基于非负张量分解的药物-药物相互作用预测方法及系统与流程
本发明涉及计算机辅助药物研究技术领域,具体是一种基于非负张量分解的药物-药物相互作用预测方法及系统。
背景技术:
在同时使用多种药物的情况下,每种药物的药效作用可能彼此并不独立的,不同药物之间在药代动力学(pharmacokinetic,pk)或药效动力学(pharmacodynamic,pd)行为方面会相互影响,这种影响被称为药物-药物相互作用(drug-druginteraction,ddi)。药物与药物之间产生的相互作用可能引发药物不良反应,继而导致复杂疾病治疗失败。因此,在开具复合药物处方之前识别ddi具有非常重要的意义。
然而,传统的ddi识别方法依赖于临床鉴定,通常需要消耗大量的金钱和时间。虽然,现有的计算方法利用药物特性(例如,化学结构等)能够大规模地预测潜在的ddi;但是,现有计算方法只能预测“有或无”ddi,故至少还存在如下不足:其一,不能预测包含药理类型(pk类型和pd类型)的ddi、药理类型ddi发生在药代动力学和/或药效动力学层次;其二、无法解释两种药物产生相互作用的原因;其三、不能为新开发的药物预测ddi。
技术实现要素:
本发明的目的在于提供基于非负张量分解的药物-药物相互作用预测方法及系统,以解决上述背景技术中提出的问题。
为实现上述目的,本发明实施例提供如下技术方案:
一种基于非负张量分解的药物-药物相互作用预测方法,包括以下步骤:
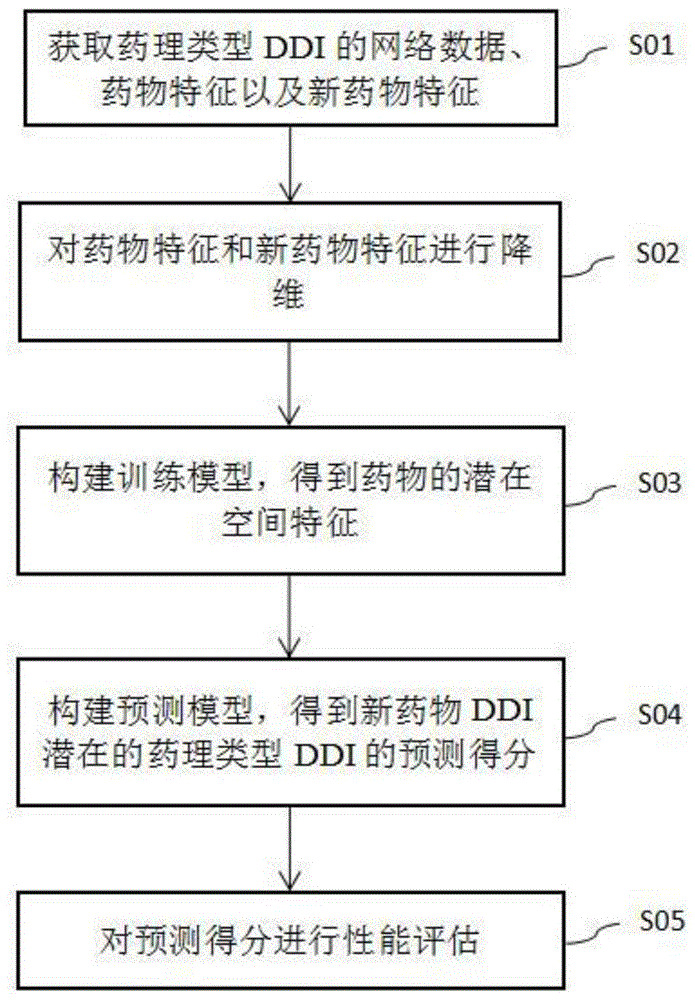
获取药理类型药物-药物相互作用的网络数据、药物特征以及新药物特征;
利用基于核函数的主成分分析方法对药物特征和新药物特征进行降维;
根据药理类型药物-药物相互作用的网络数据以及降维后的药物特征,通过非负张量分解方法构建训练模型,得到药物的潜在空间特征;
根据降维后的新药物特征以及药物的潜在空间特征,得到新药物的潜在空间特征,并通过非负张量分解方法构建预测模型,得到新药物潜在的药理类型药物-药物相互作用的预测得分。
本发明实施例提供的一种优选方案,所述的步骤中,降维后的药物特征与药物的潜在空间特征之间通过偏最小二乘法回归方法进行关联。
本发明实施例提供的另一种优选方案,所述的步骤中,根据降维后的新药物特征以及药物的潜在空间特征,通过偏最小二乘法回归映射得到新药物的潜在空间特征。
本发明实施例提供的另一种优选方案,所述的步骤中,在构建训练模型过程中存在两个可调参数,两个可调参数分别为张量分解的潜在空间因子数量以及偏最小二乘法回归的潜在因子数量。
本发明实施例提供的另一种优选方案,所述的预测方法还包括以下步骤:
利用接收器工作特征曲线下面积(areaunderthereceiveroperatingcharacteristiccurve,auroc)和精确召回曲线下面积(areaunderthepreciserecallcurve,aupr)对预测得分进行性能评估。
本发明实施例还提供了一种基于非负张量分解的药物-药物相互作用预测系统,其包括:
获取模块,用于获取药理类型药物-药物相互作用的网络数据、药物特征以及新药物特征;
降维模块,用于利用基于核函数的主成分分析方法对药物特征和新药物特征进行降维;
训练模块,用于根据药理类型药物-药物相互作用的网络数据以及降维后的药物特征,通过非负张量分解方法构建训练模型,得到药物的潜在空间特征;
预测模块,用于根据降维后的新药物特征以及药物的潜在空间特征,得到新药物的潜在空间特征,并通过非负张量分解方法构建预测模型,得到新药物潜在的药理类型药物-药物相互作用的预测得分。
本发明实施例提供的另一种优选方案,所述降维后的药物特征与药物的潜在空间特征之间通过偏最小二乘法回归方法进行关联。
本发明实施例提供的另一种优选方案,所述的预测模块根据降维后的新药物特征以及药物的潜在空间特征,通过偏最小二乘法回归映射得到新药物的潜在空间特征。
本发明实施例提供的另一种优选方案,所述的训练模块在构建训练模型过程中存在两个可调参数,两个可调参数分别为张量分解的潜在空间因子数量以及偏最小二乘法回归的潜在因子数量。
本发明实施例提供的另一种优选方案,所述的预测系统还包括:
评估模块,用于利用auroc和aupr对预测得分进行性能评估。
本发明实施例的提供的上述技术方案,相比于现有技术,具有以下技术效果:
(1)本发明提出了一种基于非负张量分解的药理类型ddi预测方法,简称为ddintd。该方法的主要思想为:通过构建药物的化学结构特征或药物结合蛋白特征(drugbindingprotein,dbp)和药物潜空间特征(通过非负张量分解提取的特征)的关联来预测新药物潜在的药理类型ddi。由于药物的自身特征数据维度较大,因此需要使用降维方法对其降维,去除药物特征中的噪音和干扰信息。本发明实施例提供的药理类型ddi预测方法只需要手动设置两个训练模型的可调参数,其整个过程快速简捷便利,消耗较少的时间达到很好的预测效果,能够适用不同规模的社交网络和生物信息网络。
(2)本发明实施例提供的药理类型ddi预测方法利用药理类型ddi网络的结构信息和药物特征的信息,可以构建药理类型ddi预测模型,在预测结果方面auroc达到了0.9183±0.0011,通过新药物的版本独立测试验证该方法具有良好的稳定性和鲁棒性;该方法模型的结果证明药物的dbp特征相对于药物的化学结构特征更能表达药物的信息,因为两个药物产生相互作用的直接原因是两个药物的dbp趋向一个或多个相同的蛋白质功能社区;另外,药理类型ddi的出现不是随机的,它们之间是相互依赖的。
附图说明
图1为实施例1提供的一种基于非负张量分解的药物-药物相互作用预测方法的流程图。
图2为实施例1在不同潜在空间因子数量下的药理类型ddi网络的预测结果图。
图3为实施例1提取的一种药物潜在空间特征和药物的度对应的关系图。
图4为实施例1提取的另一种药物潜在空间特征和药物的度对应的关系图。
图5为实施例2提供的一种基于非负张量分解的药物-药物相互作用预测系统的结构示意图。
具体实施方式
下面的具体实施例是结合本说明书中提供的附图对本申请的技术方案作出的具体、清楚的描述。其中,说明书的附图只是为了用于将本申请的技术方案呈现得更加清楚明了,并不代表实际生产或使用中的形状或大小,以及也不能将附图的标记作为所涉及的权利要求的限制。
实施例1
参照附图1,该实施例提供了一种基于非负张量分解的药物-药物相互作用预测方法,包括以下步骤:
s01、获取药理类型药物-药物相互作用(ddi)的网络数据、药物特征以及新药物特征;具体的,药理类型ddi的网络数据为一个自环的无向网络,记为g,g=(v,e,l);其中v={v1,v2,...,vm}是网络中的节点集合,每一个节点表示一个药物;e={e1,e2,...,ec}是节点之间连边的集合,每一个连边表示一个ddi,l={pk,pd}是连边集合的属性。该ddi网络数据的邻接矩阵为单类型三阶张量,记为y(广义邻接矩阵),可以被视为pk和pd类型相互作用的两个相邻矩阵的堆叠。若药物di与药物dj存在相互作用且药理类型为l,则y(i,j,l)=1,否则y(i,j,l)=0。药物ddi的数量称为药物的度,记为deg,药物pk类型ddi的数量和其pd类型ddi的数量分别称为pk度和pd度,分别记为degk和degd,另外,将它们的总和定义为degk+d=degk+degd,将它们的差值定义为degk-d=degk-degd。
该实施例是从drugbank数据库获取药理类型ddi的网络数据,包括有db_v4、db_v5_ex和药物结合蛋白特征(drugbindingprotein,dbp)信息,db_v4是drugbank4.0版本的ddi数据集,db_v5_ex是drugbank5.0版本的新出现的药物对应的ddi数据集,用来做版本独立测试,药物的dbp信息包含药物结合蛋白质的类型和数量。具体的药理类型ddi的网络数据信息如表1,表2,表3所示,表中的pkd是指药物ddi数据中存在既是pk类型也是pd类型的ddi。
表1
表2
表3
另外,药物特征以及新药物特征分别记为f和fx。其中,药物特征包括药物化学结构特征和药物dbp特征。药物化学结构特征是在drugbank中下载药物化学结构文件,并通过padel-descriptor计算得到的,该特征采用的是pubchem中定义的881种化学结构相对应的化学结构指纹(fingerprint),特征示例如表4所示。药物的dbp特征由drugbank中药物结合蛋白质信息收集整理得到的一种药物特征,特征示例如表5所示。
表4
其中,strk表示药物第k个pubchem指纹片段信息,当strk=1,药物存在该pubchem指纹信息,否则strk=0。
表5
其中,dbpk表示药物结合第k个蛋白质,当dbpk=1,药物结合该蛋白质,否则dbpk=0。
s02、利用基于核函数的主成分分析(kernelprincipalccomponentanalysis,kpca)方法对药物特征和新药物特征进行降维;具体的,kpca是由主成分分析(principalcomponentanalysis,pca)扩展得到的。当一个数据集特征维度过大时,其中往往包含着一部分噪音数据,噪音数据不仅会额外增加实验时间,而且会对实验结果产生更大的误差,因此对药物特征和新药物特征进行降维预处理是很有必要的。
s03、根据药理类型药物-药物相互作用的网络数据以及降维后的药物特征,通过非负张量分解方法构建训练模型,得到药物的潜在空间特征;具体的,通过非负张量分解y≈g×1a(1)×2a(2)×3a(3)提取药物的潜在空间特征a(1)或a(2),其中a(n)是分解得到的因子分量矩阵,g为核心张量,再利用a(1)=f×b建立药物特征和药物潜空间特征的联系,其中,建立药物的特征和药物的潜在空间特征之间关联的方法为偏最小二乘法回归,b为回归系数。另外,在构建训练模型过程中存在两个可调参数,第一个为张量分解的潜在空间因子数量r,第二个为偏最小二乘法回归的潜在因子k的数量。在实际操作中可以将两个参数设置为同样的,并在{10,20,30,40,50,60,70,80,90,100}中选择。
s04、根据降维后的新药物特征以及药物的潜在空间特征,得到新药物的潜在空间特征,并通过非负张量分解方法构建预测模型,得到新药物潜在的药理类型药物-药物相互作用的预测得分;具体的,假设现有n个新药物,它们对应的特征为fx,通过
s05、采用auroc和aupr对上述得到的预测得分进行性能评估。
参照附图2,其在不同的潜在空间因子数下对应的实验预测结果,可以发现不同的潜在空间因子对预测结果会有较大的影响,因此采用手动调参的方式确定最优参数。从图中可以看出药物化学结构特征对应的最优参数r=40,药物dbp特征对应的最优参数r=40;而且药物dbp特征对应的预测结果均优于药物化学结构对应的预测结果。
在最优参数的情况下,采用不同的随机种子将10倍交叉验证重复50次,在每次10-cv重复中,auroc和aupr都评估了ddi预测的性能,将50次重复的评估平均值作为该预测模型的最终表现,如表6所示。在药理类型ddi的预测实验中,将1121维的dbp特征与881维的药物化学结构特征(两者特征都是经过kpca降维之后的)进行比较,实验结果显示基于dbp特征的预测结果明显优于基于化学结构特征的预测结果。在药理类型ddi预测中,auroc提高了约5%,aupr提高了约12%。因此在版本独立测试阶段只采用基于dbp的特征。
表6
为了验证该实施例预测药理类型ddi的性能,采用新药物的版本独立测试来验证。首先将db_v4数据集中的药物作为训练样本,将db_v5_ex数据集中的药物作为测试样本,最后用drugbank5.1版本对预测结果进行检验。测试验证是通过前n个预测药物对的正确率来衡量的,其中n=10,20,...,100。其中pk类型ddi的命中率是通过检查统计预测得分张量矩阵的第一张切片的前n个预测的候选交互得到,pd类型ddi的命中率是通过检查统计预测得分张量矩阵的第二张切片的前n个预测的候选交互得到。如表7所示,该预测方法实现了pk类型ddi以及pd类型ddi的准确预测。基于测试结果可以发现,在pk类型ddi的前20个候选交互中,ddi预测的命中率高达80%,但在pd类型ddi的前20个候选交互中,ddi预测的命中率高达60%,这样的结果可能是由于数据的不完整性造成的。
表7
此外,参照图3,其是由上述实施例提取的药物1维潜在空间特征和degk-d生成的,可以发现与翼形相关的流行结构。但当随机改变药理类型ddi网络中的成对相互作用之后,参照图4,其并不能观察到翼形结构,这说明药理类型ddi网络不是一个随机的网络,ddi之间存在相互的关联。
需要说明的是,现实中存在两个生物学假设,首先,相似序列的蛋白质通常是同源的,因此具有相似的功能。其次,以相关方式进化的蛋白质总是在结构复合物,途径或相关途径中共同起作用。因此产生药物相互作用的两个药物可能结合相同的蛋白质或结合同源的蛋白质,经实验统计分析发现83.96%的药物符合上述规则。为了验证这个结果,分别检测验证利西拉和达氟吡啶两个药物结合的蛋白质,验证结果显示均符合上述规则。因此,产生相互作用的不同药物会结合同一个dbp或同一个dbp同源社区。
通过auroc和aupr评估的实验结果显示,该实施例提供的预测方法在药理类型ddi网络中表现优秀,在预测新药物的药理类型ddi结果中展示了优秀的性能。并且根据实验结果发现药理类型ddi网络不是一个随机的网络,更解释说明了不同药物之间产生ddi的原理。
实施例2
参照附图5,该实施例提供了一种用于实现上述实施例1提供的基于非负张量分解的药物-药物相互作用预测方法的系统,其包括:获取模块、降维模块、训练模块、预测模块和评估模块。
其中,获取模块,用于获取药理类型药物-药物相互作用的网络数据、药物特征以及新药物特征。
降维模块,用于利用基于核函数的主成分分析方法对药物特征和新药物特征进行降维。
训练模块,用于根据药理类型药物-药物相互作用的网络数据以及降维后的药物特征,通过非负张量分解方法构建训练模型,得到药物的潜在空间特征;所述降维后的药物特征与药物的潜在空间特征之间通过偏最小二乘法回归方法进行关联;所述的训练模块在构建训练模型过程中存在两个可调参数,两个可调参数分别为张量分解的潜在空间因子数量以及偏最小二乘法回归的潜在因子数量。
预测模块,用于根据降维后的新药物特征以及药物的潜在空间特征,得到新药物的潜在空间特征,并通过非负张量分解方法构建预测模型,得到新药物潜在的药理类型药物-药物相互作用的预测得分;所述的预测模块根据降维后的新药物特征以及药物的潜在空间特征,通过偏最小二乘法回归映射得到新药物的潜在空间特征。
评估模块,用于利用auroc和aupr对预测得分进行性能评估。
需要说明的是,上述实施例只是针对本申请的技术方案和技术特征进行具体、清楚的描述。而对于本领域技术人员而言,属于现有技术或者公知常识的方案或特征,在上面实施例中就不作详细地描述了。
当然,本申请的技术方案不只局限于上述的实施例,本领域技术人员应当将说明书作为一个整体,各实施例中的技术方案也可以经适当组合,从而可以形成本领域技术人员可以理解的其他实施方式。
- 还没有人留言评论。精彩留言会获得点赞!