本发明属于计算机应用技术与生物信息学的技术领域;具体涉及一种预测癌细胞株中药物反应的权重模块化映射方法。
背景技术:
相同癌症类型的病人具有相似的临床表现,但对相同的药物可能会产生不同的反应。因此,药物反应预测(drugresponseprediction,drp)成为一个研究热点。大量的生物实验和计算方法涌现而出预测药物在癌细胞株中的反应,然而,通过生物实验检测药物反应不仅费时费力,而且耗费大量资金。
二分网络投影(bipartitenetworkprojection,bnp)算法恰好适用于该问题;对于二分网络g(d,c,e),d是所有药物构成的集合,d={d1,d2,...,ds},c是所有细胞株构成的集合,c={c1,c2,...,ct},e是药物与细胞株的敏感关联,即药物与细胞株之间的边集,选择细胞株cseed为种子结点,bnp算法可以按照下述公式计算cseed与所有药物的关联得分:
bnp(d,ccseed)={score'(d1),score'(d2),...,score'(ds)}
score(di)=a]di][cseed]
aij=a[di][cj]
比较典型的三个方法是ntsmda,zhang等人提出的方法,以及hnmdrp·ntsmda通过整合网络拓扑相似度,改进bnp算法预测潜在关联关系;zhang等人提出一种应用于双层网络的权重模型;hnmdrp通过构建一个异构网络预测敏感关联对的得分,但预测精度有待提高。
技术实现要素:
本发明的目的是提供一种预测癌细胞株中药物反应的权重模块化映射方法(weight-basedmodularmappingmethodtopredictdrug-celllineassociations,wmmdca),用以解决上述问题且在预测效果上更优。
本发明通过以下技术方案实现:
一种预测癌细胞株中药物反应的权重模块化映射方法,所述癌细胞株中的药物反应计算所有药物与细胞株构成的关联对得分包括以下步骤:
步骤1:首先计算药物di和药物dj之间的关系,
dis(di,dj)=1-exp(-||a[di]-a[di]||2)
按照上述公式计算药物di和药物dj之间的距离。
步骤2:选择任意一个细胞株cseed作为种子结点,利用该细胞株cseed将药物集合划分为m个模块{m1,m2,...,mm},其中m1对应着s1个药物,m2对应着s2个药物,...mm对应着sm个药物;
其中m是cseed的度;
步骤3:通过权重系数计算细胞株cseed与模块{m1,m2,...,mm}中药物的关联得分集合
进一步的,所述步骤1中计算药物di和药物dj之间的距离为药物集合的分模块做基础。
进一步的,所述步骤2中模块划分的步骤如下:
步骤2.1:将细胞株cseed关联的m药物分别加入到m个模块中,并将细胞株cseed设置为该模块的核心点,此时每个模块仅包含一个药物;
步骤2.2:将未被分配进模块的药物加入与其距离最近的核心点所在的模块中;
步骤2.3:为了保证细胞株cseed从不同模块接收到的资源的平衡性,当sl>[s/m],将距离该模块核心点最远的sl-[s/m]个药物从该模块中移除,其中sl是模块m1中的药物个数;
步骤2.4:重复步骤2.2和步骤2.3,直到每个模块中的药物数量不超过[s/m]个。
进一步的,所述步骤3中关联得分集合下述公式,
w(ml,mj)是模块ml从模块mj(j≠l)处接收到的资源的权重系数,
是对和cseed运行bnp算法得到的关联得分集合,
细胞株cseed和模块中的药物ml之间的关联得分,细胞株cseed和模块中的药物mj之间的关联得分,
是模块ml与模块mj中药物的并集,
是与中药物关联的细胞株cseed的集合;
将步骤2中的m个模块分别执行步骤3,获得细胞株cseed与所有药物的关联得分集合依次选择每个细胞株为种子结点,可获得所有药物与细胞株构成的关联对的关联得分。
附图说明
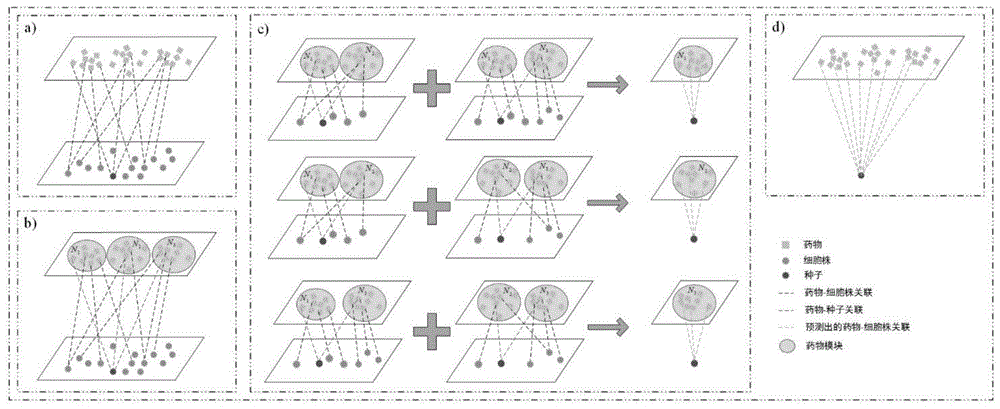
附图1本发明的(a)构建药物细胞株关联网络,选择细胞株种子,(b)根据种子将药物分成几个相同大小的模块,(c)通过根据权重贡献其他模块的资源,计算种子和每个模块之间的关联得分,(d)获得种子和所有药物之间的关联分数。
附图2本发明的(a)药物belinostat的roc曲线,(b)药物dabrafenib的roc曲线,(c)药物seliciclib的roc曲线,(d)药物talazoparib的roc曲线。
附图3本发明的(a)药物belinostat的roc曲线,(b)药物dabrafenib的roc曲线,(c)药物seliciclib的pr曲线,(d)药物talazoparib的pr曲线。
附图4本发明的(a)auroc值的箱线图,(b)auroc平均值,(c)aupr值的箱线图,(d)aupr平均值。
附图5本发明的(a)细胞株19种组织类型的分布,(b)细胞株三种主要组织类型的auroc值,(c)细胞株三种主要组织类型的aupr值。
具体实施方式
下面将结合本发明实施例中的附图对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
一种预测癌细胞株中药物反应的权重模块化映射方法,对于由s个药物构成的集合d,d={d1,d2,...,ds},t个细胞株构成的集合c,c={c1,c2,...,ct},关联矩阵a具有s行t列,若di与cj之间存在关联,a[di][cj]=1,否则a[di][cj]=0。
所述癌细胞株中的药物反应计算所有药物与细胞株构成的关联对得分包括以下步骤:
步骤1:首先计算药物di和药物dj之间的关系,
dis(di,dj)=1-exp(-||a[di]-a[di]||2)
按照上述公式计算药物di和药物dj之间的距离。
步骤2:选择任意一个细胞株cseed作为种子结点,见图1(a),利用该细胞株cseed将药物集合划分为m个模块{m1,m2,...,mm},模块m1内含s1个药物,以此类推,有分别包含s1,s2,...,sm个药物;见图1(b)
其中m是cseed的度。
步骤3:通过权重系数计算细胞株cseed与模块{m1,m2,...,mm}中药物的关联得分集合见图1(c),
进一步地,所述步骤1中计算药物di和药物dj之间的距离为药物集合的分模块做基础。
进一步地,所述步骤2中模块划分的步骤如下:
步骤2.1:将细胞株cseed关联的m药物分别加入到m个模块中,并将细胞株cseed设置为该模块的核心点,此时每个模块仅包含一个药物;
步骤2.2:将未被分配进模块的药物加入与其距离最近的核心点所在的模块中;
步骤2.3:为了保证细胞株cseed从不同模块接收到的资源的平衡性,当sl>[s/m],将距离该模块核心点最远的sl-[s/m]个药物从该模块中移除,其中sl是模块m1中的药物个数;
步骤2.4:重复步骤2.2和步骤2.3,直到每个模块中的药物数量不超过[s/m]个。
所述步骤3中关联得分集合下述公式,
w(ml,mj)是模块ml从模块mj(j≠l)处接收到的资源的权重系数,
是对和cseed运行bnp算法得到的关联得分集合,
细胞株cseed和模块中的药物ml之间的关联得分,细胞株cseed和模块中的药物mj之间的关联得分,
是模块ml与模块mj中药物的并集,
是与中药物关联的细胞株cseed的集合;
将步骤2中的m个模块分别执行步骤3,获得细胞株cseed与所有药物的关联得分集合见图1(d),依次选择每个细胞株为种子结点,可获得所有药物与细胞株构成的关联对的关联得分。
在gdsc数据库中下载药物反应数据,即标准化的ic50值。去除没有化学结构特征的药物和没有基因表达谱的细胞株,剩余184个药物和962个细胞株。将他们之间的ic50值划分为两部分:敏感关联和抵抗关系。最终构建出一个具有184个药物,962个细胞株和16910个敏感关联的网络。
采用留一交叉验证法(leave-one-outcross-validation,loocv)评估wmmdca的预测性能。依次选择每个已被证实的关联对作为测试集,其他作为训练集。绘制接受者操作特征曲线(receiveroperatingcharacteristiccurve,roc)和精确率-召回率曲线(precision–recallcurve,pr)直观表示预测效果。roc曲线线下面积(areaunderroccurve,auroc)和pr曲线线下面积(areaunderprcurve,aupr)越大,代表预测效果越好。
计算了所有药物的auroc值,并绘制箱线图,见图4.a)。在wmmdca中,药物dasatinib获得了最高的auroc值0.9661,四分之一的药物的auroc值超过0.8867,一半药物的auroc值超过0.833。所有药物的auroc平均值为0.8184,见图4.b)。同时,我们还计算了所有药物的aupr值,箱线图见图4.c)。在wmmdca中,药物belinostat获得了最高的aupr值0.8006,四分之一的药物的aupr值超过0.469,一半药物的aupr值超过0.346。所有药物的aupr平均值为0.3715,见图4.d)。
每一个细胞株都属于一种组织类型,从962个细胞株中统计出19种不同的组织类型,每种类型所占的比例如图5.a)所示。其中,非小细胞肺癌(lungnon-smallcelllungcancer,nsclc)、泌尿生殖系统癌(urogenitalsystem)和白血病(leukemia)居前三位,分别占11.3%、10.4%和8.32%。wmmdca在这三种主要组织类型中的auroc值分别为0.8028、0.7879和0.8058,超过了其他三种方法,见图5.b)。wmmdca在这三种主要组织类型中的aupr值分别为0.3077、0.3489和0.6097,也超过了其他三种方法,见图5.c)。这些结果表明wmmdca对主要组织类型有较好的预测能力。
综上,我们提出了方法wmmdca用于预测药物-细胞株关联。该方法具有创新性,体现在对药物的模块划分方法及将模块信息与bnp算法融合的方法中。实验表明,wmmdca的预测性能超出了其他三个典型预测方法,证明wmmdca具有有效性。