考虑每个因素对二指标的影响,才能达到载药量和包封率俱佳的制备效果。通过designexpert8.0数据分析软件对2个响应指标进行二次回归优化求解,得到各指标的最优参数为:cacl2浓度3.66g/l,时间1.62h,溶剂浓度0.69%,温度49.04℃。预测该条件下,姜黄素的包封率和载药量分别为69.50%和11.48%。为进一步验证此模型的可信度,同时结合实际的可控性操作,对优化参数进行修正:cacl2浓度3.7g/l,时间100min,溶剂浓度0.69%,温度50℃。在此条件下,通过三次平行实验,制备的固体分散体的包封率和载药量分别为67.17%和11.40%。实验值和预测值的结果表明,该模型的可信度良好,优化参数可用于果胶多糖制备姜黄素固体分散体。
5.7dsc分析
为了研究姜黄素固体分散体的热性能,对其进行了差示扫描量热法(dsc)分析。图9显示了姜黄素对照品在183℃时出现一个单一的强吸热峰(熔化峰),而其分散体的热图模式发生较大变化,姜黄素的熔化峰在分散体微粒体系中完全消失,只能发现75℃和138℃两个弱的无定型吸热峰。上述结果表明,姜黄素与载体分子之间相互作用形成热共存单元;姜黄素不是以晶体方式存在于固体分散体中,而是以高能无定形状态或分子状态分散于载体中。
5.8xrd分析
固体分散体中的药物一般以分子形式存在,当药物以分子形式装载于载体上时,可使具有一定晶型的药物变为无定型。xrd结果进一步证实了姜黄素在分散体中的固态性质。x射线粉末衍射图10显示,晶态姜黄素对照品在2θ为10~50°范围之间存在强的晶体衍射峰;果胶为无定型聚合物,其衍射图谱不具有明显的晶体衍射峰。在分散体的衍射图中,姜黄素晶型衍射峰消失,据此推测共沉淀物中姜黄素的分散状态可能受果胶聚合物理化性质影响,在制备分散体过程中形成无定型共沉淀物后,主药以无定型或分子形式分散于无定型的果胶聚合物中。这说明了固体分散体中的姜黄素是以非晶态形式存在,该结果与dsc结果相一致。
5.9扫描电镜分析
不同加速电压下对姜黄素固体分散体颗粒的形貌进行了×1000倍扫描电镜观察,如图11所示。姜黄素和果胶之间的强结合抑制了姜黄素组装成有序的晶体结构,从而导致非晶态的形成。因此,sem结果与上述xrd和dsc的结果相一致。sem元素分析结果(图12)证实了碱溶剂与盐酸发生中和反应形成了无机盐nacl,表明本实验方法可以有效避免有机溶剂残留给递药安全性带来的不良影响。
5.10体外释药实验
(1)不同ph介质中释放度的测定
将姜黄素固体分散体置于不同ph值的释放介质中,测定不同时间介质中姜黄素的浓度,计算药物累积释放度、绘制其与时间之间的关系曲线,如图13所示。随释放介质ph值的增大,相同时间内姜黄素的累积释放度增大;在整个释药过程中,未显示出突释行为。实验结果表明,以果胶为载体制备的姜黄素固体分散体具有ph敏感性。果胶为聚阴电解质,在低ph时,由于果胶分子中的羧基高度质子化,强烈的静电作用力的束缚,使得姜黄素不易释放,12h的累积释放度为20%左右;随释放介质ph的增大,果胶分子中羧基的质子化程度不断减弱,分子链逐渐以舒展态存在,且发生不同程度降解,载体对药物分子的静电作用力降低,使得姜黄素释放度增大。此外,在ph7.40的释放介质中,随释放时间的进一步延长,累积释放度也未能达到百分之百。这是因为果胶分子链上的羧基未完全电离,通过氢键对姜黄素的束缚作用未能完全消除,故不能完全释药。
(2)模拟胃肠液中释放度的测定
将姜黄素固体分散体置于胃液、小肠液和结肠液的模拟体液中,参照药物在胃肠液中一般作用时间,恒温震荡条件下使姜黄素分散体在各模拟体液中释药,计算累积释放度,图14为姜黄素分散体在模拟胃肠液中的释放行为。在模拟胃液2h的释药过程中,姜黄素累积释放度低于20%;在模拟小肠液3h时的释药过程中,继续部分的释药;进入结肠模拟液中,发生药物突释,7h的累积释放度几乎达到80%,显示出一定的结肠定位释放特征,其机理在于分散体中的果胶大分子对姜黄素分子的静电作用力对释放介质的ph具有敏感性。酸性条件下果胶分子强烈的静电作用,使分散体中姜黄素不易释放;随ph增大,药物骨架结构溶蚀,姜黄素释放量增大;到达结肠后,在果胶酶的作用下,果胶-钙交联网络瓦解、药物骨架进一步溶蚀,使药物大量溶出。
综上所述,本发明提出的姜黄素固体分散体的制备方法,本发明选择无机溶剂,将溶剂法和沉淀法相结合,利用分子自组装,在酸性条件下团簇、聚集,形成亮黄色沉淀,进行固体分散体的制备,避免了有机溶剂残留带来的用药毒副作用,解决了现有技术中采用有机溶剂法制备姜黄素固体分散体存在溶剂残留的问题存在溶剂残留的问题。
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明披露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!