一种具有抗肿瘤活性的前体药物及制备与应用的制作方法
本发明属于靶向药物设计和制备领域,特别涉及一种具有抗肿瘤活性的前体药物及制备与应用。
背景技术:
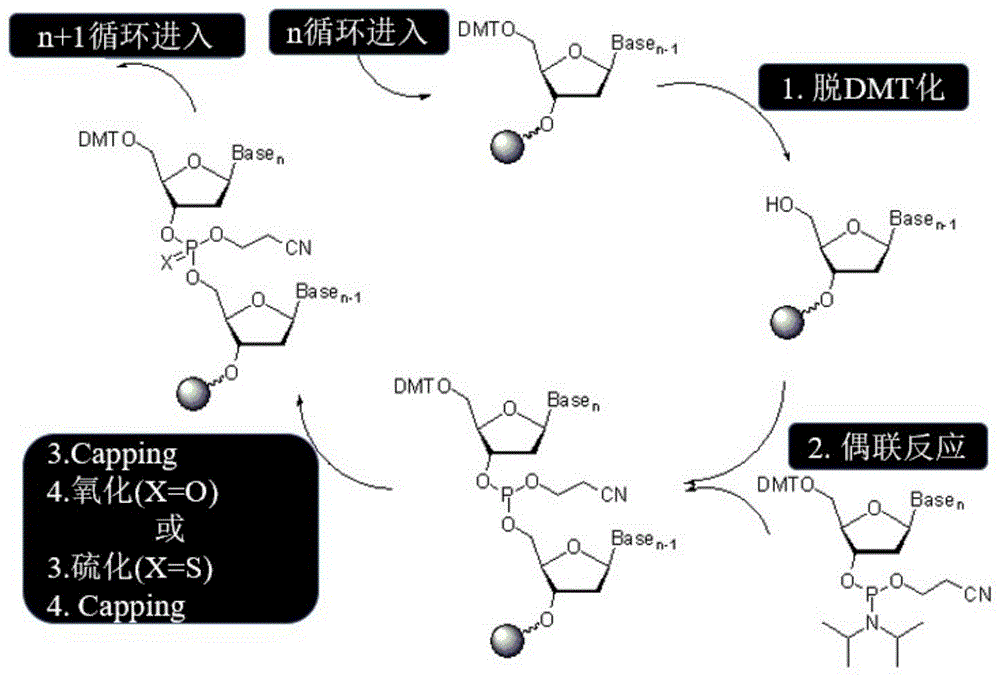
:癌症的化疗,除杀伤癌细胞/组织的作用外,其非靶向选择性会对非癌部位(骨髓造血细胞,毛囊,口腔、消化道、生殖系统的细胞等)的细胞产生毒副作用,使得化疗药物在临床上的最大耐受用药剂量非常有限,降低了化疗药物对癌症的真实治疗效果和药物经济。鉴此,寻找/设计安全、有效、低毒的抗肿瘤药物及其系统和研究其作用机理,具有重要意义。众所周知,现代癌症治疗中如何解决高效安全地将抗肿瘤药物靶向递送到肿瘤器官或组织,提高癌症治疗效率,同时降低药物的毒副作用,真正达到肿瘤靶向给药的目的,仍然是靶向给药系统的重点科学问题。靶向给药系统主要由载体及靶向基团组成;在基于靶向基团高亲和识别分子基础构建的主动靶向给药治疗系统中,主要还是基于靶向基团(dna/rna片段、单克隆抗体、多肽、糖类、脂蛋白、维生素叶酸/生物素类等)直接与药物或间接与载体(生物纳米硅、脂质体、蛋白质/多肽、糖类及plga/peg等高分子材料)携带药物模块进行修饰连接,进而达到靶向递送药物的目的。然而,现有载药系统仍然具有一些缺陷,如材料生物相容性不够好、表征不够精准、合成工艺复杂、工业纯化低效及后功能修饰等限制,严重影响了药物的靶向运输效率和临床应用。随着近年来对生命科学研究的深入,发现核酸作为生物材料,已展现了诸多优势,包括生物可降解性、生物相容性好、高稳定性、易修饰性和便于分子工程等等,且核酸碱基的生物安全性已经随着2004年美国fda对第一个核酸适体类药物macgen的审批而得到了广泛认可。核酸是由众多核苷酸通过3’,5’-磷酸二酯键聚合而成的多聚核苷酸;核苷酸由糖苷键连接的碱基(嘌呤/嘧啶)和戊糖(核糖和脱氧核糖)构成的核苷中的戊糖5’碳原子上羟基被磷酸酯化形成核苷酸。同时,在很低ph和或核酸酶作用下,dna和rna都会发生磷酸二酯键断裂,且碱基和核糖之间的糖苷键更易被水解;而肿瘤组织强代谢增殖、高氧化还原、缺氧和强酸等微环境,为核酸载体的降解及药物响应释放提供了高亲和特异性;已有相关研究证明核酸为药物载体具有很好的靶向治疗载体的前景,如本课题组前期构建的基于核酸适配体靶向传递策略构建阿霉素嵌入gc互补碱基间的纳米火车系统(procnatlacadsciusa.2013;110(20):7998-8003.)和人工合成5-fu碱基药物系统(jamchemsoc.2014;136(7):2731-4.)及zuyl课题组构建的阿霉素嵌入核酸载药系统(leukemia.2016;30(4):987-91.)等。这些现有研究基础为核酸骨架药物的设计提供了前期的分子理论支持。生物素(biotin,b)因其结构简单,分子量低,高肿瘤特异性及其生物素受体在肿瘤组织膜表面广泛高表达而被作为一种靶向配体广泛应用于各种抗癌药物的靶向递送运输。生物素又称维生素h、辅酶r,是水溶性维生素,也属于维生素b族,分子量为244.31,基本结构为双环结构:i环为咪唑酮环,是与亲和素结合的部位;ii为噻吩环,含一个戊酸侧链,其末端羧基可与生物大分子连接,形成生物素标记抗原、抗体、酶等。其化学结构中有咪唑酮环,可与亲和素(avidin,av)和链霉亲和素(streptavidin,sa)特异性结合。生物素与亲和素之间结合的亲和力高(结合常数约为1×10-15mol/l,且可视为不可逆结合)、特异性强,各自均可以与各型大小分子结合,一分子链霉亲合蛋白可与四分子生物素结合,二者“链霉亲和素-生物素”结合反应时具有多级放大作用等优越性,其相关技术被广泛应用在各种标记免疫分析技术和肿瘤靶向治疗药物领域中。同时,b的各类磷酸碱基单体的成功商业化,例如:biotin-dmt、biotin-teg-dmt、5’-biotin-dmt、5′-biotin-barbellspacerdmt等(结构式如下所示),为基于b构建癌组织靶向成像和靶向药物于癌症靶向精准治疗奠定了良好的分子基础。吉西他滨(gemcitabine,gem)是一种拮抗核苷酸代谢的核苷类抗肿瘤药物,同时,gem的亚磷酰胺单体已经商业化成熟(gem-dmt,结构式如下所示),该类药物体内经细胞内三磷酸化后,通过抑制脱氧核苷三磷酸(dntp)的合成、掺入dna或rna分子中干扰细胞复制、竞争性抑制dna聚合酶等作用,特异性干扰核酸的代谢,阻止细胞的分裂和繁殖,最终导致肿瘤细胞死亡。研究表明,吉西他滨其磷酸酯前药能够减少耐药发生(如进入临床研究的磷酸酯前药nuc-1031(slusarczykm,etal.jmedchem.2014;57(4):1531-42.和吉西他滨磷酸酯及氨基酰化前药wo2015/134334),但仍然在减少药物对非肿瘤组织的毒副作用方面不够理想。通过国内外文献和专利检索,尚未发现有将生物素以碱基单体或末端酰胺键形式共修饰在含吉西他滨药物分子的核酸骨架上,构建成吉西他滨前体药物系统实现癌症靶向给药方案的先例。技术实现要素:为了克服现有技术的缺点与不足,本发明的首要目的在于提供一种具有抗肿瘤活性的前体药物,该类化合物是通过3’,5’-磷酸二酯键将生物素和吉西他滨的亚磷酰胺单体构建在10~100个碱基核酸骨架上或通过酰胺键交联在含有吉西他滨药物的10~100个碱基核酸骨架上的前体药物,其中吉西他滨不少于3个,生物素不少于1个。本发明的另一目的在于提供上述具有抗肿瘤活性的前体药物的制备方法。本发明的再一目的在于提供上述具有抗肿瘤活性的前体药物的应用。本发明的目的通过下述技术方案实现:一种具有抗肿瘤活性的前体药物,是通过3’,5’-磷酸二酯键将生物素(b)的亚磷酰胺单体和吉西他滨(gem)的亚磷酰胺单体构建在10~100个碱基核酸骨架上的前体药物或是通过酰胺键将生物素交联在含有吉西他滨的10~100个碱基核酸骨架延伸出的带有nh2的多碳链上的前体药物,其中,吉西他滨在核酸序列的中间,生物素在吉西他滨的5’和/或3’临近两端或末端,吉西他滨不少于3个,生物素不少于1个;所述的具有抗肿瘤活性的前体药物具有如式ⅰ或ⅱ简化所示的结构:5’-bx(mix)agemn(mix)cby-3’式ⅰ;5’-(mix)abx(mix)bgemn(mix)cby(mix)d-3’式ⅱ;其中,gem为吉西他滨;b为生物素;mix为天然碱基和/或人造碱基类似物的任意组合;所述的式ⅰ中,x+y≧1;mix为天然碱基和/或人造碱基类似物的任意组合,其中,10≤a+c≤100;n≧3;所述的式ⅱ中,x+y≧1;mix为天然碱基和/或人造碱基类似物的任意组合,其中,10≤a+b+c+d≤100,且a和c≧1;n≧3;所述的碱基为a、t、c或g;所述的n优选为3~10;所述的具有抗肿瘤活性的前体药物的制备方法,包含如下步骤:将亚磷酰胺碱基单体(a/t/c/g/gem/biotin)按药物系统的设计要求和dna合成仪标准操作流程进行合成,然后去dmt、脱盐沉淀和hplc分离纯化、干燥,得到具有抗肿瘤活性的前体药物,其具有如式ⅰ/ⅱ所示的结构;或将亚磷酰胺碱基单体(a/t/c/g/gem/biotin/nh2-c链)按药物系统的设计要求和dna合成仪标准操作流程进行合成,然后去dmt、脱盐沉淀和hplc分离纯化、干燥,得到含有吉西他滨、且序列末端延伸出带有nh2的多碳链的gem核酸序列,对该序列末端进行biotin的保护连接,得到具有抗肿瘤活性的前体药物;所述的干燥优选为冷冻干燥;所述的具有抗肿瘤活性的前体药物在制备治疗癌症药物中的应用;所述的治疗癌症药物中含有治疗有效量的具有抗肿瘤活性的前体药物或其药学上可接受的盐及药学上可接受的载体;本发明的原理:基于生物素受体介导肿瘤细胞亲和靶向递送原理,本发明以核酸骨架为药物载体,生物素/吉西他滨杂合核酸序列解决吉西他滨药物的肿瘤靶向性和肿瘤细胞核酸转运载体表达量降低导致产生的耐药性及半衰期短(吉西他滨的半衰期<75min,临床上需持续的静脉给药来维持其抗癌细胞的毒性作用)等问题,同时生物素和核酸具有很好的溶解性、生物相容性和无免疫源性等优点,可以获得具有肿瘤靶向作用的吉西他滨前体药物,提高吉西他滨的药效,增加肿瘤细胞选择性,降低毒副作用,更适合抗癌临床使用。本发明在设计和实验发明中发现通过核酸骨架构建靶向分子生物素-吉西他滨前体药物具有更好的溶解性,显著增加其肿瘤细胞选择性,提高了抗癌活性和降低了核苷转运载体表达的依赖性,大大降低了原型药物的毒性。本发明提供的前体药物具有肿瘤靶向选择性和更优的抗癌活性及核酸转运载体的非依赖性。经过mtt法测试细胞毒表明,本发明提供的前体药物对具有生物素受体阳性表达的肿瘤细胞(宫颈癌hela、胰腺癌bxpc-3、乳腺癌mda-mb-231和肝癌hepg2等)体外生长抑制作用ic50比原型药物吉西他滨显著提高5~7倍。对正常肝脏细胞lo2毒性数据表明提供的前体药物比原型药物对非癌细胞具有大于3倍的ic50值,提示该类前体药物具有更低的毒性和更好的肿瘤靶向性;且核酸转运载体竞争实验表明,本发明提供的前体药物体外细胞转运作用几乎不受核酸转运载体抑制剂的影响。本发明所涉及的前体药物具有更优的抗肿瘤治疗效果。以接种乳腺癌mda-mb-231细胞的裸鼠为评价模型结果表明,与吉西他滨原形药相比,该类前体药物具有更强的体内抗肿瘤疗效。在癌细胞靶向分子生物素的介导驱动下,本发明提供的前体药物在机体内可以很快进入生物素受体高表达的肿瘤细胞,改善吉西他滨药物的抗癌生物活性,提高其细胞膜穿透性和癌组织穿透性,降低毒副作用。同时,本发明策略对吉西他滨类似结构组成的核苷药物,如扎西他滨、去氧尿苷类、阿糖腺苷、碘苷、拉米夫定、三氟胸苷和西多福韦等均可获得靶向和减毒增效的效果。与现有技术相比,本发明具有以下优点及有益效果:通过体外mtt检测实验,发现本发明制得的具有抗肿瘤活性的前体药物对肿瘤细胞株(宫颈癌hela、胰腺癌bxpc-3、乳腺癌mda-mb-231和肝癌hepg2细胞等)具有更好的抑制效果,比吉西他滨药物强5~7倍,但对正常肝细胞lo2的毒性低3倍以上。该生物素偶联的吉西他滨前体药物具有生物素受体介导靶向肿瘤的特性,可克服吉西他滨类抗癌药物选择性低、核苷转运载体蛋白表达依赖性强、被脱氧胞苷脱氨酶的快速代谢、半衰期短和易多耐药性的问题,通过靶向拮抗肿瘤细胞的代谢等途径抑制癌细胞增殖达到清除癌细胞的效果。附图说明图1是dna的合成示意图。图2是b-gem系列前体药物系统的抗肿瘤药效学评价结果分析图。具体实施方式下面结合实施例及附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。实施例1dna合成仪的操作、脱保护和浓度测定流程1)dna合成仪的操作:以亚磷酰胺寡核苷酸合成为例介绍polygen生产的dna合成仪器的操作步骤(图1):(1)准备合成试剂:保护碱基的5’-dmt,a、g、c、t亚磷酰胺单体,四唑偶联催化剂,乙酐,n-甲基咪唑封闭试剂,三氯乙酸(tca)脱保护溶液,氧化混合物,乙腈清洗溶剂,氨水切除溶液等。(2)仔细检查合成仪上所有试剂瓶中的试剂量,必要时换掉试剂瓶,特别注意无水乙腈的量,因为该溶剂用量较大。(3)将标记好的干净收集瓶装在合成仪上。(4)将要合成的dna序列输入合成仪控制电脑程序里面。(5)对照目标序列,检查选择的合成步骤是否正确。(6)选择结束方法:trityloffauto是仪器的原始状态,若打算以5,-dmt基团连接纯化寡核苷酸,将其转变为tritylonauto结束状态。(7)选择结束方法,一般为系统的原始状态。(8)在每个柱监视器的username下,输入你所希望的保存名字。(9)以代码代替相应的dna序列标记每一个收集瓶。(10)打印出每一个柱设置的详细资料。(11)检查打印出来的资料是否正确。(12)运行startcolumn步骤检查每一个柱的位置,确保合成仪上合成柱处于正确的位置。(13)检查连接在合成仪上的溶剂残留(废液瓶)是否充满。(14)如果分部收集器用来收集每个dmt脱保护步骤的流出物,确保试管充分清洁干净,并打开分部收集器,确保dmt废液管路通向分部收集器。(15)启动循环,运行开始程序清洗试剂管路,除去原来的试剂。2)dna的脱保护处理:a、将合成的dna小柱进行干燥;b、打开填充柱和转移柱颗粒到1.5ml的无菌离心管中;c、参照dna合成仪厂家推荐的方法对cpg和其修饰(染料、氨基、羧基、巯基等)进行处理;通常操作为加入1~3ml浓氨水和40%甲胺(50/50),按200nmol左右产物加入1ml,5μmol左右产物加入3ml;接着在65℃水浴15~30min;若为羧基修饰产物,即加入2~3ml甲醇/0.4mnaoh(4/1),400mgnaoh溶解于20ml甲醇和5ml的dna专用水中;然后室温孵化过夜;若为cy3、cy5或tamracpg修饰,加入2~3ml的tamra脱保护液(甲醇:叔丁胺:水=1:1:2),接着在65℃水浴3~4小时;或加入2~3ml氨水,室温孵化过夜17小时;d、待脱保护液冷却后,转移上清至1.5ml无菌离心管中;3)dna的沉淀处理:a、加入1/10体积的3mnacl(~250μl);b、加入2倍体积50%的冰乙醇(~6ml);c、充分涡旋后,冰冻30min至过夜,即获得dna沉淀;4)dna的hplc分离和脱盐处理:a、4℃条件下,4,000rpm离心15~30min;b、弃上清,加入200~600μl0.1mteaa三乙胺溶解,其中约200nmole时加入200μl0.1mteaa;当约5μmol时加入600μl0.1mteaa;c、4,000rpm离心1min,转移至无菌ep离心管中,14,000rpm离心1min;d、然后按不同的序列要求分别调整长时间或短时间的梯度洗脱序列,如20个碱基序列,可以用10~85%的梯度洗脱程序;若为40~50个碱基,则选用10~60%梯度洗脱;若为80个碱基,可以选择10~40%或10~50%的梯度程序;e、hplc分离完成后,真空干燥样品,然后用80%冰醋酸200μl切除dmt,然后用移液器溶解样品,室温放置20min;然后再沉淀hplc分离获得的目标dna,即加入1/10体积的3mnacl(~20μl),加入2.5倍体积冰乙醇(~500μl),涡旋,冰冻10~20min,dna即可沉淀析出;然后14,000rpm离心5min,真空干燥充分;若有必要可在此进行脱盐。5)dna的浓度检测:a、将dna产物溶解于合适体积的dna水溶液中,同时从网站http://www.idtdna.com/analyzer/applications/oligoanalyzer/获得目标基因序列及相关修饰的吸光系数;b、将溶解好的dna溶液吸在200μl光谱测定池中进行测定,如取1μldna稀释在199μl的dna水中;c、读吸光值和采用吸光系数按a=εbc进行计算,记住乘以稀释倍数;若采用200μl检测池,b=0.2cm;若采用中心检测池,检测体积为70~1000μl,b=1.0cm。实施例2具有抗肿瘤活性的前体药物(b-3gem-1)的制备将亚磷酰胺碱基单体(a/t/c/g/gem(z)/biotin(b))分别置入核酸合成仪相应的无水试剂瓶中,按dna合成仪的标准流程对设计序列5′-b-cttzzzccggcg-3′进行自动化合成,待合成完毕后,依次按标准流程进行dmt切割、脱盐沉淀和hplc分离纯化,纯度大于95%,产率不低于75%,经质谱鉴定,分子量为4102.1,与计算分子量4101.4一致,即得5′-b-cttzzzccggcg-3′。实施例3具有抗肿瘤活性的前体药物(b-3gem-2)的制备将亚磷酰胺碱基单体(a/t/c/g/gem(z)/biotin(b))分别置入核酸合成仪相应的无水试剂瓶中,按dna合成仪的标准流程对设计序列5′-b-tttzczczggcc-3′进行自动化合成,待合成完毕后,依次按标准流程进行dmt切割、脱盐沉淀和hplc分离纯化,纯度大于94%,产率不低于76%,经质谱鉴定,分子量为4102.0,与计算分子量4101.4一致,即得5′-b-cttzczczggcc-3′。实施例4具有抗肿瘤活性的前体药物(b-5gem-1)的制备将亚磷酰胺碱基单体(a/t/c/g/gem(z)/biotin(b))分别置入核酸合成仪相应的无水试剂瓶中,按dna合成仪的标准流程对设计序列5′-b-cttzzzcczzggcg-3′进行自动化合成,待合成完毕后,依次按标准流程进行dmt切割、脱盐沉淀和hplc分离纯化,纯度大于93%,产率不低于75%,经质谱鉴定,分子量为4148.0,与计算分子量4148.3一致,即得5′-b-cttzzzcczzggcg-3′。实施例5具有抗肿瘤活性的前体药物(b2-3gem-1)的制备将亚磷酰胺碱基单体(a/t/c/g/gem(z)/biotin(b))分别置入核酸合成仪相应的无水试剂瓶中,按dna合成仪的标准流程对设计序列5′-b-cttzczczggcc-b-3′进行自动化合成,待合成完毕后,依次按标准流程进行dmt切割、脱盐沉淀和hplc分离纯化,纯度大于96%,产率不低于70%,经质谱鉴定,分子量为4567.8,与计算分子量4568.3一致,即得5′-b-cttzczczggcc-b-3′。实施例6具有抗肿瘤活性的前体药物(b2-3gem-2)的制备将亚磷酰胺碱基单体(a/t/c/g/gem(z)/biotin(b))分别置入核酸合成仪相应的无水试剂瓶中,按dna合成仪的标准流程对设计序列5′-b-cttzczczggcbc-3′进行自动化合成,待合成完毕后,依次按标准流程进行dmt切割、脱盐沉淀和hplc分离纯化,纯度大于95%,产率不低于70%,经质谱鉴定,分子量为4567.8,与计算分子量4568.3一致,即得5′-b-cttzczczggcbc-3′。实施例7具有抗肿瘤活性的前体药物(b3-3gem-1)的制备将亚磷酰胺碱基单体(a/t/c/g/gem(z)/biotin(b))分别置入核酸合成仪相应的无水试剂瓶中,按dna合成仪的标准流程对设计序列5′-b-cttzczczbggcbc-3′进行自动化合成,待合成完毕后,依次按标准流程进行dmt切割、脱盐沉淀和hplc分离纯化,纯度大于95%,产率不低于70%,经质谱鉴定,分子量为4930.8,与计算分子量4931.3一致,即得5′-b-cttzczczbggcbc-3′。实施例8具有抗肿瘤活性的前体药物(b2-5gem-1)的制备将亚磷酰胺碱基单体(a/t/c/g/gem(z)/biotin(b))分别置入核酸合成仪相应的无水试剂瓶中,按dna合成仪的标准流程对设计序列5′-b-cttzczczggzgczbc-3′进行自动化合成,待合成完毕后,依次按标准流程进行dmt切割、脱盐沉淀和hplc分离纯化,纯度大于90%,产率不低于70%,经质谱鉴定,分子量为5475.1,与计算分子量5475.9一致,即得5′-b-cttzczczggzgczbc-3′。实施例9具有抗肿瘤活性的前体药物(b2-5gem-2)的制备将亚磷酰胺碱基单体(a/t/c/g/gem(z)/biotin(b))分别置入核酸合成仪相应的无水试剂瓶中,按dna合成仪的标准流程对设计序列5′-b-cttzczczggzgczbc-3′进行自动化合成,待合成完毕后,依次按标准流程进行dmt切割、脱盐沉淀和hplc分离纯化,纯度大于90%,产率不低于70%,经质谱鉴定,分子量为5475.5,与计算分子量5475.9一致,即得5′-b-cttzczczggzgczcb-3′。实施例10具有抗肿瘤活性的前体药物(b3-5gem-1)的制备将亚磷酰胺碱基单体(a/t/c/g/gem(z)/biotin(b))分别置入核酸合成仪相应的无水试剂瓶中,按dna合成仪的标准流程对设计序列5′-b-cttzczczbgzgczbc-3′进行自动化合成,待合成完毕后,依次按标准流程进行dmt切割、脱盐沉淀和hplc分离纯化,纯度大于90%,产率不低于70%,经质谱鉴定,分子量为5581.5,与计算分子量5581.7一致,即得5′-b-cttzczczbgzgczbc-3′。实施例11具有抗肿瘤活性的前体药物(b3-5gem-2)的制备将亚磷酰胺碱基单体(a/t/c/g/gem(z)/biotin(b))分别置入核酸合成仪相应的无水试剂瓶中,按dna合成仪的标准流程对设计序列5′-b-cttzczczbgzgczbc-3′进行自动化合成,待合成完毕后,依次按标准流程进行dmt切割、脱盐沉淀和hplc分离纯化,纯度大于90%,产率不低于70%,经质谱鉴定,分子量为5581.2,与计算分子量5581.7一致,即得5′-b-cttzczczbgzgczcb-3′。实施例12细胞毒性检测评价实验gem和实施例1~8制得的具有抗肿瘤活性的前体药物的体外抗癌效应评价比较。鉴于吉西他滨类核苷药物是广谱抗癌细胞毒剂,本实施例中采用购自于美国atcc和或上海细胞库的多种癌组织来源的生物素受体阳性表达的肿瘤细胞(hela、hepg2、bxpc-3、mda-mb-231和sk-ov-3)对实施例制备的相应偶联产物及其原型化合物进行药效评价,同时lo2肝脏细胞(上海细胞库)对其进行正常细胞的毒实验测试。取对数生长期的细胞,根据细胞的大小接种2~10×103个于96孔板上,待生长24h后,弃上清,然后按以下分组给药:癌细胞设不加药组和加药组(浓度0.05~50μm对癌细胞,浓度0.5~100μm对lo2细胞;其中生物素浓度设置0.5~100μm),吉西他滨作为阳性药物和对照比较,对实施例制备的相应产物进行细胞毒性检测。每组设4~6个复孔,培养72h后,弃上清,加入100μl含0.5mg/ml的mtt(四氮唑盐)无血清培养液培养4h,加入100μldmso(二甲亚砜),放置于微型振荡仪上振荡10min,再置于酶标仪上570nm处检测od值。正常人细胞系lo2做对照。每次实验均重复3次。结果显示(表1),随着药物浓度增加,与非处理对照组比较,细胞增殖活性分别下降,说明实施例1~8制得的具有抗肿瘤活性的前体药物呈浓度依赖性抑制癌细胞的生长增殖;且前药显著增加抗增殖效应,ic50浓度显著降低2~6倍。而对正常肝细胞系lo2细胞的增殖活性未有明显变化,ic50浓度值增加3倍以上,显示出该前药策略对正常细胞具有低毒特性(表1)。表1不同细胞的ic50值(72h)及不同化合物ic50比值注:b在最大检测浓度下,尚未发现具有抑制细胞增殖的毒性。实施例13具有抗肿瘤活性的前体药物体外细胞膜转运机制评价核苷酸类药物是平衡核苷转运蛋白和浓缩核苷转运蛋白(含ent和cnt)的底物,具体通过人ent1、ent2、cnt1和cnt3转运,但不通过嘌呤选择性浓缩转运蛋白cnt2转运(mackeyjr,etal.,cancerres.1998;58:4349-57;j.natl.cancerinst.1999;91:1876-81)。吉西他滨在人细胞内摄取转运载体主要是通过人平衡型核苷转运蛋白1(hent1),hent1转运载体表达量的调控或抑制,在临床上会引起吉西他滨耐药性的产生。通常在体外可通过加入核酸转运载体抑制剂来模拟其耐药性,而本发明制得的具有抗肿瘤活性的前体药物对核酸转运载体抑制剂不敏感。本试验采用两种核苷转运载体抑制剂进行底物抑制竞争验证实验(100μmnbmpr(s-(4-硝基苄基)-6-硫肌苷,s-(4-nitrobenzyl)-6-thioinosine)和5μg/mldpam(双嘧达莫,dipyridamole))。在加入各种浓度的前体药物或原形药物之前,预先用核苷转运体抑制剂处理靶细胞30~60min,然后继续完成体外细胞毒性实验的评价程序。研究结果表明(表2),两种抑制剂处理后的mda-mb-231细胞,对吉西他滨的细胞毒活性分别降低了约21倍和20倍,而对b-3gem-1的细胞毒活性只分别降低约1.4倍和1.3倍,b2-3gem-1的细胞毒活性只分别降低约1.2倍和1.3倍;bxpc-3细胞则对吉西他滨的细胞毒活性分别降低约35倍和35倍,而对b-3gem-1的细胞毒活性只分别降低了1.3倍和1.2倍,b2-3gem-1的细胞毒活性只分别降低了1.2倍和1.1倍。这说明吉西他滨的转运需要hent1载体,而b-3gem-1和b2-3gem-1的跨细胞转运可不依赖于hent1载体,这可减少吉西他滨的耐药发生。表2不同核苷抑制剂对肿瘤细胞的ic50值(72h)的改变实施例14肝s9组分进行体外代谢转化释放试验在通过组织匀浆及高速离心(10000~12000×g/min,20min)获得的鼠肝s9组分中,分别平行将原药和具有抗肿瘤活性的前体药物化合物以10μm浓度与肝s9组分混合均匀,于37℃水浴振荡器中进行孵育,一式两份。在0min和60min取样;阴性对照(无s9组分)和阳性对照(7-乙氧基香豆素和7-羟基香豆素)对本测试进行确证。采用安捷伦uplc-ms/ms分析全部样本,监控母体化合物的消耗和吉西他滨的形成(与等摩尔浓度的吉西他滨药物进行百分比计算)。研究结果表明(表3),经过肝脏s9组分处理后的b-3gem-1和b2-3gem-1及b2-5gem-1前药可以很好地释放出吉西他滨原型系列药物,证明了本发明设计的有效性和潜在转化价值。表3吉西他滨前药的肝脏转化及释放备注:“—”为尚未检测出。实施例15前体药物gem-b体内静脉注射后的药物动力学研究将具有抗肿瘤活性的前体药物溶解在peg-200和10%乙醇(v/v)的混合溶媒(1:4,v/v)中,用生盐水稀释配制成注射液;同时,将盐酸吉西他滨用生理盐水中配制成注射液。在雄性sd大鼠(市购)尾静脉按10mg/kg注射,给药后眼眶取血,用高效液相法测定血浆中药物浓度。研究结果表明(表4),与原形药相比,静脉注射gem-b前体药物后血浆中的游离吉西他滨浓度显著较高,并且血浆中药物半衰期和auc分别增加了3~4倍。表4静脉注射b-gem系列前药和盐酸吉西他滨注射后血浆中游离吉西他滨药物动力学参数(每组4只sd大鼠)药动学参数b-3gem-1b2-3gem-1b3-3gem-1b2-5gem-1盐酸吉西他滨t1/2(h)6.98±1.158.43±1.119.27±1.058.37±1.222.39±0.17auc0-24(mg*h/l)21.54±2.7138.67±3.5244.58±3.7234.05±3.077.45±1.08mrt0-24(h)4.98±0.556.38±0.517.15±0.726.15±0.452.11±0.34cl(l/h/kg)0.49±0.110.32±0.080.27±0.090.34±0.071.12±0.20实施例16b-gem系列前体药物系统的抗肿瘤药效学评价将b-3gem-1和b2-3gem-1系列溶解在peg-200和10%乙醇(v/v)的混合溶媒(1:4,v/v)中,用生盐水稀释配制成注射液,将盐酸吉西他滨直接溶解在生理盐水中配制成注射液。雌性balb/c裸鼠(初始重量18g左右,上海动物实验中心提供),接种5×106/只裸鼠的bxpc-3细胞后形成实体瘤,待长成50~100mm3后分组,按20mg/kg吉西他滨及等摩尔量的前药进行3天/次进行尾静脉注射给药6次,末次给药后72h进行裸鼠牺牲,采用游标卡尺测定瘤体尺寸,并计算瘤体大小。研究结果表明(图2),与生理盐水对照组相比,b-3gem-1和b2-3gem-1系列与盐酸吉西他滨注射液均具有良好地抗裸鼠移植瘤增殖效应,其中静脉注射前药组b-3gem-1和b2-3gem-1的抗肿瘤效应更为优,且统计具有显著性(##,p<0.01);模型组比较,前药组和吉西他滨组裸鼠的体重均未呈降低趋势(##,p<0.01),且前药组的体重比吉西他滨组要偏重(#,p<0.05),可能与前药组改善和降低吉西他滨在裸鼠体内的毒副作用有关。上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1