一种miRNA数据与转录组数据贯穿分析的方法及系统与流程
本发明涉及mirna和转录组学技术领域,特别是涉及一种mirna数据与转录组数据贯穿分析的方法及系统。
背景技术:
转录组被定义为特定物种、组织或细胞类型转录的所有rna(转录本)的集合,包括mrna和非编码rna(non-codingrna);非编码rna主要是指microrna(mirna)。
microrna(mirna)是一类长度约为19-25nt的内源性非编码rna,广泛参与基因转录后调控活动,具有高度序列保守性、表达时序性和组织特异性。研究表明mirna参与各种各样的调节途径,包括发育、病毒防御、造血过程、脂肪代谢等,具有重要的基因表达调控作用。mrna翻译成肽链的过程是受调控因子调控的,研究表明,多数mirna具有调控特定的mrna翻译成蛋白质的作用;成熟的mirna通过以碱基配对的方式结合到与它互补的mrna位点上去,影响mrna的翻译过程,从而实现对基因表达的调控。mirna及其调控的靶基因的表达,构成了一个非常严密的调控网络,这个网络的精确运行确保了生物体各种生理过程的正常运行;因而,mirna与mrna之间相互关系的分析研究,对人类遗传学、生物学、疾病学等都具有重要的参考意义。
随着研究的不断深入,单一组学数据反应的生物学信息具有一定的局限性,不能全面深入地达到预期的研究目的;因此,需要将多组学技术结合进行数据整合,来全面深入地进行科学探究;公开号为cn104911261b的中国专利公开了一种研究水稻与病原物互作模式的方法,综合应用了转录组学、蛋白组学及mirna组学来研究水稻响应病原物侵染的互作模式的方法。目前还缺乏一种系统完整的能够应用于任何物种的mirna和转录组学贯穿分析的方法。
技术实现要素:
为克服上述现有技术存在的不足,本发明之目的在于提供一种mirna测序和转录组测序数据的贯穿分析方法及系统,以实现一种可以适用于任何生物体的mirna测序和转录组学测序数据的贯穿分析技术。
为达上述及其它目的,本发明提出一种mirna测序和转录组学测序数据的贯穿分析方法,包括如下步骤:
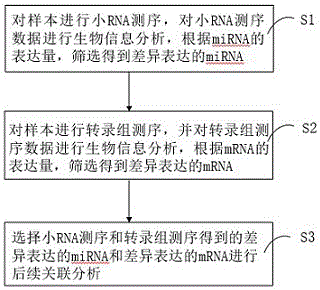
步骤s1,对样本进行小rna测序,对小rna测序数据进行生物信息分析,根据mirna的表达量,筛选得到差异表达的mirna;
步骤s2,对样本进行转录组测序,并对转录组测序数据进行生物信息分析,根据mrna的表达量,筛选得到差异表达的mrna;
步骤s3,选择小rna测序和转录组测序得到的差异表达的mirna和差异表达的mrna进行后续关联分析。
优选地,所述的对样本进行小rna测序并对小rna测序数据进行生物信息分析。得到样本中mirna的表达量,根据p<0.05且|log2fc|>1筛选出差异表达的mirna。对于两组样本,是直接根据上述标准筛选得到差异表达的mirna;对于多组样本,是取多个比较组差异表达的mirna并集。
优选地,步骤1中不仅限于根据mirna表达量筛选出差异表达的mirna,还可以对mirna进行靶基因预测,并且对差异表达的mirna的靶基因进行go或者kegg功能富集分析等。
优选地,所述的对样本进行转录组测序并对转录组数据进行生物信息分析,得到样本中mrna的表达量,根据fdr<0.05且|log2fc|>1筛选出差异表达的mrna。对于两组样本,是直接根据上述标准筛选得到差异表达的mrna。对于多组样本,是取多个比较组差异表达的mrna并集。
优选地,步骤2中不仅限于筛选出差异表达的mrna,还可以对差异表达的mrna进行go或kegg功能富集分析等。
优选地,所述的选择小rna测序和转录组测序得到的差异表达的mirna和差异表达的mrna进行关联分析。mirna与mrna存在相互调控关系需满足两个条件:1)mirna和mrna存在靶向关系;2)mirna和mrna表达量负相关。具体包括如下步骤:
步骤s3.1筛选出满足mirna与mrna存在靶向关系,根据mirna和靶基因的差异表达结果及两者的靶向关系结果,筛选出表达量负相关的mirna-靶基因对;
步骤s3.2mirna靶向能力分析,mirna靶向基因的数量可在一定程度上反映出mirna的调控能力,对mirna靶向基因数量进行统计。结果可以通过以mirna靶基因数量为横坐标,以靶向特定数量靶基因的mirna数量为纵坐标的mirna靶基因数量分布图进行结果展示;
步骤s3.3基因结合mirna能力分析,基因结合mirna的数量可在一定程度上反映该基因参与调控的能力,对基因结合mirna的数量进行统计。分析结果以基因结合mirna数量分布图进行展示,横坐标为被靶向mirna数量,纵坐标为mrna数量;
步骤s3.4mirna负相关靶基因富集分析,对上述步骤中筛选出的mirna负相关的靶基因进行go富集分析、kegg富集分析;
步骤s3.5cytoscape分析,根据得到的负相关的mirna-靶基因对,利用cytoscape构建调控网络图;
优选地,所述的筛选mirna与mrna存在靶向关系,且表达量负相关的mirna-靶基因对。包括对两组样本和多组样本间进行筛选。对于两组样本,即筛选出样本中差异表达的mirna出现上调,其对应靶基因出现下调;或者样本中差异表达的mirna出现下调,其对应的靶基因出现上调的mirna-靶基因对。对于多组样本,结合mirna和靶基因的相关性及两者靶向关系分析结果,计算pearson相关系数,根据“相关系数小于-0.7且p值小于0.05”的标准筛选出表达量负相关的mirna-靶基因对。
为达到上述目的,本发明还提供一种mirna测序和转录组学测序数据的贯穿分析系统,包括:
小rna测序分析单元,用于对实验样本进行转录组测序,对小rna测序数据进行生物信息分析,根据mirna的表达量,筛选得到差异表达的mirna,并且对差异表达的mirna进行靶基因预测,可以对预测的靶基因进行go/kegg功能富集分析;
转录组测序分析单元,用于对实验样本进行转录组测序,并对转录组测序数据进行生物信息分析,根据mrna的表达量,筛选得到差异表达的mrna,可以对差异表达的mrna进行go/kegg功能富集分析;
贯穿分析单元,用于将所述小rna测序分析单元获得的差异表达的mirna测序数据和所述转录组测序分析单元获得的差异表达的rna测序数据进行贯穿分析。
与现有技术相比,本发明一种mirna测序和转录组学测序数据的贯穿分析方法及系统通过分别对样本进行小rna测序和转录组测序,将得到的差异表达的mirna和差异表达的mrna进行贯穿分析,筛选出负调控的mirna-靶基因对,同时构建mirna-靶基因调控网络,并对mirna靶基因进行go/kegg功能富集分析,为后续挑选目标基因提供线索。本发明关联分析有效提高了测序数据利用率,充分深入挖掘了mirna和小rna的数据信息,提高了小rna靶基因预测准确性;直接分析差异表达的mirna和差异表达的mrna负性调控关系,能够得到多个mirna与多个基因的调控网络关系,从不同的层面去挖掘数据之间的潜在联系和有重要研究价值的生物学信息,达到了数据整合的目的。
附图说明
图1为本发明一种mirna测序和转录组测序数据关联分析方法的步骤流程图;
图2为本发明一种mirna测序和转录组测序数据关联分析系统的系统架构图。
具体实施方式
以下通过特定的具体实例并结合附图说明本发明的实施方式,本领域技术人员可由本说明书所揭示的内容轻易地了解本发明的其它优点与功效;本发明亦可通过其它不同的具体实例加以施行或应用,本说明书中的各项细节亦可基于不同观点与应用,在不背离本发明的精神下进行各种修饰与变更。
图1为本发明一种mirna数据和转录组学测序数据的贯穿分析方法的步骤流程图。如图1所示,本发明一种mirna数据和转录组学测序数据的贯穿分析方法,包括如下步骤:
步骤s1,对样本进行小rna测序,对小rna测序数据进行生物信息分析,根据mirna的表达量,筛选得到差异表达的mirna;
步骤s2,对样本进行转录组测序,并对转录组测序数据进行生物信息分析,根据mrna的表达量,筛选得到差异表达的mrna;
步骤s3,选择小rna测序和转录组测序得到的差异表达的mirna和差异表达的mrna进行后续关联分析。
其中,步骤s1包括从两组样本和多组样本中筛选出差异表达的mirna。对于两组样本而言,以p<0.05且|log2fc|>1为标准,对两组样本的mirna表达量进行筛选得到差异表达的mirna。对于多组样本而言,则是以p<0.05且|log2fc|>1为标准进行筛选,取多组样本差异表达的mirna并集。
其中,步骤s2也包括从两组样本和多组样本中筛选出差异表达的mrna;对于两组样本而言,以fdr<0.05且|log2fc|>1为标准,对两组样本的mrna表达量进行筛选得到差异表达的mrna;对于多组学样本而言,则是以fdr<0.05且|log2fc|>1为标准进行筛选,取多组样本差异表达的mrna并集。
在本发明具体实施例中,所述贯穿分析包括五个部分:第一、mirna-靶基因对筛选;第二、mirna靶向能力分析;第三、基因结合mirna能力分析;第四、mirna负相关靶基因富集分析;第五、cytoscape分析。
1.mirna-靶基因对筛选:mirna与mrna存在相互调控关系需满足两个条件:1)mirna和mrna存在靶向关系;2)mirna和mrna表达量负相关;根据mirna和靶基因的差异表达结果及两者的靶向关系结果,筛选出表达量负相关的mirna-靶基因对。对于两组样本而言,直接根据mirna和靶基因的差异表达结果及两者的靶向关系结果进行筛选。即即筛选出样本中差异表达的mirna出现上调,其对应靶基因出现下调;或者样本中差异表达的mirna出现下调,其对应的靶基因出现上调的mirna-靶基因对。对于多组样本(三组及以上)而言,具体步骤为结合mirna和靶基因的相关性及两者的靶向关系分析结果,计算皮尔逊相关系数(pearsoncorrelationcoefficient),皮尔逊相关系数可用来度量两个变量之间的相互关系,代表了两个变量共变性的强弱,取值范围为[-1,+1]。当皮尔逊相关系数大于0时,表明两个变量正相关;当皮尔逊相关系数小于0时,表明两个变量负相关。相关系数的绝对值越大则表明相关性越强。以相关系数小于-0.7且p值小于0.05为标准筛选出表达量负相关的mirna-靶基因对。
2.mirna靶向能力分析:mirna可以靶向1个基因或者多个基因,
mirna靶向基因的数量可在一定程度上反映出mirna的调控能力,对mirna靶向基因数量进行统计。mirna靶向统计表中清晰地展示出了mirnaid、样品和比较组tpm表达量、log2处理差异倍数、检验p值、靶基因数量及mirna对应的靶基因列表等信息;根据统计表信息,可以将mirna靶基因数量为横坐标,以靶向特定数量靶基因的mirna数量为纵坐标,绘制出mirna靶基因数量分布图进行结果展示;可以通过mirna靶向基因的数量直观看出mirna的调控能力。
3.基因结合mirna能力分析:同样地,一个基因也可以与1个或者多个mirna进行结合。基因结合mirna的数量可在一定程度上反映该基因参与调控的能力;因此,我们对基因结合mirna的数量进行统计,从而对基因结合mirna能力进行分析。根据mrna被靶向统计表中的mrna、靶向该基因的mirna的数量等信息,对基因结合mirna的能力进行分析,结果以基因结合mirna数量分布图进行展示,横坐标为被靶向mirna数量,纵坐标为mrna数量。
4.mirna负相关靶基因富集分析:主要包括对上述步骤中筛选出来的mirna负相关的靶基因进行go富集分析和kegg富集分析。
go总共有三个ontology(本体),分别描述转录本的分子功能(molecularfunction)、细胞组分(cellularcomponent)、参与的生物过程(biologicalprocess)。go富集分析,一方面给出差异表达转录本的go功能分类注释;另一方面给出差异表达转录本的go功能显著性富集分析。首先,将差异表达转录本向go数据库(http://www.geneontology.org/)的各term映射,并计算每个term的转录本数,从而得到具有某个go功能的转录本及转录本数目统计。然后应用超几何检验,找出与整个转录本组背景相比,在差异表达转录本中显著富集的go条目,该假设检验的p-value计算公式为:
其中,n为所有unigene中具有go注释的转录本数目;n为n中差异表达转录本的数目;m为所有unigene中注释为某特定goterm的转录本数目;m为注释到某特定的goterm的差异表达转录本数目。计算得到的p-value通过fdr校正之后,以q-value小于等于0.05为阈值,满足该条件的goterm定义为在差异表达转录本中显著富集的goterm;通过go功能显著性富集分析能确定差异表达转录本行使的主要生物学功能;go富集分析结果可以用表格的形式进行概括展示,列明了所有进行比较的基因集名称、细胞组分、分子功能、生物学过程及go分类表;细胞组分、分子功能及生物学过程结果是以html的形式进行保存,go分类表是以excel表格的形式进行保存;具体结果设置的了超链接,点开表格中的具体的某一比较组之间的细胞组分、分子功能和生物学过程富集分析的html结果,就看到详细的富集结果。go功能富集分析结果以表格的形式进行展现,表格的第一列为goterm的id,可以显示这个goterm包含的所有基因,再点击goid,就可以自动链接到http://amigo.geneontology.org官网,可以查看go的具体信息;第二列为goterm的功能描述;第三列为差异表达基因总数(表头数字)、注释到某个goterm的基因数以及占总差异基因数目的百分比;第四列为所有基因总数(表头数字)、注释到某个goterm的基因数以及占总基因数目的百分比;第五列为p-value;第六列为多重检验校正后的p.adjust(即qvalue)。
同时,也可以绘制go分类柱状图,横坐标代表go三个ontology:
分子功能(molecularfunction)、细胞组分(cellularcomponent)
及参与的生物过程(biologicalprocess)的更细以及分类。纵
坐标代表每个分类条目对应的基因数目;还可以绘制go富集分析热图等进行结果展示。
kegg富集分析:kegg是有关pathway的主要公共数据库。pathway显著性富集分析是以keggpathway为单位,应用超几何检验,找出与整个转录本组背景相比,在差异基因表达转录本中显著富集的pathway;p-value计算公式同上述go功能显著性分析相似。经过多重检验校正后,选择qvalue小于等于0.05的pathway定义为在差异表达转录本中显著富集的pathway。q-value和fdr类似,都是对p-value的一种校正;某一个假设检验的q-value就是fdr的最小值,在这些fdr下该假设检验可以被认为是最显著的;kegg富集结果可以以一个表格概括的形式进行呈现,表格中列明了所有进行比较的基因集名称信息、对应比较组之间的pathway富集结果html格式文件名称以及相应的pathway注释表xls格式文件名称。可以点击所关注的比较组pathway富集结果html格式文件名称直接链接到富集结果详细信息,列明了所有的pathway名称,差异表达基因总数(表头数字)、注释到某个pathway的差异基因数及占总差异基因数目的比例,所有基因总数(表头数字)、注释到某个pathway的基因数及占总基因数目的比例,p-value,q-value以及pathwayid等信息。同样点击pathway注释表xls格式文件名称也可以直接查看pathway注释的详细信息;kegg富集分析结果还可以用气泡图的形式进行展示。可以根据详细的kegg富集分析结果选出显著富集的前20的pathway来进行作图。横坐标为富集因子(richfactor),纵坐标为pathway名称;richfactor是指差异表达的转录本中位于该pathway条目的转录本数目与所有转录本中我与该pathway条目的转录本总数的比值,richfactor越大,则代表富集程度越高。q-value是经过多重假设检验校正之后的p-value,范围为0到1,约接近于0,表示显著越富集。
5.cytoscape分析:对于筛选出的负相关的mirna-靶基因对,利用
cytoscape软件通过输入能够识别的格式文件进行相关操作设置绘制出mirna与其负相关靶基因之间的调控网络图。
图2为一种mirna测序和转录组学测序数据的贯穿分析系统的系统架构图。如图2所示,一种mirna测序和转录组学测序数据的贯穿分析系统,包括:
小rna测序分析单元201,用于对实验样本进行转录组测序,对小rna测序数据进行生物信息分析,根据mirna的表达量,筛选得到差异表达的mirna,并对差异表达的mirna进行靶基因预测,可进一步对差异表达的mirna靶基因进行go/kegg功能富集分析;
具体地,包括从两组样本和多组样本中筛选出差异表达的mirna。对于两组样本而言,以p<0.05且|log2fc|>1为标准,对两组样本的mirna表达量进行筛选得到差异表达的mirna;对于多组样本而言,则是以p<0.05且|log2fc|>1为标准进行筛选,取多组样本差异表达的mirna并集;此部分不仅限于筛选得到差异表达的mirna,还包含对差异表达的mirna进行靶基因预测,并对差异表达的mirna的靶基因进行go或者kegg功能富集分析等。
转录组测序分析单元202,用于对实验样本进行转录组测序,并对转录组测序数据进行生物信息分析,根据mrna的表达量,筛选得到差异表达的mrna,可以对差异表达的mrna进行go/kegg功能富集分析;
具体也包括从两组样本和多组样本中筛选出差异表达的mrna。对于两组样本而言,以fdr<0.05且|log2fc|>1为标准,对两组样本的mrna表达量进行筛选得到差异表达的mrna;对于多组学样本而言,则是以fdr<0.05且|log2fc|>1为标准进行筛选,取多组样本差异表达的mrna并集。不仅限于通过mrna表达量筛选得到差异表达的mrna,还可以对差异表达的mrna进行go或者kegg功能富集分析等。
贯穿分析单元203,用于将小rna测序分析单元201获得的差异表达的mirna测序数据和转录组测序分析单元202获得的差异表达的rna测序数据进行贯穿分析。
在本发明具体实施例中,所述贯穿分析包括五个部分:第一、mirna-靶基因对筛选;第二、mirna靶向能力分析;第三、基因结合mirna能力分析;第四、mirna负相关靶基因富集分析;第五、cytoscape分析。
1.mirna-靶基因对筛选:mirna与mrna存在相互调控关系需满足两个条件:1)mirna和mrna存在靶向关系;2)mirna和mrna表达量负相关;根据mirna和靶基因的差异表达结果及两者的靶向关系结果,筛选出表达量负相关的mirna-靶基因对。对于两组样本而言,直接根据mirna和靶基因的差异表达结果及两者的靶向关系结果进行筛选。即即筛选出样本中差异表达的mirna出现上调,其对应靶基因出现下调;或者样本中差异表达的mirna出现下调,其对应的靶基因出现上调的mirna-靶基因对。对于多组样本(三组及以上)而言,具体步骤为结合mirna和靶基因的相关性及两者的靶向关系分析结果,计算皮尔逊相关系数(pearsoncorrelationcoefficient),皮尔逊相关系数可用来度量两个变量之间的相互关系,代表了两个变量共变性的强弱,取值范围为[-1,+1];当皮尔逊相关系数大于0时,表明两个变量正相关;当皮尔逊相关系数小于0时,表明两个变量负相关;相关系数的绝对值越大则表明相关性越强。以相关系数小于-0.7且p值小于0.05为标准筛选出表达量负相关的mirna-靶基因对。
2.mirna靶向能力分析:mirna可以靶向1个基因或者多个基因,mirna靶向基因的数量可在一定程度上反映出mirna的调控能力,对mirna靶向基因数量进行统计;mirna靶向统计表中清晰地展示出了mirnaid、样品和比较组tpm表达量、log2处理差异倍数、检验p值、靶基因数量及mirna对应的靶基因列表等信息;根据统计表信息,可以将mirna靶基因数量为横坐标,以靶向特定数量靶基因的mirna数量为纵坐标,绘制出mirna靶基因数量分布图进行结果展示;可以通过mirna靶向基因的数量直观看出mirna的调控能力。
3.基因结合mirna能力分析:同样地,一个基因也可以与1个或者多个mirna进行结合。基因结合mirna的数量可在一定程度上反映该基因参与调控的能力;因此,我们对基因结合mirna的数量进行统计,从而对基因结合mirna能力进行分析。根据mrna被靶向统计表中的mrna、靶向该基因的mirna的数量等信息,对基因结合mirna的能力进行分析,结果以基因结合mirna数量分布图进行展示,横坐标为被靶向mirna数量,纵坐标为mrna数量。
4.mirna负相关靶基因富集分析:主要包括对上述步骤中筛选出来的mirna负相关的靶基因进行go富集分析和kegg富集分析;go总共有三个ontology(本体),分别描述转录本的分子功能(molecularfunction)、细胞组分(cellularcomponent)、参与的生物过程(biologicalprocess);go富集分析,一方面给出差异表达转录本的go功能分类注释;另一方面给出差异表达转录本的go功能显著性富集分析。首先,将差异表达转录本向go数据库(http://www.geneontology.org/)的各term映射,并计算每个term的转录本数,从而得到具有某个go功能的转录本及转录本数目统计;然后应用超几何检验,找出与整个转录本组背景相比,在差异表达转录本中显著富集的go条目,计算p-value,得到的p-value通过fdr校正之后,以q-value小于等于0.05为阈值,满足该条件的goterm定义为在差异表达转录本中显著富集的goterm。通过go功能显著性富集分析能确定差异表达转录本行使的主要生物学功能。go富集分析结果可以用表格的形式进行概括展示,列明了所有进行比较的基因集名称、细胞组分、分子功能、生物学过程及go分类表。细胞组分、分子功能及生物学过程结果是以html的形式进行保存,go分类表是以excel表格的形式进行保存。具体结果设置的了超链接,点开表格中的具体的某一比较组之间的细胞组分、分子功能和生物学过程富集分析的html结果,就看到详细的富集结果;go功能富集分析结果以表格的形式进行展现,表格的第一列为goterm的id,可以显示这个goterm包含的所有基因,再点击goid,就可以自动链接到http://amigo.geneontology.org官网,可以查看go的具体信息;第二列为goterm的功能描述;第三列为差异表达基因总数(表头数字)、注释到某个goterm的基因数以及占总差异基因数目的百分比;第四列为所有基因总数(表头数字)、注释到某个goterm的基因数以及占总基因数目的百分比;第五列为p-value;第六列为多重检验校正后的p.adjust(即qvalue)。同时,也可以绘制go分类柱状图,横坐标代表go三个ontology:分子功能(molecularfunction)、细胞组分(cellularcomponent)及参与的生物过程(biologicalprocess)的更细以及分类。纵坐标代表每个分类条目对应的基因数目;还可以绘制go富集分析热图等进行结果展示。
kegg富集分析:kegg是有关pathway的主要公共数据库。pathway显著性富集分析是以keggpathway为单位,应用超几何检验,找出与整个转录本组背景相比,在差异基因表达转录本中显著富集的pathway。p-value计算公式同上述go功能显著性分析相似。经过多重检验校正后,选择qvalue小于等于0.05的pathway定义为在差异表达转录本中显著富集的pathway。q-value和fdr类似,都是对p-value的一种校正;某一个假设检验的q-value就是fdr的最小值,在这些fdr下该假设检验可以被认为是最显著的;kegg富集结果可以以一个表格概括的形式进行呈现,表格中列明了所有进行比较的基因集名称信息、对应比较组之间的pathway富集结果html格式文件名称以及相应的pathway注释表xls格式文件名称。可以点击所关注的比较组pathway富集结果html格式文件名称直接链接到富集结果详细信息,列明了所有的pathway名称,差异表达基因总数(表头数字)、注释到某个pathway的差异基因数及占总差异基因数目的比例,所有基因总数(表头数字)、注释到某个pathway的基因数及占总基因数目的比例,p-value,q-value以及pathwayid等信息;同样点击pathway注释表xls格式文件名称也可以直接查看pathway注释的详细信息。kegg富集分析结果还可以用气泡图的形式进行展示。可以根据详细的kegg富集分析结果选出显著富集的前20的pathway来进行作图。横坐标为富集因子(richfactor),纵坐标为pathway名称。richfactor是指差异表达的转录本中位于该pathway条目的转录本数目与所有转录本中我与该pathway条目的转录本总数的比值,richfactor越大,则代表富集程度越高;q-value是经过多重假设检验校正之后的p-value,范围为0到1,约接近于0,表示显著越富集。
5.cytoscape分析:对于筛选出的负相关的mirna-靶基因对,利用cytoscape软件通过输入能够识别的格式文件进行相关操作设置绘制出mirna与其负相关靶基因之间的调控网络图。
综上所述,本发明一种mirna测序和转录组学测序数据的贯穿分析方法及系统通过对样本进行小rna测序和转录组测序,将得到的差异表达的mirna和差异表达的mrna进行贯穿分析,筛选出负调控的mirna-靶基因对,同时构建mirna-靶基因调控网络,并对mirna靶基因进行go/kegg功能富集分析,为后续挑选目标基因提供线索。本发明关联分析针对性强,在小rna测序及转录组测序常规分析的基础之上,针对性地对mirna与其存在负相关的靶基因进行深入分析。有效提高了测序数据利用率,充分深入挖掘了mirna和小rna的数据信息,提高了小rna靶基因预测准确性。直接分析差异表达的mirna和差异表达的mrna负性调控关系,能够得到多个mirna与多个基因的调控网络关系,从不同的层面去挖掘数据之间的潜在联系和有重要研究价值的生物学信息,达到了数据整合的目的。利用cytoscape软件将检测结果进行可视化展示;另外,本发明可将贯穿分析结果以网页版的形式进行展示,使复杂的分析结果条理清晰,便于进行查阅,同一分析结果可以有多种形式进行展示,用户可以根据需要自主选择。同时还可设置超链接,能够帮助客户更加透彻理解分析结果,可以查看关注的结果详细信息。
上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何本领域技术人员均可在不违背本发明的精神及范畴下,对上述实施例进行修饰与改变。因此,本发明的权利保护范围,应如权利要求书所列。
- 还没有人留言评论。精彩留言会获得点赞!