一种蒙花苷包合物及其制备方法和应用与流程
本发明涉及包合
技术领域:
,具体而言,涉及一种蒙花苷包合物及其制备方法和应用。
背景技术:
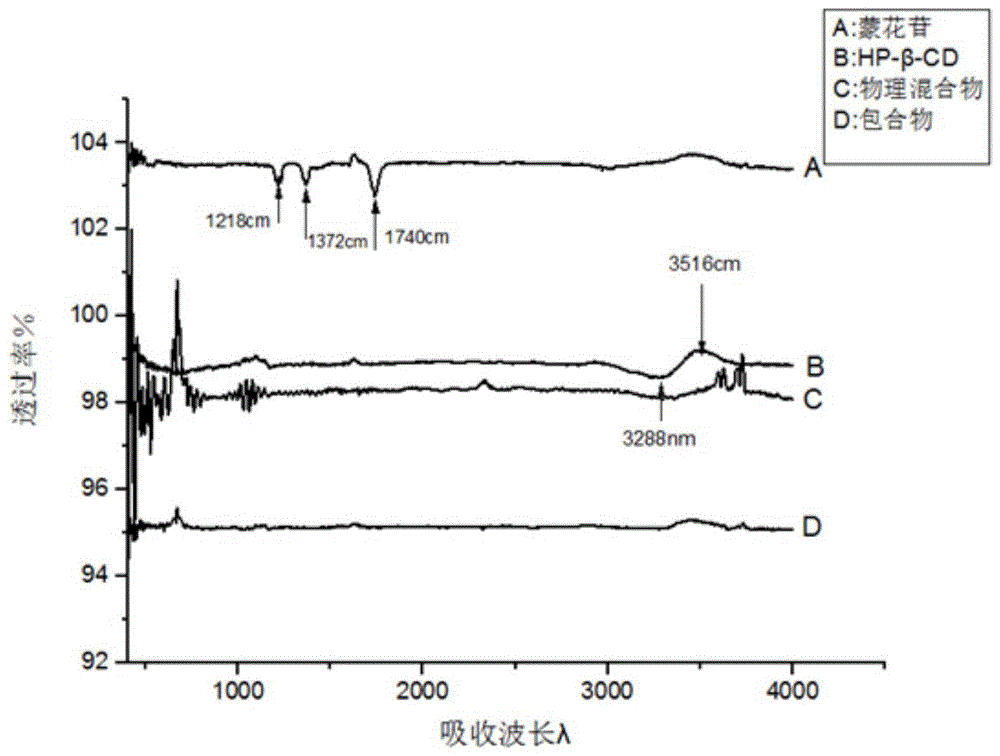
:蒙花苷是一种广泛存在于菊科野菊花、密蒙花等植物中的天然黄酮苷类成分。其具有广泛的生理活性和药理活性,可预防糖尿病和改善糖尿病,并且还具有抑制醛糖还原酶、磷酸二酯酶、金黄色葡萄球菌、乙型溶血链球菌等作用,能保护肝脏,并对慢性气管炎有一定疗效。还具有抗衰老、抗缺氧作用、抗不良刺激作用、对机体双向调节作用、抗疲劳作用。但是蒙花苷属于热敏性化合物,易受热降解,不溶于水,脂溶性差。因此,限制其在药物中的应用,其制备的药物的生物利用度较低。有鉴于此,特提出本发明。技术实现要素:本发明的第一目的在于提供一种蒙花苷包合物的制备方法,包括以下步骤:a)将蒙花苷溶于有机溶剂制成蒙花苷溶液;b)将包合材料溶于水制成包合材料溶液;c)将所述蒙花苷溶液与包合材料溶液加热搅拌进行包合;步骤a)与b)无先后顺序;所述包合材料选自环糊精和环糊精衍生物中的至少一种;所述蒙花苷和所述包合材料的质量比为0.5-1.6:8-32;所述加热的温度为40-60℃;所述搅拌的时间为2-9h,搅拌的转速为300-600rpm。本发明的蒙花苷包合物的制备方法可以解决因蒙花苷溶解性较差限制其制备药物的生物利用度的问题。本发明的蒙花苷包合物的制备方法将蒙花苷、有机溶剂、包合材料和水混合,采用特定的投料比,通过特定的加热温度、搅拌转速及搅拌时间,将蒙花苷包覆于包合材料中。该方法操作简单,通过该方法可得到较高载药量的包合物。根据本发明的一个方面,本发明还涉及一种如上所述的蒙花苷包合物的制备方法制备得到的蒙花苷包合物。该蒙花苷包合物具有较高的载药量及优异的抗炎和抗过敏效果。根据本发明的另一个方面,本发明还涉及一种抗炎和/或抗过敏药物,包含如上所述的蒙花苷包合物。本发明的蒙花苷包合物可用于制备具有抗炎和/或抗过敏作用的药物,具有较好的抗炎抗过敏效果,提高药物的生物利用度。与现有技术相比,本发明的有益效果为:(1)本发明将蒙花苷、有机溶剂、包合材料和水混合,通过采用特定的蒙花苷和包合材料的配比,特定的加热温度、搅拌转速及搅拌时间,利用溶解度较好的环糊精和/或环糊精衍生物作为包合材料去包覆蒙花苷,利用旋蒸的方式去除溶剂,通过特定的冷冻干燥条件冷冻成粉末,得到蒙花苷包合物。(2)本发明采用包合技术,利用水溶性较好的环糊精和/或环糊精衍生物作为包合材料去包覆蒙花苷,可提高蒙花苷药物的稳定性,增大药物的溶解度,降低药物的刺激性与毒副作用,调节药物的释放速度,提高药物的生物利用度。通过蒙花苷和包合材料的配合作用,可获得具有较高蒙花苷负载量的包合物,该包合物具有较好的抗炎和抗过敏作用。(3)本发明中的蒙花苷包合物可用于制备具有抗炎、抗过敏功效的药物,具有较好的抗炎和抗过敏效果,提高药物的生物利用度。附图说明图1为本发明中蒙花苷、羟丙基-β-环糊精、实施例1蒙花苷包合物、蒙花苷与羟丙基-β-环糊精物理混合物的红外光谱图;图2为本发明蒙花苷的dsc图;图3为本发明羟丙基-β-环糊精的dsc图;图4为本发明蒙花苷和羟丙基-β-环糊精的物理混合物dsc图;图5为本发明实施例1中蒙花苷和羟丙基-β-环糊精包合物的dsc图;图6为本发明实施例1中蒙花苷和羟丙基-β-环糊精包合物的xrd图;图7为本发明蒙花苷的xrd图;图8为本发明羟丙基-β-环糊精的xrd图;图9为本发明蒙花苷和羟丙基-β-环糊精物理混合物的xrd图;图10为本发明蒙花苷包合物的体外释放的测试结果;图11为本发明中蒙花苷甲醇溶液的标准曲线图。具体实施方式下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限制本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。一种蒙花苷包合物的制备方法,包括以下步骤:a)将蒙花苷溶于有机溶剂制成蒙花苷溶液;b)将包合材料溶于水制成包合材料溶液;c)将所述蒙花苷溶液与包合材料溶液加热搅拌进行包合;步骤a)与b)无先后顺序;所述包合材料选自环糊精和环糊精衍生物中的至少一种;所述蒙花苷和所述包合材料的质量比为0.5-1.6:8-32;所述加热的温度为40-60℃;所述搅拌的时间为2-9h,搅拌的转速为300-600rpm。本发明为了改善蒙花苷溶解度差的问题,采用包合技术,包合技术是指一种分子被包藏于另一种分子的空穴结构内,形成包合物的技术。这种包合物是由主分子和客分子两种组分组成,主分子即是包合材料,具有较大的空穴结构,足以将客分子容纳在内,形成分子囊。通过采用特定的蒙花苷和包合材料的配比,特定的加热温度、搅拌转速及搅拌时间,利用溶解度较好的环糊精衍生物作为包合材料去包覆蒙花苷,利用蒸发的方式去除溶剂,通过特定的冷冻干燥条件冷冻成粉末,得到蒙花苷包合物。本发明利用水溶性较好的环糊精和环糊精衍生物中的至少一种物质作为包合材料包覆蒙花苷,进而改善蒙花苷溶解度。在一些实施方式中,所述蒙花苷和所述包合材料的质量比为0.5-1.6:8-32,还可以选择0.5:10、0.5:15、0.5:20、0.5:30、1:10、1:15、1:20、1:25、1.6;10、1.6:20或1.6:30。在一些实施方式中,所述加热的温度为40-60℃,还可以选择42℃、45℃、47℃、50℃、52℃、55℃、57℃或59℃。在一些实施方式中,所述搅拌的时间为1-9h,还可以选择2h、3h、4h、5h、6h、7h或8h。在一些实施方式中,所述搅拌的转速为300-600rpm,还可以选择350rpm、400rpm、450rpm、500rpm、550rpm或590rpm。优选地,所述包合材料选自甲基-β-环糊精、羟丙基-β-环糊精、β-环糊精和羟乙基-β-环糊精中的至少一种。环糊精的一个重要特点就是既能在水溶液中也能在固体状态下将客分子包合在疏水性的空腔而形成包合物。在环糊精的衍生物中,甲基-β-环糊精、羟丙基-β-环糊精和羟乙基-β-环糊精具有较高的水溶性。可利用这些物质作为包合物进行包覆蒙花苷,提高蒙花苷的水溶性。其中,羟丙基-β-环糊精刺激性小,优选使用羟丙基-β-环糊精。优选地,所述蒙花苷和所述包合材料的质量比为0.8-1.2:10-25。通过进一步优选蒙花苷和所述包合材料的质量比,可得到载药量更高的包合物。优选地,所述搅拌之后静置12-24h。通过静置可进一步将未反应的蒙花苷进行分离,并通过过滤进行去除。在一些实施方式中,所述搅拌之后静置12-24h,还可以选择静置13h、14h、15h、16h、17h、18h、19h、20h、21h、22h或23h。优选地,所述包合后采用蒸发的方式去除溶剂,所述蒸发后静置1-4h,所述蒸发优选为旋蒸。本发明去除溶剂后,静置1-4h,使溶液冷却到室温。在一些实施方式中,所述旋蒸后静置1-4h,还可以选择静置2h、2.5h、3h或3.5h。优选地,所述有机溶剂选自甲醇、乙醇和吡啶中的至少一种。优选地,所述有机溶剂为乙醇。通过采用甲醇、乙醇和吡啶中的至少一种物质作为有机溶剂,可更好的溶解蒙花苷。优选使用乙醇,其毒性较低,并且可更好的溶解蒙花苷。优选地,所述蒙花苷包合物的制备方法还包括冻干的操作,所述冻干的温度为冻干的时间为24-48h。本发明进一步采用特定的冻干温度和时间可更好的去除水分,得到包合物。在一些实施方式中,所述冻干的温度为还可以选择-76℃、-77℃、-78℃或-79℃。在一些实施方式中,所述冻干的时间为24-48h,还可以选择25h、27h、30h、32h、35h、37h、40h、42h、45h或47h。在一种优选地实施方式中,所述蒙花苷包合物的制备方法,包括以下步骤:(a)将蒙花苷溶于有机溶剂中得到蒙花苷溶液;(b)将包合材料溶于水中的到包合材料溶液;(c)将步骤(a)和步骤(b)的溶液混合,在温度为40-60℃的条件下搅拌1-9h,搅拌转速为300-600rpm,室温下静置12-24h,采用旋蒸的方式去除溶剂,静置1-4h冷却到室温,在的条件下冷冻24-48h,得到蒙花苷包合物;所述包合材料选自环糊精和环糊精衍生物中的至少一种;所述蒙花苷和所述包合材料的质量比为0.5-1.6:8-32。如上所述的蒙花苷包合物的制备方法制备得到的蒙花苷包合物。该包合物具有较高的载药量,具有较好的抗炎和抗过敏效果。一种抗炎和/或抗过敏药物,包含如上所述的蒙花苷包合物。优选地,所述药物为眼用药物;优选的,所述眼用药物包括0.5-3重量份的所述蒙花苷包合物和10-30重量份的人工泪液。所述人工泪液包括如下重量份数的组分:碳酸氢钠1.5-3份、氯化钠5-7份、二水氯化钙0.05-0.1份和氯化钾1-2份。所述人工泪液的ph为7.2-7.6。所述所述人工泪液的制备方法,包括以下步骤:将碳酸氢钠、氯化钠、二水氯化钙、氯化钾和水混合,定容,得到人工泪液。该眼用制剂可以改变角膜的通透性及药物与眼组织的结合性,同时通过对药物的包合,能减少药物对眼的刺激及副作用。下面将结合具体的实施例、对比例及附图,对本发明作进一步说明。实施例1一种蒙花苷包合物的制备方法,包括如下步骤:(a)将蒙花苷溶于乙醇中得到蒙花苷溶液;(b)将羟丙基-β-环糊精溶于水中得到羟丙基-β-环糊精的水溶液;(c)将步骤(a)的蒙花苷溶液滴加入步骤(b)的羟丙基-β-环糊精的水溶液中,在温度为60℃的条件下搅拌4h,搅拌转速为400rpm,室温下静置12h,采用旋蒸的方式去除溶剂,静置1.5h冷却到室温,在-80℃的条件下冷冻24h,得到蒙花苷包合物;所述蒙花苷、乙醇份、羟丙基-β-环糊精和水的质量比为0.5:10:24:10。实施例2一种所述蒙花苷包合物的制备方法,包括如下步骤:(a)将羟丙基-β-环糊精溶于水中得到羟丙基-β-环糊精水溶液;(b)将蒙花苷溶于乙醇得到蒙花苷溶液;(c)将步骤(a)的蒙花苷溶液滴加入步骤(b)的羟丙基-β-环糊精的水溶液中,在温度为50℃的条件下搅拌6h,搅拌转速为500rpm,室温下静置12h,采用旋蒸的方式去除溶剂,静置1h冷却到室温,在-80℃的条件下冷冻24h,得到蒙花苷包合物;所述蒙花苷、乙醇份、羟丙基-β-环糊精和水的质量比为0.5:10:15.5:20。实施例3一种蒙花苷包合物的制备方法,包括如下步骤:(a)将蒙花苷溶于甲醇中得到蒙花苷溶液;(b)将羟丙基-β-环糊精溶于水中得到羟丙基-β-环糊精水溶液;(c)将步骤(a)的蒙花苷溶液滴加入步骤(b)的羟丙基-β-环糊精的水溶液中,在温度为50℃的条件下搅拌3h,搅拌转速为450rpm,室温下静置12h,采用旋蒸的方式去除溶剂,静置2h冷却到室温,在-80℃的条件下冷冻24h,得到蒙花苷包合物;所述蒙花苷、甲醇、羟丙基-β-环糊精和水的质量比为0.5:10:24:10。实施例4一种蒙花苷包合物的制备方法,包括如下步骤:(a)将蒙花苷溶于乙醇中得到蒙花苷溶液;(b)将羟乙基-β-环糊精溶于水中得到羟乙基-β-环糊精水溶液;(c)将步骤(a)的蒙花苷溶液滴加入步骤(b)的羟乙基-β-环糊精的水溶液中,在温度为45℃的条件下搅拌9h,搅拌转速为400rpm,室温下静置24h,采用旋蒸的方式去除溶剂,静置2h冷却到室温,在-80℃的条件下冷冻48h,得到蒙花苷包合物;所述蒙花苷、乙醇、羟乙基-β-环糊精和水的质量比为1.6:20:32:30。实施例5一种蒙花苷包合物的制备方法,包括如下步骤:(a)将蒙花苷溶于吡啶中得到蒙花苷溶液;(b)将β-环糊精溶于水中得到β-环糊精水溶;(c)将步骤(a)的蒙花苷溶液滴加入步骤(b)的β-环糊精的水溶液中,在温度为55℃的条件下搅拌2h,搅拌转速为300rpm,室温下静置18h,采用旋蒸的方式去除溶剂,静置4h冷却到室温,在-80℃的条件下冷冻30h,得到蒙花苷包合物;所述蒙花苷、吡啶、β-环糊精和水的质量比为0.5:5:8:10。实施例6一种蒙花苷包合物的制备方法,除蒙花苷、吡啶、β-环糊精和水的质量比为0.8:8:10:13以外,其他操作步骤与实施例1相同。实施例7一种蒙花苷包合物的制备方法,除蒙花苷、吡啶、β-环糊精和水的质量比为1.2:15:25:22以外,其他操作步骤与实施例1相同。实施例8一种蒙花苷包合物的制备方法,除反应温度为50℃以外,其他操作步骤与实施例2相同。实施例9一种眼用药物,包括如下重量分数的组分:2重量份蒙花苷包合物和20重量份的人工泪液;所述人工泪液的制备方法,包括以下步骤:将碳酸氢钠2.18g、氯化钠6.78g、二水氯化钙0.084g和氯化钾1.38g与1l水混合,得到人工泪液,所述人工泪液的ph为7.4;所述蒙花苷包合物为实施例1制备得到的蒙花苷包合物。实施例10一种眼用药物,包括如下重量分数的组分:0.5重量份蒙花苷包合物和30重量份的人工泪液;所述人工泪液的制备方法,包括以下步骤:将碳酸氢钠2.5g、氯化钠6.5g、二水氯化钙0.096g和氯化钾1.54g与1l水混合,得到人工泪液,所述人工泪液的ph为7.3;所述蒙花苷包合物为实施例1制备得到的蒙花苷包合物。实施例11一种眼用药物,包括如下重量分数的组分:1重量份蒙花苷包合物和10重量份的人工泪液;所述人工泪液的制备方法,包括以下步骤:将碳酸氢钠2.8g、氯化钠5.59g、二水氯化钙0.075g和氯化钾1.18g与1l水混合,得到人工泪液,所述人工泪液的ph为7.4;所述蒙花苷包合物为实施例1制备得到的蒙花苷包合物。对比例1一种蒙花苷包合物的制备方法,所述蒙花苷和所述包合材料的质量比为0.2:40,其他操作步骤与实施例1相同。对比例2一种蒙花苷包合物的制备方法,所述蒙花苷和所述包合材料的质量比为2:7.5,其他操作步骤与实施例1相同。试验例1.利用红外光谱分别检测蒙花苷、羟丙基-β-环糊精、实施例1蒙花苷包合物、蒙花苷与羟丙基-β-环糊精物理混合物的图谱,测试结果如图1所示。由图1的红外光谱可以看出,通过本发明的制备方法成功利用包合材料将蒙花苷包覆其中,得到蒙花苷包合物。2.利用差示扫描量热法(dsc)验证包合物形成,其中,图2为蒙花苷的dsc图,图3为羟丙基-β-环糊精的dsc图,图4为蒙花苷和羟丙基-β-环糊精的物理混合dsc图,图5为实施例1中蒙花苷和羟丙基-β-环糊精包合物的dsc图。当药物分子部分或全部地包合到环糊精腔内时,药物分子的熔点、沸点和升华点都会移至不同的温度,甚至消失。在dsc热分析图可以看出,蒙花苷在272.9℃处有一个尖锐的分解峰,羟丙基-β-环糊精的分解峰在356.1℃,形成包合物的后,蒙花苷特征吸热峰消失,包合物最大分解峰移至了更低的温度334.8℃处,说明蒙花苷和羟丙基-β-环糊精发生了作用,药物分子取代了环糊精腔内的水分子。在此基础上,研究结果表明,该药物的疏水性部分已成功进入到环糊精的腔内,形成了无定型的包合物。3.利用x射线衍射(xrd)进行分析,其中,图6表示实施例1中蒙花苷和羟丙基-β-环糊精包合物的xrd图,图7表示蒙花苷的xrd图,图8表示羟丙基-β-环糊精的xrd图,图9表示蒙花苷和羟丙基-β-环糊精物理混合物的xrd图。设置实验条件为cukα(k=0.15460nm),40ma,45kv,测试温度为室温,扫描速率6°/min,步长0.02°,扫描范围从5°~90°。由xrd图可以看出,蒙花苷具有良好的晶型结构:在12.2°、15.6°、19.8°、22.4°、25°处有较强的衍射峰。羟丙基-β-环糊精无定形结构,在2θ:14.00°~25.00°出现宽大的特征峰,从物理混合物的图显示了物理混合物的衍射峰基本上是蒙花苷与羟丙基-β-环糊精的简单叠加,蒙花苷较强的衍射峰与羟丙基-β-环糊精较宽的衍射峰都清晰可见。蒙花苷与羟丙基-β-环糊精形成包合后的衍射峰如图6所示,蒙花苷在2θ:10°~30°的大部分晶体衍射峰都已基本消失,蒙花苷与羟丙基-β-环糊精形成包合物后,其结构在某种程度上重新排列,比起单一的蒙花苷具有更显著的不定形结构,说明了蒙花苷与羟丙基-β-环糊精之间生成了包合物。4.蒙花苷包合物的体外释放的测试:透析袋:分子截留量为1000;释放介质:超纯水、ph=7.2-7.4的pbs、人工泪液;精密称取实施例1制得的蒙花苷包合物1.4mg,分别溶于5ml超纯水、ph=7.2-7.4的pbs、人工泪液中,置于透析袋中,扎紧透析袋两端,将透析袋置于200ml释放介质中,在37±1℃水浴中恒温振荡,转速为100r·min-1,于10、20、30、60、120、180、240、300、360、420、480、600min及720min精密移取3ml外相介质,并同时补充3ml新鲜介质,样品经0.22μm微孔滤膜过滤后,在326nm处测定样品吸光度,以包合物的累积释放量对时间作曲线图,绘制体外释放曲线,如图10所示。(为达到漏槽条件,以含有1%吐温-80的人工泪液为溶出介质)。根据此公式累计释放计算累积释放量,结果如图10所示,包合物在超纯水、pbs、人工泪液中2h的累计释放率分别为45.72%、72.56%、32.87%,包合物在人工泪液中的累积释放量在2h时最小,说明包合物作用于眼睛中能更好的充当药物释放的“栅栏”作用,保持药物持续缓慢的释放,达到良好的治疗效果。5.载药量的测定:将实施例1-8和对比例制备得到的蒙花苷包合物进行载药量的测定,测试结果如表1所示。载药量测定的具体步骤:(a)精密称取1mg的蒙花苷溶于10ml甲醇置于容量瓶中,制成0.1mg/ml的蒙花苷甲醇溶液,稀释一定倍数后,得到0.01mg/ml-0.1mg/ml的蒙花苷甲醇溶液。高效液相色谱法在326nm波长处测得各个浓度的峰面积,绘制峰面积-浓度的标准曲线,如图11所示,得到的标准曲线方程为:y=17518522.47x-12723.8274,r2=0.9992,其中,x表示蒙花苷甲醇溶液的浓度,y表示峰面积。(b)取0.5mg-1.5mg实施例1-8制备得到的蒙花苷包合物,用甲醇进行复溶,用高效液相色谱法及步骤(a)的标准曲线方程,测得包进包合材料的蒙花苷含量。表1实施例1-8和对比例制备得到的蒙花苷包合物的载药量百分比实施例和对比例载药量百分比(%)实施例13.9实施例21.3实施例32.8实施例41.2实施例51.1实施例64.2实施例74.5实施例81.8对比例10.4对比例20.6由表1可以看出,本发明通过特定的组分、配比及特定的制备方法得到的蒙花苷包合物具有较高的载药量,载药量最高可达到4.5%,可更好的发挥药物的生物利用度。6.蒙花苷包合物的细胞毒性测试,测试结果如表2所示。药物的细胞毒性研究是考察药物体外抗炎作用必不可少的环节。药物可通过影响细胞通路抑制炎症细胞因子或者炎性介质的产生,从而发挥抗炎作用。然而,药物如果对细胞的毒性作用存在,会抑制细胞的增殖,导致细胞的数量和功能下降,从而表现出抑制炎症细胞因子或炎性介质分泌的作用,误导人们对药物药效的有效判断。因此,考察并筛选药物的无毒剂量是进一步研究药物抗炎作用的前提和基础。蒙花苷的细胞毒性实验(cck-8法):将巨噬细胞培养至合适的状态,制备细胞悬液铺于96孔板中,培养24h至细胞贴壁→制备梯度浓度的蒙花苷溶液,每孔加入100μl,放入培养箱培养24h→每孔加入10μlcck-8液,于培养箱中继续培养4h→酶标仪检测450nm处的吸光度。吸光度相关数据如下:样品组:含细胞悬液,培养基、药物、cck-8,蒙花苷包合物的药物浓度分别为:5μm、10μm、20μm、30μm、40μm、50μm;空白组:含细胞悬液、培养基、cck-8;调零组:含培养基、cck-8;表2蒙花苷包合物的细胞毒性测试由表2可以看出,本发明中蒙花苷的无毒浓度为小于30μm,当蒙花苷的浓度在30μm以上会抑制细胞的增值,具有较高的毒性。因此,本发明在利用蒙花苷包合物制备具有抗炎和/或抗过敏药物时,要控制蒙花苷的释放浓度不超过30μm。7.抗炎活性实验在诱发炎症反应的诸多因素中,内毒素是激发炎症反应的一类重要的触发剂,它是一种革兰氏阴性菌细胞壁的组成成分,其化学本质为脂多糖(lps)。肿瘤坏死因子tnf-a是炎症反应过程中产生的最重要细胞因子之一,具有很强的炎症损伤作用。因此,通过选取细胞因子tnf-a作为抗炎活性研究指标,可判断药物抗炎作用效果。蒙花苷包合物的抗炎活性实验方法,包括以下步骤:a)使用1μg/ml脂多糖(lps)刺激raw264.7小鼠巨噬细胞系,构建炎症细胞模型;b)将raw264.7细胞按细胞数1.5×104/孔接种于96-孔板内,培养24h后,弃去培养液;c)用含不同浓度的蒙花苷的蒙花苷包合物共同刺激24h后,收集细胞上清液进行elisa实验,测定肿瘤坏死因子(tnf-α)水平,蒙花苷包合物采用实施例1制备得到的。抗炎活性测试结果如表3所示。表3蒙花苷包合物抗炎活性测试结果由表3实验结果显示,脂多糖(lps)可以较好刺激细胞并使细胞因子tnf-α的分泌水平増加,与lps对照组比较,蒙花苷包合物在5-20μm时能够显著抑制tnf-α的分泌,达到较好的抗炎效果。利用本发明实施例2-8得到的蒙花苷包合物进行抗炎活性实验,同样能够显著抑制tnf-α的分泌,具有优异的抗炎效果。最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,但本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1