α螺旋跨膜蛋白质拓扑结构预测方法及装置与流程
本发明属于生物检测技术领域,特别涉及一种基于多尺度深度学习的α螺旋跨膜蛋白质拓扑结构预测方法及装置。
背景技术:
细胞膜是细胞的屏障,能够隔绝细胞内部环境和外部环境。细胞膜由磷脂双分子层以及嵌在其上的大量膜蛋白组成。膜蛋白在细胞信号传导、离子传导性、细胞凝聚、细胞识别和细胞间通信等一系列生物过程中发挥了重要的作用。因此,很多药物被设计成与膜蛋白结合,进而影响生物过程。
在所有的膜蛋白中,α螺旋跨膜蛋白质占了大部分。据估计,人体中27%的蛋白质是α螺旋跨膜蛋白质。它们通常分布在真核生物的质膜,细菌细胞的内膜,甚至外膜中。蛋白质的跨膜α螺旋拓扑信息可以帮助科学家识别结合位点,设计新的药物。但是由于膜蛋白难以溶解,纯化和结晶,并且对于nmr来说又太大,通过实验的方法确定膜蛋白的结构非常具有挑战性。据报道,膜蛋白结构仅占pdb数据库中所有结构的1%。因此,领域内十分需要一种能够精确预测膜蛋白拓扑结构的计算预测方法。
在过去的三十年中,领域内已经开发了许多预测方法。这些方法可以分为三类:
第一类预测方法仅使用亲水指数来预测tmh。这些方法使用长度为19个氨基酸残基的滑动窗口作为模型的输入。19个氨基酸残基的平均亲水指数即为中心残基的亲水指数。然后使用一个固定的阈值来确定这个氨基酸残基是否位于tmh上。此外,著名的positive-inside规则也在这个阶段被提出。规则的内容是位于细胞内侧的短的loop主要由lys和arg残基组成。这个规则后续的工作具有长远的影响;
第二类方法使用机器学习算法和统计模型得到了更精确的预测结果,例如隐马尔可夫模型,支持向量机器和k-近邻模型。同时,除了亲水指数外,这些模型还采用了更加强大的进化信息特征;
第三类预测算法是融合方法。这些方法的主要思想是通过融合几种拓扑结构预测的方法得到最终的结果。实验表明,对于可靠性高的蛋白质,这种方法可以显著提升性能。
尽管在该领域已经有了大量的研究工作,这些工作大多数仅仅预测了完全埋在膜内的α螺旋区域。这意味着这些工作认为tmh指完全埋在细胞膜内的螺旋片段。例如在图3中,只有helix的区域被认为是跨膜α螺旋区域,而其余的tail区域并没有被考虑进去。但是,据报道,这些tail区域在细胞间通讯,细胞识别等生物过程中起着至关重要的作用。并且其位置信息还能够帮助科学家更好地理解蛋白质的功能。另外,随着评价标准越来越严格,以往的预测算法在精度上还存在提升的空间。因此,设计一种能够精确预测出helix区域和tail区域位置的算法就显得尤为重要。
技术实现要素:
本发明实施例提供了一种α螺旋跨膜蛋白质拓扑结构预测方法。
本发明实施例的基于多尺度深度学习模型来预测α螺旋跨膜蛋白质拓扑结构的算法。算法主要分成两个部分:预测tmh区域和预测non-tmh区域位置。在预测tmh区域中,使用了基于整条序列和基于固定滑动窗口的两种尺度不同的深度残差网络,从pssm,hmm和结构信息特征中提取更高级的特征来预测tmh。在使用深度学习的同时,又结合了机器学习模型。针对过分割和欠分割问题,设计了动态阈值算法,进一步提升了深度模型的预测精度。在预测non-tmh区域位置算法中,由于训练样本较少,算法采用了支持向量机模型,使用hmm和亲水指数作为模型的输入特征。考虑到预测过程中可能存在不准确的问题,算法使用了集成方法。对于一个non-tmh区域,共提取了10个与tmh区域的交界区域作为输入。经过支持向量机模型,共得到10个预测分数。取这10个分数的平均值作为最终的预测分数。最后对于一条蛋白质中所有的non-tmh区域,使用最大最小分配法得到最终的预测结果。结合两个部分的预测结果就可以得到α螺旋跨膜蛋白质的拓扑结构。
本发明具有如下有益效果:
1.本发明中使用的tmh定义不同于领域内其他工作。如图3,本发明中tmh区域既包括了完全埋在细胞膜中的helix区域,也包括在细胞膜外,与helix区域相连的tail区域。这些tail区域对于理解蛋白质的生物功能具有重要的作用。
2.本发明使用了多尺度的深度残差网络。具体来讲,既包括了基于整条序列的网络,又包括了基于固定长度的滑动窗口的网络。两种网络的预测结果之间存在一定的互补性。通过集成这两种网络的预测结果,能够进一步提升模型的预测精度。
3.本发明将深度学习与机器学习相结合。在预测tmh位置模型中,使用动态阈值模型处理深度学习模型的预测结果,成功解决了预测过程中的过分割和欠分割问题,提升了模型的效果。
4.本发明在模型的搭建过程中广泛使用了集成的思想。在预测tmh算法中,集成了两个不同尺度的深度学习模型。在预测non-tmh区域位置的算法中,集成了10个交界区域的预测结果,减小了预测tmh位置的不精确性带来的影响,保证了预测精度。
5.本发明在一些较难预测的tmh上也取得了较好的表现。
附图说明
通过参考附图阅读下文的详细描述,本发明示例性实施方式的上述以及其他目的、特征和优点将变得易于理解。在附图中,以示例性而非限制性的方式示出了本发明的若干实施方式,其中:
图1是根据本发明实施例之一的tmh预测流程图。
图2是根据本发明实施例之一的non-tmh区域位置预测流程图。
图3是α螺旋跨膜蛋白质示意图。
图4是根据本发明实施例之一中动态阈值算法在过分割和欠分割问题上的效果示意图。
具体实施方式
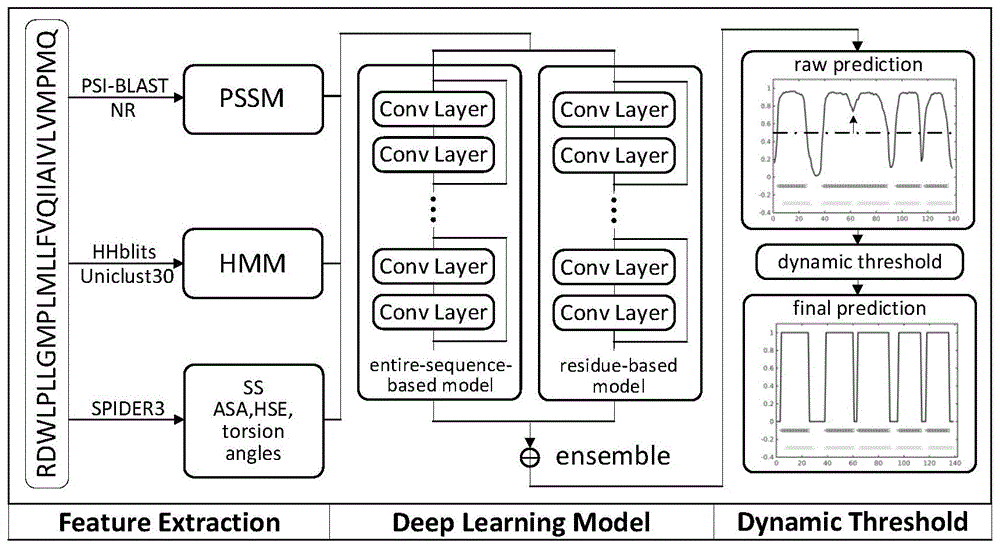
本发明涉及α螺旋跨膜蛋白质生物学领域,具体涉及一种基于多尺度深度学习的α螺旋跨膜蛋白质拓扑结构预测算法(membrain2.1)。算法主要分为两个部分:跨膜α螺旋区域(tmh)预测和其他区域(non-thm)位置预测。在第一部分中,本发明采用了两种不同尺度的深度学习模型以及动态阈值算法。第一种模型基于整条序列预测tmh位置,第二种模型基于固定长度的滑动窗口预测tmh位置。这两种模型因为尺度不同而具有较好的互补性,通过将这两种模型融合,可以提高tmh位置预测的精度。动态阈值算法能够检测到过分割和欠分割现象,纠正深度学习的预测结果。第二部分中,本发明采用了支持向量机模型配合最大最小分配法预测non-tmh区域的位置。支持向量机模型使模型更多关注起决定性作用的训练样本,最大最小分配法使模型更多的关注预测值的相对大小而非绝对大小。两者都可以提高模型的鲁棒性。结合两部分的预测结果,可以得到α螺旋跨膜蛋白质的拓扑结构。
根据一个或者多个实施例,如图1和图2所示,一种基于多尺度深度学习的α螺旋跨膜蛋白质拓扑结构预测算法,包括以下步骤:
s1、根据tmh的定义组织训练集、验证集和测试集;
s2、使用psi-blast,hhblits,spider3工具对训练集、验证集和测试集中的序列提取位置特异性打分矩阵(pssm)、hmm、水溶性、二级结构、扭转角和亲水指数特征;
s3、使用训练集训练基于整条序列的深度残差网络模型和基于滑动窗口的深度残差网络模型。将两种网络的输出取平均值集成后,采用动态阈值算法得到tmh预测结果;
s4、使用训练集训练支持向量机模型。模型的输入是non-tmh区域和tmh区域的交界部分,输出为0到1之间的实数,表示当前的non-tmh区域倾向于位于外部(outside)还是内部(inside)。然后采用最大最小分配法确定最终的预测结果;
s5、对于一条待预测的蛋白质,首先预测蛋白质中的tmh位置,然后预测non-tmh区域的位置,结合两部分的预测结果,就可以得到蛋白质的最终拓扑结构。
进一步的,所述步骤s1根据tmh新的定义,组织训练集、验证集和测试集的具体步骤如下:
s11、从opm数据库中提取全部的α螺旋跨膜蛋白质结构,总共有1783个pdb文件。根据文件中蛋白质链的编号,将这1783个分割成以蛋白质链为单位的pdb文件;
s12、选取tmseg工作中使用的40个测试蛋白质作为本实施例的测试集。对于剩下的pdb文件,如果蛋白质链断开,或者蛋白质长度小于20个氨基酸,或者蛋白质中没有跨膜α螺旋,直接将其剔除。这样共得到5741条蛋白质链;
s13、使用uniqueprot软件以hval>0为标准去除5741条蛋白质与测试集之间的冗余性,然后再对自身去冗余,总共得到318条蛋白质。随机选出其中的39条蛋白质作为验证集,剩下的279条蛋白质作为训练集。
s14、根据pdb文件得到蛋白质中每一氨基酸残基是否属于tmh,以及每个non-tmh区域的位置。在本实施例中,氨基酸残基属于tmh需要满足以下要求:残基位于一段α螺旋上;这段α螺旋有部分在细胞膜中。
进一步的,所述步骤s2根据蛋白质序列信息,使用blast,hhblits和spider3软件提取pssm,hmm,二级结构、水溶性、扭转角等结构信息,同时获取亲水指数信息。具体如下:
位置特异性打分矩阵(pssm)是生物序列中常用的一种表征模体。它含有丰富的进化信息,并在以往的tmh预测工作中被证明是非常有用的特征。要获得pssm矩阵,首先需要生成一个多重序列比对文件。在本实施例中使用blast软件搜索nr(non-redundant)数据库获得。具体执行命令及参数为:
psiblast-querysequence.fasta-dbnr-out_ascii_pssmpssm.matrix-save_pssm_after_last_round-evalue1e-3-max_target_seqs10000-num_iterations3-num_threads6
pssm矩阵可以通过下式从多重序列比对结果中提取出来:
其中i=1,…,l,l表示蛋白质序列的长度,j=1,…,20,表示20种氨基酸。ppm指位置概率矩阵,ppmi,j表示第j种氨基酸出现在多重序列比对的第i列的概率。bj表示第j种氨基酸的背景频率。对于一个氨基酸残基来说,pssm矩阵共20维。
hmm特征是另外一种包含进化信息的特征。它由hhblits序列比对工具生成。与blast相比,hhblits使用hmm-hmm比对算法得到同源序列,灵敏度更高,结果更加准确。对于一个氨基酸残基来说,hmm特征共30维。在本发明中,使用hhblits软件搜索uniclust30数据库得到hmm特征。具体执行命令及参数为:
hhblits–isequence.fasta-n3-e0.001-duniclust30_2017_10-cpu6-ohhmsequence.hmm-diffinf-id99-cov50
结构信息特征包含扭转角、水溶性和二级结构。这些特征通过spider3软件预测得到。对于一个氨基酸残基来说,结构信息特征共14维。
亲水指数描述了氨基酸支链的亲水性或疏水性程度。亲水指数越大,这种氨基酸的疏水性就越强。本发明实施例使用kyte-doolittle亲水指数。对于一个氨基酸残基来说,亲水指数特征共1维。
在预测tmh算法中,本发明实施例使用pssm,hmm和结构信息特征。在预测non-tmh区域位置算法中,本发明实施例使用hmm和亲水指数特征。
进一步的,所述步骤s3使用训练集训练基于整条序列的深度残差网络模型和基于滑动窗口的深度残差网络模型。将两种网络的输出取平均值集成后,采用动态阈值算法得到tmh预测结果。具体如下:
s31、根据模型在验证集上的效果,确定基于整条序列的深度残差模型的层数、正则项系数、学习率、批大小等参数。训练集共279条序列;
s32、根据模型在验证集上的效果,确定基于滑动窗口的深度残差模型的层数、正则项系数、学习率、批大小、滑动窗口大小等参数。训练集共17437个正样本(滑动窗口中心的氨基酸残基在tmh上)和20003个负样本(滑动窗口中心的氨基酸残基在non-tmh上);
s33、对于一条α螺旋跨膜蛋白质序列,使用s31和s32两个步骤中训练出的两个深度残差模型得到两个预测结果。取平均集成两种不同尺度的深度学习模型的预测结果。根据模型在验证集上的效果,调整动态阈值模型中的参数,如初始阈值、合并标准、分裂标准等,以此解决预测中的过分割和欠分割问题。动态阈值算法内容如下:
i.使用长度为5个残基的滑动窗口对预测分数做均值滤波。滤波过程中,去掉滑动窗口中的最大值和最小值。使用值为0.55的初始阈值获得最初的tmh预测结果。
ii.对于两个相邻的tmh,如果他们之间的间隙不大于5个残基,并且两个tmh的长度和不大于24个残基,那么将这两个tmh合并成一个tmh。
iii.对于每一个tmh,如果其长度大于33个残基,那么就用初始值为0.55,增量为0.05的阈值检测其中的tmh。如果有多于一个tmh被识别出,并且他们不满足合并条件,那么将这个tmh分裂开。
进一步的,所述步骤s4使用训练集训练支持向量机模型,具体如下:
s41、tmh区域与non-tmh区域的交界处,对于预测non-tmh的位置有较大的影响。本发明中,这样的交界处指由6个在tmh区域的氨基酸残基和7个在non-tmh区域的氨基酸残基组成的窗口。对于一段non-tmh区域来说,共有前后两种与tmh区域交界的部分。由于这两种交界相差较大,本实施例训练两种支持向量机模型。通过集成这两种模型的预测结果,得到最终的预测分数。利用网格搜索方法训练多个支持向量机模型,根据模型在验证集上的效果,确定最终的模型。训练集包括646个inside的样本和613个outside的样本。
s42、使用最大最小分配法,根据预测分数得到最终的预测效果。首先在所有的预测分数中选择预测分数最大的作为inside,最小的为outside。对于其余分数,如果离最大分数近,则为inside,反之为outside。最大最小分配法更多的关注预测分数的相对大小而不是绝对大小,因此可以避免误分情况。
进一步的,所述步骤s5预测一条蛋白质的拓扑结构,具体如下:
给定一条待预测的蛋白质序列,首先使用预测其中的tmh,如果没有tmh被检测到,那么认为这条蛋白质为水溶性蛋白质。如果有至少一个tmh被检测到,那么认为这条蛋白质是α螺旋跨膜蛋白质。然后预测其中non-tmh区域的位置。由于第一步预测tmh区域的结果可能不准确,会导致预测non-tmh区域位置受到较大的影响。因此,本实施例采用了集成的方法,将由10,8,6,4,2个在tmh区域的氨基酸残基和3,5,7,9,11个在non-tmh区域的氨基酸残基组成的共5个交界区域提取出来。由于一个non-tmh区域有前后两种交界区域,所以总共提取了10个交界区域作为支持向量机模型的输入。通过这种集成的方法,大大提升了模型的鲁棒性。
根据一个或者多个实施例,一种α螺旋跨膜蛋白质拓扑结构预测装置,其特征在于,所述预测装置包括存储器;以及耦合到所述存储器的处理器,该处理器被配置为执行存储在所述存储器中的指令,所述处理器执行以下rpa操作:
s1、根据tmh的定义组织训练集、验证集和测试集;
s2、获取蛋白质的亲水指数信息。使用psi-blast,hhblits,spider3工具对整理数据集中的序列分别提取pssm,hmm以及水溶性、二级结构、扭转角等蛋白质结构信息;
s3、使用训练集训练基于整条序列的深度残差网络模型和基于滑动窗口的深度残差网络模型。将两种网络的输出取平均值集成后,采用动态阈值算法得到tmh的预测结果;
s4、使用训练集训练支持向量机模型。模型的输入是non-tmh区域和tmh区域的交界部分,输出为0到1之间的实数,表示当前的non-tmh区域倾向于位于外部(outside)还是内部(inside)。然后采用最大最小分配法确定最终的预测结果;
s5、对于一条待预测的蛋白质,首先预测蛋白质中的tmh,然后预测non-tmh区域的位置,结合两部分的预测结果,就可以得到蛋白质的最终拓扑结构。
rpa,即roboticprocessautomation(软件流程自动化),是指用软件自动化方式实现在各个行业中本来是人工操作计算机完成的业务。
根据一个或者多个实施例,根据新的tmh定义,从opm数据库中提取了279条蛋白质作为训练数据。基于整条序列和基于固定长度滑动窗口的深度网络具有相同的网络结构,含有6层卷积层,优化器为adam。在基于整条序列的模型中,训练数据为279条蛋白质,batch_size为11,epoch数目为100。基于滑动窗口的模型中,训练数据有17437个正样本和20003个负样本,batch_size为40,滑动窗口大小为17,epoch数目为100。在预测non-tmh区域位置模型中,共从279条蛋白质中提取出646个正样本和613个负样本。样本为在tmh区域中的6个氨基酸残基和在non-tmh域中的7个氨基酸残基组成的长度为13个残基的交界区域。
采用的评价指标如下:
其中,一段tmh被正确预测的标准为:预测的tmh的端点不能偏离真实的tmh端点±5个残基;预测的和真实的tmh重叠部分的长度,既要占预测的tmh长度的一半以上,又要占真实的tmh长度的一半以上。一条α螺旋跨膜蛋白质的tmh被正确预测指的是:预测的tmh数目和真实的tmh数目相同;每个真实的tmh都被正确预测出。一条α螺旋跨膜蛋白质的拓扑结构被正确预测指的是:tmh被正确预测;所有的non-tmh区域位置被正确预测。
将本发明实施例提出的算法与领域中已有的算法在测试集上进行比较。对比结果如表1所示。本发明实施例提出的算法在几个较为重要的指标上(preh,rech,vp,vtop)都明显优于领域内其他算法。
表1.不同算法与领域内已有算法在测试集上的效果
表2显示了使用动态阈值和固定阈值在测试集上的效果。固定阈值指得到不同尺度深度模型的集成结果后,根据模型在验证集上的效果确定一个固定的阈值处理预测分数。可以看出,使用了动态阈值后,preh和rech指标分别提升了4.6%和4.7%。图4给出了过分割和欠分割的例子,使用了动态阈值算法后,这两个问题都被成功解决。实验结果证明了动态阈值的有效性。
表2.动态阈值和固定阈值在测试集上的效果
表3显示了使用固定阈值和最大最小分配算法在测试集上的效果。其中mcc指的是在已知真实的tmh位置后,模型在预测non-tmh区域位置上的马修斯系数。vtop指的是在已知真实的tmh位置后,有多少蛋白质的拓扑结构被正确预测出。mccpred与vtop_pred和mcc与vtop类似,不同的是前面两个指标是在未知真实的tmh位置情况下指标。从表3可以看出,最大最小分配法要优于固定阈值方法。尤其是在未知真实tmh位置的情况下。
表3.最大最小分配法和固定阈值方法在测试集上的效果
表4显示了集成不同尺度的深度模型后的效果。这三种对比方法的结果都经过了动态阈值方法的处理。可以看出,不同尺度的深度学习模型之间存在互补性。集成后的效果明显提升。
表4.集成不同尺度的深度模型的效果
表5显示了在未知真实tmh位置时,集成多个交界区域方法在预测non-tmh区域位置上的效果。junction2_11表示当前使用的交界区域由2个在tmh内的氨基酸残基和11个在non-tmh区域内的氨基酸残基组成。其他名字类似。可以看出,通过集成多个交界区域的预测结果,能够减小tmh位置预测不准确带来的影响。这6种对比方法的结果都经过了最大最小分配法的处理。
表5.集成多个交界区域的预测结果在测试集上的效果
表6显示了本发明在较难预测的tmh上的表现。具体有两类tmh。一类是半跨膜α螺旋,这类tmh只跨过一半细胞膜,并且它前后的两个non-tmh区域的位置相同。第二类是近跨膜α螺旋,这类跨膜α螺旋指一对tmh,这对tmh中间的间隙不大于3个氨基酸残基。一对近跨膜α螺旋被成功预测指的是其中的两个tmh均被预测正确。在测试集种,共有11对近跨膜α螺旋和6个半跨膜α螺旋。从表6可以看出,本发明取得的效果要好于领域内其他算法。
表6.本发明在较难预测的tmh上的效果
所述集成的单元如果以软件功能单元的形式实现并作为独立的产品销售或使用时,可以存储在一个计算机可读取存储介质中。基于这样的理解,本发明的技术方案本质上或者说对现有技术做出贡献的部分,或者该技术方案的全部或部分可以以软件产品的形式体现出来,该计算机软件产品存储在一个存储介质中,包括若干指令用以使得一台计算机设备(可以是个人计算机,服务器,或者网络设备等)执行本发明各个实施例所述方法的全部或部分步骤。而前述的存储介质包括:u盘、移动硬盘、只读存储器(rom,read-onlymemory)、随机存取存储器(ram,randomaccessmemory)、磁碟或者光盘等各种可以存储程序代码的介质。
以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到各种等效的修改或替换,这些修改或替换都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应以权利要求的保护范围为准。
- 还没有人留言评论。精彩留言会获得点赞!