本发明涉及药物设计领域,尤其是涉及一种蛋白质、水分子和配体的对接方法。
背景技术:
传统分子对接方法多是通过忽略水分子,或者将其作为受体的一部分仅考虑其静止状态这些方式对水分子进行隐式或显式地考虑。虽然这些策略能够不同程度地简化计算模型进而降低计算复杂度,然而均会对受体-配体的结合模式预测以及二者结合亲和力计算的精确性产生影响。而在蛋白质与配体对接过程中,综合考虑参与其相互作用的水分子的数量变化或动态运动等多个因素,进而能够逼近真实的结合过程。
考虑水分子参与蛋白质-配体对接的多体对接方法主要包括以下三类:
1)系统搜索方法。该方法从配体和蛋白质的起始构象出发,按照预定义的步长,对整个结合构象的构型空间进行系统遍历性采样,进而获得配体-蛋白质最佳的结合模式。典型的系统搜索方法如片段生长法、构象搜索法、构象库法。如,dock引入“锚优先”(anchorfirst)构型搜索方法,通过形状匹配首先将锚最大的刚性片段对接到受体结合位点,柔性侧链则分层依次加上。然后对二面角的自由度进行分析并经过最小化,采用修剪算法过滤不合理的构象。最后采用单纯型优化算法对结合模式进行优化。在此基础上,dock在模拟配体与蛋白质相互作用时设置一个开关(on/off),可以自由选择是否保留水分子。
2)随机搜索方法。该方法采用不同的随机算法对起始构象进行随机扰动从而生成新的构象,然后对新生成的构象进行能量优化进而得到其附近的局部极小点。与系统搜索方法相比,随机搜索方法不仅采样效率高,能够较快地找到全局最优的结合模式,并且构型空间的增长对随机搜索方法的优化效率制约性较小。典型的随机搜索方法如遗传算法、蒙特卡洛法、禁忌搜索。
gold在传统遗传算法中引入子种群(sub-population)策略,将初始种群划分为多个子种群。然后引入迁移岛(migrationislands)策略,允许各个子种群之间的个体进行迁移,从而可以预测水分子介导蛋白质-配体之间的结合模式。特别地,可以选择是否考虑水分子参与对接以及是否绕着它的三个主轴旋转。若考虑水分子参与,则采用一个代表刚性熵损失的固定罚值来奖赏水分子的取代。autodock采用拉马克遗传算法进行构象搜索,其中遗传算法用于全局搜索,而局部优化只用于优化遗传选择操作产生的解,当且仅当映射函数逆转时,将局部优化结果的表现型转化为相应的基因型,并代替原来的基因型。此外,autodock将预测得到的水分子作为配体的一部分固定,通过计算蛋白质与配体之间的交互能,并采用含水的分子力场理论计算游离的水分子与配体结合时的熵和焓值,从而提高结合模式的预测精度及对接性能。essex等采用蒙特卡洛模拟方法通过复型交换热力学积分来计算水分子的结合自由能。研究发现,保守水分子一般比游离水分子结合的更加紧密。此外,通过采用贝叶斯统计方法计算水分子被配体取代的概率,进而可以定量评价某一给定的水分子。
3)确定性搜索方法。该方法的求解过程为从同一始态出发,最终到达同一终态。其中,分子动力学模拟是一种典型的确定性搜索方法。它建立在经典力学的基础上,通过计算各种分子体系在不同外界(如温度、压力、电场等)条件影响下,分子呈现各种状态时的能量分布,由此推算分子在真实实验体系中出现的最大概率状态(最低能量状态)和可能出现的状态或过渡态(能量高于最低能量态时的状态)。
watermap采用分子动力学模拟及不均匀溶解理论计算配体与蛋白结合时水分子的熵和焓值。在此基础上,wscore采用watermap获得水分子的位置信息及热力学信息,同时采用一个计算配体与蛋白相互作用能的模型,并将这些能量项与mm-gbsa(molecularmechanical-generalizedbornsurfacearea)打分函数结合,进而可以对显式考虑水分子参与的对接结果进行评估。watsite采用分子动力学模拟的方法,根据自由能分布曲线图计算每个水分子的熵和焓值,进而确定水分子可能存在的位置。在此基础上,watsite3.0通过结合watsite的分子动力学模拟方法与基于3d-rism的水分子预测方法,提出成为一种精确而高效的水分子位点预测方法。olson等将水分子作为配体的一部分参与对接,然后采用分子动力学模拟的方法计算每个水分子结合配体时的熵和焓值,从而能够区分晶体结构中的游离水分子和结合水分子。
系统搜索方法由于加入预判规则进行过滤操作,一般适用于研究柔性尺度较小的分子;对于一些柔性尺度较大的分子,考虑到柔性尺度与构象空间的尺寸呈指数型函数关系,柔性单元的增长将会使构象空间急速膨胀,从而导致计算复杂度提升或系统搜索任务无法完成。
随机搜索方法出于随机性的本质,其执行效率比较低,并且容易受算法稳定性的影响,从而导致计算容易陷入局部最优。此外,由于随机搜索方法其搜索和优化方向由打分函数直接决定,因此该方法对打分函数的准确性和敏感性要求比较高。
确定性搜索方法可以跨越较大的能量壁垒,因此被认为是一种有效的构象搜索方法。然而这类方法仍然存在计算复杂度高,且容易陷入局部最小值中等缺陷。因此,这类方法不适合化合物库特别是大规模化合物库进行虚拟筛选。
技术实现要素:
本发明的目的就是为了克服上述现有技术存在的缺陷而提供一种适用于不确定分子个数、可避免陷入局部最优且计算复杂度小的蛋白质、水分子和配体的对接方法。
本发明的目的可以通过以下技术方案来实现:
一种蛋白质、水分子和配体的对接方法,该方法包括以下步骤:
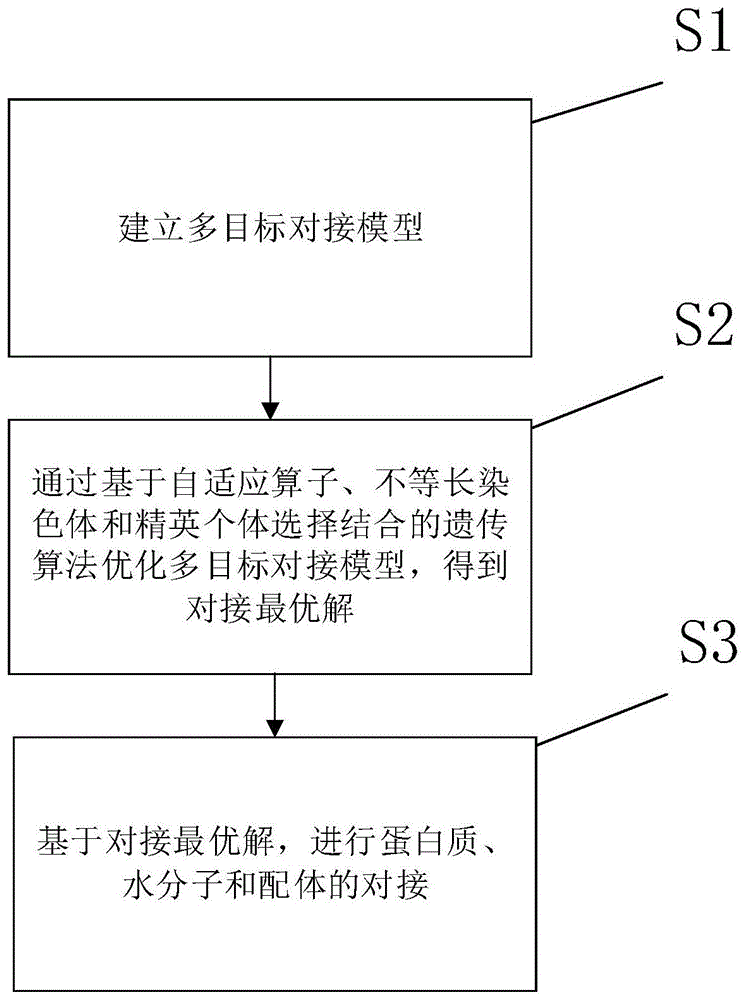
步骤s1:建立多目标对接模型;
步骤s2:通过基于自适应算子、不等长染色体和精英个体选择结合的遗传算法优化多目标对接模型,得到对接最优解;
步骤s3:基于对接最优解,进行蛋白质、水分子和配体的对接。
所述多目标对接模型的目标函数为:
miny=f(x)=(f1(x),f2(x))
其中,f1(x)为蛋白质与配体、蛋白质与水分子之间的相互作用能,f2(x)为配体内部、配体与水分子,以及水分子与水分子之间的相互作用能。
所述多目标对接模型的约束条件为:
e(x)=(e1(x),e2(x),...,ek(x))≤0
x={x1,x2,...,xm}t∈s
其中,e(x)为模型的约束集合,k为约束个数,s为决策向量的范围,x为决策向量且表示为:
x={x1,x2,…,xm}t={p1,p2,tx,ty,tz,rx,ry,rz,rb1,…,rbn,wm,wx1,wy1,wz1,…,wxm,wym,wzm}t
其中,p1、p2分别为交叉概率和变异概率的自由度,(tx,ty,tz)和(rx,ry,rz)分别为配体的平动自由度和转动自由度,(rb1,…,rbn)为配体的n个旋转键的旋转角度,wm为标识位自由度,(wx1,wy1,wz1,…,wxm,wym,wzm)为m个水分子的平动自由度,且满足下式:
plow≤p1,p2≤pup
xlow≤tx,wx1,...,wxm≤xup
ylow≤ty,wy1,...,wym≤yup
zlow≤tz,wz1,...,wzm≤zup
0≤rx,ry,rz,rb1,...,rbn≤2π
其中,pup为交叉概率或变异概率自由度的上限,plow为交叉概率或变异概率自由度的下限,xup,yup和zup为配体和水分子的平动自由度的上限,xlow,ylow,zlow为配体和水分子的平动自由度的下限。
所述的自适应算子包括交叉概率和变异概率,所述交叉概率和变异概率与多目标对接模型的目标函数相关。
所述的精英个体选择基于排挤机制和循环拥挤距离。
所述的不等长染色体包括遗传算子基因、配体基因、水分子基因和标记水分子数量的标识位基因。
所述的步骤s2包括:
步骤s21:得到初始参数、初始蛋白质基因、初始水分子基因和初始配体基因;
步骤s22:基于初始参数、初始蛋白质基因、初始水分子基因和初始配体基因,得到初始父代种群;
步骤s23:对初始父代种群依次进行选择、不等长染色体交叉、变异和合并后,计算目标函数值;
步骤s24:基于目标函数值,通过精英个体选择得到子代种群;
步骤s25:子代种群代替初始父代种群,重复步骤s23-步骤s25,直至满足终止条件,得到对接最优解。
所述的不等长染色体交叉包括:
比较参与交叉的两条父代染色体,在基因个数少的染色体上产生交叉位点;
判断交叉位点的位置,若交叉位点位于遗传算子基因或配体基因,交换两条父代染色体的交叉位点之后的所有基因,若交叉位点位于水分子基因或标记水分子数量的标识位基因,变换两条父代染色体的标识位基因和交叉位点之后的所有基因。
与现有技术相比,本发明具有以下优点:
(1)通过采用自适应算子策略为多目标优化算法中的控制参数赋值,进而可以避免反复调试参数,从而降低计算的复杂度。
(2)通过采用不等长染色体设计与交叉策略,进而可以解决多体对接过程中水分子的个数不确定问题。
(3)通过采用基于排挤机制与循环拥挤距离结合的精英个体选择策略,从而可以增加精英个体数量和多样性,并且能够避免种群陷入局部最优,进而提高算法的搜索性能。
(4)通过引入多目标对接模型,开展蛋白质-水分子-配体三者之间的多体优化问题研究,为进一步开发精确度良好的结合模式预测的药物设计方法奠定基础,同时也为多学科之间的交叉互补提供一种合理可行的研究策略。
(5)通过自适应算子、不等长染色体交叉和基于排挤机制与循环拥挤距离结合的精英个体选择策略的三者的有机结合,从而实现蛋白质、水分子和配体的最优对接。
附图说明
图1为本发明的流程图;
图2(a)为本发明的交叉位点位于遗传算子基因或配体基因时不等长染色体交叉示意图;
图2(b)为本发明的交叉位点位于水分子基因或标识位基因时不等长染色体交叉示意图;
图3(a)为选取每一级非支配前沿的全部个体作为下一代进化父代种群时的精英个体选择示意图;
图3(b)为本发明的选取每一级非支配前沿的部分个体作为下一代进化父代种群时的精英个体选择示意图;
图4为本发明的遗传算法优化流程图。
具体实施方式
下面结合附图和具体实施例对本发明进行详细说明。本实施例以本发明技术方案为前提进行实施,给出了详细的实施方式和具体的操作过程,但本发明的保护范围不限于下述的实施例。
实施例
本实施例提供一种适用于不确定分子个数、可避免陷入局部最优且计算复杂度小的蛋白质、水分子和配体的对接方法,如图1所示,包括以下步骤:
步骤s1:建立多目标对接模型;
步骤s2:通过基于自适应算子、不等长染色体和精英个体选择结合的遗传算法优化多目标对接模型,得到对接最优解;
步骤s3:基于对接最优解,进行蛋白质、水分子和配体的对接。
具体而言:
1)自适应算子
对于遗传算法中交叉概率和变异概率这两个重要的控制参数,让二者作为优化设计变量参与整个进化过程,使二者的变化与多目标对接模型的目标函数直接相关。假设原优化设计变量个数为n,则实际参与处理的变量个数变为n+2。在进化过程中,对于参与交叉的染色体个体对,可以采用随机策略选择其中一个个体的交叉概率进行交叉,而每个个体均根据自己的变异概率进行变异。这样只需要凭借传统经验给定的控制参数的大致范围即可避免反复调试。
2)不等长染色体的设计与交叉
遗传算法中染色体的组成包括遗传算子基因、配体基因、水分子基因,以及用来标记水分子数量的标识位基因。根据基因数量变化情况及所表示的含义可以将基因分成以下两部分:(i)固定长度的基因序列:包括染色体中基因数量固定的遗传算子基因(包括交叉概率基因和变异概率基因两个基因),以及配体基因;(ii)变长度的基因序列:包括优化过程中数量可变的水分子基因,以及标记水分子数量的标识位基因。
采用实数编码方案进行不等长染色体交叉操作如图2所示:
(i)对于参与交叉的两条父代染色体,首先比较其基因的个数。为了防止溢出,选择其中基因个数较少的染色体(若两条染色体的基因个数相等,则任意选择其中一条染色体)在其基因序列上随机产生一个交叉位点。
(ii)若交叉位点位于固定长度的基因序列上,如图2(a),则直接交换两条父代染色体的交叉位点之后的所有基因,从而生成两个新的子代。
(iii)若交叉位点位于变长度的基因序列上,如图2(b),则交换两条父代染色体的标识位基因以及交叉位点之后的基因,从而生成两个新的子代。
采用上述不等长染色体交叉的操作不仅可以确保标识位基因中标记的个数与变长度基因序列的个数相匹配,同时还能保证父代种群向子代种群正常进化。
3)基于排挤机制与循环拥挤距离的精英个体选择
采用排挤机制与循环拥挤距离结合的精英个体选择策略主要包括:
(i)排挤机制:主要通过淘汰原种群中相似度极高的个体,进而使种群中个体之间保持一定的距离。通过引入排挤机制,从而对所有个体进行分类,即形成各类不同的生存环境,这样优化得到的pareto解具有良好的分布性,并且能够保持种群的多样性。其具体步骤如下:
①设置一个较小的数l,l的设定由不同个体所对应的同一目标函数的目标函数值之差决定,即不同个体对应的目标函数值允许的最小误差。
②在目标函数空间中随机选取一个目标函数fm(m=1,2,…,r)作为评价指标,r为目标函数的个数,通过将父代种群pt(t为进化代数)和子代种群qt合并为rt,再从rt的2n个个体中随机选择2个个体i和j,分别计算其对应的目标函数值。
③若||fm(i)|-|fm(j)||≤l,则重新选取另一目标函数fn(n=1,2,…,r),且n≠m,进行比较;
④比较|fm(i)|与|fm(j)|的大小,淘汰较差个体。若两者相等,则采取随机策略。
⑤重复步骤②至④,直至rt中所有个体均参与比较。
(ii)循环拥挤距离:从每一非支配前沿等级选择个体进入下一代父代种群pt+1时,若需要从该前沿|fi|总个体中选择ni个个体,则首先计算该前沿|fi|个个体的拥挤距离,剔除拥挤距离最小的个体;然后重新计算剩余(|fi|-1)个个体的拥挤距离,重复以上步骤,直至选出ni个个体为止。采用这种循环拥挤距离策略筛选得到的个体具有良好的分布性。
(iii)精英个体选择:首先对合并后的种群rt中的个体进行非支配排序,得到各级pareto前沿f1,f2,...,fn;再按照式(1)所示的分布函数选取每一级非支配前沿的部分个体作为下一代进化父代种群pt+1,如图2(b),而不是将前面各个前沿的所有个体均填充至pt+1集合中,如图2(a)。优化后的精英个体选择策略只保留各级pareto前沿中的大多数精英个体,同时丢弃少部分的精英个体,这样能够避免最优个体以很大概率一直保留到最后进而影响种群的多样性。
ni=|fi|*ri(1)
其中,ni为从第i级非支配前沿选取进入下一代父代种群pt+1的个体数,|fi|为第i级非支配前沿上的个体数,ri为0至1之间的一个随机数,即ri∈(0,1)。
4)多目标对接模型
将仿真模拟蛋白质-水分子-配体三者之间的求解描述成多目标对接模型,涉及:(i)标记交叉概率和变异概率的两个自由度。(ii)配体的对接构象采用三部分自由度来描述,即3个平动自由度、3个转动自由度和n个旋转键自由度。其中,平动自由度用于标记配体的位置信息;转动自由度用于标记配体的取向;旋转键自由度用于标记配体中旋转键的变化。(iii)水分子的对接构象采用两部分自由度来描述,即1个标识位自由度和3m个平动自由度。其中,标识位自由度用于标记参与对接的水分子的个数m;平动自由度用于标记m个水分子的位置信息。由于考虑蛋白质柔性尤其是侧链柔性将大大增加算法的计算量,因此在设计中,蛋白质被假定为刚性,而在对接过程中只考虑配体及水分子的构象变化。
具体描述如下:
其中,f1(x)为蛋白质与配体、蛋白质与水分子之间的相互作用能,f2(x)为配体内部、配体与水分子,以及水分子与水分子之间的相互作用能,e(x)为模型的约束集合,k为约束个数,s为决策向量的范围,x={x1,x2,…,xm}t={p1,p2,tx,ty,tz,rx,ry,rz,rb1,…,rbn,wm,wx1,wy1,wz1,…,wxm,wym,wzm}t为决策向量(decisionvector),p1、p2分别为交叉概率和变异概率自由度;(tx,ty,tz)和(rx,ry,rz)分别为配体的平动自由度和转动自由度;(rb1,…,rbn)为配体的n个旋转键的旋转角度;wm为标识位自由度;(wx1,wy1,wz1,…,wxm,wym,wzm)为m个水分子的平动自由度。多目标对接模型的约束条件如下式(3):
式中pup(plow)为交叉概率或变异概率自由度的上(下)限;xup(xlow),yup(ylow)和zup(zlow)分别为配体和水分子的平动自由度的上(下)限。
5)遗传算法优化流程
考虑水分子参与分子对接的遗传算法优化流程的整体流程如图4所示,主要分为以下三个模块:(i)初始化模块:包括初始化打分函数的参数文件,对接所需要的蛋白质、配体和水分子文件,以及父代种群与精英档案种群等。(ii)对接优化模块:包括选择、不等长染色体交叉、变异、合并及优化的精英个体选择等。根据优化算法与优化参数的设置从而可以搜索最优解,即最优的结合模式。(iii)结果输出模块:将多体-多目标优化方法的最优解输出,包括各项打分值、最优解个数以及对接结合模式中配体和水分子的构象文件输出等。
本实施例的方法具有以下优势:
1、为考虑水分子参与蛋白质-配体结合提供新的解决途径。水分子参与蛋白质-配体结合的数量为不确定因素,因此,综合考虑其个数为0或非0,以及个数为固定或变化等不同情况建立多体相互作用模型,这样更加逼近真实的结合过程。
2、为研究蛋白质-水分子-配体三者之间的多体优化提供新的研究方案。采用非支配排序遗传算法算法、自适应算子、不等长染色体设计与交叉及优化的精英个体选择策略相结合的算法作为优化引擎,建立多目标对接模型,进而发展考虑水分子参与对接的多体-多目标优化方法。对于拓宽药物研究的手段和策略,以及寻找新的解决思路都具有重要的现实意义。
3、利用多学科之间的交叉,为开发精确预测结合模式的药物设计方法奠定基础。通过引入多目标优化算法,开展蛋白质-水分子-配体三者之间的多体优化问题研究,为进一步开发精确度良好的结合模式预测的药物设计方法奠定基础,同时也为多学科之间的交叉互补提供一种合理可行的研究策略。