抗-EGFR抗体及抗体药物偶联物的制作方法
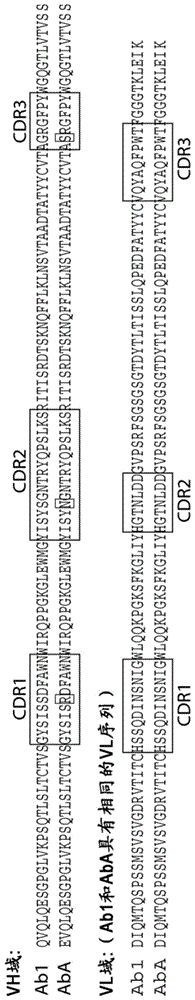
本申请为国际申请pct/us2015/021849进入中国国家阶段的中国专利申请(申请号为201580026453.4,申请日为2015年3月20日,发明名称为“抗-egfr抗体及抗体药物偶联物”)的分案申请。相关申请本申请要求2014年3月21日提交的美国临时申请no.61/968,819的优先权权益。前述优先权申请的内容通过引用全文合并于此。序列表本申请包含以ascii格式电子提交的并由此通过引用全文并入的序列表。2015年3月12日生成的所述ascii拷贝名称为117813-06202_sl.txt且大小为110,863字节。
背景技术:
:人表皮生长因子受体(也称为her-1或erb-b1,且在本文中称为“egfr”)是由c-erbb原癌基因编码的170kda跨膜受体,且表现出固有的酪氨酸激酶活性(modjtahedi等人,br.j.cancer73:228-235(1996);herbst和shin,cancer94:1593-1611(2002))。swissprot数据库登录项p00533提供了人egfr的序列。egfr通过酪氨酸激酶介导的信号转导途径调节多种细胞过程,包括但不限于控制细胞增殖、分化、细胞存活、凋亡、血管生成、有丝分裂发生和转移的信号转导途径的激活(atalay等人,ann.oncology14:1346-1363(2003);tsao和herbst,signal4:4-9(2003);herbst和shin,cancer94:1593-1611(2002);modjtahedi等人,br.j.cancer73:228-235(1996))。egfr的已知配体包括egf、tgfa/tgf-α、双向调节因子、epigen/epgn、btc/β细胞素、表皮调节素/ereg和hbegf/肝素结合egf。通过egfr的配体结合引发受体同二聚化和/或异二聚化及关键胞质残基的自体磷酸化。磷酸化的egfr募集衔接蛋白如grb2,其随之激活复杂的下游信号传导级联,包括至少以下主要下游信号传导级联:ras-raf-mek-erk、pi3激酶-akt、plcγ-pkc和stats模块。这种自体磷酸化也通过几种其它蛋白质(其通过自身的磷酸酪氨酸结合sh2结构域与磷酸化酪氨酸结合)诱导下游激活和信号传导。这些下游信号传导蛋白启动几种信号转导级联,主要为mapk、akt和jnk途径,从而导致细胞增殖。通过egfr的配体结合也可以激活nf-κ-b信号传导级联。配体结合也直接磷酸化其它蛋白质如rgs16,从而激活其gtp酶活性和潜在地将egf受体信号传导与g蛋白偶联的信号传导偶联。配体结合也磷酸化muc1并增加其与src和ctnnb1/β-连环蛋白的相互作用。egfr的过表达已在许多人类恶性病症中报道,包括膀胱、脑、头和颈、胰腺、肺、乳腺、卵巢、结肠、前列腺和肾的癌症(atalay等人,ann.oncology14:1346-1363(2003);herbst和shin,cancer94:1593-1611(2002),及modjtahedi等人,br.j.cancer73:228-235(1996))。在众多的这些病症中,egfr的过表达与患者的不良预后相关或关联(herbst和shin,cancer94:1593-1611(2002),及modjtahedi等人,br.j.cancer73:228-235(1996))。egfr也在正常组织的细胞中表达,特别是皮肤、肝和胃肠道的上皮组织,虽然一般以低于恶性细胞中的水平表达(herbst和shin,cancer94:1593-1611(2002))。包含egfr基因的扩增(即,多个拷贝的egfr基因)的大部分肿瘤也过度表达受体的截短形式(wikstrand等人,(1998)j.neurovirol.4,148-158),称为de2-7egfr、δegfr、egfrviii或δ2-7(在本文中可互换使用的术语)(olapade-olaopa等人,(2000)br.j.cancer.82,186-94)。de2-7egfr中看到的重排导致缺乏跨外显子2-7的801个核苷酸的框内成熟mrna(wong等人,(1992)proc.natl.acad.sci.u.s.a.89,2965-9;yamazaki等人,(1990)jpn.j.cancerres.81,773-9;yamazaki等人,(1988)mol.cell.biol.8,1816-20,及sugawa等人,(1990)proc.natl.acad.sci.u.s.a.87,8602-6)。相应的egfr蛋白具有包含细胞外域的残基6-273的267氨基酸缺失及在融合接合处的新的甘氨酸残基(sugawa等人,1990)。这种缺失与甘氨酸残基的插入一起在缺失界面处产生独特的接合肽(sugawa等人,1990)。egfrviii已在多种肿瘤类型中报道,包括神经胶质瘤、乳腺肿瘤、肺肿瘤、卵巢肿瘤和前列腺肿瘤(wikstrand等人,(1997)cancerres.57,4130-40;olapade-olaopa等人,(2000)br.j.cancer.82,186-94;wikstrand等人,(1995)cancerres.55,3140-8;garciadepalazzo等人,(1993)cancerres.53,3217-20)。尽管这种截短的受体不结合配体,但其具有低组成性活性并对作为肿瘤异种移植物在裸鼠中生长的神经胶质瘤细胞赋予显著的生长益处(nishikawa等人(1994)proc.natl.acad.sci.u.s.a.91,7727-31)和能够转化nih3t3细胞(batra等人,(1995)cellgrowthdiffer.6,1251-9)和mcf-7细胞。神经胶质瘤细胞中de2-7egfr所利用的细胞机制并未完全确定,但被报道包括凋亡的降低(nagane等人,(1996)cancerres.56,5079-86)和小的增殖增强(nagane等人,1996)。由于这种截短的受体的表达限于肿瘤细胞,其代表抗体疗法的高度特异性的靶标。抗体药物偶联物(adc)代表一类新的治疗剂,其包含通过化学接头与细胞毒性药物偶联的抗体。adc的治疗概念是将抗体的结合能力与药物相结合,其中抗体用于通过与靶表面抗原的结合递送药物到肿瘤细胞。因此,本领域中仍然存在对于可以用于癌症治疗的治疗性目的的抗-egfr抗体和adc的需要。技术实现要素:在某些方面,本发明提供特异性地结合egfrviii的抗-egfr抗体和抗体药物偶联物(adc)。在一个实施方式中,本发明特征在于结合氨基酸序列cgadsyemeedgvrkc(seqidno:45)内的表位或在竞争结合分析中与第二抗-hegfr抗体竞争结合表皮生长因子受体变体iii(egfrviii)(seqidno:33)的抗-人受体表皮生长因子(抗-hegfr)抗体或其抗原结合部分,其中该第二抗-egfr抗体包含含有seqidno:1中所示氨基酸序列的重链可变域和含有seqidno:5中所示氨基酸序列的轻链可变域;以约1x10-6m或更低的解离常数(kd)结合egfr(1-525)(seqidno:47),如通过表面等离子体共振测定的;且在体内人非小细胞肺癌(nsclc)异种移植分析中相对于非egfr特异性的人igg抗体以至少约50%的肿瘤生长抑制%(tgi%)抑制肿瘤生长,其中所述人igg抗体在nsclc异种移植分析中以与抗-hegfr抗体或其抗原结合部分相同的剂量和频率施用。在本发明的某些实施方式中,抗体或其抗原结合部分以约1x10-6m和约1x10-10m之间的kd结合egfr(1-525)(seqidno:47),如通过表面等离子体共振测定的。在本发明的其它实施方式中,抗体或其抗原结合部分以约1x10-6m和约1x10-7m之间的kd结合egfr(1-525)(seqidno:47),如通过表面等离子体共振测定的。在某些实施方式中,本发明的抗体或其抗原结合部分以约8.2x10-9m或更低的kd结合egfrviii(seqidno:33),如通过表面等离子体共振测定的。在进一步的实施方式中,抗体或其抗原结合部分以约8.2x10-9m和约6.3x10-10m之间的kd结合egfrviii(seqidno:33),如通过表面等离子体共振测定的。在一些实施方式中,抗体或其抗原结合部分以约8.2x10-9m和约2.0x10-9m之间的kd结合egfrviii(seqidno:33),如通过表面等离子体共振测定的。在本发明的再其它的实施方式中,抗体或其抗原结合部分在体内人非小细胞肺癌(nsclc)异种移植分析中相对于非egfr特异性的人igg抗体抑制肿瘤生长至少约60%。在某些实施方式中,本发明特征在于抗体或其抗原结合部分,其在体内人非小细胞肺癌(nsclc)异种移植分析中相对于非egfr特异性的人igg抗体抑制肿瘤生长至少约70%。在某些实施方式中,抗体或其抗原结合部分在体内人非小细胞肺癌(nsclc)异种移植分析中相对于非egfr特异性的人igg抗体抑制肿瘤生长至少约80%。在一些实施方式中,抗体或其抗原结合部分包含含有seqidno:12中所示的氨基酸序列的重链cdr3域、含有seqidno:11中所示的氨基酸序列的重链cdr2域和含有seqidno:10中所示的氨基酸序列的重链cdr1域,及含有seqidno:8中所示的氨基酸序列的轻链cdr3域、含有seqidno:7中所示的氨基酸序列的轻链cdr2域和含有seqidno:6中所示的氨基酸序列的轻链cdr1域。在又一实施方式中,抗体或其抗原结合部分包含含有seqidno:9中所示的氨基酸序列的重链可变区和含有seqidno:5中所示的氨基酸序列的轻链可变区。在其它实施方式中,抗体或其抗原结合部分包含含有seqidno:41中所示的氨基酸序列的重链恒定区和/或含有seqidno:43中所示的氨基酸序列的轻链恒定区。在进一步的实施方式中,抗体或其抗原结合部分包含含有seqidno:15中所示的氨基酸序列的重链和含有seqidno:13中所示的氨基酸序列的轻链。在另一实施方式中,抗体或其抗原结合部分与阿里他汀偶联。在某些实施方式中,本发明还提供编码抗体或其抗原结合部分(如本文中描述的)的分离的核酸。在某些实施方式中,本发明还包括抗-hegfr抗体或其抗原结合部分,其包含含有seqidno:40中所示的氨基酸序列的轻链cdr3域、含有seqidno:39中所示的氨基酸序列的轻链cdr2域和含有seqidno:38中所示的氨基酸序列的轻链cdr1域,及含有seqidno:37中所示的氨基酸序列的重链cdr3域、含有seqidno:36中所示的氨基酸序列的重链cdr2域和含有seqidno:35中所示的氨基酸序列的重链cdr1域。在某些实施方式中,本发明特征在于抗-hegfr抗体或其抗原结合部分,其包含含有选自50、52、54、56、58、60、62、64、66、68、70、72、74、76和78的氨基酸序列的重链可变区及含有选自51、53、55、57、59、61、63、65、67、69、71、73、75、77和79的氨基酸序列的轻链可变区。在其它实施方式中,本发明包括抗-hegfr抗体或其抗原结合部分,其包含选自seqidno:10、11和12,seqidno:16、17和18,seqidno:10、11和19,seqidno:20、11和12,seqidno:21、3和22,seqidno:16、17和19,seqidno:2、3和4,seqidno:10、3和12,seqidno:80、11和18,seqidno:80、3和18,seqidno:20、3和12,seqidno:80、11和12及seqidno:81、11和22的重链cdr组(cdr1,cdr2和cdr3);以及选自seqidno:6、7和8,seqidno:23、24和25,seqidno:26、27和28,seqidno:29、30和31,seqidno:6、7和84,seqidno:82、83和31及seqidno:82、27和85的轻链cdr组(cdr1,cdr2和cdr3),其中该抗体或其抗原结合部分不同时包含seqidno:2、3和4的重链cdr组及seqidno:6、7和8的轻链cdr组。在一些实施方式中,抗体或其抗原结合部分包含含有seqidno:41中所示的氨基酸序列的重链恒定区和/或含有seqidno:43中所示的氨基酸序列的轻链恒定区。在本发明的一些实施方式中,抗体或其抗原结合部分包含选自人igg恒定域、人igm恒定域、人ige恒定域和人iga恒定域的重链免疫球蛋白恒定域。在一些实施方式中,igg恒定域选自选自igg1恒定域、igg2恒定域、igg3恒定域和igg4恒定域。在其它实施方式中,抗体是多特异性抗体。在本发明的其它实施方式中,抗体或其抗原结合部分包括fab、fab’、f(ab’)2、fv、二硫键连接的fv、scfv、单域抗体和双抗体(diabody)。在本发明的再其它的实施方式中,抗体或其抗原结合部分与成像剂偶联。在本发明的某些实施方式中,成像剂选自放射标记、酶、荧光标记、发光标记、生物发光标记、磁性标记和生物素。在本发明的其它实施方式中,放射标记是铟。在再其它的实施方式中,本发明包括包含该抗体或其抗原结合部分和药学上可接受的载体的药物组合物。在一些实施方式中,本发明还包括包含与至少一种药物偶联的本文所述抗体或其抗原结合部分的抗体药物偶联物(adc)。在某些实施方式中,抗体是与氨基酸序列cgadsyemeedgvrkc(seqidno:45)内的表位结合或在竞争结合分析中与第二抗-hegfr抗体竞争结合表皮生长因子受体变体iii(egfrviii)(seqidno:33)的抗-人受体表皮生长因子(抗-hegfr)抗体或其抗原结合部分,其中该第二抗-egfr抗体包含含有seqidno:1中所示的氨基酸序列的重链可变域和含有seqidno:5中所示的氨基酸序列的轻链可变域;如通过表面等离子体共振测定的,以约1x10-6m或更低的解离常数(kd)结合egfr(1-525)(seqidno:47);且在体内人非小细胞肺癌(nsclc)异种移植分析中相对于非egfr特异性的人igg抗体以至少约50%的肿瘤生长抑制%(tgi%)抑制肿瘤生长,其中该人igg抗体在nsclc异种移植分析中以与抗-hegfr抗体或其抗原结合部分相同的剂量和频率施用。在本发明的一个实施方式中,至少一种药物选自抗-细胞凋亡剂、有丝分裂抑制剂、抗-肿瘤抗生素、免疫调节剂、用于基因疗法的核酸、烷化剂、抗-血管生成剂、抗-代谢物、含硼剂、化疗保护剂、激素剂、抗-激素剂、皮质类固醇、光敏治疗剂、寡核苷酸、放射性核素剂、放射致敏剂、拓扑异构酶抑制剂和酪氨酸激酶抑制剂。在某些实施方式中,有丝分裂抑制剂是多拉司他汀、阿里他汀、类美登素和植物生物碱。在某些实施方式中,药物是多拉司他汀、阿里他汀、类美登素和植物生物碱。阿里他汀的实例是单甲基阿里他汀f(mmaf)或单甲基阿里他汀e(mmae)。类美登素的实例包括,但不限于dm1、dm2、dm3和dm4。在某些实施方式中,抗-肿瘤抗生素选自放射菌素、蒽环类、卡奇霉素和倍癌霉素。在某些实施方式中,放射菌素是吡咯并苯并二氮(pyrrolobenzodiazepine)(pbd)。在一些实施方式中,本发明还包括包含与阿里他汀偶联的抗-egfr抗体的adc,其中该抗体包含重链可变区和轻链可变区,该重链可变区包含含有seqidno:12的氨基酸序列的cdr3域、含有seqidno:11的氨基酸序列的cdr2域和含有seqidno:10的氨基酸序列的cdr1域;和该轻链可变区包含含有seqidno:8的氨基酸序列的cdr3域、含有seqidno:7的氨基酸序列的cdr2域和含有seqidno:6的氨基酸序列的cdr1域。在一个实施方式中,抗体包含含有seqidno:9中所示的氨基酸序列的重链可变区和含有seqidno:5的氨基酸序列的轻链可变区。在又一实施方式中,本发明包括抗体或其抗原结合部分,其包含含有seqidno:15中所示的氨基酸序列的重链和含有seqidno:13的氨基酸序列的轻链。在一些实施方式中,本发明还包括包含与至少一种药物(包括,但不限于mmae)偶联的抗-egfr抗体的adc,其中1-8个药物分子与抗体偶联。在一个实施方式中,1-4个药物分子与adc的抗体偶联。在一个实施方式中,2-4个药物分子与adc的抗体偶联。在一些实施方式中,本发明还包括包含与至少一种药物偶联的抗-egfr抗体的adc,其中该药物通过马来酰亚胺基己酰基,缬氨酸-瓜氨酸接头偶联。在进一步的实施方式中,药物通过马来酰亚胺基己酰基,缬氨酸-瓜氨酸,p-氨基苄氧羰基(paba)接头与抗体偶联。在一些实施方式中,本发明还包括包含通过接头(例如,马来酰亚胺基己酰基,缬氨酸-瓜氨酸)与单甲基阿里他汀e(mmae)共价连接的抗-egfrigg1抗体的adc。在某些实施方式中,抗体包含含有seqidno:9中所示的氨基酸序列的重链可变区,且包含含有seqidno:5中所示的氨基酸序列的轻链可变区。在某些实施方式中,1-4个分子的mmae与抗体连接。在一些实施方式中,本发明还包括包含与马来酰亚胺基己酰基,缬氨酸-瓜氨酸,p-氨基苄氧基氨甲酰基-单甲基阿里他汀e(mc-vc-paba-mmae)共价连接的抗-egfrigg1抗体,其中抗体包含含有seqidno:9中所示的氨基酸序列的重链可变区,和包含含有seqidno:5中所示的氨基酸序列的轻链可变区,且其中1-4个分子的mmae与抗体连接。在某些实施方式中,抗体包含含有seqidno:15中所示的氨基酸序列的重链,且包含含有seqidno:13中所示的氨基酸序列的轻链。在某些实施方式中,2-4个分子的mmae与抗体连接。在某些实施方式中,egfr抗体如图11中所示的与mc-vc-paba-mmae连接。在一些实施方式中,本发明还包括含有对于人egfr特异性的igg1抗体、mmae和将mmae共价连接于抗体的接头的针对egfr的adc。在某些实施方式中,抗体包含重链可变区和轻链可变区,该重链可变区包含含有seqidno:12的氨基酸序列的cdr3域、含有seqidno:11的氨基酸序列的cdr2域和含有seqidno:10的氨基酸序列的cdr1域,且该轻链可变区包含含有seqidno:8的氨基酸序列的cdr3域、含有eqidno:7的氨基酸序列的cdr2域和含有seqidno:6的氨基酸序列的cdr1域。在一个实施方式中,抗体包含含有seqidno:9中所示的氨基酸序列的重链可变区和含有seqidno:5的氨基酸序列的轻链可变区。在又一实施方式中,本发明包括包含重链和轻链的抗体或其抗原结合部分,该重链包含seqidno:15中所示的氨基酸序列和该轻链包含seqidno:13的氨基酸序列。在再其它的实施方式中,本发明包括包含adc混合物的药物组合物,该adc混合物包含多种本文所述的adc和药学上可接受的载体。在某些实施方式中,adc混合物具有2-4的平均药物-抗体比率(dar)。在其它实施方式中,adc混合物包含各具有2-8的dar的adc。在某些实施方式中,adc混合物具有约2.4-约3.6的平均-抗体比率(dar)。在某些实施方式中,本发明包括用于治疗患有癌症的受试者的方法,包括对受试者施用本文所述的药物组合物,使得患有癌症的受试者被治疗。在一个实施方式中,癌症选自乳腺癌、肺癌、成胶质细胞瘤、前列腺癌、胰腺癌、结肠癌、头颈癌和肾癌。在一个实施方式中,癌症选自乳腺癌、肺癌、成胶质细胞瘤、前列腺癌、胰腺癌、结肠癌、结肠直肠癌、头颈癌、间皮瘤、肾癌、鳞状细胞癌、三阴性乳腺癌和非小细胞肺癌。在一个实施方式中,癌症是乳腺癌。在一个实施方式中,癌症是肺癌。在一个实施方式中,癌症是前列腺癌。在一个实施方式中,癌症是胰腺癌。在一个实施方式中,癌症是结肠癌。在一个实施方式中,癌症是头颈癌。在一个实施方式中,癌症是肾癌。在一个实施方式中,癌症是结肠直肠癌。在一个实施方式中,癌症是间皮瘤。在一个实施方式中,癌症是鳞状细胞癌。在一个实施方式中,癌症是三阴性乳腺癌。在一个实施方式中,癌症是非小细胞肺癌。在某些实施方式中,鳞状细胞癌是肺鳞癌或头颈鳞状细胞癌。在又一实施方式中,癌症包含egfr的扩增或过表达egfr。在某些实施方式中,癌症特征为具有egfr过表达。在某些实施方式中,癌症特征为具有egfr扩增。在某些实施方式中,本发明进一步包括用于抑制或降低患有实体肿瘤的受试者中的实体肿瘤生长的方法,包括对患有实体肿瘤的受试者施用本文所述的药物组合物,使得实体肿瘤生长被抑制或降低。在某些实施方式中,实体肿瘤特征为具有egfr过表达。在某些实施方式中,实体肿瘤特征为具有egfr扩增。在本发明的一个实施方式中,本发明提供用于抑制或降低患有实体肿瘤的受试者中的实体肿瘤生长的方法,包括对患有实体肿瘤的受试者施用有效量的本文所述的抗体或adc,使得实体肿瘤生长被抑制或降低。在某些实施方式中,实体肿瘤是表达egfr的实体肿瘤或egfrviii阳性实体肿瘤。在其它实施方式中,实体肿瘤是非小细胞肺癌或成胶质细胞瘤。在其它实施方式中,实体肿瘤是鳞状细胞癌。在本发明的一个实施方式中,本发明提供用于治疗患有癌症的受试者的方法,包括施用有效量的包含与至少一个阿里他汀偶联的抗-egfr抗体或其抗原结合部分的adc,其中抗-egfr抗体或其抗原结合部分是igg同种型的;包含重链可变区和轻链可变区,该重链可变区包含含有seqidno:12中所示的氨基酸序列的cdr3域、含有seqidno:11中所示的氨基酸序列的cdr2域和含有seqidno:10中所示的氨基酸序列的cdr1域,且该轻链可变区包含含有seqidno:8中所示的氨基酸序列的cdr3域、含有seqidno:7中所示的氨基酸序列的cdr2域和含有seqidno:6中所示的氨基酸序列的cdr1域。在某些实施方式中,抗体或其抗原结合部分与mc-vc-paba-mmae连接。在某些实施方式中,本发明包括用于治疗患有癌症的受试者的方法,包括与另外的药剂或另外的疗法组合施用本文所述的药物组合物于受试者。在某些实施方式中,另外的药剂选自抗-pd1抗体(例如,帕母单抗(pembrolizumab)或纳武单抗(nivolumab))、抗-ctla-4抗体(例如,依匹木单抗(ipilimumab))、依鲁替尼、duvelisib、idelalisib、venetoclax和替莫唑胺。在某些实施方式中,另外的疗法是放疗。在某些实施方式中,另外的药剂是抗-pd1抗体(例如,帕母单抗或纳武单抗)。在某些实施方式中,另外的药剂是抗-ctla-4抗体(例如,依匹木单抗)。在某些实施方式中,另外的药剂是依鲁替尼。在某些实施方式中,另外的药剂是duvelisib。在某些实施方式中,另外的药剂是idelalisib。在某些实施方式中,另外的药剂是venetoclax。在某些实施方式中,另外的药剂是替莫唑胺。在某些实施方式中,本发明还提供了编码抗体或其抗原结合部分(如本文所述的抗体或其抗原结合部分)的分离的核酸。进一步地,本发明包括包含该核酸的载体和包含该载体的宿主细胞,例如,原核细胞或真核细胞(例如,动物细胞、protest细胞、植物细胞和真菌细胞)。在本发明的实施方式中,动物细胞选自哺乳动物细胞、昆虫细胞和鸟类细胞。在一个实施方式中,哺乳动物细胞选自cho细胞、cos细胞和sp2/0细胞。在某些实施方式中,本发明特征在于包含与阿里他汀偶联的抗-hegfr抗体的抗-hegfr抗体药物偶联物(adc),其中抗体包含含有seqidno:12的氨基酸序列的重链cdr3域、含有seqidno:11的氨基酸序列的重链cdr2域和含有seqidno:10的氨基酸序列的重链cdr1域,及含有seqidno:8的氨基酸序列的轻链cdr3域、含有seqidno:7的氨基酸序列的轻链cdr2域和含有seqidno:6的氨基酸序列的轻链cdr1域。在一个实施方式中,抗体包含含有seqidno:9中所示的氨基酸序列的重链可变区和含有seqidno:5的氨基酸序列的轻链可变区。在又一实施方式中,抗体包含igg重链免疫球蛋白恒定域。在再另一实施方式中,igg是igg1或igg4重链免疫球蛋白恒定域。在一个实施方式中,本发明包括adc,其中阿里他汀是单甲基阿里他汀f(mmaf)或单甲基阿里他汀e(mmae)。在一个实施方式中,本发明包括adc,其中阿里他汀是单甲基阿里他汀f(mmaf)。在一个实施方式中,本发明包括adc,其中阿里他汀是单甲基阿里他汀e(mmae)。在进一步的实施方式中,本发明包括含有seqidno:15的氨基酸序列的重链,且包含含有seqidno:13的氨基酸序列的轻链。在本发明的再另一实施方式中,抗-egfr抗体通过包含马来酰亚胺基己酰基,缬氨酸-瓜氨酸,p-氨基苯甲醇的接头(mc-vc-paba)与阿里他汀共价连接。在一个实施方式中,本发明包括包含抗-egfr抗体和放射标记(例如,铟)的adc。在一个实施方式中,本文所述的抗-egfr抗体与至少一个吡咯并苯并二氮(pbd)共价连接。在某些实施方式中,本文公开的抗-egfr抗体如图12中所述与pbd连接(即,sgd-1882)。在一些实施方式中,本发明特征在于包含本文所述的adc和药学上可接受的载体的药物组合物。在某些实施方式中,本发明特征在于包含含有本文所述的adc的adc混合物的药物组合物,其中adc混合物的平均药物-抗体比率(dar)范围是2-4。在某些实施方式中,adc混合物的平均药物-抗体比率(dar)范围是2.4-3.6。在一个实施方式中,本发明特征在于包含含有抗-hegfr抗体药物偶联物(adc)的adc混合物和药学上可接受的载体的药物组合物,其中adc混合物具有2-4的平均药物-抗体比率(dar),且其中所述adc包含与抗-hegfr抗体偶联的单甲基阿里他汀e(mmae),该抗-hegfr抗体包含含有seqidno:12的氨基酸序列的重链cdr3域、含有seqidno:11的氨基酸序列的重链cdr2域和含有seqidno:10的氨基酸序列的重链cdr1域,及含有seqidno:8的氨基酸序列的重链cdr3域、含有seqidno:7的氨基酸序列的重链cdr2域和含有seqidno:6的氨基酸序列的轻链cdr3域。在一个实施方式中,抗体的重链可变区包含seqidno:9中所示的氨基酸序列和抗-egfr抗体的轻链可变区包含seqidno:5中所示的氨基酸序列。在本发明的其它实施方式中,抗体包含igg重链免疫球蛋白恒定域。在进一步的实施方式中,本发明包括具有igg1或igg4重链免疫球蛋白恒定域的抗体。在一个实施方式中,本发明包括igg1同种型的抗体。在又一实施方式中,本发明包括包含重链和轻链的抗体,重链包含seqidno:15中所示的氨基酸序列和轻链包含seqidno:13的氨基酸序列。在一个实施方式中,本发明特征在于具有通过马来酰亚胺基己酰基,val-cit,paba接头与抗体偶联的mmae。在本发明的一个实施方式中,本发明提供用于治疗患有癌症的受试者的方法,包括施用包含本文所述的抗体或adc的药物组合物于受试者,使得患有癌症的受试者被治疗。在一个实施方式中,癌症选自乳腺癌、肺癌、成胶质细胞瘤、前列腺癌、胰腺癌、结肠癌、头颈癌和肾癌。在一个实施方式中,癌症选自乳腺癌、肺癌、成胶质细胞瘤、前列腺癌、胰腺癌、结肠癌、结肠直肠癌、头颈癌、间皮瘤、肾癌、鳞状细胞癌、三阴性乳腺癌和非小细胞肺癌。在又一实施方式中,癌症包含egfr的扩增或过表达egfr。在一个实施方式中,鳞状细胞癌是肺鳞癌或头颈鳞状细胞癌。在一个实施方式中,癌症是egfr过表达的癌症。在一个实施方式中,癌症特征为扩增的egfr。在一个实施方式中,癌症是乳腺癌。在一个实施方式中,癌症是肺癌。在一个实施方式中,癌症是前列腺癌。在一个实施方式中,癌症是胰腺癌。在一个实施方式中,癌症是结肠癌。在一个实施方式中,癌症是头颈癌。在一个实施方式中,癌症是肾癌。在一个实施方式中,癌症是结肠直肠癌。在一个实施方式中,癌症是间皮瘤。在一个实施方式中,癌症是鳞状细胞癌。在一个实施方式中,癌症是三阴性乳腺癌。在一个实施方式中,癌症是非小细胞肺癌。在某些实施方式中,鳞状细胞癌是肺鳞癌或头颈鳞状细胞癌。另外,在某些实施方式中,本发明提供用于抑制或降低患有实体肿瘤的受试者中的实体肿瘤生长的方法,所述方法包括对患有实体肿瘤的受试者施用本文所述的药物组合物,使得实体肿瘤的生长被抑制或降低。在一个实施方式中,实体肿瘤是非小细胞肺癌或成胶质细胞瘤。在又一实施方式中,实体肿瘤是egfrviii阳性肿瘤或表达egfr的实体肿瘤。在又一实施方式中,实体肿瘤是egfr过表达的实体肿瘤。在又一实施方式中,实体肿瘤是egfr扩增的肿瘤。在一个实施方式中,实体肿瘤是具有扩增的egfr的非小细胞肺癌。在一个实施方式中,实体肿瘤是具有egfr过表达的非小细胞肺癌。在一个实施方式中,实体肿瘤是具有扩增的egfr的成胶质细胞瘤。在一个实施方式中,实体肿瘤是具有egfr过表达的成胶质细胞瘤。在某些实施方式中,本发明提供了组合疗法,由此本文所述的药物组合物施用于需要的受试者(例如,患有癌症或实体肿瘤的受试者)。本文所述的药物组合物可以与另外的药剂或另外的疗法的施用同时、在其之前或之后施用。在某些实施方式中,另外的药剂选自抗-pd1抗体、抗-ctla-4抗体、替莫唑胺、bcl-x1抑制剂和烟酰胺磷酸核糖基转移酶(nampt)抑制剂。在再其它的实施方式中,另外的药剂是化疗剂。在某些实施方式中,另外的疗法是放疗。在其它实施方式中,另外的药剂是依鲁替尼(pharmacyclics)。在其它实施方式中,另外的药剂是duvelisib。在其它实施方式中,另外的药剂是idelalisib(gileadsciences,inc.)。在其它实施方式中,另外的药剂是venetoclax(abt-199/gdc-0199,abbvie,inc.)。在某些实施方式中,另外的药剂是抗-pd1抗体(例如,帕母单抗或纳武单抗)。在某些实施方式中,另外的药剂是抗-ctla-4抗体(例如,依匹木单抗)。在某些实施方式中,另外的药剂是替莫唑胺。在某些实施方式中,本发明特征在于包含本文所述的抗体的抗原结合区(例如,cdr)或本文所述的scfv的嵌合抗原受体(car)。在某些实施方式中,本发明特征在于包含轻链可变区和重链可变区的car,轻链可变区包含含有seqidno:40中所示的氨基酸序列的cdr3域、含有seqidno:39中所示的氨基酸序列的cdr2域和含有seqidno:38中所示的氨基酸序列的cdr1域,和重链可变区包含含有seqidno:37中所示的氨基酸序列的cdr3域、含有seqidno:36中所示的氨基酸序列的cdr2域和含有seqidno:35中所示的氨基酸序列的cdr1域。在某些实施方式中,本发明特征在于包含重链可变区和轻链可变区的car,重链可变区包含含有seqidno:12中所示的氨基酸序列的cdr3域、含有seqidno:11中所示的氨基酸序列的cdr2域和含有seqidno:10中所示的氨基酸序列的cdr1域,和轻链可变区包含含有seqidno:8中所示的氨基酸序列的cdr3域、含有seqidno:7中所示的氨基酸序列的cdr2域和含有seqidno:6中所示的氨基酸序列的cdr1域。附图说明图1提供ab1的可变重链(vh)和可变轻链(vl)区氨基酸序列(seqidno:1和5)和aba的可变重链(vh)和可变轻链(vl)区氨基酸序列(seqidno:9和5)。vh和vl区内的cdr序列加框,且ab1vh序列和abavh序列之间的差异加轮廓。图2描述了ab1的全长轻链和重链(seqidno:13和14)及aba的全长轻链和重链(seqidno:13和15)。重链中ab1序列和aba序列之间的差异突出显示。图3提供了总结多个ab1变体抗体与ab1和ab2相比的biacore结合分析亲和力测量的表格。egfr(1-525)和egfrviii用于结合分析中。如图3中所述的ka(m-1s-1)是指抗体与抗原结合以形成抗体/抗原复合体的速率常数,kd(s-1)是指抗体从抗体/抗原复合体解离的解离速率常数,且kd(m)是指平衡解离常数速率。图4提供了facs分析的图形总结,其显示aba相对于ab1具有改善的与a431肿瘤细胞(人鳞状细胞癌细胞)的结合,但与ab2相比具有较低的结合亲和力。图5描绘了来自facs竞争分析的结果,其表明ab1变体抗体识别与ab1相同的egfr表位。图6提供了ab1和ab1变体抗体与egfr(1-525)的结合的总结。实心圆代表ab1或ab2(对照)且空心圆代表ab1变体抗体。圆圈指示组1和组2,总结了图7中提供的数据。图7提供了来自在体外检验ab1和ab1变体抗体对不同细胞系的活性的western印迹分析的结果。来自scc-15的细胞(图7a)和h292细胞暴露于如实施例4中描述的条件,并利用抗-磷酸酪氨酸egfr(pyegfr)、抗-egfr(totegfr(总egfr))和抗-肌动蛋白(肌动蛋白)抗体使用western印迹分析进行分析。图7a提供的结果显示ab1、ab2和ab1变体抗体在scc-15细胞中抑制egf-介导的egfr的酪氨酸磷酸化的能力。图7b提供的结果显示ab1、ab2和ab1变体抗体在h292细胞中抑制egf-介导的egfr的酪氨酸磷酸化的能力。图8用图形描绘了包括ab1、ab2和ab1变体的pegfrelisa分析的结果(图8a)和来自a431抑制研究的ab1相比于ab2和abp的抑制水平(图8b)。图8a的y-轴是450nm处的光密度(od)。图9用图形描绘了使用facs结合分析测量的ab1、ab2和ab1变体抗体与表达野生型egfr的正常人表皮角质细胞的结合。图10用图形描绘了小鼠异种移植物抑制分析的结果,其比较aba、abg、abk、abm和abp与ab1、ab2和人igg(huigg)对照相比在人nsclc癌异种移植物中抑制肿瘤生长的能力。箭头指示各种抗体施用的时间点。图11提供了aba-马来酰亚胺基己酰基(malemidocaproyl)-vc-paba-mmaeadc(在本文中称为“aba-vcmmae”)的结构。图12-1和12-2提供了来自aba-vcmmae纯化的疏水相互作用色谱(hic)分析的结果。图13-1和13-2提供了来自aba-vcmmae的尺寸排阻色谱(sec)分析的结果。图14用图形描绘了来自使用抗-egfradc的两个小鼠异种移植物抑制分析的结果。图14a描绘了来自小鼠异种移植物抑制分析的结果,其比较了来自人nsclc癌异种移植物的nci-h1703细胞中肿瘤生长的抑制,证明aba-vcmmae与ab1和ab1-mcmmafadc相比增强的抑制作用。图14b描绘了来自小鼠异种移植物抑制分析的结果,其比较了来自人nsclc癌异种移植物的ebc-1细胞中肿瘤生长的抑制,证明aba-vcmmae与ab1和ab1-mcmmafadc相比增强的抑制作用。箭头指示抗体施用的时间点。图15用图形描绘了来自使用抗-egfradc的小鼠异种移植物抑制分析的结果。图15a显示小鼠异种移植物抑制分析的结果,其比较了nci-h292细胞中肿瘤生长的抑制,证明纯化的aba-vcmmae(aba-vcmmaep)和aba-vcmmae与纯化的ab1-vcmmae(ab1-vcmmaep)、ab1-vcmmae、纯化的ab1-mcmmafadc(ab1-mcmmafp)和ab1-mcmmaf(相对于三个对照)相比增强的抑制作用。图15b显示小鼠异种移植物抑制分析的结果,其比较了nci-h292细胞中肿瘤生长的抑制,证明aba-vcmmae相比于纯化的aba-vcmmae(aba-vcmmaep)和aba-vcmmae相比于纯化的ab1-vcmmae(ab1-vcmmaep)、ab1-vcmmae、ab1-mcmmaf和ab1-mcmmafp增强的抑制活性。图15a和b中分子的剂量显示在括号中,即,3mg/kg或6mg/kg。箭头指示抗体或adc施用的时间点。图15中的对照2代表阴性对照,其是不结合egfr的抗-破伤风毒素抗体。图16提供了ab1变体可变重链(vh)文库设计(图16a)和ab1变体可变轻链(vl)文库设计(图16b)的氨基酸序列。图17显示egfr及由ab1和ab2结合的区域的示意图。图18用图形描绘了来自采用抗-egfradc的小鼠异种移植物抑制分析(使用nci-h292(nsclc)细胞)的结果。分子的剂量显示在括号中,即,3mg/kg或6mg/kg。箭头指示抗体或adc施用的时间点。图19用图形描绘了来自采用抗-egfrmmae和mmafadc的小鼠成胶质细胞瘤异种移植物抑制分析的结果。图19中分子的剂量显示在括号中,即,1mg/kg。箭头指示抗体或adc施用的时间点。图19中的对照2代表阴性对照,其是不结合egfr的抗-破伤风毒素抗体。图20a和b用图形描绘了来自单光子发射计算机断层摄影(spect)成像分析的结果,其比较了使用用111in标记的aba、ab1或对照抗体在两个肿瘤模型(分别地sw48(图20a)和ebc1(图20b)肿瘤模型)中egfr表达的肿瘤的抗体摄取效率。图21描绘了通过马来酰亚胺基己酰基-缬氨酸-丙氨酸接头(统称为sgd-1910)与抗体(ab)偶联的pbd二聚体(sgd-1882)的结构。具体实施方式本发明的各个方面涉及抗-egfr抗体和抗体片段、抗-egfradc及其药物组合物以及用于制备这样的抗体和片段的核酸、重组表达载体和宿主细胞。使用本文所述的抗体和adc检测人egfr、抑制人egfr活性(体外或体内)和治疗癌症如上皮癌、乳腺癌、结肠直肠癌、头颈癌(例如,成胶质细胞瘤)、肺癌、肾癌、胰腺癌、间皮瘤、鳞状细胞癌(例如,肺鳞癌或头颈鳞状细胞癌)、三阴性乳腺癌、非小细胞肺癌和前列腺癌的方法也包括在本发明中。i.定义为了使得本发明可以更容易地理解,首先定义某些术语。另外,应当注意,无论何时提及参数的值或值的范围,所述值之间的值和范围也意图是本发明的部分。本文中可互换使用的术语“抗-表皮生长因子(egf)受体抗体”或“抗-egfr抗体”是指特异性结合egfr的抗体。“结合”目标抗原(即,egfr)的抗体是能够以足够的亲和力结合该抗原以使得该抗体可用于靶向表达该抗原的细胞的抗体。在优选的实施方式中,抗体特异性地结合人egfr(hegfr)。抗-egfr抗体的实例公开于以下实施例1中。除非另外说明,术语“抗-egfr抗体”意思是指结合野生型egfr或egfr的任何变体如egfrviii的抗体。野生型人egfr的氨基酸序列以下提供为seqidno:32,其中信号肽(氨基酸残基1-24)加下划线,且细胞外域(ecd,氨基酸残基25-645)的氨基酸残基以黑体突出显示。egfr的截短的野生型ecd(在本文中也称为egfr(1-525))对应于seqidno:47且等同于seqidno:32的氨基酸1-525。野生型egfr的成熟形式对应于没有信号肽的蛋白质,即,seqidno:32的氨基酸残基25-1210。人egfr的ecd的氨基酸序列以下提供为seqidno:34,且包括信号序列(加下划线的)。图17中描述了egfr的总体结构。egfr的ecd具有四个结构域(cochran等人,(2004)j.immunol.methods,287,147-158)。结构域i和iii表明有助于形成对于配体的高亲和力结合位点。结构域ii和iv是富半胱氨酸的层粘连蛋白样区域,其使蛋白质折叠稳定并包含可能的egfr二聚化界面。egfr变体可以由通过egfr基因扩增完成的基因重排产生。egfrviii是人类癌症中最常出现的egfr变体(kuan等人,endocrrelatcancer.8(2):83-96(2001))。在基因扩增的过程中,267个氨基酸缺失出现在egfr的细胞外域中,且甘氨酸残基在融合接合处插入。因此,egfrviii缺乏野生型egfr细胞外域的氨基酸6-273并包括接合处的甘氨酸残基插入。egfr的egfrviii变体包含细胞外域中267个氨基酸残基的缺失,其中甘氨酸残基在缺失接合处插入。egfrviii氨基酸序列以下显示为seqidno:33(ecd以黑体突出显示并对应于seqidno:46,信号序列加下划线)。egfrviii造成以非配体依赖性的方式通过组成性信号传导的肿瘤进展。正常组织中egfrviii的表达是未知的(wikstrand等人,cancerresearch55(14):3140-3148(1995);olapade-olaopa等人,brjcancer.82(1):186-94(2000)),但在肿瘤细胞中显示显著的表达,包括乳腺癌、神经胶质瘤、nscl癌、卵巢癌和前列腺癌(wikstrand等人,cancerresearch55(14):3140-3148(1995);ge等人,intjcancer.98(3):357-61(2002);wikstrand等人,cancerresearch55(14):3140-3148(1995);moscatello等人,cancerres.55(23):5536-9(1995);garciadepalazzo等人,cancerres.53(14):3217-20(1993);moscatello等人,cancerres.55(23):5536-9(1995)及olapade-olaopa等人,2(1):186-94(2000))。如本文中使用的“egfr的生物活性”是指egfr的所有内在生物学性质,包括但不限于与表皮生长因子(egf)的结合、与肿瘤生长因子α(tgfα)的结合、同型二聚化、jak2激酶活性的激活、mapk激酶活性的激活和跨膜受体蛋白酪氨酸激酶活性的激活。如本文中使用的涉及抗体或adc与第二化学物质的相互作用的术语“特异性结合”或“特异性地结合”意思是该相互作用依赖于该化学物质上特定结构(例如,抗原决定簇或表位)的存在;例如,抗体识别和结合特定蛋白质结构而不是总体地结合于蛋白质。如果抗体或adc对于表位“a”是特异性的,在包含标记的“a”和抗体的反应中包含表位a的分子(或游离的未标记的a)的存在将减小与抗体或adc结合的标记的a的量。如本文中使用的短语“与hegfr特异性地结合”或“特异性地结合于hegfr”是指抗-egfr抗体或adc以等于或大于ab1或ab1adc的亲和力与hegfr相互作用的能力。如本文中使用的术语“与egfr(1-525)特异性结合”或“特异性地结合于egfr(1-525)”是指抗体或adc结合egfr(1-525)且如通过表面等离子体共振测定的,具有2.3x10-6m或更低的解离常数(kd)。术语“抗体”广义地指免疫球蛋白(ig)分子(一般由四个多肽链构成,两个重链(h)和两个轻链(l))或者其任何功能片段、突变体、变体或衍生物,其保留ig分子的关键的靶结合特征。这样的突变体、变体或衍生物抗体形式是本领域中已知的。其非限制性实施方式在下面讨论。在全长抗体中,各重链由重链可变区(本文中缩写为hcvr或vh)和重链恒定区构成。重链恒定区由三个结构域ch1、ch2和ch3构成。各轻链由轻链可变区(本文中缩写为lcvr或vl)和轻链恒定区构成。轻链恒定区由一个结构域cl构成。vh和vl区可以进一步细分成高变区(称为互补决定区(cdr)),其间插有更保守的区域(称为框架区(fr))。各vh和vl由三个cdr和四个fr构成,其从氨基端至羧基端按以下顺序排列:fr1、cdr1、fr2、cdr2、fr3、cdr3、fr4。免疫球蛋白分子可以是任何类型(例如,igg、ige、igm、igd、iga和igy)和类别(例如,igg1、igg2、igg3、igg4、iga1和iga2)或亚类。如本文中使用的术语抗体的“抗原结合部分”(或简称为“抗体部分”)是指保留与抗原(例如,hil-13)特异性结合的能力的抗体的一个或多个片段。已经证明抗体的抗原结合功能可以通过全长抗体的片段执行。这样的抗体实施方式也可以是双特异性的、双重特异性的或多特异性的形式;特异性地结合两个或更多个不同抗原。术语抗体的“抗原结合部分”内涵盖的结合片段的实例包括(i)fab片段,由vl、vh、cl和ch1结构域组成的单价片段;(ii)f(ab′)2片段,包含由铰链区的二硫桥连接的两个fab片段的双价片段;(iii)由vh和ch1结构域组成的fd片段;(iv)由抗体单臂的vl和vh结构域组成的fv片段;(v)dab片段(ward等人,(1989)nature341:544-546,winter等人,pct公开wo90/05144a1,通过引用并入本文),其包含单一可变域,及(vi)分离的互补决定区(cdr)。此外,虽然fv片段的两个结构域,vl和vh,通过单独的基因编码,但它们可以使用重组方法通过使得它们能够作为单一蛋白质链生成的合成接头接合,其中vl和vh区配对以形成单价分子(称为单链fv(scfv);参见,例如,bird等人,(1988)science242:423-426,及huston等人,(1988)proc.natl.acad.sci.usa85:5879-5883)。这样的单链抗体也旨在包括在术语抗体的“抗原结合部分”内。在本发明的某些实施方式中,scfv分子可以合并到融合蛋白中。也涵盖其它形式的单链抗体如双抗体。双抗体是二价的双特异性的抗体,其中vh和vl结构域在单一多肽链上表达,但使用过短而不允许相同链上的两个结构域之间配对的接头,从而迫使结构域与另一链的互补结构域配对且形成两个抗原结合位点(参见,例如,holliger,p.等人,(1993)proc.natl.acad.sci.usa90:6444-6448;poljak,r.j.等人,(1994)structure2:1121-1123)。这样的抗体结合部分是本领域中已知的(kontermann和dubel编辑,antiodyengineering(2001)springer-verlag.newyork.790pp.(isbn3-540-41354-5)。如本文中使用的术语“抗体构建体”是指包含连接于接头多肽或免疫球蛋白恒定域的一个或多个本发明的抗原结合部分的多肽。接头多肽包含通过肽键接合的两个或更多个氨基酸残基并用于连接一个或多个抗原结合部分。这样的接头多肽是本领域中公知的(参见,例如,holliger,p.等人,(1993)proc.natl.acad.sci.usa90:6444-6448;poljak,r.j.等人,(1994)structure2:1121-1123)。免疫球蛋白恒定域是指重链或轻链恒定域。示意性的人igg重链和轻链恒定域氨基酸序列是本领域已知的并显示如下。人igg重链恒定域和轻链恒定域的序列再进一步地,抗体或其抗原结合部分可以是通过抗体或抗体部分与一个或多个其它蛋白质或肽的共价或非共价结合形成的较大免疫吸附分子的部分。这样的免疫吸附分子的例子包括使用链霉亲和素核心区域形成四聚的scfv分子(kipriyanov,s.m.等人,(1995)humanantibodiesandhybridomas6:93-101)和使用半胱氨酸残基、标志肽和c-末端聚组氨酸标签以形成二价和生物素化的scfv分子(kipriyanov,s.m.等人,(1994)mol.immunol.31:1047-1058)。抗体部分如fab和f(ab′)2片段可以使用常规技术(如全抗体的分别木瓜蛋白酶或胃蛋白酶消化)从全抗体制备。而且,抗体、抗体部分和免疫吸附分子可以使用标准重组dna技术获得,如本文所述的。如本文中使用的“分离的抗体”意图是指基本上不含具有不同抗原特异性的其它抗体的抗体(例如,特异性地结合egfr的分离的抗体基本上不合特异性地结合egfr以外的抗原的抗体)。但是,特异性地结合egfr的分离的抗体可以具有对来自其它物种的其它抗原(如egfr分子)的交叉反应性。而且,分离的抗体可以基本上不含其它细胞物质和/或化学物质。术语“人源化抗体”是指包含来自非人物种(例如,小鼠)的重链和轻链可变区序列的抗体,但其中vh和/或vl序列的至少一部分被改变以成为更“类人的”,即,更类似于人种系可变序列。特别地,术语“人源化抗体”是抗体或其变体、衍生物、类似物或片段,其与目标抗原免疫特异性地结合并包含基本上具有人抗体的氨基酸序列的框架区(fr)和基本上具有非人抗体的氨基酸序列的互补决定区(cdr)。如本文中使用的,关于cdr的术语“基本上”是指具有与非人抗体cdr的氨基酸序列至少80%,优选至少85%、至少90%、至少95%、至少98%或至少99%相同的氨基酸序列的cdr。人源化抗体包含基本上全部的至少一个和通常两个可变域(fab、fab′、f(ab′)2、fabc、fv),其中全部或基本上全部的cdr区对应于非人免疫球蛋白(即,供体抗体)的cdr区且全部或基本上全部的框架区是人免疫球蛋白共有序列的框架区。优选地,人源化抗体还包含免疫球蛋白恒定区的至少一部分(fc),通常人免疫球蛋白的免疫球蛋白恒定区的至少一部分。在一些实施方式中,人源化抗体包含轻链以及至少重链的可变域两者。抗体也可以包括重链的ch1、铰链、ch2、ch3和ch4区。在一些实施方式中,人源化抗体仅包含人源化轻链。在其它实施方式中,人源化抗体仅包含人源化重链。在特定的实施方式中,人源化抗体仅包含轻链的人源化可变域和/或人源化重链。人源化抗体可以选自任何类别的免疫球蛋白,包括igm、igg、igd、iga和ige,及任何同种型,包括但不限于igg1、igg2、igg3和igg4。人源化抗体可以包含来自超过一个类别或同种型的序列,且可以选择特定恒定域以利用本领域中公知的技术优化所需的效应子功能。术语“kabat编号”、“kabat定义”和“kabat标记”在本文中可互换使用。本领域中公认的这些术语是指对抗体或其抗原结合部分的重链和轻链可变区中比其它氨基酸残基更可变的(即,高变的)氨基酸残基编号的系统(kabat等人,(1971)ann.nyacad,sci.190:382-391和kabat,e.a.等人,(1991)sequencesofproteinsofimmunologicalinterest,fifthedition,u.s.departmentofhealthandhumanservices,nih公开no.91-3242)。对于重链可变区,高变区范围对于cdr1为氨基酸位置31-35,对于cdr2为氨基酸位置50-65及对于cdr3为氨基酸位置95-102。对于轻链可变区,高变区范围对于cdr1为氨基酸位置24-34,对于cdr2为氨基酸位置50-56及对于cdr3为氨基酸位置89-97。如本文中使用的,术语“cdr”是指抗体可变序列中的互补决定区。重链(hc)和轻链(lc)的各可变区中具有三个cdr,其对于各可变区命名为cdr1、cdr2和cdr3(或具体地hccdr1、hccdr2、hccdr3、lccdr1、lccdr2和lccdr3)。如本文中使用的术语“cdr组”是指单一可变区中存在的能够结合抗原的三个cdr的组。这些cdr的精确边界按照不同系统不同地定义。kabat描述的系统(kabat等人,sequencesofproteinsofimmunologicalinterest(nationalinstitutesofhealth,bethesda,md.(1987)和(1991))不仅提供可应用于抗体的任何可变区的明确的残基编号系统,而且提供确定三个cdr的准确残基边界。这些cdr可以称为kabatcdr。chothia和同事(chothia&lesk,j.mol.biol.196:901-917(1987)和chothia等人,nature342:877-883(1989))发现,kabatcdr内的某些子部分采取近乎相同的肽骨架构象,尽管在氨基酸序列的水平上具有大的多样性。这些子部分命名为l1、l2和l3或者h1、h2和h3,其中“l”和“h”分别指明轻链和重链区域。这些区域可以称为chothiacdr,其具有与kabatcdr重叠的边界。限定与kabatcdr重叠的cdr的其它边界由padlan(fasebj.9:133-139(1995))和maccallum(jmolbiol262(5):732-45(1996))描述。再其它的cdr边界定义可能不严格地遵循上述系统之一,但仍然与kabatcdr重叠,虽然它们根据特定残基或残基组或者甚至整个cdr不显著影响抗原结合的预测或实验发现而缩短或延长。本文中使用的方法可以采用按照这些系统中任一种定义的cdr,虽然优选的实施方式使用kabat或chothia定义的cdr。如本文中使用的,术语“框架”或“框架序列”是指可变区减去cdr的其余序列。因为cdr序列的精确定义可以通过不同系统确定,框架序列的含义进行相应的不同解释。六个cdr(轻链的cdr-l1、cdr-l2和cdr-l3及重链的cdr-h1、cdr-h2和cdr-h3)也在各链上将轻链和重链上的框架区分割成四个子区域(fr1、fr2、fr3和fr4),其中cdr1位于fr1和fr2之间,cdr2位于fr2和fr3之间且cdr3位于fr3和fr4之间。在未具体指明特定子区域为fr1、fr2、fr3或fr4的情况下,另外所称的框架区表示单一的天然存在免疫球蛋白链的可变区内的组合fr。如本文中使用的,fr表示四个子区域之一,且frs表示构成框架区的四个子区域中的两个或更多个。人源化抗体的框架和cdr区不需要精确地对应于亲本序列,例如,供体抗体cdr或共有框架可以通过至少一个氨基酸残基的置换、插入和/或删除进行诱变,使得该位点处的cdr或框架残基不对应于供体抗体或共有框架。但是在优选的实施方式中,这样的突变不是广泛的。通常,至少80%,优选至少85%,更优选至少90%,且最优选至少95%的人源化抗体残基对应于亲本fr和cdr序列的那些残基。如本文中使用的,术语“共有框架”是指共有免疫球蛋白序列中的框架区。如本文中使用的,术语“共有免疫球蛋白序列”是指由相关免疫球蛋白序列家族中最常出现的氨基酸(或核苷酸)形成的序列(参见,例如,winnaker,fromgenestoclones(verlagsgesellschaft,weinheim,germany1987)。在免疫球蛋白家族中,共有序列中的各位置被家族中最常出现于该位置处的氨基酸占据。如果两个氨基酸以等同频率出现,则其任一可以包括在共有序列中。关于肽或多肽序列的“百分(%)氨基酸序列同一性”定义为在比对序列和引入空位(如果必要)以获得最大百分序列同一性且不考虑任何保守性置换作为序列同一性的部分后,候选序列中与特定肽或多肽序列中的氨基酸残基相同的氨基酸残基的百分比。为测定百分氨基酸序列同一性的目的的比对可以例如使用公众可得的计算机软件如blast、blast-2、align或megalign(dnastar)软件以本领域技能内的各种方式实现。本领域技术人员可以确定用于测量比对的适宜参数,包括在所比较的序列全长上实现最大比对需要的任何算法。在一个实施方式中,本发明包括与seqidno:1-31、35-40或50-85任一中所示的氨基酸序列具有至少80%、至少85%、至少90%、至少95%、至少96%、至少97%、至少98%或至少99%同一性的氨基酸序列。术语“多价抗体”本文中用于表示包含两个或更多个抗原结合位点的抗体。在某些实施方式中,多价抗体可以工程化以具有三个或更多个抗原结合位点,且一般不是天然存在的抗体。术语“多特异性抗体”是指能够结合两个或更多个不相关的抗原的抗体。在一个实施方式中,多特异性抗体是能够结合两个不相关的抗原的双特异性抗体(例如,双特异性抗体)或其抗原结合部分,其结合egfr(例如,egfrviii)和cd3。本文中可互换使用的术语“双重可变域”或“dvd”是包含两个或更多个抗原结合位点且是四价或多价结合蛋白的抗原结合蛋白。这样的dvd可以是单特异性的,即,能够结合一种抗原,或者是多特异性的,即能够结合两种或更多种抗原。包含两个重链dvd多肽和两个轻链dvd多肽的dvd结合蛋白称为dvdig。dvdig的各一半包含重链dvd多肽和轻链dvd多肽及两个抗原结合位点。各结合位点包含重链可变域和轻链可变域,每抗原结合位点具有总共6个参与抗原结合的cdr。在一个实施方式中,本文所述的cdr用于抗-egfrdvd中。术语“嵌合抗原受体”或“car”是指至少包含(1)抗原结合区,例如,抗体的可变重链或轻链,(2)锚定car到t细胞中的跨膜结构域,和(3)一个或多个细胞内信号传导域的重组蛋白质。术语“活性”包括如抗体或adc(例如,结合hegfr抗原的抗-hegfr抗体)对于抗原的结合特异性/亲和力和/或抗体(例如,抗-hegfr抗体,其与hegfr的结合抑制hegfr的生物活性)的中和效能的活性,例如,egfr表达细胞系(例如,人肺癌细胞系h292)中egfr磷酸化的抑制或egfr表达细胞系(例如,人h292肺癌细胞、人h1703肺癌细胞或人ebc1肺癌细胞)增殖的抑制。如本文中使用的术语“非小细胞肺癌(nsclc)异种移植分析”是指用于确定抗-egfr抗体或adc是否能够抑制肿瘤生长(例如,进一步生长)和/或降低由nsclc细胞移植到免疫缺陷小鼠中导致的肿瘤生长的体内分析。nsclc异种移植分析包括nsclc细胞移植到免疫缺陷小鼠中以使得肿瘤生长到所需大小,例如,200-250mm3,此时抗体或adc施用于小鼠以确定抗体或adc是否能够抑制和/或降低肿瘤生长。在某些实施方式中,抗体或adc的活性按照相对于对照抗体(例如,不特异性结合肿瘤细胞的人igg抗体(或其集合),例如,针对不与癌症相关的或从非癌性的来源(例如,正常人血清)获得的抗原)的百分肿瘤生长抑制(%tgi)确定。在这样的实施方式中,抗体(或adc)和对照抗体以相同剂量和相同的频率且通过相同的途径施用于小鼠。在一个实施方式中,nsclc异种移植分析中使用的小鼠是严重联合免疫缺陷(scid)小鼠和/或无胸腺cd-1裸鼠。可以用于nsclc异种移植分析中的nsclc细胞的实例包括,但不限于h292细胞(例如,ncih292[h292](crl1848tm))。术语“表位”是指被抗体或adc结合的抗原区域。在某些实施方式中,表位决定簇包括分子的化学活性表面群组如氨基酸、糖侧链、磷酰基或磺酰基,且在某些实施方式中,可以具有特定的三维结构特征和/或特定的电荷特征。在某些实施方式中,当抗体优先识别蛋白质和/或大分子的复杂混合物中的其靶抗原时,抗体可以说是特异性地与抗原结合。在特定实施方式中,本发明的抗体结合由氨基酸序列cgadsyemeedgvrkc(seqidno:45)(其对应于hegfr成熟形式的氨基酸残基287-302)限定的表位。如本文中使用的术语“表面等离子体共振”是指允许通过检测生物感测器(biosensor)矩阵内蛋白质浓度的改变,例如使用biacore系统(pharmaciabiosensorab,uppsala,sweden和piscataway,nj),来分析实时生物特异性相互作用的光学现象。进一步的说明参见u.等人,(1993)ann.biol.clin.51:19-26;u.等人,(1991)biotechniques11:620-627;johnsson,b.等人,(1995)j.mol.recognit.8:125-131及johnnson,b.等人,(1991)anal.biochem.198:268-277。在一个实施方式中,表面等离子体共振按照实施例2中描述的方法测定。如本文中使用的术语“kon”或“ka”意图是指抗体与抗原结合以形成抗体/抗原复合物的结合速率常数。术如本文中使用的语“koff”或“kd”意图是指抗体从抗体/抗原复合物解离的解离速率常数。如本文中使用的术语“kd”意图是指特定的抗体-抗原相互作用(例如,aba抗体和egfr)的平衡解离常数。kd通过ka/kd计算。如本文中使用的术语“竞争结合”是指其中第一抗体与第二抗体竞争第三分子(例如,抗原)上的结合位点的情况。在一个实施方式中,两个抗体之间的竞争结合使用facs分析测定。术语“竞争结合分析”是用于确定两个或更多个抗体是否结合相同表位的分析法。在一个实施方式中,竞争结合分析是竞争荧光活化细胞分选(facs)分析,其用于通过测定标记抗体的荧光信号是否由于非标记抗体的引入而降低来确定两个或更多个抗体是否结合相同表位,其中对于相同表位的竞争将降低荧光水平。竞争结合facs分析的实例提供在实施例3中,其中竞争facs分析利用u87mg细胞(其表达egfrviii)进行描述。如本文中使用的术语“标记抗体”是指具有整合的标记的抗体或其抗原结合部分,该标记用于鉴别结合蛋白,例如,抗体。优选地,标记是可检测标志物,例如,掺入放射标记的氨基酸或将可以通过标记的抗生物素蛋白检测的生物素基部分(例如,包含可通过光学或比色方法检测的荧光标记或酶活性的链霉亲和素)附接于多肽。用于多肽的标记的实例包括,但不限于以下:放射性同位素或放射性核素(例如,3h、14c、35s、90y、99tc、111in、125i、131i、177lu、166ho或153sm)、荧光标记(例如,fitc、罗丹明、稀土磷光体)、酶标记(例如,辣根过氧化物酶、萤光素酶、碱性磷酸酶)、化学发光标记、生物素基团、次级报告体识别的预定多肽表位(例如,亮氨酸拉链对序列、第二抗体的结合位点、金属结合结构域、表位标签)及磁性剂如钆螯合物。术语“抗体-药物偶联物”或“adc”是指与一个或多个化学药物(在本文中也称为药剂)化学连接的结合蛋白,如抗体或其抗原结合片段,该化学药物可以任选地为治疗剂或细胞毒性剂。在优选的实施方式中,adc包括抗体、细胞毒性或治疗药物及使得药物能够与抗体附接或偶联的接头。adc通常具有从1至8的任何数目的与抗体偶联的药物,包括2、4、6或8的载药种类(drugloadedspecies)。可以包括在adc中的药物的非限制性实例是有丝分裂抑制剂、抗肿瘤抗生素、免疫调节剂、用于基因疗法的载体、烷化剂、抗血管生成剂、抗代谢物、含硼剂、化疗保护剂、激素、抗激素剂、皮质类固醇、光敏治疗剂、寡核苷酸、放射性核素剂、拓扑异构酶抑制剂、酪氨酸激酶抑制剂和放射致敏剂。本文中可互换使用的术语“抗-表皮生长因子抗体药物偶联物”、“抗-egfr抗体药物偶联物”或“抗-egfradc”是指包含与egfr特异性地结合的抗体的adc,由此抗体与一个或多个化学剂偶联。在一个实施方式中,抗-egfradc是与阿里他汀(例如,mmae或mmaf)偶联的抗体aba。对应于抗体aba的轻链和重链的氨基酸序列分别提供在seqidno:13和seqidno:15中。如本文中使用的术语“阿里他汀”是指一个抗有丝分裂剂的家族。阿里他汀衍生物也包括在术语“阿里他汀”的定义内。阿里他汀的实例包括,但不限于阿里他汀e(ae)、单甲基阿里他汀e(mmae)、单甲基阿里他汀f(mmaf)和多拉司他汀的合成类似物。在一个实施方式中,本文所述的抗-egfr抗体与阿里他汀偶联以形成抗-egfradc。如本文中使用的术语“aba-vcmmae”用于指包含通过马来酰亚胺基己酰基缬氨酸瓜氨酸p-氨基苄氧基氨甲酰基(paba)接头与单甲基阿里他汀e(mmae)偶联的抗体aba的adc。aba-vcmmae如图11中所述。如本文中使用的术语“mcmmaf”用于指马来酰亚胺基己酰基-单甲基阿里他汀f(mmaf)的接头/药物组合。术语“药物-抗体比率”或“dar”是指与adc的抗体连接的药物(例如,阿里他汀)的数目。adc的dar范围可以为1-8,虽然更高载量(例如,10)也是可能的,取决于抗体上连接位点的数目。术语dar可以用于指负载到单个抗体上的药物的数目,或者可选地,可以用于指一组adc的平均或均值dar。如本文中使用的术语“不希望的adc种类”是指将从具有不同药物载量的adc种类分离的任何载药种类。在一个实施方式中,术语不希望的adc种类可以指6或更高的载药种类,即具有6或更高的dar的adc,包括dar6、dar7、dar8和大于8的dar(即,6、7、8或大于8的载药种类)。在单独的实施方式中,术语不希望的adc种类可以指8或更高的载药种类,即,具有8或更高的dar的adc,包括dar8和大于8的dar(即,8或高于8的载药种类)。如本文中使用的术语“adc混合物”是指包含adc的多相dar分布的组合物。在一个实施方式中,adc混合物包含dar分布为1-8的adc,例如,2、4、6和8(即,2、4、6和8的载药种类)。值得注意的是,可能产生降解产物,使得1、3、5和7的dar也可能包含于混合物中。此外,混合物中adc也可以具有超过8的dar。adc混合物由链间二硫键还原然后偶联获得。在一个实施方式中,adc混合物包含dar为4或更低的adc(即,4或更低的载药种类)和dar为6或更高的adc(即,6或更高的载药种类)两者。术语“癌症”意指或描述哺乳动物中通常特征是不受调节的细胞生长的生理状况。癌症的实例包括但不限于癌瘤、淋巴瘤、胚细胞瘤、肉瘤、白血病或淋巴样恶性肿瘤。此类癌症的更具体的实例包括成胶质细胞瘤、非小细胞肺癌、肺癌、结肠癌、结肠直肠癌、头颈癌、乳腺癌(例如,三阴性乳腺癌)、胰腺癌、鳞状细胞肿瘤、鳞状细胞癌(例如,肺鳞癌或头颈鳞状细胞癌)、肛门癌、皮肤癌和外阴癌。在一个实施方式中,向患有包含egfr基因扩增的肿瘤的患者施用本发明的抗体或adc,从而肿瘤表达截短形式的egfr,egfrviii。在一个实施方式中,本发明的抗体或adc向患有很可能过表达egfr的实体肿瘤的受试者施用。在一个实施方式中,本发明的抗体或adc向患有鳞状细胞非小细胞肺癌(nsclc)的患者施用。在一个实施方式中,本发明的抗体或adc向患有实体肿瘤(包括晚期实体肿瘤)的患者施用。如本文中使用的术语“表达egfr的肿瘤”是指表达egfr蛋白的肿瘤。在一个实施方式中,肿瘤中的egfr表达使用肿瘤细胞膜的免疫组织化学染色测定,其中肿瘤样品中高于背景水平的任何免疫组织化学染色表明该肿瘤是表达egfr的肿瘤。用于检测肿瘤中的egfr表达的方法是本领域已知的,例如,egfrpharmdxtmkit(dako)。相反,“egfr阴性肿瘤”定义为肿瘤样品中不存在高于背景的egfr膜染色的肿瘤,如通过免疫组织化学技术测定的。如本文中使用的术语“egfrviii阳性肿瘤”是指表达egfrviii蛋白的肿瘤。在一个实施方式中,肿瘤中的egfrviii表达使用肿瘤细胞膜的免疫组织化学染色测定,其中肿瘤样品中高于背景水平的任何免疫组织化学染色表明该肿瘤是表达egfrviii的肿瘤。用于检测肿瘤中的egfr表达的方法是本领域已知的,且包括免疫组织化学染色分析。相反,“egfrviii阴性肿瘤”定义为肿瘤样品中不存在高于背景的egfrviii染色的肿瘤,如通过免疫组织化学技术测定的。可互换的术语“过表达”、“超表达”或“过表达的”是指与正常细胞相比(通常在癌细胞中)以可检测的较大水平转录或翻译的基因。因此过表达是指蛋白质和rna的过表达两者(由于增加的转录、转录后加工、翻译、翻译后加工、改变的稳定性和改变的蛋白质降解导致)以及由于改变的蛋白质运输模式(增加的核定位)和增强的功能活性(例如,在提高的底物酶水解中)导致的局部过表达。因此,过表达是指蛋白质或rna水平。过表达相比于正常细胞或对比细胞也可以是50%、60%、70%、80%、90%或更高。在某些实施方式中,本发明的抗-egfr抗体或adc用于治疗很可能过表达egfr的实体肿瘤。如本文中使用的术语“施用”意思是指物质(例如,抗-egfr抗体或adc)的递送以实现治疗目的(例如,治疗egfr-相关障碍)。施用的模式可以是肠胃外、经肠和局部。肠胃外施用通常是通过注射,且包括,但不限于静脉内、肌肉内、动脉内、鞘内、囊内、眶内、心内、皮内、腹膜内、经气管、皮下、表皮下、关节内、包膜下、蛛网膜下、脊椎内和胸骨内注射和输注。如本文中使用的术语“组合疗法”是指施用两种或更多种治疗物质,例如,抗-egfr抗体或adc和另外的治疗剂。另外的治疗剂可以与抗-egfr抗体或adc的施用同时、在抗-egfr抗体或adc的施用之前或之后施用。如本文中使用的术语“有效量”或“治疗有效量”是指足以减轻或缓解障碍(例如,癌症)或者其一种或多种症状的严重性和/或持续时间,防止障碍的进展,导致障碍的消退,防止与障碍相关的一种或多种症状的复发、发生、发作或进展,检测障碍或者增强或改善另一疗法(例如,预防或治疗剂)的预防或治疗效果的药物(例如,抗体或adc)的量。有效量的抗体或adc可以,例如,抑制肿瘤生长(例如,抑制肿瘤体积的增大)、降低肿瘤生长(例如,减小肿瘤体积)、减少癌症细胞数量和/或一定程度上缓解与癌症相关的一种或多种症状。有效量可以,例如,改善无疾病存活(dfs)、改善总体存活(os)或减小复发的可能性。本发明的各个方面更详细地描述在以下小节中。ii.抗-egfr抗体本发明的一个方面提供了抗-egfr抗体或其抗原结合部分,其具有改善的特性,例如,相对于ab1和本领域中已知的其它抗体提高的对egfr的结合亲和力。本发明的另一方面特征在于包含本文所述的抗-egfr抗体和至少一种药物(例如,但不限于阿里他汀)的抗体药物偶联物(adc)。本发明的抗体或adc具有包括,但不限于与表达egfrviii的肿瘤细胞结合、与表达egfr的肿瘤细胞上的野生型egfr结合、识别egfr上的表位cgadsyemeedgvrkc(seqidno:45)、与正常人表皮角质细胞上的egfr结合和降低或抑制小鼠模型中的异种移植肿瘤生长的特性。ab1(抗体1)是人源化抗-egfr抗体。ab1的轻链和重链序列分别描述于seqidno:13和seqidno:14中(也参见美国专利申请公开no.20120183471,其通过引用并入本文)。ab1的轻链可变区描述于seqidno:5中,且包含seqidno:6中所示的cdr1氨基酸序列、seqidno:7中所示的cdr2氨基酸序列和seqidno:8中所示的cdr3氨基酸序列。ab1的重链可变区描述于seqidno:1中,且包含seqidno:2中所示的cdr1氨基酸序列、seqidno:3中所示的cdr2氨基酸序列和seqidno:4中所示的cdr3氨基酸序列。一般地,本发明的ab1变体抗体保留亲本抗体ab1的表位特异性。因此,在一个实施方式中,本发明的抗-egfr抗体能够结合由seqidno:45限定的egfr中的表位和/或能够与ab1竞争结合egfr。在各种不同实施方式中,结合可以按照以下实施例3中给出的方案测定。在本发明的优选实施方式中,抗-egfr抗体与ab1竞争并对于egfr的1-525(seqidno:47)具有改善的结合亲和力,例如,约1x10-6m至约1x10-10m的解离常数(kd),如通过表面等离子体共振测定的。在一个实施方式中,本发明特征在于作为ab1的变体并具有改善的特性的抗-egfr抗体,例如,改善的结合亲和力和在体内抑制nsclc肿瘤细胞增殖的能力,如以下实施例中所描述的。总的说来,这些新抗体在本文中称为“ab1变体抗体”。一般地,ab1变体抗体保留与ab1相同的表位特异性。因此,在一个实施方式中,本发明的抗-egfr抗体或其抗原结合部分结合seqidno:45中所示的氨基酸序列内的表位并在竞争结合分析中与包含含有seqidno:1中所示的氨基酸序列的重链可变域和含有seqidno:5中所示的氨基酸序列的轻链可变域的抗-egfr抗体竞争结合egfrviii。与ab1相反,本发明的抗-egfr抗体能够在裸鼠的h292人非小细胞肺癌(nsclc)异种移植分析中体内抑制或降低肿瘤生长和/或结合正常人表皮角质细胞上的野生型egfr。在各种不同实施方式中,本发明的抗-egfr抗体或其抗原结合片段能够调节egfr的生物学功能。在前述方面的其它实施方式中,抗-egfr抗体或其抗原结合片段结合egfrviii,结合过表达egfr的细胞上的egfr且识别egfr上的表位cgadsyemeedgvrkc(seqidno:45)。在进一步的实施方式中,抗-egfr抗体或其抗原结合片段在与egfrviii连接肽不同的表位处结合egfrviii。在前述方面的另外的实施方式中,抗-egfr抗体或其抗原结合片段不与西妥昔单抗竞争结合egfr。以下实施例中描述的aba抗体和ab1变体具有前述特性。因此,本发明包括在竞争结合分析中可以与ab1竞争但更有效地抑制或降低肿瘤生长的抗-egfr抗体或其抗原结合部分。在一个实施方式中,本发明的抗-egfr抗体或其抗原结合部分能够结合氨基酸序列cgadsyemeedgvrkc(seqidno:45)内的表位并在竞争结合分析中与ab1(或包含含有seqidno:1中所示的氨基酸序列的重链可变域和含有seqidno:5中所示的氨基酸序列的轻链可变域的抗-egfr抗体)竞争结合表皮生长因子受体变体iii(egfrviii)(seqidno:33)。在一个实施方式中,本发明的抗-egfr抗体或其抗原结合部分以约1x10-6m或更低的解离常数(kd)结合egfr(1-525)(seqidno:47),如通过表面等离子体共振测定的。或者,抗体或其抗原结合部分以约1x10-6m至约1x10-10m的kd结合egfr(1-525)(seqidno:47),如通过表面等离子体共振测定的。在进一步的替代方式中,抗体或其抗原结合部分以约1x10-6m至约1x10-7m的kd结合egfr(1-525)(seqidno:47),如通过表面等离子体共振测定的。或者,本发明的抗体或其抗原结合部分以约1x10-6m至约5x10-10m的kd、约1x10-6m至约1x10-9m的kd、约1x10-6m至约5x10-9m的kd、约1x10-6m至约1x10-8m的kd、约1x10-6m至约5x10-8m的kd、约5.9x10-7m至约1.7x10-9m的kd、约5.9x10-7m至约2.2x10-7m的kd结合egfr(1-525)(seqidno:47),如通过表面等离子体共振测定的。在某些实施方式中,本发明的抗体和抗原-结合片段的解离常数(kd)在一个实施方式中低于ab1的解离常数但高于抗-egfr抗体西妥昔单抗的速率。本发明的抗-egfr抗体及其其抗原结合部分的一个优势是该抗体能够结合表达egfrviii的肿瘤细胞。尽管egfrviii与某些类型的癌症相关,但本领域中已知的许多抗-egfr抗体,例如,西妥昔单抗,不能有效地抑制或降低egfrviii表达肿瘤中的肿瘤生长。因此,在一个实施方式中,本发明的抗体或其抗原结合部分以约8.2x10-9m或更低的kd结合egfrviii(seqidno:33),如通过表面等离子体共振测定的。或者,本发明的抗体或其抗原结合部分以约8.2x10-9m至约6.3x10-10m的kd、约8.2x10-9m至约2.0x10-9m的kd、约2.3x10-9m至约1.5x10-10m的kd结合egfrviii(seqidno:33),如通过表面等离子体共振测定的。在一个实施方式中,本发明的抗体能够在体内异种移植小鼠模型中抑制或降低肿瘤生长。例如,本发明的抗体或其抗原结合部分能够在体内人非小细胞肺癌(nsclc)异种移植分析中相对于非egfr特异性的人igg抗体抑制肿瘤生长至少约50%。在某些实施方式中,当以相同的剂量和给药周期施用时,本发明的抗体或其抗原结合部分能够在体内人非小细胞肺癌(nsclc)异种移植分析中相对于非egfr特异性的人igg抗体抑制或降低肿瘤生长至少约55%、至少约60%、至少约65%、至少约70%、至少约75%或至少约80%。在某些实施方式中,当以相同的剂量和给药周期施用时,本发明的抗体或其抗原结合部分能够在体内人非小细胞肺癌(nsclc)异种移植分析中相对于非egfr特异性的人igg抗体抑制或降低肿瘤生长约80%至约90%,或约84%至约90%,或约88%至约90%。如本文中使用的术语“异种移植分析”是指人肿瘤异种移植分析,其中人肿瘤细胞移植到皮肤下或移植到肿瘤起源的器官类型中、到不排斥人细胞的免疫功能受损小鼠中。应当注意,具有前述特征的组合的抗-egfr抗体或其抗原结合部分也考虑是本发明的实施方式。例如,本发明的抗体可以以约1x10-6m或更低的解离常数(kd)结合egfr(1-525)(seqidno:47),如通过表面等离子体共振测定的,且结合氨基酸序列cgadsyemeedgvrkc(seqidno:45)内的表位和在竞争结合分析中与ab1(或包含含有seqidno:1中所示的氨基酸序列的重链可变域和含有seqidno:5中所示的氨基酸序列的轻链可变域的抗-egfr抗体)竞争结合表皮生长因子受体变体iii(egfrviii)(seqidno:331。在某些实施方式中,抗-egfr抗体或其抗原结合部分结合氨基酸序列cgadsyemeedgvrkc(seqidno:45)内的表位且在竞争结合分析中与ab1(或包含含有seqidno:1中所示的氨基酸序列的重链可变域和含有seqidno:5中所示的氨基酸序列的轻链可变域的抗-egfr抗体)竞争结合表皮生长因子受体变体iii(egfrviii)(seqidno:33),且以约8.2x10-9m或更低的kd结合egfrviii(seqidno:33),如通过表面等离子体共振测定的。在某些实施方式中,抗-egfr抗体或其抗原结合部分结合氨基酸序列cgadsyemeedgvrkc(seqidno:45)内的表位并在竞争结合分析中与ab1(或包含含有seqidno:1中所示的氨基酸序列的重链可变域和含有seqidno:5中所示的氨基酸序列的轻链可变域的抗-egfr抗体)竞争结合表皮生长因子受体变体iii(egfrviii)(seqidno:33),且在体内异种移植小鼠模型中抑制或降低肿瘤生长。更具体地,当以相同的剂量和给药周期施用时,本发明的抗体或其抗原结合部分能够在体内人非小细胞肺癌(nsclc)异种移植分析中相对于非egfr特异性的人igg抗体抑制肿瘤生长至少约50%。或者,当以相同的剂量和给药周期施用时,本发明的抗体或其抗原结合部分能够在体内人非小细胞肺癌(nsclc)异种移植分析中相对于非egfr特异性的人igg抗体抑制肿瘤生长至少约55%、至少约60%、至少约65%、至少约70%、至少约75%或至少约80%。在某些实施方式中,当以相同的剂量和给药周期施用时,本发明的抗体或其抗原结合部分能够在体内人非小细胞肺癌(nsclc)异种移植分析中相对于非egfr特异性的人igg抗体抑制或降低肿瘤生长约80%至约90%、或约84%至约90%、或约88%至约90%。具有任何前述特性的组合的抗体设想为本发明的方面。在下面更详细描述的本发明的adc也可以具有任何前述特性。在一个实施方式中,本发明包括抗-hegfr抗体或其抗原结合部分,其包含含有seqidno:40中所示的氨基酸序列的lccdr3域、含有seqidno:39中所示的氨基酸序列的lccdr2域和含有seqidno:38中所示的氨基酸序列的lccdr1域,及含有seqidno:37中所示的氨基酸序列的hccdr3域、含有seqidno:36中所示的氨基酸序列的hccdr2域和含有seqidno:35中所示的氨基酸序列hccdr1域。在一个实施方式中,本发明包括抗-hegfr抗体或其抗原结合部分,其包含含有选自50、52、54、56、58、60、62、64、66、68、70、72、74、76和78的氨基酸序列的重链可变区及含有选自51、53、55、57、59、61、63、65、67、69、71、73、75、77和79的氨基酸序列的轻链可变区。在一个实施方式中,本发明包括抗-hegfr抗体或其抗原结合部分,其包含选自seqidno:10,11和12;seqidno:16,17和18;seqidno:10,11和19;seqidno:20,11和12;seqidno:21,3和22;seqidno:16,17和19;seqidno:2,3和4;seqidno:10,3和12;seqidno:80,11和18;seqidno:80,3和18;seqidno:20,3和12;seqidno:80,11和12及seqidno:81,11和22的hccdr组(cdr1、cdr2和cdr3);及选自seqidno:6,7和8;seqidno:23,24和25;seqidno:26,27和28;seqidno:29,30和31;seqidno:6,7和84;seqidno:82,83和31及seqidno:82,27和85的lc轻链cdr组(cdr1,cdr2和cdr3),其中抗体或其抗原结合部分不同时包含hccdr组seqidno:2,3和4及lccdr组seqidno:6,7和8。优选地,本发明的抗-egfr抗体表现出降低或中和egfr活性的高能力,例如,如通过本领域中已知的几种体外和体内分析的任何一种评价的。例如,可以测量egfr表达细胞系(例如,h292细胞系)中egfr磷酸化的抑制。在某些实施方式中,分离的抗体或其抗原结合部分结合人egfr,其中抗体或其抗原结合部分以约5.9x10-7m或更低的kd速率常数与人egfr(egfr1-525)解离,如通过表面等离子体共振测定的。或者,抗体或其抗原结合部分可以以约4.2x10-7m的kd速率常数与人egfr(1-525)解离,如通过表面等离子体共振测定的。或者,抗体或其抗原结合部分可以以约2.5x10-7m的大致kd速率常数与人egfr(1-525)解离,如通过表面等离子体共振测定的。在某些实施方式中,本发明的抗-egfr抗体或其抗原结合部分具有5.9x10-7m至5x10-9m的kd速率常数。或者,抗体或其抗原结合部分可以以约6.1x10-9m或更低的kd速率常数与人egfrviii解离,如通过表面等离子体共振测定的。或者,抗体或其抗原结合部分可以以约3.9x10-9m或更低的kd速率常数与人egfrviii解离,如通过表面等离子体共振测定的。或者,抗体或其抗原结合部分可以以约2.3x10-9m或更低的kd速率常数与人egfrviii解离,如通过表面等离子体共振测定的。在一个实施方式中,本发明特征在于抗-egfr抗体或其抗原结合部分,其为抗体aba。aba相对于ab1具有对于egfr的改善的结合亲和力,且也相对于ab1表现出独特的体外和体内特性。aba在体外角质细胞结合分析中以比ab1高得多的亲和力结合egfr。进一步地,aba能够在异种移植h292细胞分析中抑制或降低肿瘤生长。值得注意的是,aba具有与其它ab1变体抗体(其具有比aba高的结合亲和力)相当的改善的体外和体内特性。尽管与其它ab1变体抗体相比具有较低的结合亲和力(参见,例如图3中的abp和abqvs.aba),但aba在体内分析中抑制细胞生长方面是相当的。术语“aba”意图包括具有至少aba的六个cdr的igg抗体。aba抗体具有与ab1相同的轻链,但包含相对于亲本抗体ab1具有六个氨基酸序列改变的重链(重链可变区中的四个氨基酸改变和恒定区中的两个改变)。aba抗体包含重链可变区和轻链可变区,该重链可变区包含含有seqidno:12的氨基酸序列的cdr3域、含有seqidno:11的氨基酸序列的cdr2域和含有seqidno:10的氨基酸序列的cdr1域,且轻链可变区包含含有seqidno:8的氨基酸序列的cdr3域、含有seqidno:7的氨基酸序列的cdr2域和含有seqidno:6的氨基酸序列的cdr1域。aba的重链可变区通过seqidno:9中所示的氨基酸序列限定和轻链可变区包含seqidno:5的氨基酸序列。抗体aba的全长重链在seqidno:15中描述的氨基酸序列中给出,而抗体aba的全长轻链在seqidno:13中描述的氨基酸序列中给出(参见图2)。aba重链的核酸序列提供如下:aba轻链的核酸序列提供如下:在一个实施方式中,本发明特征在于抗-egfr抗体或其抗原结合部分,其是抗体abb。abb抗体包含重链可变区和轻链可变区,该重链可变区包含含有seqidno:19的氨基酸序列的cdr3域、含有seqidno:17的氨基酸序列的cdr2域和含有seqidno:16的氨基酸序列的cdr1域,和轻链可变区包含含有seqidno:8的氨基酸序列的cdr3域、含有seqidno:7的氨基酸序列的cdr2域和含有seqidno:6的氨基酸序列的cdr1域。在进一步的实施方式中,本发明提供具有包含seqidno:64的氨基酸序列的重链可变区和包含seqidno:65的氨基酸序列的轻链可变区的抗体。在一个实施方式中,本发明特征在于抗-egfr抗体或其抗原结合部分,其是抗体abc。abc抗体包含重链可变区和轻链可变区,该重链可变区包含含有seqidno:4的氨基酸序列的cdr3域、含有seqidno:3的氨基酸序列的cdr2域和含有seqidno:2的氨基酸序列的cdr1域,和轻链可变区包含含有seqidno:84的氨基酸序列的cdr3域、含有seqidno:7的氨基酸序列的cdr2域和含有seqidno:6的氨基酸序列的cdr1域。在进一步的实施方式中,本发明提供具有包含seqidno:66的氨基酸序列的重链可变区和包含seqidno:67的氨基酸序列的轻链可变区的抗体。在一个实施方式中,本发明特征在于抗-egfr抗体或其抗原结合部分,其是抗体abd。abd抗体包含重链可变区和轻链可变区,该重链可变区包含含有seqidno:4的氨基酸序列的cdr3域、含有seqidno:3的氨基酸序列的cdr2域和含有seqidno:2的氨基酸序列的cdr1域,和轻链可变区包含含有seqidno:31的氨基酸序列的cdr3域、含有seqidno:83的氨基酸序列的cdr2域和含有seqidno:82的氨基酸序列的cdr1域。在进一步的实施方式中,本发明提供具有包含seqidno:68的氨基酸序列的重链可变区和包含seqidno:69的氨基酸序列的轻链可变区的抗体。在一个实施方式中,本发明特征在于抗-egfr抗体或其抗原结合部分,其是抗体abe。abe抗体包含重链可变区和轻链可变区,该重链可变区包含含有seqidno:4的氨基酸序列的cdr3域、含有seqidno:3的氨基酸序列的cdr2域和含有seqidno:2的氨基酸序列的cdr1域,和轻链可变区包含含有seqidno:85的氨基酸序列的cdr3域、含有seqidno:27的氨基酸序列的cdr2域和含有seqidno:82的氨基酸序列的cdr1域。在进一步的实施方式中,本发明提供具有包含seqidno:50的氨基酸序列的重链可变区和包含seqidno:51的氨基酸序列的轻链可变区的抗体。在一个实施方式中,本发明特征在于抗-egfr抗体或其抗原结合部分,其是抗体abf。abf抗体包含重链可变区和轻链可变区,该重链可变区包含含有seqidno:12的氨基酸序列的cdr3域、含有seqidno:3的氨基酸序列的cdr2域和含有seqidno:10的氨基酸序列的cdr1域,和轻链可变区包含含有seqidno:8的氨基酸序列的cdr3域、含有seqidno:7的氨基酸序列的cdr2域和含有seqidno:6的氨基酸序列的cdr1域。在进一步的实施方式中,本发明提供具有包含seqidno:52的氨基酸序列的重链可变区和包含seqidno:53的氨基酸序列的轻链可变区的抗体。在一个实施方式中,本发明特征在于抗-egfr抗体或其抗原结合部分,其是抗体abg。abg抗体包含重链可变区和轻链可变区,该重链可变区包含含有seqidno:18的氨基酸序列的cdr3域、含有seqidno:17的氨基酸序列的cdr2域和含有seqidno:16的氨基酸序列的cdr1域,和轻链可变区包含含有seqidno:25的氨基酸序列的cdr3域、含有seqidno:24的氨基酸序列的cdr2域和含有seqidno:23的氨基酸序列的cdr1域。在进一步的实施方式中,本发明提供具有包含seqidno:72的氨基酸序列的重链可变区和包含seqidno:73的氨基酸序列的轻链可变区的抗体。在一个实施方式中,本发明特征在于抗-egfr抗体或其抗原结合部分,其是抗体abh。abh抗体包含重链可变区和轻链可变区,该重链可变区包含含有seqidno:18的氨基酸序列的cdr3域、含有seqidno:11的氨基酸序列的cdr2域和含有seqidno:80的氨基酸序列的cdr1域,和轻链可变区包含含有seqidno:25的氨基酸序列的cdr3域、含有seqidno:24的氨基酸序列的cdr2域和含有seqidno:23的氨基酸序列的cdr1域。在进一步的实施方式中,本发明提供具有包含seqidno:54的氨基酸序列的重链可变区和包含seqidno:55的氨基酸序列的轻链可变区的抗体。在一个实施方式中,本发明特征在于抗-egfr抗体或其抗原结合部分,其是抗体abj。abj抗体包含重链可变区和轻链可变区,该重链可变区包含含有seqidno:18的氨基酸序列的cdr3域、含有seqidno:3的氨基酸序列的cdr2域和含有seqidno:80的氨基酸序列的cdr1域,和轻链可变区包含含有seqidno:25的氨基酸序列的cdr3域、含有seqidno:24的氨基酸序列的cdr2域和含有seqidno:23的氨基酸序列的cdr1域。在进一步的实施方式中,本发明提供具有包含seqidno:56的氨基酸序列的重链可变区和包含seqidno:57的氨基酸序列的轻链可变区的抗体。在一个实施方式中,本发明特征在于抗-egfr抗体或其抗原结合部分,其是抗体abk。abk抗体包含重链可变区和轻链可变区,该重链可变区包含含有seqidno:19的氨基酸序列的cdr3域、含有seqidno:11的氨基酸序列的cdr2域和含有seqidno:10的氨基酸序列的cdr1域,和轻链可变区包含含有seqidno:28的氨基酸序列的cdr3域、含有seqidno:27的氨基酸序列的cdr2域和含有seqidno:26的氨基酸序列的cdr1域。在进一步的实施方式中,本发明提供具有包含seqidno:74的氨基酸序列的重链可变区和包含seqidno:75的氨基酸序列的轻链可变区的抗体。在一个实施方式中,本发明特征在于抗-egfr抗体或其抗原结合部分,其是抗体abl。abl抗体包含重链可变区和轻链可变区,该重链可变区包含含有seqidno:18的氨基酸序列的cdr3域、含有seqidno:11的氨基酸序列的cdr2域和含有seqidno:80的氨基酸序列的cdr1域,和轻链可变区包含含有seqidno:28的氨基酸序列的cdr3域、含有seqidno:27的氨基酸序列的cdr2域和含有seqidno:26的氨基酸序列的cdr1域。在进一步的实施方式中,本发明提供具有包含seqidno:58的氨基酸序列的重链可变区和包含seqidno:59的氨基酸序列的轻链可变区的抗体。在一个实施方式中,本发明特征在于抗-egfr抗体或其抗原结合部分,其是抗体abm。abm抗体包含重链可变区和轻链可变区,该重链可变区包含含有seqidno:12的氨基酸序列的cdr3域、含有seqidno:11的氨基酸序列的cdr2域和含有seqidno:20的氨基酸序列的cdr1域,和轻链可变区包含含有seqidno:28的氨基酸序列的cdr3域、含有seqidno:27的氨基酸序列的cdr2域和含有seqidno:26的氨基酸序列的cdr1域。在进一步的实施方式中,本发明提供具有包含seqidno:76的氨基酸序列的重链可变区和包含seqidno:77的氨基酸序列的轻链可变区的抗体。在一个实施方式中,本发明特征在于抗-egfr抗体或其抗原结合部分,其是抗体abn。abn抗体包含重链可变区和轻链可变区,该重链可变区包含含有seqidno:12的氨基酸序列的cdr3域、含有seqidno:3的氨基酸序列的cdr2域和含有seqidno:20的氨基酸序列的cdr1域,和轻链可变区包含含有seqidno:28的氨基酸序列的cdr3域、含有seqidno:27的氨基酸序列的cdr2域和含有seqidno:26的氨基酸序列的cdr1域。在进一步的实施方式中,本发明提供具有包含seqidno:60的氨基酸序列的重链可变区和包含seqidno:61的氨基酸序列的轻链可变区的抗体。在一个实施方式中,本发明特征在于抗-egfr抗体或其抗原结合部分,其是抗体abo。abo抗体包含重链可变区和轻链可变区,该重链可变区包含含有seqidno:12的氨基酸序列的cdr3域、含有seqidno:11的氨基酸序列的cdr2域和含有seqidno:80的氨基酸序列的cdr1域,和轻链可变区包含含有seqidno:28的氨基酸序列的cdr3域、含有seqidno:27的氨基酸序列的cdr2域和含有seqidno:26的氨基酸序列的cdr1域。在进一步的实施方式中,本发明提供具有包含seqidno:62的氨基酸序列的重链可变区和包含seqidno:63的氨基酸序列的轻链可变区的抗体。在一个实施方式中,本发明特征在于抗-egfr抗体或其抗原结合部分,其是抗体abp。abp抗体包含重链可变区和轻链可变区,该重链可变区包含含有seqidno:22的氨基酸序列的cdr3域、含有seqidno:3的氨基酸序列的cdr2域和含有seqidno:21的氨基酸序列的cdr1域,和轻链可变区包含含有seqidno:31的氨基酸序列的cdr3域、含有seqidno:30的氨基酸序列的cdr2域和含有seqidno:29的氨基酸序列的cdr1域。在进一步的实施方式中,本发明提供具有包含seqidno:78的氨基酸序列的重链可变区和包含seqidno:79的氨基酸序列的轻链可变区的抗体。在一个实施方式中,本发明特征在于抗-egfr抗体或其抗原结合部分,其是抗体abq。abq抗体包含重链可变区和轻链可变区,该重链可变区包含含有seqidno:22的氨基酸序列的cdr3域、含有seqidno:11的氨基酸序列的cdr2域和含有seqidno:81的氨基酸序列的cdr1域,和轻链可变区包含含有seqidno:31的氨基酸序列的cdr3域、含有seqidno:30的氨基酸序列的cdr2域和含有seqidno:29的氨基酸序列的cdr1域。在进一步的实施方式中,本发明提供具有包含seqidno:70的氨基酸序列的重链可变区和包含seqidno:71的氨基酸序列的轻链可变区的抗体。如在以下给出实施例的表1中所述的,ab1变体抗体序列提供代表导致改善的与ab1egfr表位结合的cdr域的氨基酸共有序列。因此,在一个实施方式中,本发明特征在于抗-egfr抗体或其抗原结合部分,其包含含有seqidno:40中所示的氨基酸序列的cdr3域、含有seqidno:39中所示的氨基酸序列的cdr2域和含有seqidno:38中所示的氨基酸序列的cdr1域的轻链可变区,及含有seqidno:37中所示的氨基酸序列的cdr3域、含有seqidno:36中所示的氨基酸序列的cdr2域和含有seqidno:35中所示的氨基酸序列的cdr1域的重链可变区。在一个实施方式中,抗-表皮生长因子受体(抗-egfr)抗体或其抗原结合部分包含含有选自50、52、54、56、58、60、62、64、66和68的氨基酸序列的重链可变区及含有选自51、53、55、57、59、61、63、65、67和69的氨基酸序列的轻链可变区。在进一步的实施方式中,本发明的抗-egfr抗体或其抗原结合部分包含重链可变区和轻链可变区,该重链可变区包含含有seqidno:12、18、19和22中所示的氨基酸序列的cdr3域、含有seqidno:11或17中所示的氨基酸序列的cdr2域和含有seqidno:10、16、20和21中所示的氨基酸序列的cdr1域;和轻链可变区包含含有seqidno:8、25、28和31中所示的氨基酸序列的cdr3域、含有seqidno:7、24、27和30中所示的氨基酸序列的cdr2域和含有seqidno:6、23、26和29中所示的氨基酸序列的cdr1域。磷酸化和增殖分析表明本文所述的抗体抑制egfr介导的磷酸化和肿瘤细胞生长。例如,如实施例6中所示的,本发明的egfr抗体(如所测试的)显示为在体内抑制肿瘤细胞生长。前述抗-egfr抗体cdr序列建立了新的egfr结合蛋白家族,其根据本发明分离且包含包括以下表1-3中所列的cdr序列的多肽。为生成和选择具有优选的egfr结合和/或对于hegfr的中和活性的cdr,可以使用本领域中已知用于生成抗体或其抗原结合部分和评估这些抗体或其抗原结合部分的egfr结合和/或中和特性的标准方法,包括但不限于本文中具体描述的那些。在某些实施方式中,抗体包含重链恒定区如igg1、igg2、igg3、igg4、iga、ige、igm或igd恒定区。在某些实施方式中,抗-egfr抗体或其抗原结合部分包含选自人igg恒定域、人igm恒定域、人ige恒定域和人iga恒定域的重链免疫球蛋白恒定域。在进一步的实施方式中,抗体或其抗原结合部分具有igg1重链恒定区、igg2重链恒定区、igg3重链恒定区或igg4重链恒定区。优选地,重链恒定区是igg1重链恒定区或igg4重链恒定区。此外,抗体可以包含轻链恒定区,或者κ轻链恒定区或者λ轻链恒定区。优选地,抗体包含κ轻链恒定区。或者,抗体部分可以是例如fab片段或单链fv片段。在某些实施方式中,抗-egfr抗体结合部分是fab、fab’、f(ab’)2、fv、二硫键连接的fv、scfv、单域抗体或双抗体。在某些实施方式中,抗-egfr抗体或其抗原结合部分是多特异性抗体,例如,双特异性抗体。在某些实施方式中,抗-egfr抗体或其抗原结合部分包含含有seqidno:41中所示的氨基酸序列的重链恒定区和/或含有seqidno:43中所示的氨基酸序列的轻链恒定区。替换fc部分中氨基酸残基以改变抗体效应子功能已经被描述(winter等人。美国专利no.5,648,260和5,624,821,其通过引用并入本文)。抗体的fc部分介导几种重要的效应子功能,例如细胞因子诱导、adcc、胞吞作用、补体依赖性细胞毒性(cdc)及抗体和抗原-抗体复合物的半衰期/清除率。在一些情况中,这些效应子功能是治疗性抗体所需的,但在其它情况中,可能是不必要的或甚至是有害的,这取决于治疗目的。某些人igg同种型,特别是igg1和igg3,通过分别与fcγr和补体c1q结合来介导adcc和cdc。新生fc受体(fcrn)是决定抗体的循环半衰期的关键成分。在再另一实施方式中,至少一个氨基酸残基在抗体的恒定区(例如抗体的fc区)中被替换,使得抗体的效应子功能被改变。本发明的一个实施方式包括重组嵌合抗原受体(car),其包含本文所述的抗体的结合区,例如,aba的重链和/或轻链cdr。本文所述的重组car可以用于以人白细胞抗原(hla)-依赖性的方式重定向t细胞对抗原的特异性。因此,本发明的car可以用于免疫疗法中以帮助对人类受试者自身的免疫细胞工程化而识别和攻击受试者的肿瘤(参见,例如,美国专利no.6,410,319;8,389,282;8,822,647;8,906,682;8,911,993;8,916,381;8,975,071,及美国专利申请公开no.us20140322275,其关于car技术的内容各自通过引用并入本文)。这种类型的免疫疗法称为过继细胞转移(act),且可以用于治疗需要的受试者的癌症。本发明的抗-egfrcar优选含有对于egfr(例如,egfrviii)特异性的细胞外抗原结合域、跨膜结构域(其用于将car锚定到t细胞中)和一个或多个细胞间信号传导结构域。在本发明的一个实施方式中,car包括跨膜结构域,其包括选自t-细胞受体的α、β或ζ链,cd28,cd3ε,cd45,cd4,cd5,cd8,cd9,cd16,cd22,cd33,cd37,cd64,cd80,cd86,cd134,cd137和cd154的蛋白质的跨膜结构域。在本发明的一个实施方式中,car包含共刺激结构域,例如,包含选自ox40、cd2、cd27、cd28、cd5、icam-1、lfa-1(cd11a/cd18)、icos(cd278)和4-1bb(cd137)的蛋白质的功能性信号传导结构域的共刺激结构域。在本发明的某些实施方式中,car包含含有本文所述的cdr或可变区(例如,来自aba抗体的cdr或可变区)的scfv、跨膜结构域、共刺激结构域(例如,来自cd28或4-1bb的功能性信号传导结构域)和包含来自cd3(例如,cd3-ζ)的功能性信号传导结构域的信号传导结构域。在某些实施方式中,本发明包括包含car的t细胞(也称为cart细胞),其包含本文所述抗体的抗原结合区(例如,cdr)或本文所述的scfv。在本发明的某些实施方式中,car包含轻链可变区及重链可变区,该轻链可变区包含含有seqidno:40中所示的氨基酸序列的cdr3域、含有seqidno:39中所示的氨基酸序列的cdr2域和含有seqidno:38中所示的氨基酸序列的cdr1域,和重链可变区包含含有seqidno:37中所示的氨基酸序列的cdr3域、含有seqidno:36中所示的氨基酸序列的cdr2域和含有seqidno:35中所示的氨基酸序列的cdr1域。在本发明的某些实施方式中,car包含重链可变区及轻链可变区,该重链可变区包含含有seqidno:12中所示的氨基酸序列的cdr3域、含有seqidno:11中所示的氨基酸序列的cdr2域和含有seqidno:10中所示的氨基酸序列的cdr1域,和轻链可变区包含含有seqidno:8中所示的氨基酸序列的cdr3域、含有seqidno:7中所示的氨基酸序列的cdr2域和含有seqidno:6中所示的氨基酸序列的cdr1域。本发明的一个实施方式包括标记的抗-egfr抗体或其抗体部分,其中该抗体被衍生化或连接于一个或多个功能分子(例如,另一种肽或蛋白质)。例如,标记的抗体可以通过将本发明的抗体或抗体部分功能性地连接(通过化学偶联、遗传融合、非共价缔合或其它方式)于一个或多个其它分子实体如另一抗体(例如,双特异性抗体或双抗体)、可检测试剂、药物剂、可以介导抗体与抗体部分与另一分子(如链霉亲和素核心区或聚组氨酸标签)的结合的蛋白质或肽和/或细胞毒性剂或治疗剂(选自有丝分裂抑制剂、抗肿瘤抗生素、免疫调节剂、用于基因疗法的载体、烷化剂、抗血管生成剂、抗代谢物、含硼剂、化疗保护剂、激素、抗激素剂、皮质类固醇、光敏治疗剂、寡核苷酸、放射性核素剂、拓扑异构酶抑制剂、酪氨酸激酶抑制剂、放射致敏剂及其组合)来衍生。抗体或其抗体部分可以用以衍生化的有用的可检测试剂包括荧光化合物。示例性的荧光可检测试剂包括荧光素、异硫氰酸荧光素、罗丹明、5-二甲基胺-1-萘磺酰氯、藻红蛋白等等。抗体也可以用可检测的酶衍生化,如碱性磷酸酶、辣根过氧化物酶、葡糖氧化酶等等。当抗体用可检测的酶衍生化时,其通过添加该酶用来产生可检测的反应产物的另外的试剂进行检测。例如,当可检测试剂辣根过氧化物酶存在时,添加过氧化氢和二氨基联苯胺导致有色的反应产物,其是可检测的。抗体也可以用生物素衍生化,并通过间接测量抗生物素蛋白或链霉亲和素结合来检测。在一个实施方式中,本发明的抗体与成像剂偶联。可以用于本文所述的组合物和方法中的成像剂的实例包括,但不限于放射标记(例如,铟)、酶、荧光标记、发光标记、生物发光标记、磁性标记和生物素。在一个实施方式中,抗体或adc连接于放射标记,例如但不限于铟(111in)。111铟可以用于标记本文所述的抗体和adc而用来鉴别egfr阳性肿瘤。在一个特定实施方式中,本文所述的抗-egfr抗体(或adc)通过双功能螯合剂用111in标记,该双功能螯合剂是双功能环己基二亚乙基三胺五乙酸(dtpa)螯合物(参见美国专利no.5,124,471;5,434,287和5,286,850,其各自通过引用并入本文)。本发明的另一实施方式提供糖基化的结合蛋白,其中抗-egfr抗体或其抗原结合部分包含一个或多个糖残基。新生的体内蛋白质产生可以经历进一步加工,称为翻译后修饰。特别地,糖(糖基)残基可以酶促添加,这一过程称为糖基化。所得的携带共价连接的寡糖侧链的蛋白质称为糖基化蛋白质或糖蛋白。抗体是在fc域以及可变域中具有一个或多个糖残基的糖蛋白。fc域中的糖残基对于fc域的效应子功能具有重要影响,而对抗体的抗原结合或半衰期具有最小影响(r.jefferis,biotechnol.prog.21(2005),pp.11-16)。相反,可变域的糖基化对抗体的抗原结合活性可能有影响。可变域中的糖基化可能对抗体结合亲和力具有负面影响,很可能是由于空间位阻(co,m.s.等人,mol.immunol.(1993)30:1361-1367),或者导致对于抗原的提高的亲和力(wallick,s.c.等人,exp.med.(1988)168:1099-1109;wright,a.等人,emboj.(1991)10:2717-2723)。本发明的一个方面涉及生成糖基化位点突变体,其中结合蛋白的o-或n-连接的糖基化位点发生突变。本领域技术人员可以使用标准的公知技术生成这种突变体。保留生物活性但具有提高或降低的结合活性的糖基化位点突变体是本发明的另一目的。在再另一实施方式中,本发明的抗-egfr抗体或抗原结合部分的糖基化被改变。例如,可以制备非糖基化的抗体(即,抗体缺乏糖基化)。糖基化可以被改变以,例如,提高抗体对于抗原的亲和力。这样的糖修饰可以通过,例如,改变抗体序列内的一个或多个糖基化位点来完成。例如,可以进行一个或多个氨基酸置换,其导致一个或多个可变区糖基化位点的消除,从而消除该位点处的糖基化。这种非糖基化可以提高抗体对于抗原的亲和力。这种途径更详细地描述于pct公开wo2003016466a2及美国专利no.5,714,350和6,350,861中,其各自通过引用全文并入本文。另外地或可选地,可以制备本发明的修饰的抗-egfr抗体,其具有改变的糖基化类型,如具有减小的岩藻糖残基量的低岩藻糖基化抗体或具有增加的两分型glcnac结构的抗体。这些改变的糖基化模式已经证明提高抗体的adcc能力。这样的糖修饰可以通过,例如,在具有改变的糖基化机构的宿主细胞中表达抗体而完成。具有改变的糖基化机构的细胞已经在本领域中描述且可以用作在其中表达本发明的重组抗体的宿主细胞而由此产生具有改变的糖基化的抗体。参见,例如,shields,r.l.等人,(2002)j.biol.chem.277:26733-26740;umana等人,(1999)nat.biotech.17:176-1以及欧洲专利no:ep1,176,195;pct公开wo03/035835;wo99/5434280,其各自通过引用全文并入本文。蛋白质糖基化取决于目标蛋白质的氨基酸序列以及蛋白质在其中表达的宿主细胞。不同生物体可以产生不同的糖基化酶(例如,糖基转移酶和糖苷酶),且具有不同的可用底物(核苷酸糖)。由于这些因素,蛋白质糖基化模式和糖基残基的组成可能根据在其中表达特定蛋白质的宿主系统而不同。可用于本发明中的糖基残基可以包括,但不限于葡萄糖、半乳糖、甘露糖、岩藻糖、n-乙酰萄糖胺和唾液酸。优选地,糖基化的结合蛋白包含糖基残基以使得糖基化模式是人的。不同的蛋白质糖基化可以产生不同的蛋白质特性。例如,在微生物宿主如酵母中产生且利用酵母内源途径糖基化的治疗蛋白质的效能与在哺乳动物细胞如cho细胞系中表达的相同蛋白质相比可能降低。这样的糖蛋白在人体中也可能是免疫原性的并在施用后显示出降低的体内半衰期。人体和其它动物中的特定受体可以识别特定的糖基残基并促进蛋白质从血流的快速清除。其它不良反应可以包括蛋白质折叠、溶解性、对蛋白酶的易感性、运输、转运、区室化、分泌、其它蛋白质或因子的识别、抗原性或变应原性的变化。因此,实施者可能更喜欢具有特定糖基化组成和模式的治疗蛋白质,例如与人细胞或预期受试动物的物种特异性细胞中产生的糖基化组成和模式相同或至少相似的组成和模式。表达与宿主细胞的糖基化蛋白不同的糖基化蛋白可以通过遗传修饰宿主细胞以表达异源糖基化酶来实现。使用重组技术,实施者可以产生表现出人蛋白质糖基化的抗体或其抗原结合部分。例如,酵母菌株已经进行遗传修饰以表达非天然存在的糖基化酶,使得在这些酵母菌株中产生的糖基化蛋白质(糖蛋白)表现出与动物细胞(特别是人细胞)相同的蛋白质糖基化(美国专利公开no.20040018590和20020137134及pct公开wo2005100584a2)。抗体可以通过多种技术中的任何一种产生。例如,从宿主细胞表达,其中编码重链和轻链的表达载体通过标准技术转染到宿主细胞中。术语“转染”的各种形式旨在涵盖通常用于将外源dna引入原核宿主细胞或真核宿主细胞中的广泛的技术,例如,电穿孔、磷酸钙沉淀、deae-萄聚糖转染等等。虽然有可能在原核宿主细胞或真核宿主细胞中表达抗体,但真核细胞中抗体的表达是优选的,且最优选在哺乳动物宿主细胞中,因为这样的真核细胞(且特别是哺乳动物细胞)与原核细胞相比更可能组装和分泌正确折叠的和免疫活性的抗体。用于表达本发明的重组抗体的优选哺乳动物宿主细胞包括中国仓鼠卵巢(cho细胞)(包括dhfr-cho细胞,描述于urlaub和chasin,(1980)proc.natl.acad.sci.usa77:4216-4220中,其与dhfr选择标记一起使用,例如,如r.j.kaufman和p.a.sharp(1982)mol.biol.159:601-621中所述的)、ns0骨髓瘤细胞、cos细胞和sp2细胞。当编码抗体基因的重组表达载体引入哺乳动物宿主细胞中时,抗体通过培养宿主细胞足以允许在宿主细胞中表达抗体,或更优选地,分泌抗体到宿主细胞在其中生长的培养基中的一段时间而产生。抗体可以使用标准蛋白质纯化方法从培养基回收。宿主细胞也可以用于产生功能性抗体片段,如fab片段或scfv分子。应理解上述过程的变型在本发明的范围内。例如,可能需要的是用编码本发明抗体的轻链和/或重链的功能性片段的dna转染宿主细胞。重组dna技术也可以用于除去一部分或全部编码轻链和重链任一或两者(其不是与目标抗原的结合必需的)的dna。从这样的截短dna分子表达的分子也涵盖在本发明的抗体内。另外,也可以利用标准化学交联方法通过将本发明的抗体与第二抗体交联来产生双功能抗体,其中一个重链和一个轻链是本发明抗体的而另一重链和轻链对于目标抗原以外的抗原是特异性的。在用于重组表达抗体或其抗原结合部分的优选系统中,编码抗体重链和抗体轻链两者的重组表达载体通过磷酸钙-介导的转染引入dhfr-cho细胞中。在该重组表达载体内,抗体重链和轻链基因各自可操作地连接于cmv增强子/admlp启动子调控元件以驱动基因的高水平转录。重组表达载体还携带dhfr基因,其允许使用甲氨蝶呤选择/扩增来选择已经被载体转染的cho细胞。选择的转化体宿主细胞进行培养以允许表达抗体重链和轻链且完整抗体从培养基回收。标准分子生物学技术用于制备重组表达载体、转染宿主细胞、选择转化体、培养宿主细胞和从培养基回收抗体。更进一步地,本发明提供通过在适宜培养基中培养宿主细胞直至重组抗体被合成而合成本发明的重组抗体的方法。本发明的重组抗体可以使用对应于本文公开的氨基酸序列的核酸分子来产生。在一个实施方式中,seqidno:86和/或87中给出的核酸分子用于产生重组抗体。该方法可以进一步包括从培养基分离重组抗体。iii.抗-egfr抗体药物偶联物(adc)本文所述的抗-egfr抗体可以与药物部分偶联以形成抗-egfr抗体药物偶联物(adc)。由于抗体-药物偶联物(adc)选择性地递送一个或多个药物部分到靶组织(如肿瘤相关抗原,例如,egfr表达肿瘤)的能力,adc可以提高抗体治疗疾病(例如,癌症)的治疗效力。因此,在某些实施方式中,本发明提供抗-egfradc用于治疗性应用,例如,治疗癌症。本发明的抗-egfradc包含与一个或多个药物部分连接的抗-egfr抗体,即,与egfr特异性地结合的抗体。adc的特异性通过抗体(即,抗-egfr)的特异性确定。在一个实施方式中,抗-egfr抗体连接于一个或多个细胞毒性药物,其被内部递送至表达egfr的转化癌细胞。可以用于本发明的抗-egfradc中的药物的实例在以下提供,以及可以用于将抗体与一个或多个药物偶联的接头。术语“药物”、“药剂”和“药物部分”在本文中可互换使用。术语“连接的”和“偶联的”在本文中也可互换使用并表明抗体和部分共价连接。在一些实施方式中,adc具有下式(式i):ab-(l-d)n(i)其中ab是抗体,例如,抗-egfr抗体aba,且(l-d)是接头-药物部分。接头-药物部分由作为接头的l-和作为药物部分的-d构成,该药物部分具有例如针对靶细胞(例如,表达egfr的细胞)的细胞生长抑制性、细胞毒性或其它方面的治疗活性,且n是1-20的整数。在一些实施方式中,n的范围为1-8、1-7、1-6、1-5、1-4、1-3、1-2或是1。adc的dar等同于式i中所称的“n”。在一个实施方式中,adc具有式ab-(l-d)n,其中ab是抗-egfr抗体,例如aba,l是接头,例如,缬氨酸瓜氨酸(vc),d是药物,例如,阿里他汀如mmaf或mmae,且n是2-4(等同于2-4的dar)。关于可以用于本发明的adc中的药物(式i的d)和接头(式i的l)的另外的细节以及可选的adc结构描述如下。a.抗-egfradc:用于偶联的示例性药物抗-egfr抗体可以用于adc中以将一个或多个药物靶向于目标细胞,例如,表达egfr的癌细胞。本发明的抗-egfradc提供靶向治疗,其可以例如减轻通常在抗癌疗法中看到的副作用,因为一个或多个药物被递送到特定细胞。阿里他汀类本发明的抗-egfr抗体,例如,aba抗体,可以与至少一个阿里他汀偶联。阿里他汀类代表一组多拉司他汀类似物,其总体上证明通过干扰微管动力学和gtp水解,由此抑制细胞分裂而具有抗癌活性。例如,阿里他汀e(美国专利no.5,635,483)是海洋天然产物多拉司他汀10(一种通过结合微管蛋白上与抗癌药物长春新碱相同的位点而抑制微管蛋白聚合的化合物)的合成类似物(g.r.pettit,prog.chem.org.nat.prod,70:1-79(1997))。多拉司他汀10、阿里他汀pe和阿里他汀e是具有四个氨基酸的线性肽,其中三个对于多拉司他汀类化合物是独特的。阿里他汀亚类有丝分裂抑制剂的示例性实施方式包括,但不限于单甲基阿里他汀d(mmad或阿里他汀d衍生物)、单甲基阿里他汀e(mmae或阿里他汀e衍生物)、单甲基阿里他汀f(mmaf或阿里他汀f衍生物)、阿里他汀f苯二胺(afp)、阿里他汀eb(aeb)、阿里他汀efp(aefp)和5-苯甲酰基戊酸-ae酯(aevb)。阿里他汀衍生物的合成和结构描述于美国专利申请公开no.2003-0083263,2005-0238649和2005-0009751;国际专利申请公开no.wo04/010957,国际专利申请公开no.wo02/088172及美国专利no.6,323,315;6,239,104;6,034,065;5,780,588;5,665,860;5,663,149;5,635,483;5,599,902;5,554,725;5,530,097;5,521,284;5,504,191;5,410,024;5,138,036;5,076,973;4,986,988;4,978,744;4,879,278;4,816,444和4,486,414中,其各自通过引用并入本文。在一个实施方式中,本发明的抗-egfr抗体,例如,aba,与至少一个mmae(单甲基阿里他汀e)偶联。单甲基阿里他汀e(mmae,vedotin)通过阻断微管蛋白的聚合来抑制细胞分裂。由于其超高毒性,不可能将其本身用作药物。在最近的癌症治疗进展中,单甲基阿里他汀e连接于识别癌细胞中的特定标志物表达并将mmae指引到癌细胞的单克隆抗体(mab)。在一个实施方式中,将mmae与抗-egfr抗体连接的接头在细胞外液(即,细胞外部的介质和环境)中是稳定的,但一旦adc结合于特定癌细胞抗原并进入癌细胞中,其被组织蛋白酶切割,因此释放毒性mmae并激活强力的抗有丝分裂机制。在一个实施方式中,本文所述的抗-egfr抗体,例如,aba,与至少一个mmaf(单甲基阿里他汀f)偶联。单甲基阿里他汀f(mmaf)通过阻断微管蛋白的聚合抑制细胞分裂。它具有带电的c-末端苯丙氨酸残基,这减弱了其毒性毒性活性(与其不带电的对应物mmae相比)。由于其超高毒性,不可能将其本身用作药物,但其可以与将其指引到癌细胞的单克隆抗体(mab)连接。在一个实施方式中,连接于抗-egfr抗体的接头在细胞外液中是稳定的,但一旦偶联物进入肿瘤细胞中,其被组织蛋白酶切割,因此激活抗有丝分裂机制。mmaf和mmae的结构提供如下。aba-vcmmae的实例也提供在图11中。值得注意的是,图11描述了其中抗体(例如,aba)与单个药物偶联且因此具有1的dar的情况。在某些实施方式中,adc的dar为2-8,或可选地为2-4。用于偶联的其它药物可以用于adc中的药物(即,可以与本发明的抗-egfr抗体偶联的药物)的实例在下文提供,且包括有丝分裂抑制剂、抗肿瘤抗生素、免疫调节剂、基因治疗载体、烷化剂、抗血管生成剂、抗代谢物、含硼剂、化疗保护剂、激素剂、糖皮质激素、光敏治疗剂、寡核苷酸、放射性同位素、放射致敏剂、拓扑异构酶抑制剂、酪氨酸激酶抑制剂及其组合。1.有丝分裂抑制剂在一个方面,抗-egfr抗体可以与一个或多个有丝分裂抑制剂偶联以形成用于治疗癌症的adc。如本文中使用的术语“有丝分裂抑制剂”是指阻断有丝分裂或细胞分裂的细胞毒性剂和/或治疗剂,有丝分裂或细胞分裂是对于癌细胞特别重要的生物学过程。有丝分裂抑制剂破坏微管以使得细胞分裂被阻止,通常是通过影响微管聚合或微管解聚。因此,在一个实施方式中,本发明的抗-egfr抗体与通过抑制微管蛋白聚合破坏微管形成的一个或多个有丝分裂抑制剂偶联。在一个实施方式中,本发明的adc中使用的有丝分裂抑制剂是ixempra(伊沙匹隆)。可以用于本发明的抗-egfradc中的有丝分裂抑制剂的实例在下面提供。有丝分裂抑制剂的类中包括如上所述的阿里他汀类。a.多拉司他汀本发明的抗-egfr抗体可以与至少一个多拉司他汀偶联以形成adc。多拉司他汀类是从印度洋海兔短头海兔(dolabellaauricularia)分离的短肽化合物(参见pettit等人,j.am.chem.soc.,1976,98,4677)。多拉司他汀类的实例包括多拉司他汀10和多拉司他汀(dolatstin)15。多拉司他汀15是一种源自短头海兔的七亚基缩肽,且是结构上与抗微管蛋白剂多拉司他汀10(从相同生物体获得的五亚基的肽)相关的强效抗有丝分裂剂。因此,在一个实施方式中,本发明的抗-egfradc包含如本文所述的抗-egfr抗体和至少一个多拉司他汀。如上所述的阿里他汀类是多拉司他汀10的合成衍生物。b.类美登素类本发明的抗-egfr抗体可以与至少一个类美登素偶联以形成adc。类美登素是最初从卫矛科、鼠李科和大戟科高等植物的成员以及一些苔藓物种分离的强力抗肿瘤剂(kupchan等人,j.am.chem.soc.94:1354-1356[1972];wani等人,j.chem.soc.chem.commun.390:[1973];powell等人,j.nat.prod.46:660-666[1983];sakai等人,j.nat.prod.51:845-850[1988]及suwanborirux等人,experientia46:117-120[1990])。证据表明类美登素类通过抑制微管蛋白的聚合,从而防止微管形成来抑制有丝分裂(参见,例如,美国专利no.6,441,163和remillard等人,science,189,1002-1005(1975))。类美登素类已使用细胞培养模型在体外和使用实验室动物系统在体内证明抑制肿瘤细胞生长。而且,类美登素类的细胞毒性比常规化疗剂(如,例如,甲氨蝶呤、道诺霉素和长春新碱)高1,000倍(参见,例如,美国专利no.5,208,020)。类美登素类包括美登素、美登醇、美登醇的c-3酯及其它美登醇类似物和衍生物(参见,例如,美国专利no.5,208,020和6,441,163,其各自通过引用并入本文)。美登醇的c-3酯可以是天然存在的或合成得到的。而且,天然存在和合成的c-3美登醇酯都可以分类为与简单羧酸的c-3酯或与n-甲基-l-丙氨酸衍生物的c-3酯,后者比前者细胞毒性更高。合成的类美登素类似物描述于例如,kupchan等人,j.med.chem.,21,31-37(1978)中。用于本发明的adc中的合适类美登素可以从天然来源分离、合成产生或半合成产生。而且,类美登素可以以任何合适的方式修饰,只要在最终偶联物分子中保持足够的细胞毒性。就此而言,类美登素类缺乏抗体可以与之连接的合适官能团。希望的是利用连接部分来将类美登素与抗体连接以形成偶联物,且连接部分更详细地描述于iii.b节中。示例性类美登素,mertansine(dm1),的结构提供如下。类美登素类的代表性实例包括,但不限于dm1(n2′-去乙酰基-n2′-(3-巯基-1-氧代丙基)-美登素(也称为mertansine,药物类美登素1;immunogen,inc.;也参见chari等人(1992)cancerres52:127)、dm2、dm3(n2′-去乙酰基-n2′-(4-巯基-1-氧代戊基)-美登素)、dm4(4-甲基-4-巯基-1-氧代戊基)-美登素)和美登醇(一种合成的类美登素类似物)。类美登素类的其它实例描述于美国专利no.8,142,784中,其通过引用并入本文。安丝菌素类是从各种细菌来源分离的一组类美登素抗生素。这些化合物具有强抗肿瘤活性。代表性实例包括,但不限于安丝菌素p1、安丝菌素p2、安丝菌素p3和安丝菌素p4。在本发明的一个实施方式中,抗-egfr抗体与至少一个dm1偶联。在一个实施方式中,抗-egfr抗体与至少一个dm2偶联。在一个实施方式中,抗-egfr抗体与至少一个dm3偶联。在一个实施方式中,抗-egfr抗体与至少一个dm4偶联。d.植物生物碱本发明的抗-egfr抗体可以与至少一个植物生物碱偶联,例如,紫杉烷或长春花生物碱。植物生物碱从某些类型的植物产生的化疗治疗。长春花生物碱由长春花植物(catharanthusrosea)产生,而紫杉烷类由太平洋紫杉(紫杉(taxus))树皮制得。长春花生物碱和紫杉烷类两者也称为抗微管剂,且更详细地描述如下。紫杉烷类本文所述的抗-egfr抗体可以与至少一个紫杉烷偶联。如本文中使用的术语“紫杉烷”是指具有微管作用机制且具有包括紫杉烷环结构和立体定向侧链(其是细胞抑制活性所需的)的抗肿瘤剂类别。术语“紫杉烷”还包括多种已知衍生物,包括亲水性衍生物和疏水性衍生物。紫杉烷衍生物包括,但不限于国际专利申请no.wo99/18113中所描述的半乳糖和甘露糖衍生物;wo99/14209中所描述的哌嗪和其它衍生物;wo99/09021,wo98/22451和美国专利no.5,869,680中所描述的紫杉烷衍生物;wo98/28288中所描述的6-巯基衍生物;美国专利no.5,821,263中所描述的亚磺酰胺衍生物和美国专利no.5,415,869中所描述的紫杉醇衍生物,其各自通过引用并入本文。紫杉烷化合物之前也描述于美国专利no.5,641,803,5,665,671,5,380,751,5,728,687,5,415,869,5,407,683,5,399,363,5,424,073,5,157,049,5,773,464,5,821,263,5,840,929,4,814,470,5,438,072,5,403,858,4,960,790,5,433,364,4,942,184,5,362,831,5,705,503和5,278,324中,其全部通过引入明示并入。紫杉烷类的进一步实例包括,但不限于多西他赛(泰素帝;sanofiaventis)、紫杉醇(abraxane或泰素;abraxisoncology)和纳米颗粒紫杉醇(abi-007/abraxene;abraxisbioscience)。在一个实施方式中,本发明的抗-egfr抗体与至少一个多西他赛偶联。在一个实施方式中,本发明的抗-egfr抗体与至少一个紫杉醇偶联。长春花生物碱在一个实施方式中,抗-egfr抗体与至少一个长春花生物碱偶联。长春花生物碱是一类通过作用于微管蛋白并阻止微管形成而抑制癌细胞分裂的能力来发挥作用的细胞周期特异性药物。可以用于本发明的adc中的长春花生物碱实例包括,但不限于硫酸长春地辛、长春新碱、长春花碱和长春瑞滨。2.抗肿瘤抗生素本发明的抗-egfr抗体可以与一个或多个抗肿瘤抗生素偶联用于治疗癌症。如本文中使用的术语“抗肿瘤抗生素”意思是通过干扰dna来阻断细胞生长并由微生物产生的抗肿瘤药物。通常,抗肿瘤抗生素破坏dna链或者减缓或停止dna合成。可以包括在本发明的抗-egfradc中的抗肿瘤抗生素的实例包括,但不限于放射菌素类(例如,吡咯并[2,1-c][1,4]苯并二氮类)、蒽环类、卡奇霉素类和倍癌霉素类,在下面更详细地描述。a.放射菌素类本发明的抗-egfr抗体可以与至少一个放射菌素偶联。放射菌素类是从链霉菌属的细菌分离的抗肿瘤抗生素的一个亚类。放射菌素类的代表性实例包括,但不限于放射菌素d(cosmegen[也称为放射菌素、更生霉素、放射菌素iv、放射菌素c1],lundbeck,inc.)、氨茴霉素、奇卡霉素a、dc-81、甲基氨茴霉素、新茴霉素a、新茴霉素b、porothramycin、prothracarcinb、sg2285、sibanomicin、西伯利亚霉素(sibiromycin)和茅层霉素(tomaymycin)。在一个实施方式中,本发明的抗-egfr抗体与至少一个吡咯并苯并二氮(pbd)偶联。pbd类的实例包括,但不限于氨茴霉素、奇卡霉素a、dc-81、甲基氨茴霉素、新茴霉素a、新茴霉素b、porothramycin、prothracarcinb、sg2000(sjg-136)、sg2202(zc-207)、sg2285(zc-423)、sibanomicin、西伯利亚霉素和茅层霉素。因此,在一个实施方式中,本发明的抗-egfr抗体与至少一个放射菌素偶联,例如,放射菌素d,或与至少一个pbd偶联,例如,吡咯并苯并二氮(pbd)二聚体。pbd的结构可以在例如美国专利申请公开no.2013/0028917和2013/0028919及wo2011/130598a1中找到,其各自通过引入全文并入本文中。pbd的通式结构提供如下。pbd类在其芳香a环和吡咯并c环两者中取代基的数目、类型和位置上及在c环的饱和度上不同。在b-环中,一般在n10-c11位置处存在亚胺(n=c)、甲醇胺(nh-ch(oh))或甲醇胺甲基醚(nh-ch(ome)),该位置为负责烷基化dna的亲电子中心。所有已知的天然产物在手性c11α位置处具有(s)-构型,当从c环朝向a环观察时,这为它们提供了右手扭曲(right-handedtwist)。本文提供的pbd实例可以与本发明的抗-egfr抗体偶联。可以与本发明的抗-egfr抗体偶联的pbd类的进一步实例可以在例如,美国专利申请公开no.2013/0028917a1和2013/0028919a1,美国专利no.7,741,319b2及wo2011/130598a1和wo2006/111759a1中找到,其各自通过引用全文并入本文。具有以下式ii的代表性pbd二聚体可以与本发明的抗-egfr抗体偶联:其中:r2具有式iii:其中a是c5-7芳基,x是与选自-o-、-s-、-c(o)o-、-c(o)-、-nh(c=o)-和-n(rn)-的接头单元偶联的基团,其中rn选自h、c1-4烷基和(c2h4o)mch3,其中m是1-3,且具有以下任一:(i)q1是单键,且q2选自单键和-z-(ch2)n-,其中z选自单键、o、s和nh且n是1-3;或(ii)q1是-ch=ch-,且q2是单键;r12是c5-10芳基,任选地被选自卤素、硝基、氰基、c1-12烷氧基、c3-20杂环烷氧基、c5-20芳氧基、杂芳氧基、烷基烷氧基、芳烷氧基、烷基芳氧基、杂芳基烷氧基、烷基杂芳氧基、c1-7烷基、c3-7杂环基和双-氧基-c1-3亚烷基的一个或多个取代基取代;r6和r9独立地选自h、r、oh、or、sh、sr、nh2、nhr、nrr′、硝基、me3sn和卤素;其中r和r′独立地选自任选取代的c1-12烷基、c3-20杂环基和c5-20芳基;r7选自h、r、oh、or、sh、sr、nh2、nhr、nhrr′、硝基、me3sn和卤素;以下任一:(a)r10是h,且r11是oh、ora,其中ra是c1-4烷基;(b)r10和r11在它们结合的氮和碳原子之间形成氮-碳双键;或(c)r10是h和r11是sozm,其中z是2或3;r″是c3-12亚烷基,(该链可以被选自o、s、nh的一个或多个杂原子打断)和芳香环;y和y′选自o、s和nh;r6′、r7′、r9′分别选自与r6、r7和r9相同的组,且r10′和r11′与r10和r11相同,和各个m是单价的药学上可接受的阳离子或者两个m基团一起是二价的药学上可接受的阳离子。如本文中使用的短语“任选取代的”涉及可以是未取代的或可以是取代的母体基团。除非另外说明,如本文中使用的术语“取代的”涉及携带一个或多个取代基的母体基团。术语“取代基”在本文中以常规的意义使用且是指与母体基团共价连接,或者如果适宜的话,与母体基团融合的化学部分。广泛的取代基是公知的,且用于其形成和引入多种母体基团中的方法也是公知的。c1-12烷基:如本文中使用的术语“c1-12烷基”涉及通过从具有1-12个碳原子的烃化合物(其可以是脂族的或脂环族的,且其可以是饱和的或不饱和的(例如,部分不饱和的、完全不饱和的))的碳原子除去氢原子获得的单价的部分。因此,术语“烷基”包括亚类烯基、炔基、环烷基等,如下所述。饱和烷基的实例包括,但不限于甲基(c1)、乙基(c2)、丙基(c3)、丁基(c4)、戊基(c5)、己基(c6)和庚基(c7)。饱和线性烷基的实例包括,但不限于甲基(c1)、乙基(c2)、正-丙基(c3)、正-丁基(c4)、正-戊基(戊基)(c5)、正-己基(c6)和正-庚基(c7)。饱和支链烷基的实例包括异-丙基(c3)、异-丁基(c4)、仲-丁基(c4)、叔-丁基(c4)、异-戊基(c5)和新戊基(c5)。c3-20杂环基:如本文中使用的术语“c3-20杂环基”涉及通过从杂环化合物(该部分具有3-20个环原子,其中1-10个是环杂原子)的环原子除去氢原子获得的单价的部分。优选地,各环具有3-7个环原子,其中1-4个是环杂原子。在这种情况中,前缀(例如c3-20、c3-7、c5-6等)表示环原子的数目或者环原子数的范围,无论是碳原子还是杂原子。例如,如本文中使用的术语“c5-6杂环基”涉及具有5-6个环原子的杂环基。单环杂环基的实例包括,但不限于源自以下的那些:n1:氮杂环丙烷(c3)、氮杂环丁烷(c4)、吡咯烷(四氢吡咯)(c5)、吡咯啉(例如,3-吡咯啉、2,5-二氢吡咯)(c5)、2h-吡咯或3h-吡咯(异吡咯,异唑)(c5)、哌啶(c6)、二氢吡啶(c6)、四氢吡啶(c6)、吖庚因(c7);o1:氧杂环丙烷(c3)、氧杂环丁烷(c4)、氧杂环戊烷(四氢呋喃)(c5)、氧杂环戊二烯(二氢呋喃)(c5)、氧杂环己烷(四氢吡喃)(c6)、二氢吡喃(c6)、吡喃(c6)、噁庚因(c7);s1:硫杂环丙烷(c3)、硫杂环丁烷(c4)、硫杂环戊烷(四氢噻吩)(c5)、硫杂环己烷(四氢噻喃)(c6)、硫杂环庚烷(c7);o2:二氧戊环(c5)、二氧六环(c6)和二氧杂环庚烷(c7);o3:三氧杂环己烷(c6);n2:咪唑烷(c5)、吡唑烷(diazolidine)(c5)、咪唑啉(c5)、吡唑啉(二氢吡唑)(c5)、哌嗪(c6);n1o1:四氢噁唑(c5)、二氢噁唑(c5)、四氢异噁唑(c5)、二氢异噁唑(c5)、吗啉(c6)、四氢噁嗪(c6)、二氢噁嗪(c6)、噁嗪(c6);n1s1:二氢噻唑(c5)、四氢噻唑(c5)、硫代吗啉(c6);n2o1:噁二嗪(c6);o1s1:氧硫杂环戊二烯(oxathiole)(c5)和氧硫杂环己烷(噻噁烷)(c6);及n1o1s1:氧杂噻嗪(c6)。取代的单环杂环基的实例包括源自环状形式的糖类的那些,例如,呋喃糖(c5)如阿拉伯呋喃糖、来苏呋喃糖、呋喃核糖和呋喃木糖,及吡喃糖(c6)如别吡喃糖、阿卓吡喃糖、吡喃葡萄糖、吡喃甘露糖、古洛吡喃糖、艾杜吡喃糖、吡喃半乳糖和塔罗吡喃糖。c5-20芳基:如本文中使用的术语“c5-20芳基”涉及通过从芳香化合物的芳香环原子除去氢原子获得的单价的部分,该部分具有3-20个环原子。优选地,各环具有5-7个环原子。在这种情况中,前缀(例如,c3-20、c5-7、c5-6等)表示环原子的数目或者环原子数的范围,无论是碳原子还是杂原子。例如,如本文中使用的术语“c5-6芳基”涉及具有5-6个环原子的芳基。在一个实施方式中,本发明的抗-egfr抗体可以与具有下式的pbd二聚体偶联:其中以上结构描述了pbd二聚体sg2202(zc-207),并通过接头l与本发明的抗-egfr抗体偶联。sg2202(zc-207)公开于例如,美国专利申请公开no.2007/0173497中,其通过引用全文并入本文。在另一实施方式中,pbd二聚体,sgd-1882,通过药物接头与本发明的抗-egfr抗体偶联,如图21中所示的。sgd-1882公开于sutherland等人,(2013)blood122(8):1455和美国专利申请公开no.2013/0028919中,其通过引用全文并入本文。如图21中所描述的,pbd二聚体sgd-1882可以通过mc-val-ala-二肽接头(图21中统称为sgd-1910)与抗体偶联。在特定的实施方式中,本文公开的抗-egfr抗体与图21中所描述的pbd二聚体偶联。因此,在进一步的实施方式中,本发明包括通过mc-val-ala-二肽接头与pbd二聚体偶联的本文公开的抗-egfr抗体,如图21中所描述的。在某些实施方式中,本发明包括与pbd(包括,但不限于图21中描述的pbd二聚体)偶联的包含重链可变区和轻链可变区的抗-egfr抗体,该重链可变区包含含有seqidno:12的氨基酸序列的cdr3域、含有seqidno:11的氨基酸序列的cdr2域和含有seqidno:10的氨基酸序列的cdr1域,和该轻链可变区包含含有seqidno:8的氨基酸序列的cdr3域、含有seqidno:7的氨基酸序列的cdr2域和含有seqidno:6的氨基酸序列的cdr1域。在某些实施方式中,本发明包括包含由seqidno:9中所示的氨基酸序列定义的aba的重链可变区和包含seqidno:5的氨基酸序列的轻链可变区的抗-egfr抗体,其中抗体与pbd偶联,例如,但不限于图21的示例性pbd二聚体。b.蒽环类本发明的抗-egfr抗体可以与至少一个蒽环类偶联。蒽环类是从链霉菌属的细菌分离的抗肿瘤抗生素亚类。代表性实例包括,但不限于道诺霉素(cerubidine,bedfordlaboratories)、阿霉素(亚德里亚霉素,bedfordlaboratories;也称为阿霉素盐酸盐、羟基柔红霉素和rubex)、表柔比星(ellence,pfizer)和依达比星(idamycin;pfizerinc.)。因此,在一个实施方式中,本发明的抗-egfr抗体与至少一个蒽环类,例如阿霉素,偶联。c.卡奇霉素类本发明的抗-egfr抗体可以与至少一个卡奇霉素偶联。卡奇霉素类是源自土壤生物体棘孢小单孢菌(micromonosporaechinospora)的烯二炔类抗生素家族。卡奇霉素类结合dna的小沟并诱导双链dna断裂,从而导致相对于其它化疗剂提高100倍的细胞死亡(damle等人,(2003)curropinpharmacol3:386)。可以用作本发明的药物偶联物的卡奇霉素的制备已经被描述,参见美国专利no.5,712,374;5,714,586;5,739,116;5,767,285;5,770,701;5,770,710;5,773,001和5,877,296。可以使用的卡奇霉素的结构类似物包括,但不限于γ11、α21、α3i、n-乙酰基-γ1i、psag和θi1(hinman等人,cancerresearch53:3336-3342(1993),lode等人,cancerresearch58:2925-2928(1998)及前述美国专利no.5,712,374;5,714,586;5,739,116;5,767,285;5,770,701;5,770,710;5,773,001和5,877,296)。因此,在一个实施方式中,本发明的抗-egfr抗体与至少一个卡奇霉素偶联。d.倍癌霉素类本发明的抗-egfr抗体可以与至少一个倍癌霉素偶联。倍癌霉素类是从链霉菌属的细菌分离的抗肿瘤抗生素亚类(参见nagamura和saito(1998)chemistryofheterocycliccompounds,vol.34,no.12)。倍癌霉素类结合于dna的小沟并使n3位置的核碱基腺嘌呤烷基化(boger(1993)pureandapplchem65(6):1123,及boger和johnson(1995)pnasusa92:3642)。倍癌霉素类的合成类似物包括,但不限于阿多来新、比折来新和卡折来新。因此,在一个实施方式中,本发明的抗-egfr抗体与至少一个倍癌霉素偶联。e.其它抗肿瘤抗生素除前述之外,可以用于本发明的抗-egfradc中的另外的抗肿瘤抗生素包括博莱霉素(blenoxane,bristol-myerssquibb)、丝裂霉素和光辉霉素(也称为光神霉素)。3.免疫调节剂在一个方面,本发明的抗-egfr抗体可以与至少一个免疫调节剂偶联。如本文中使用的术语“免疫调节剂”是指可以刺激或改变免疫反应的药剂。在一个实施方式中,免疫调节剂是增强受试者的免疫反应的免疫刺激剂。在另一实施方式中,免疫调节剂是防止或降低受试者的免疫反应的免疫抑制剂。免疫调节剂可以调节髓样细胞(单核细胞、巨噬细胞、树突状细胞、巨核细胞和粒细胞)或淋巴样细胞(t细胞、b细胞和自然杀伤(nk)细胞)及其任何进一步分化的细胞。代表性实例包括,但不限于卡介苗(bcg)和左旋咪唑(ergamisol)。可以用于本发明的adc中的免疫调节剂的其它实例包括,但不限于癌症疫苗、细胞因子和免疫调节基因疗法。a.癌症疫苗本发明的抗-egfr抗体可以与癌症疫苗偶联。如本文中使用的术语“癌症疫苗”是指引起肿瘤特异性免疫反应的组合物(例如,肿瘤抗原和细胞因子)。该反应通过施用癌症疫苗,或者在本发明的情况中,施用包含抗-egfr抗体和癌症疫苗的adc从受试者的自身免疫系统诱发。在优选的实施方式中,免疫反应导致身体中肿瘤细胞的根除(例如,原发或转移的肿瘤细胞)。癌症疫苗的使用一般包括施用特定抗原或抗原组,其例如存在于特定癌细胞的表面上或存在于证明促进癌症形成的特定传染剂的表面上。在一些实施方式中,癌症疫苗的使用用于预防目的,而在其它实施方式中该用途是用于治疗目的。可以用于本发明的抗-egfradc中的癌症疫苗的非限制性实例包括重组双价人乳头状瘤病毒(hpv)疫苗16和18型疫苗(cervarix,glaxosmithkline)、重组四价人乳头状瘤病毒(hpv)6、11、16和18型疫苗(gardasil,merck&company)及sipuleucel-t(provenge,dendreon)。因此,在一个实施方式中,本发明的抗-egfr抗体与作为免疫刺激剂或免疫抑制剂的至少一个癌症疫苗偶联。b.细胞因子本发明的抗-egfr抗体可以与至少一个细胞因子偶联。术语“细胞因子”一般是指由一个细胞群体释放的蛋白质,其作为细胞内介质作用于另一细胞。细胞因子直接刺激肿瘤部位的免疫效应细胞和基质细胞并增强细胞毒性免疫效应细胞的肿瘤细胞识别(lee和margolin(2011)cancers3:3856)。许多动物肿瘤模型研究证明细胞因子具有广泛的抗-肿瘤活性且这可以转用为多种基于细胞因子的癌症治疗途径(lee和margoli,同上)。最近几年看到多种细胞因子(包括gm-csf、il-7、il-12、il-15、il-18和il-21)进入晚期癌症患者的临床试验(lee和margoli,同上)。可以用于本发明的adc中的细胞因子的实例包括,但不限于甲状旁腺激素,甲状腺素,胰岛素,胰岛素原,松弛素,松弛素原,糖蛋白激素如促滤泡激素(fsh)、促甲状腺素(tsh)和促黄体激素(lh),肝生长因子,成纤维细胞生长因子,催乳素,胎盘催乳激素,肿瘤坏死因子,缪勒管抑制物质,小鼠促性腺激素相关肽,抑制素(inhibin),活化素(activin),血管内皮生长因子,整联蛋白,促血小板生成素(tpo),神经生长因子如ngf,血小板-生长因子,转化生长因子(tgf),胰岛素样生长因子-i和-ii,促红细胞生成素(epo),骨诱导因子,干扰素如干扰素α、β和γ,集落刺激因子(csf),粒细胞-巨噬细胞-c-sf(gm-csf)及粒细胞-csf(g-csf),白介素(il)如il-1、il-1α、il-2、il-3、il-4、il-5、il-6、il-7、il-8、il-9、il-11、il-12,肿瘤坏死因子,及其它多肽因子包括lif和kit配体(kl)。如本文中使用的,术语细胞因子包括来自天然来源或来自重组细胞培养物的蛋白质和天然序列细胞因子的生物活性等同物。因此,在一个实施方式中,本发明提供包含本文所述的抗-egfr抗体和细胞因子的adc。c.集落刺激因子(csf)本发明的抗-egfr抗体可以与至少一个集落刺激因子(csf)偶联。集落刺激因子(csf)是帮助骨髓产生红细胞的生长因子。因为一些癌症治疗(例如,化疗)可以影响白细胞(其帮助对抗感染),因此可以引入集落刺激因子以帮助支持白细胞水平和加强免疫系统。集落刺激因子也可以在骨髓移植后用于帮助新骨髓开始产生白细胞。可以用于本发明的抗-egfradc中的csf的代表性实例包括,但不限于红细胞生成素(epoetin)、非格司亭(neopogen(也称为粒细胞集落刺激因子(g-csf);amgen,inc.)、沙莫司亭(白细胞素(leukine)(粒细胞-巨噬细胞集落刺激因子和gm-csf),genzymecorporation)、promegapoietin和奥普瑞白介素(重组il-11;pfizer,inc.)。因此,在一个实施方式中,本发明提供包含本文所述的抗-egfr抗体和csf的adc。4.基因疗法本发明的抗-egfr抗体可以与至少一个核酸偶联(直接地或通过载体间接地)用于基因疗法。基因疗法一般是指将遗传物质引入细胞中,由此遗传物质设计为治疗疾病。在其涉及免疫调节剂时,基因疗法用于刺激受试者抑制癌细胞增殖或杀死癌细胞的天然能力。在一个实施方式中,本发明的抗-egfradc包含编码功能性、治疗性基因(其用于替代与癌症相关的突变的或另外地功能失调的(例如,截短的)基因)的核酸。在其它实施方式中,本发明的抗-egfradc包含编码或者以其它方式用于产生治疗蛋白质以治疗癌症的核酸。编码治疗性基因的核酸可以直接地与抗-egfr抗体偶联,或者可选地,可以通过载体与抗-egfr抗体偶联。可以用于递送用于基因疗法的核酸的载体的实例包括,但不限于病毒载体或脂质体。5.烷化剂本发明的抗-egfr抗体可以与一个或多个烷化剂偶联。烷化剂是将烷基附接于dna的抗肿瘤化合物类别。可以用于本发明的adc中的烷化剂的实例包括,但不限于烷基磺酸酯类、乙烯亚胺类、甲胺衍生物、环氧化物类、氮芥类、亚硝基脲类、三嗪类和肼类。a.烷基磺酸酯类本发明的抗-egfr抗体可以与至少一个烷基磺酸酯偶联。烷基磺酸酯是具有通式:r-so2-o-r1的烷化剂的亚类,其中r和r1通常是烷基或芳基。烷基磺酸酯的代表性实例包括,但不限于白消安(myleran,glaxosmithkline;busulfexiv,pdlbiopharma,inc.)。b.氮芥类本发明的抗-egfr抗体可以与至少一个氮芥偶联。抗癌化合物的这一亚类的代表性实例包括,但不限于苯丁酸氮芥(瘤可宁,glaxosmithkline)、环磷酰胺(癌得星,bristol-myerssquibb;neosar,pfizer,inc.)、雌氮芥(雌氮芥磷酸钠或艾去适,pfizer,inc.)、异环磷酰胺(ifex,bristol-myerssquibb)、二氯甲基二乙胺(mustargen,lundbeckinc.)和美法仑(alkeran或l-pam或苯丙氨酸氮芥;glaxosmithkline)。c.亚硝基脲类本发明的抗-egfr抗体可以与至少一个亚硝基脲偶联。亚硝基脲类是脂溶性的烷化剂亚类。代表性实例包括,但不限于卡莫司丁(bcnu[也称为bicnu,n,n-双(2-氯乙基)-n-亚硝基脲或1,3-双(2-氯乙基)-l-亚硝基脲],bristol-myerssquibb)、福莫司汀(也称为muphoran)、罗莫司丁(ccnu或1-(2-氯-乙基)-3-环己基-1-亚硝基脲,bristol-myerssquibb)、尼莫司丁(也称为acnu)和链佐星(zanosar,tevapharmaceuticals)。d.三嗪类和肼类本发明的抗-egfr抗体可以与至少一个三嗪或肼偶联。三嗪类和肼类是含氮的烷化剂的亚类。在一些实施方式中,这些化合物自发地分解或可以被代谢以产生烷基重氮盐中间体,其促进烷基转移到核酸、肽和/或多肽,从而引起诱变、致癌或细胞毒性效应。代表性实例包括,但不限于达卡巴嗪(dtic-dome,bayerhealthcarepharmaceuticalsinc.)、甲基苄肼(mutalane,sigma-taupharmaceuticals,inc.)和替莫唑胺(temodar,scheringplough)。e.其它烷化剂本发明的抗-egfr抗体可以与至少一个乙烯亚胺、甲胺衍生物或环氧化物偶联。乙烯亚胺类是通常包含至少一个氮丙啶环的烷化剂的亚类。环氧化物类表示特征为仅具有三个环原子的环状醚的烷化剂的亚类。乙烯亚胺类的代表性实例包括,但不限于噻替派(thiopeta)(thioplex,amgen)、地吖醌(也称为氮丙啶基苯醌(azq))和丝裂霉素c。丝裂霉素c是包含氮丙啶环并表现为通过交联dna而诱导细胞毒性的天然产物(dorrrt等人,cancerres.1985;45:3510;kennedyka等人,cancerres.1985;45:3541)。甲胺衍生物及其类似物的代表性实例包括,但不限于六甲蜜胺(hexalen,mgipharma,inc.),其也称为六甲铵和六甲氰胺。这一抗癌化合物类别的环氧化物的代表性实例包括,但不限于二去水卫矛醇。二去水卫矛醇(1,2:5,6-二去水己六醇)与氮丙啶类化学相关且通常通过与如上所述相似的机制促进烷基的转移。二溴卫矛醇水解为二去水卫矛醇且因此是环氧化物的前药(selleic等人,cancerchemotherrep.1969;53:377)。6.抗血管生成剂在一个方面,本文所述的抗-egfr抗体与至少一个抗血管生成剂偶联。抗血管生成剂抑制新血管的生长。抗血管生成剂以多种方式发挥其作用。在一些实施方式中,这些药剂干扰生长因子到达其靶标的能力。例如,血管内皮生长因子(vegf)是通过与细胞表面上的特定受体结合参与启动血管生成的主要蛋白质之一。因此,阻碍vegf与其同源受体的相互作用的某些抗血管生成剂阻止vegf启动血管生成。在其它实施方式中,这些药剂干扰细胞内信号传导级联。例如,一旦细胞表面上的特定受体被触发,其它化学信号的级联被启动以促进血管的生长。因此,已知促进导致例如细胞增殖的细胞内信号传导级联的某些酶,例如,一些酪氨酸激酶,是癌症治疗的靶标。在其它实施方式中,这些药剂干扰细胞间信号传导级联。还在其它实施方式中,这些药剂使得激活和促进细胞生长的特定靶标失能或者通过直接干扰血管细胞的生长。血管生成抑制性质已经在具有众多直接和间接抑制性作用的超过300种物质中发现。可以用于本发明的adc中的抗血管生成剂的代表性实例包括,但不限于血管抑素、abxegf、c1-1033、pki-166、egf疫苗、ekb-569、gw2016、icr-62、emd55900、cp358、pd153035、ag1478、imc-c225(爱必妥,zd1839(易瑞沙),osi-774,厄洛替尼(它塞瓦)、血管生成抑制素、阻抑素、内皮抑素、bay12-9566和w/氟尿嘧啶或阿霉素、血管能抑素(canstatin)、羧酰胺三唑(carboxyamidotriozole)和与紫杉醇联用、emd121974、s-24、vitaxin、二甲基氧杂蒽酮乙酸、im862、白介素-12、白介素-2、nm-3、humv833、ptk787、rhumab、angiozyme(核酶)、imc-1c11、新伐司他、marimstat、普啉司他、bms-275291、col-3、mm1270、su101、su6668、su11248、su5416,与紫杉醇联用、与吉西他滨和顺铂联用,及与依立替康和顺铂联用和与放疗、替可加兰(tecogalan)、替莫唑胺和peg干扰素α2b联用、四硫钼酸盐、tnp-470、沙利度胺(thalidomide)、cc-5013和与泰索帝联用、肿瘤抑素(tumstatin)、2-甲氧基雌二醇、vegftrap、mtor抑制剂(deforolimus,依维莫司(afinitor,novartispharmaceuticalcorporation)和坦西莫司(temsirolimus)(torisel,pfizer,inc.))、酪氨酸激酶抑制剂(例如,埃罗替尼(特罗凯,genentech,inc.)、伊马替尼(格列卫,novartispharmaceuticalcorporation)、吉非替尼(易瑞沙,astrazenecapharmaceuticals)、达沙替尼(sprycel,brystol-myerssquibb)、舒尼替尼(索坦,pfizer,inc.)、尼罗替尼(泰息安,novartispharmaceuticalcorporation)、拉帕替尼(泰克博,glaxosmithklinepharmaceuticals)、索拉非尼(蕾莎瓦,bayer和onyx)、磷酸肌醇3-激酶(pi3k)。7.抗代谢物本发明的抗-egfr抗体可以与至少一个抗代谢物偶联。抗代谢物是非常类似于细胞内的正常物质的化疗治疗类型。当细胞将抗代谢物整合到细胞代谢中时,结果对于细胞是负面的,例如,细胞不能分裂。抗代谢物按照它们干扰的物质分类。可以用于本发明的adc中的抗代谢物的实例包括,但不限于叶酸拮抗剂(例如,甲氨蝶呤)、嘧啶拮抗剂(例如,5-氟尿嘧啶、氟尿苷(foxuridine)、阿糖胞苷、卡培他滨和吉西他滨)、嘌呤拮抗剂(例如,6-巯基嘌呤和6-硫鸟嘌呤)和腺苷脱氨酶抑制剂(例如,克拉屈滨、氟达拉滨、奈拉滨和喷司他丁),如以下更详细地描述的。a.抗叶酸剂本发明的抗-egfr抗体可以与至少一个抗叶酸剂偶联。抗叶酸剂是结构上与叶酸类似的抗代谢物亚类。代表性实例包括,但不限于甲氨蝶呤、4-氨基-叶酸(也称为氨基蝶呤和4-氨基蝶酸)、洛美曲索(lmtx)、培美曲塞(alimpta,elilillyandcompany)和曲美沙特(neutrexin,benvenuelaboratories,inc.)。b.嘌呤拮抗剂本发明的抗-egfr抗体可以与至少一个嘌呤拮抗剂偶联。嘌呤类似物是结构上与称为嘌呤的化合物组相似的抗代谢物的亚类。嘌呤拮抗剂的代表性实例包括,但不限于咪唑硫嘌呤(azasan,salix;imuran,glaxosmithkline)、克拉屈滨(leustatin[也称为2-cda],janssenbiotech,inc.)、巯基嘌呤(purinethol[也称为6-巯基乙醇],glaxosmithkline)、氟达拉滨(fludara,genzymecorporation)、喷司他丁(nipent,也称为2′-去氧助间霉素(dcf))、6-硫鸟嘌呤(lanvis[也称为硫鸟嘌呤],glaxosmithkline)。c.嘧啶拮抗剂本发明的抗-egfr抗体可以与至少一个嘧啶拮抗剂偶联。嘧啶拮抗剂是结构上与称为嘧啶的化合物组相似的抗代谢物的亚类。嘧啶拮抗剂的代表性实例包括,但不限于阿扎胞苷(vidaza,celgenecorporation)、卡培他滨(xeloda,rochelaboratories)、阿糖胞苷(也称为胞嘧啶阿拉伯糖苷和阿糖胞嘧啶,bedfordlaboratories)、地西他滨(达克金,eisaipharmaceuticals)、5-氟尿嘧啶(adrucil,tevapharmaceuticals;efudex,valeantpharmaceuticals,inc)、5-氟-2’-脱氧尿苷5’-磷酸酯(fdump)、5-氟尿苷三磷酸酯和吉西他滨(健择,elilillyandcompany)。8.含硼剂本发明的抗-egfr抗体可以与至少一个含硼剂偶联。含硼剂包含干扰细胞增殖的一类癌症治疗化合物。含硼剂的代表性实例包括,但不限于borophycin和硼替佐米(velcade,milleniumpharmaceuticals)。9.化疗保护剂本发明的抗-egfr抗体可以与至少一个化疗保护剂偶联。化疗保护药物是帮助保护身体对抗化疗的特定毒性效应的一类化合物。化疗保护剂可以与各种化疗一起施用以保护健康细胞免于化疗药物的毒性作用,而同时允许癌细胞被施用的化疗剂处理。代表性的化疗保护剂包括,但不限于阿米福汀(ethyol,medimmune,inc.)(其用于降低与累积剂量的顺铂相关的肾毒性)、用于治疗由施用蒽环类引起的外渗(totect)及用于治疗由施用抗肿瘤抗生素阿霉素引起的心脏相关并发症(zinecard)的右雷佐生(totect,apricuspharma;zinecard)和美司钠(mesnex,bristol-myerssquibb)(其用于预防ifocfamide化疗过程中的出血性膀胱炎)。10.激素剂本发明的抗-egfr抗体可以与至少一个激素剂偶联。激素剂(包括合成激素)是干扰内分泌系统内源产生激素的产生或活性的化合物。在一些实施方式中,这些化合物干扰细胞生长或产生细胞毒性效应。非限制性实例包括雄激素类、雌激素类、醋酸甲羟孕酮(provera,pfizer,inc.)和孕激素类。11.抗激素剂本发明的抗-egfr抗体可以与至少一个抗激素剂偶联。“抗激素”剂是抑制某些内源激素的产生和/或阻碍其功能的药剂。在一个实施方式中,抗激素剂干扰选自雄激素类、雌激素类、孕酮和促性腺激素释放激素的激素的活性,从而干扰各种癌细胞的生长。抗激素剂的代表性实例包括,但不限于氨鲁米特、阿那曲唑(arimidex,astrazenecapharmaceuticals)、比卡鲁胺(casodex,astrazenecapharmaceuticals)、醋酸环丙孕酮(cyprostat,bayerplc)、地加瑞克(firmagon,ferringpharmaceuticals)、依西美坦(aromasin,pfizerinc.)、氟他胺(drogenil,schering-ploughltd)、氟维司群(faslodex,astrazenecapharmaceuticals)、戈舍瑞林(zolodex,astrazenecapharmaceuticals)、来曲唑(femara,novartispharmaceuticalscorporation)、亮丙瑞林(prostap)、利普安、醋酸甲羟孕酮(provera,pfizerinc.)、醋酸甲地孕酮(megace,bristol-myerssquibbcompany)、它莫西芬(nolvadex,astrazenecapharmaceuticals)和曲普瑞林(decapetyl,ferring)。12.皮质类固醇本发明的抗-egfr抗体可以与至少一个皮质类固醇偶联。皮质类固醇可以用于本发明的adc中以减少炎症。皮质类固醇的实例包括,但不限于糖皮质激素,例如,强的松(deltasone,pharmacia&upjohncompany,pfizer,inc.的分部)。13.光敏治疗剂本发明的抗-egfr抗体可以与至少一个光敏治疗剂偶联。光敏治疗剂包括可以被布置用于在暴露于特定波长的电磁辐射时杀死处理的细胞的化合物。治疗相关化合物吸收穿透组织的波长的电磁辐射。在优选的实施方式中,化合物以非毒性的形式施用,其能够在充分激活时产生对于细胞或组织为毒性的光化学效应。在其它优选的实施方式中,这些化合物被癌性组织保留并容易地从正常组织清除。非限制性实例包括各种发色团和染料。14.寡核苷酸本发明的抗-egfr抗体可以与至少一个寡核苷酸偶联。寡核苷酸由短的核酸链制成,其通过干扰遗传信息的加工而发挥作用。在一些实施方式中,用于adc中的寡核苷酸是未修饰的单链和/或双链dna或rna分子,而在其它实施方式中,这些治疗性寡核苷酸是化学修饰的单链和/或双链dna或rna分子。在一个实施方式中,用于adc中的寡核苷酸是相对短的(19-25个核苷酸)并与细胞中存在的总核酸靶标池中独特的核酸序列杂交。一些重要的寡核苷酸技术包括反义寡核苷酸(包括rna干扰(rnai))、适体、cpg寡核苷酸和核酶。a.反义寡核苷酸本发明的抗-egfr抗体可以与至少一个反义寡核苷酸偶联。反义寡核苷酸设计为通过watson-crick杂交结合于rna。在一些实施方式中,反义寡核苷酸与编码egfr的区域、域、部分或片段的多核苷酸互补。在一些实施方式中,反义寡核苷酸包含约5-约100个核苷酸、约10-约50个核苷酸、约12-约35和约18-约25个核苷酸。在一些实施方式中,寡核苷酸与egfr基因的区域、部分、域或片段至少50%、至少60%、至少70%、至少80%、至少90%、至少95%、至少96%、至少97%、至少98%、至少99%或至少100%同源。在一些实施方式中,在egfr基因的至少15、20、25、30、35、40、50或100个连续核苷酸上存在实质的序列同源性。在优选的实施方式中,这些反义寡核苷酸大小的范围为12-25个核苷酸长,其中大多数反义寡核苷酸为18-21个核苷酸长。具有多种可以在寡核苷酸结合于靶rna时用于抑制rna的功能的机制(crookest.(1999).biochim.biophys.acta,1489,30-42)。最佳表征的反义机制导致所靶向的rna被内源细胞核酸酶如rnaseh或者与rna干扰机制相关的核酸酶切割。但是,通过非催化机制(如剪接的调节或翻译停滞)抑制靶基因表达的寡核苷酸也可以是基因功能的强力和选择性的调节剂。最近受到许多关注的另一rnase依赖性的反义机制是rnai(fire等人(1998).nature,391,806-811.;zamorepd.(2002).science,296,1265-1269)。rna干扰(rnai)是其中双链rna以序列特异性的方式抑制基因表达的转录后过程。在一些实施方式中,rnai效应通过引入相对较长的双链rna(dsrna)实现,而在优选的实施方式中,这种rnai效应通过引入较短的双链rna(例如小干扰rna(sirna)和/或微rna(mirna))实现。在又一实施方式中,rnai也可以通过引入产生与靶基因互补的dsrna的质粒来实现。在各前述实施方式中,双链rna设计为干扰细胞内特定靶序列的基因表达。一般地,该机制涉及dsrna转化成将核糖核酸酶指引到同源mrna靶标的短rna(概括的,ruvkun,science2294:797(2001)),其然后使对应的内源mrna降解,从而导致基因表达的调节。值得注意的是,dsrna已报告为具有抗增殖性质,这使得其也可能考虑用于治疗性应用(aubel等人,proc.natl.acad.sci.,usa88:906(1991))。例如,合成dsrna已证明抑制小鼠中的肿瘤生长(levy等人,proc.nat.acad.sci.usa,62:357-361(1969)),在白血病小鼠的治疗中是有效的(zeleznick等人,proc.soc.exp.biol.med.130:126-128(1969))和抑制小鼠皮肤中化学诱导的肿瘤发生(gelboin等人,science167:205-207(1970))。因此,在优选的实施方式中,本发明提供adc中的反义寡核苷酸治疗乳腺癌的用途。在其它实施方式中,本发明提供用于启动反义寡核苷酸治疗的组合物和方法,其中dsrna在mrna水平干扰egfr的靶细胞表达。以上所用的dsrna是指天然存在的rna、部分纯化的rna、重组产生的rna、合成rna以及通过包括非标准核苷酸、非核苷酸物质、核苷酸类似物(例如,锁核酸(lna))、脱氧核糖核苷酸及其任意组合而与天然存在的rna不同的改变的rna。本发明的rna仅需要与具有介导本文所述的基于反义寡核苷酸的调节的能力的天然rna足够相似。b.适体本发明的抗-egfr抗体可以与至少一个适体偶联。适体是基于其结合其它分子的能力从随机池选择的核酸分子。与抗体相似,适体可以以特别的亲和力和特异性结合靶分子。在许多实施方式中,适体采取复杂的、序列依赖性的三维形状,这使得它们与靶蛋白相互作用,从而与抗体-抗原相互作用类似地产生紧密结合的复合体,由此干扰所述蛋白质的功能。适体紧密和特异性结合其靶蛋白的特殊能力是它们作为靶向分子治疗的潜能的基础。c.cpg寡核苷酸本发明的抗-egfr抗体可以与至少一个cpg寡核苷酸偶联。细菌和病毒dna已知是人体中先天和特异性免疫两者的强激活剂。这些免疫特性与细菌dna中发现的未甲基化cpg二核苷酸基序相关。由于这些基序在人体中很少见的原因,人免疫系统发展出了识别这些基序为感染的早期征兆并随后启动免疫反应的能力。因此,包含这种cpg基序的寡核苷酸可以用于启动抗肿瘤免疫反应。d.核酶本发明的抗-egfr抗体可以与至少一个核酶偶联。核酶是长度范围约40-155个核苷酸的催化性rna分子。核酶识别和切割特定rna分子的能力使得它们成为治疗剂的潜在候选者。代表性实例包括核酶(angiozyme)。15.放射性核素剂(放射性同位素)本发明的抗-egfr抗体可以与至少一个放射性核素剂偶联。放射性核素剂包括特征在于能够发生放射性衰变的不稳定核的试剂。成功的放射性核素治疗的基础依赖于足够的浓度和癌细胞对放射性核素的延长保持。考虑的其它因素包括放射性核素半衰期、发射的粒子的能量和发射的粒子可以行进的最大范围。在优选的实施方式中,治疗剂是选自111in、177lu、212bi、213bi、211at、62cu、64cu、67cu、90y、i25i、i31i、32p、33p、47sc、111ag、67ga、142pr、153sm、161tb、166dy、166ho、186re、188re、189re、212pb、223ra、225ac、59fe、75se、77as、89sr、99mo、105rh、109pd、143pr、149pm、169er、194ir、198au、199au和211pb的放射性核素。还优选的是实质上随俄歇发射粒子衰变的放射性核素。例如,co-58、ga-67、br-80m、tc-99m、rh-103m、pt-109、in-1111、sb-119、i-125、ho-161、os-189m和ir-192。可用的β粒子发射核素的衰变能量优选是dy-152、at-211、bi-212、ra-223、rn-219、po-215、bi-211、ac-225、fr-221、at-217、bi-213和fm-255。可用的α粒子发射核素的衰变能量优选是2,000-10,000kev,更优选3,000-8,000kev,且最优选4,000-7,000kev。使用的另外的潜在放射性同位素包括11c、13n、15o、75br、198au、224ac、126i、133i、77br、113min、95ru、97ru、103ru、105ru、107hg、203hg、121mte、122mte、125mte、165tm、167tm、168tm、197pt、109pd、105rh、142pr、143pr、161tb、166ho、199au、57co、58co、51cr、59fe、75se、201tl、225ac、76br、169yb等等。16.放射致敏剂本发明的抗-egfr抗体可以与至少一个放射致敏剂偶联。如本文中使用的术语“放射致敏剂”定义为以治疗有效量施用于动物以提高放射致敏的细胞对于电磁辐射的敏感性和/或促进用电磁辐射可治疗的疾病的治疗的分子,优选低分子量分子。放射致敏剂是使得癌细胞对于放疗更敏感而同时通常对于正常细胞具有低得多的效果的药剂。因此,放射致敏剂可以与放射标记的抗体或adc组合使用。添加放射致敏剂与仅使用放射标记的抗体或抗体片段的治疗相比产生增强的疗效。放射致敏剂描述于d.m.goldberg(ed.),cancertherapywithradiolabeledantibodies,crcpress(1995)中。放射致敏剂的实例包括吉西他滨、5-氟尿嘧啶、紫杉烷和顺铂。放射致敏剂可以通过x-射线的电磁辐射激活。x-射线激活的放射致敏剂的代表性实例包括,但不限于以下:甲硝哒唑、米索硝唑、去甲基醚醇硝唑、哌莫硝唑、依他硝唑、尼莫拉唑、丝裂霉素c、rsu1069、sr4233、e09、rb6145、烟酰胺、5-溴脱氧尿苷(budr)、5-碘脱氧尿苷(iudr)、溴脱氧胞苷、氟脱氧尿苷(fudr)、羟基脲、顺铂及其治疗有效的类似物和衍生物。或者,放射致敏剂可以使用光动力治疗(pdt)激活。光动力学放射致敏剂的代表性实例包括,但不限于血卟啉衍生物、光卟啉(r)、苯并卟啉衍生物、npe6、初卟啉锡(tinetioporphyrin)(snet2)、pheoborbidea、细菌叶绿素a、萘菁类、酞菁类、酞菁锌及其治疗有效的类似物和衍生物。17.拓扑异构酶抑制剂本发明的抗-egfr抗体可以与至少一个拓扑异构酶抑制剂偶联。拓扑异构酶抑制剂是设计为干扰拓扑异构酶(拓扑异构酶i和ii)的作用的化疗剂,拓扑异构酶是在正常细胞周期中通过催化然后断裂和重接dna链的磷酸二酯骨架来控制dna结构的变化的酶。dna拓扑异构酶i抑制剂的代表性实例包括,但不限于喜树碱类及其衍生物依立替康(cpt-11,camptosar,pfizer,inc.)和托泊替康(hycamtin,glaxosmithklinepharmaceuticals)。dna拓扑异构酶ii抑制剂的代表性实例包括,但不限于安吖啶、道诺霉素、阿霉素、表鬼臼毒素类、玫瑰树碱类、表柔比星、依托泊苷、雷佐生和替尼泊苷。18.酪氨酸激酶抑制剂本发明的抗-egfr抗体可以与至少一个酪氨酸激酶抑制剂偶联。酪氨酸激酶是细胞内的酶,其功能是将磷酸基团连接于氨基酸酪氨酸。通过阻断蛋白质酪氨酸激酶发挥功能的能力,肿瘤生长可以被抑制。可以用于本发明的adc上的酪氨酸激酶的实例包括,但不限于阿西替尼(axitinib)、博舒替尼(bosutinib)、西地尼布(cediranib)、达沙替尼、厄洛替尼、吉非替尼、伊马替尼(imatinib)、拉帕替尼、来他替尼、尼罗替尼、semaxanib、舒尼替尼和凡德他尼。19.其它药剂可以用于本发明的adc中的其它药剂的实例包括,但不限于相思豆毒素(例如,相思豆毒素a链)、α毒素、油桐蛋白、蝇蕈毒素、巴豆毒素、麻疯树毒素、石竹素蛋白、白喉毒素(例如,白喉a链和白喉毒素的非结合活性片段)、脱氧核糖核酸酶(dnase)、白树毒素、mitogellin、蒴莲根毒素a链、苦瓜抑制剂、新霉素、豹蛙酶(onconase)、酚霉素、美洲商陆(phytolacaamericana)蛋白(papi、papii和pap-s)、商陆抗病毒蛋白、假单胞菌内毒素、假单胞菌外毒素(例如,外毒素a链(来自绿脓杆菌))、局限曲霉素、蓖麻毒蛋白a链、核糖核酸酶(rnase)、石碱草(sapaonariaofficinalis)抑制剂、皂草素、α-sarcin、金黄色葡萄球菌肠毒素-a、破伤风毒素、顺铂、卡铂和奥沙利铂(eloxatin,sanofiaventis)、蛋白酶体抑制剂(例如,ps-341[硼替佐米或velcade])、hdac抑制剂(伏立诺他(zolinza,merck&company,inc.))、贝利司他、恩替诺特、mocetinostat和帕比司他)、cox-2抑制剂、取代的脲类、热休克蛋白抑制剂(例如,格尔德霉素及其多种类似物)、肾上腺皮质抑制剂和单端孢霉烯类(参见,例如,wo93/21232)。其它药剂还包括天冬酰胺酶(espar,lundbeckinc.)、羟基脲、左旋咪唑、米托坦(lysodren,bristol-myerssquibb)和维甲酸(renova,valeantpharmaceuticalsinc.)。应当注意,可以用于本发明的抗-egfradc中的药物部分的前述组不是排他的,因为某些药物的实例可以在超过一个类别中发现,例如,安丝菌素类是有丝分裂抑制剂和抗肿瘤抗生素两者。对于本发明的化合物设想上述药物部分的所有立体异构体,即,在d的手性碳处r和s构型的任何组合。上述药剂(即,未与抗体偶联的裸药剂)也可以与本文所述的抗-egfr抗体一起用于组合疗法中。在一个实施方式中,抗-egfr抗体或adc与任何前述药剂一起用于组合疗法中以治疗癌症,其中该药剂在抗-egfr抗体或adc施用于受试者之前、同时或之后施用。b.抗-egfradc:示例性接头抗-egfradc包含抗-egfr抗体和至少一个药物,由此抗体和该至少一个药物通过接头偶联。如本文中使用的术语“接头”是指可以为双功能或多功能的且用于将抗体连接于药物部分的化学部分。接头可以包括一个偶联成分或可以包括多个偶联成分。例如,接头可以包括间隔体(spacer),其是延伸药物连接以避免例如屏蔽抗体的活性位点或提高adc的溶解性的部分。接头成分的其它实例包括延伸体(stretcher)单元和氨基酸单元。两种方法通常用于将药物与抗体偶联:还原的链间半胱氨酸二硫化物通过非可酶促切割的马来酰亚胺基或者简单和可切割的二硫化物接头的烷基化,及赖氨酸通过可切割的线性氨基酸的酰化。在一个方面,接头将抗体共价连接于药物部分。adc使用具有用于将抗体与药物结合的反应性官能团的接头制备。例如,半胱氨酸巯基或胺(例如,抗体的n-末端或氨基酸侧链如赖氨酸)可以与接头的官能团形成键。在一个方面,接头具有能够与抗体上存在的游离半胱氨酸反应以形成共价键的官能团。非限制性的和示例性的这类反应性官能团包括马来酰亚胺、卤代乙酰胺类、α-卤代乙酰基、活性酯如琥珀酰亚胺酯、4-硝基苯基酯、五氟苯基酯、四氟苯基酯、酸酐、酰基氯、磺酰氯、异氰酸酯和异硫氰酸酯。参见,例如,klussman等人,(2004),bioconjugatechemistry15(4):765-773中第766页上的偶联方法。在一些实施方式中,接头具有能够与抗体上存在的亲电子基团反应的官能团。示例性的这类亲电子基团包括,但不限于醛和酮羰基基团。在一些实施方式中,接头的反应性官能团的杂原子可以与抗体上的亲电子基团反应并与抗体单元形成共价键。非限制性的和示例性的这类反应性官能团包括,但不限于酰肼、肟、氨基、肼、缩氨基硫脲、肼羧酸酯和芳基酰肼。示例性的接头成分包括6-马来酰亚胺基己酰基、马来酰亚胺基丙酰基(“mp”)、缬氨酸-瓜氨酸(“val-cit”或“vc”)、丙氨酸-苯丙氨酸(“ala-phe”)、p-氨基苄氧羰基(“pab”)、n-琥珀酰亚胺基4-(2-吡啶基硫代)戊酸酯(“spp”)和4-(n-马来酰亚胺基甲基)环己烷-1-羧酸酯(“mcc”)。在一个方面,抗-egfr抗体通过包含马来酰亚胺基己酰基(“mc”)、缬氨酸瓜氨酸(val-cit或“vc”)和paba的接头(称为“mc-vc-paba接头”)与阿里他汀,例如,mmae偶联。马来酰亚胺基己酰基作为与抗-egfr抗体的接头发挥作用且是不可切割的。val-cit是作为接头的氨基酸单元的二肽并允许通过蛋白酶(特别是蛋白酶组织蛋白酶b)切割接头。因此,接头的val-cit成分提供了在暴露于细胞内环境时从adc释放阿里他汀的手段。在接头内,p-氨基苯甲醇(paba)作为间隔体发挥作用并且是自切割的(selfimmolative),从而允许释放mmae。mc-vc-paba-mmae接头的结构提供在图11中。合适的接头包括,例如,可切割的和不可切割的接头。接头可以是“可切割的接头”,从而利于药物的释放。非限制性和示例性的可切割的接头包括酸不稳定的接头(例如,包含腙)、蛋白酶敏感的(例如,肽酶敏感的)接头、光不稳定的接头或含二硫化物的接头(chari等人,cancerresearch52:127-131(1992);美国专利no.5,208,020)。可切割的接头通常在细胞内条件下易发生切割。合适的可切割的接头包括例如,可通过细胞内蛋白酶如溶酶体蛋白酶或胞内体蛋白酶切割的肽接头。在示例性的实施方式中,接头可以是二肽接头如缬氨酸-瓜氨酸(val-cit)或苯丙氨酸-赖氨酸(phe-lys)接头。接头在细胞外优选以足够治疗有效的方式稳定的。在转运或递送到细胞中之前,adc优选是稳定的并保持完整,即,抗体保持与药物部分偶联。在靶细胞外稳定的接头一旦在细胞内则可以以某些有效的速率切割。因此,有效的接头将:(i)保持抗体的特异性结合性质;(ii)允许递送,例如,细胞内递送药物部分,且(iii)保持药物部分的治疗效果,例如,细胞毒性效应。在一个实施方式中,接头在细胞内条件下是可切割的,使得接头的切割在细胞内环境中充分地从抗体释放药物而是治疗有效的。在一些实施方式中,可切割的接头是ph-敏感的,即,对某些ph值下的水解敏感。通常,ph-敏感的接头可在酸性条件下水解。例如,可以使用在溶酶体中可水解的酸不稳定的接头(例如,腙、缩氨基脲、缩氨基硫脲、顺式-乌头酰胺、原酸酯、缩醛、缩酮等等)(参见,例如,美国专利no.5,122,368;5,824,805;5,622,929;dubowchik和walker,1999,pharm.therapeutics83:67-123;neville等人,1989,biol.chem.264:14653-14661)。这样的接头在中性ph条件(如在血液中的那些条件)下是相对稳定的,但在低于ph5.5或5.0(大致溶酶体的ph)下是不稳定的。在某些实施方式中,可水解的接头是硫醚接头(如,例如,通过酰腙键连接于治疗剂的硫醚)(参见,例如,美国专利no.5,622,929)。在其它实施方式中,接头在还原条件下是可切割的(例如,二硫化物接头)。多种二硫化物接头是本领域已知的,包括,例如,可以使用sata(n-琥珀酰亚胺基-5-乙酰基硫代乙酸酯)、spdp(n-琥珀酰亚胺基-3-(2-吡啶二硫代)丙酸酯)、spdb(n-琥珀酰亚胺基-3-(2-吡啶二硫代)丁酸酯)和smpt(n-琥珀酰亚胺氧基羰基-α-甲基-α-(2-吡啶基-二硫代)甲苯)、spdb和smpt形成的那些(参见,例如,thorpe等人,1987,cancerres.47:5924-5931;wawrzynczak等人,inimmunoconjugates:antibodyconjugatesinradioimageryandtherapyofcancer(c.w.vogeled.,oxfordu.press,1987)。也参见美国专利no.4,880,935)。在一些实施方式中,接头是可通过细胞内环境中(例如,溶酶体或胞内体或胞膜窖(caveolea)内)存在的切割剂(例如,酶)切割的。接头可以是,例如,通过细胞内肽酶或蛋白酶(包括,但不限于溶酶体或胞内体蛋白酶)切割的肽基接头。在一些实施方式中,肽基接头是至少两个氨基酸长或至少三个氨基酸长。切割剂可以包括组织蛋白酶b和d及血纤维蛋白溶酶,其全部已知水解二肽药物衍生物,导致活性药物在靶细胞内释放(参见,例如,dubowchik和walker,1999,pharm.therapeutics83:67-123)。最典型的是可通过egfr-表达细胞中存在的酶切割的肽基接头。这样的接头的实例描述于,例如,美国专利no.6,214,345中,其通过引用以其全文和出于所有目的并入本文。在特定的实施方式中,可通过细胞内蛋白酶切割的肽基接头是val-cit接头或phe-lys接头(参见,例如,美国专利no.6,214,345,其描述了用val-cit接头合成阿霉素)。采用细胞内蛋白水解释放治疗剂的一种优势是药物在偶联时通常是减弱的且偶联物的血清稳定性通常是高的。在其它实施方式中,接头是丙二酸酯接头(johnson等人,1995,anticancerres.15:1387-93)、马来酰亚胺基苯甲酰基接头(lau等人,1995,bioorg-med-chem.3(10):1299-1304)或3′-n-酰胺类似物(lau等人,1995,bioorg-med-chem.3(10):1305-12)。在再其它的实施方式中,接头单元是不可切割的且药物,例如,通过抗体降解释放。参见u.s.公开no.20050238649,其通过引用全文并入本文。包含不可切割的接头的adc可以设计为使得adc基本上保持在细胞外并与靶细胞表面上的某些受体相互作用,从而adc的结合启动(或阻止)特定细胞信号传导途径。在一些实施方式中,接头实质上是亲水性接头(例如,peg4mal和磺基-spdb)。亲水性接头可以用于降低药物可能通过mdr(多药耐药性)或功能上类似的转运蛋白泵送到抗性癌细胞外的程度。在其它实施方式中,在切割时,接头起到直接或间接地抑制细胞生长和/或细胞增殖的功能。例如,在一些实施方式中,接头在切割时可以作为嵌入剂发挥作用,从而抑制大分子生物合成(例如,dna复制、rna转录和/或蛋白质合成)。在其它实施方式中,接头设计为通过接头-药物和/或单独的药物扩散到邻近细胞而促进旁观者杀伤(邻近细胞的杀伤)。在其它实施方式中,接头促进细胞内化。空间位阻二硫化物的存在可以提高特定二硫键的稳定性,从而增强adc的效力。因此,在一个实施方式中,接头包括空间位阻的二硫键。空间位阻的二硫化物是指在特定分子环境内存在的二硫键,其中该环境特征在于原子的特定空间排列或定向(通常在相同分子或化合物内),其阻止或至少部分地抑制二硫键的还原。因此,接近于二硫键的大体积(空间位阻的)化学部分和/或大体积氨基酸侧链的存在阻止或至少部分地抑制二硫键发生会导致二硫键还原的潜在相互作用。值得注意的是,前述接头类型不是互斥的。例如,在一个实施方式中,本文所述的抗-egfradc中使用的接头是促进细胞内化的不可切割的接头。在一些实施方式中,adc具有下式(式i):ab-(l-d)n(i)或其药学上可接受的盐或溶剂合物;其中ab是抗体,例如,抗-egfr抗体aba,且(l-d)是接头-药物部分。接头-药物部分由作为接头的l-和作为具有例如针对靶细胞(例如,表达egfr的细胞)的细胞抑制性、细胞毒性或另外的治疗活性的药物部分的-d制成,且n是1-20的整数。在一些实施方式中,n范围为1-8、1-7、1-6、1-5、1-4、1-3、1-2或是1。在一些实施方式中,-d部分相同。在又一实施方式中,-d部分不同。如上所述,接头可以是单一部分或可以包括两个或更多个成分。如此,在一些实施方式中,adc具有下式(ii):ab-(aa-ww-yy-d)n(ii)或其药学上可接受的盐或溶剂合物,其中ab是抗体,例如,抗-egfr抗体aba,和-aa-ww-yy-是包含三个或更多个成分的接头(l),其包括-a-(其是任选的延伸体单元),a是0或l,各-w-独立地是氨基酸单元(或在一些实施方式中,葡萄糖苷酸单元,也参见us公开no.2012/0107332a1),w是0-12范围的整数,-y-是自切割的间隔体单元,y是0、1或2;-d是具有例如针对靶细胞(例如,表达egfr的细胞)的细胞抑制性、细胞毒性或另外的治疗活性的药物部分,且n是1-20的整数。在一些实施方式中,接头成分包括将抗体连接于另一接头或连接于药物部分的“延伸体单元”(a)。非限制性和示例性的延伸体单元显示如下(其中波纹线表示与抗体、药物或另外的接头成分的共价连接位点):延伸体单元(a)当存在时能够将抗体连接于氨基酸单元(-w-)(如果存在)、连接于间隔体单元(-y-)(如果存在)或连接于药物(-d)(参见式ii)。可以存在于本文所述的抗-egfr抗体上(天然地或通过化学操作)的可用官能团包括,但不限于巯基、氨基、羟基、碳水化合物的端羟基和羧基。合适的官能团是巯基和氨基。在一个实例中,巯基可以通过抗-egfr抗体的分子内二硫键的还原生成。在另一实施方式中,巯基可以通过抗-egfr抗体的赖氨酸部分的氨基用2-亚氨基硫杂环戊烷(iminothiolane)(traut′s试剂)或其它巯基生成试剂进行还原来生成。在某些实施方式中,抗-egfr抗体是重组抗体且经工程化以携带一个或多个赖氨酸部分。在其它某些实施方式中,重组抗-egfr抗体经工程化以携带另外的巯基,例如,另外的半胱氨酸。在一个实施方式中,延伸体单元与抗体的硫原子形成键。硫原子可以来源于抗体的巯基。这一实施方式的代表性的延伸体单元描绘于如下所示的式iiia和iiib的方括号内:其中l-、-w-、-y-、-d、w和y定义如上,且r17选自-c1-c10亚烷基-、-c1-c10亚烯基-、-c1-c10亚炔基-、碳环基-、-o-(c1-c8亚烷基)-、o-(c1-c8亚烯基)-、-o-(c1-c8亚炔基)-、-亚芳基-、-c1-c10亚烷基-亚芳基-、-c2-c10亚烯基-亚芳基、-c2-c10亚炔基-亚芳基、亚芳基-c1-c10亚烷基-、-亚芳基-c2-c10亚烯基-、-亚芳基-c2-c10亚炔基-、-c1-c10亚烷基-(碳环基)-、-c2-c10亚烯基-(碳环基)-、c2-c10亚炔基-(碳环基)-、-(碳环基)-c1-c10亚烷基-、-(碳环基)-c2-c10亚烯基-、-(碳环基)-c2-c10亚炔基、-杂环基-、-c1-c10亚烷基-(杂环基)-、-c2-c10亚烯基-(杂环基)-、-c2-c10亚炔基-(杂环基)-、-(杂环基)-c1-c10亚烷基-、-(杂环基)-c2-c10亚烯基-、-(杂环基)-c1-c10亚炔基-、-(ch2ch2o)r-或-(ch2ch2o)r-ch2-,且r是1-10的整数,其中所述烷基、烯基、炔基、亚烷基、亚烯基、亚炔基(alkynyklene)、芳基、碳环、碳环基、杂环基和亚芳基基团(无论单独或作为另一基团的部分)是任选取代的。在一些实施方式中,所述烷基、烯基、炔基、亚烷基、亚烯基、亚炔基、芳基、碳环、碳环基、杂环基和亚芳基基团(无论单独或作为另一基团的部分)是未取代的。在一些实施方式中,r17选自-c1-c10亚烷基-、-碳环基-、-o-(c1-c8亚烷基)-、-亚芳基-、-c1-c10亚烷基-亚芳基-、-亚芳基-c1-c10亚烷基-、-c1-c10亚烷基-(碳环基)-、-(碳环基)-c1-c10亚烷基-、-c3-c8杂环基-、-c1-c10亚烷基-(杂环基)-、-(杂环基)-c1-c10亚烷基-、-(ch2ch2o)r-和-(ch2ch2o)r-ch2-,且r是1-10的整数,其中所述亚烷基是未取代的且其余基团是任选取代的。说明性的延伸体单元是式iiia的延伸体单元,其中r17是-(ch2)5-,如下所示(也参见u.s.8,309,093)。另一说明性的延伸体单元是式iiia的延伸体单元,其中r17是-(ch2ch2o)r-ch2-,且r是2,如下所示(也参见u.s.8,309,093,其通过引用并入本文)。另一说明性的延伸体单元是式iiia的延伸体单元,其中r17是亚芳基-或亚芳基-c1-c10亚烷基-。在一些实施方式中,芳基是未取代的苯基。再另一说明性的延伸体单元是式iiib的延伸体单元,其中r17是-(ch2)5-,如下所示(也参见u.s.8,309,093,其通过引用并入本文)。在某些实施方式中,延伸体单元通过抗-egfr抗体单元的硫原子和延伸体单元的硫原子之间的二硫键连接于抗-egfr抗体。这一实施方式的代表性延伸体单元描绘于式iv的方括号中(参见以下,且也参见u.s.8,309,093,其通过引用并入本文),其中r17、l-、-w-、-y-、-d、w和y定义如上。应当注意,以下所示式中的s部分(也参见u.s.8,309,093,其通过引用并入本文)是指抗体的硫原子,除非通过上下文指明另外的情况。在再其它的实施方式中,延伸体包含可以与抗体的伯氨基或仲氨基形成键的反应性位点。这些反应性位点的实例包括,但不限于活性酯如琥珀酰亚胺酯、4-硝基苯基酯、五氟苯基酯、四氟苯基酯、酸酐、酰基氯、磺酰氯、异氰酸酯和异硫氰酸酯。这一实施方式的代表性延伸体单元描绘于式va和vb的方括号中(参见以下)(也参见u.s.8,309,093,其通过引用并入本文),其中r17、l-、-w-、-y-、-d、w和y定义如上。在一些实施方式中,延伸体包含对于可能存在于抗体上修饰的碳水化合物的(-cho)基团为反应性的反应性位点。例如,碳水化合物可以使用试剂如高碘酸钠轻度氧化且氧化的碳水化合物的所得(-cho)单元可以与包含如酰肼、肟、伯胺或仲胺、肼、缩氨基硫脲、羧酸肼和芳基酰肼的官能团的延伸体缩合,如kaneko等人,1991,bioconjugatechem.2:133-41中描述的那些。这一实施方式的代表性延伸体单元描绘在式via、vib和vic的方括号内(参见以下)(也参见u.s.8,309,093,其通过引用并入本文),其中-r17-、l-、-w-、-y-、-d、w和v如上定义。在一些实施方式中,接头成分包含“氨基酸单元”(w)。在这样的一些实施方式中,氨基酸单元允许通过蛋白酶切割接头,从而在暴露于细胞内蛋白酶如溶酶体酶时促进药物从免疫偶联物释放(doronina等人,(2003)nat.biotechnol.21:778-784)。示例性的氨基酸单元包括,但不限于二肽、三肽、四肽和五肽。示例性的二肽包括,但不限于缬氨酸-瓜氨酸(vc或val-cit)、丙氨酸-苯丙氨酸(af或ala-phe)、苯丙氨酸-赖氨酸(fk或phe-lys)、苯丙氨酸-同型赖氨酸(phe-homolys)和n-甲基-缬氨酸-瓜氨酸(me-val-cit)。示例性的三肽包括,但不限于甘氨酸-缬氨酸-瓜氨酸(gly-val-cit)和甘氨酸-甘氨酸-甘氨酸(gly-gly-gly)。氨基酸单元可以包含天然存在的氨基酸残基和/或次要氨基酸和/或非天然存在的氨基酸类似物如瓜氨酸。氨基酸单元可以针对通过特定酶(例如,肿瘤相关蛋白酶、组织蛋白酶b、c和d或者血纤维蛋白溶酶蛋白酶)的酶促切割进行设计和优化。在一个实施方式中,w氨基酸单元是缬氨酸-瓜氨酸(vc或val-cit)。在另一方面,氨基酸单元是苯丙氨酸-赖氨酸(即,fk)。在氨基酸单元的再另一方面,氨基酸单元是n-甲基缬氨酸-瓜氨酸。在再另一方面,氨基酸单元是5-氨基戊酸、高苯丙氨酸赖氨酸、四异喹啉羧酸赖氨酸、环己基丙氨酸赖氨酸、六氢异烟酸赖氨酸、β-丙氨酸赖氨酸、甘氨酸丝氨酸缬氨酸谷氨酰胺和六氢异烟酸。或者,在一些实施方式中,-w-是连接延伸体单元与间隔体单元(如果延伸体和间隔体单元存在)、连接延伸体单元与药物部分(如果间隔体单元不存在)和连接接头单元与药物(如果延伸体和间隔体单元不存在)的葡萄糖苷酸单元。葡萄糖苷酸单元包括可以被β-葡糖苷酸酶切割的位点(也参见us2012/0107332,其通过引用并入本文)。在一些实施方式中,葡萄糖苷酸单元包含如下所示式的通过糖苷键(-o′-)连接于自切割基团(self-immolativegroup)(z)的糖部分(su)(也参见us2012/0107332,其通过引用并入本文)。糖苷键(-o′-)通常是β-葡糖苷酸酶-切割位点,如可被人溶酶体β-葡糖苷酸酶切割的键。在葡萄糖苷酸单元的情况中,术语“自切割基团”指能够将两个或三个间隔的化学部分(即,糖部分(通过糖苷键)、药物部分(直接地或通过间隔体单元间接地))和,在一些实施方式中,接头(直接地或通过延伸体单元间接地)共价连接在一起形成稳定分子的双功能或三功能的化学部分。如果其与糖部分的键被切割,自切割基团自发地与第一化学部分(例如,间隔体或药物单元)分离。在一些实施方式中,糖部分(su)是环状己糖如吡喃糖或环状戊糖如呋喃糖。在一些实施方式中,吡喃糖是葡萄糖苷酸或己糖。糖部分通常为β-d构型。在特定的实施方式中,吡喃糖是β-d-葡萄糖苷酸部分(即,通过可被β-葡糖苷酸酶切割的糖苷键与自切割基团-z-连接的β-d-葡糖醛酸)。在一些实施方式中,糖部分是未取代的(例如,天然存在的环状己糖或环状戊糖)。在其它实施方式中,糖部分可以是取代的β-d-葡萄糖苷酸(即,被一个或多个基团如氢、羟基、卤素、硫、氮或低级烷基取代的葡糖醛酸)。在一些实施方式中,葡萄糖苷酸单元具有如下所示的式之一(也参见us2012/0107332,其通过引用并入本文):其中su是糖部分,糖苷键包含su和自切割基团z之间的氧键,且各r独立地是氢、卤素(例如,氯、溴、氟等)、-cn、-no2或者其它吸电子基团或供电子基团,条件是葡萄糖苷酸单元(和特别地z)在糖苷键切割时自切割。在一些实施方式中,各r独立地是氢、卤素(例如,氯、溴、氟等)、-cn或-no2。在一些实施方式中,葡萄糖苷酸单元具有如下所示的式之一(也参见us2012/0107332,其通过引用并入本文):其中su是糖部分,糖苷键(-o′-)包含su和自切割基团z之间的氧键,且各r独立地是氢。在一些实施方式中,自切割基团(z)共价连接于糖部分、连接于药物(直接地或通过间隔体单元间接地)和连接于接头(直接地或通过延伸体单元间接地)。在一些实施方式中,药物接头偶联物具有如下所示的式(也参见us2012/0107332,其通过引用并入本文):其中su、o′、z、y、y、d、a和a如本文中定义。通常1-20个这样的药物-接头偶联物可以连接于接头。在一些实施方式中,包含葡萄糖苷酸单元的adc具有如下所示的式之一(也参见us2012/0107332,其通过引用并入本文),其中su、y、y、d、a、a和l如本文中定义。在一些实施方式中,包含葡萄糖苷酸单元的adc具有如下所示的式(也参见us2012/0107332,其通过引用并入本文),其中y、y、d、a、a和l如本文中定义。在一些实施方式中,包含葡萄糖苷酸单元的adc具有如下所示的式(也参见us2012/0107332,其通过引用并入本文),其中y、y、d和l如本文中定义。在一些实施方式中,包含葡萄糖苷酸单元的adc具有如下所示的式(也参见us2012/0107332,其通过引用并入本文),其中y、y、d和l如本文中定义。在一些实施方式中,包含葡萄糖苷酸单元的adc具有如下所示的式(也参见us2012/0107332a1),其中d如本文中所述且mab是单克隆抗体。间隔体单元(-y-)(当存在时)将氨基酸单元(或葡萄糖苷酸单元,也参见us2012/0107332,其通过引用并入本文)连接于药物部分(当氨基酸单元存在时)。或者,间隔体单元将延伸体单元连接于药物部分(当氨基酸单元不存在时)。当氨基酸单元和延伸体单元两者都不存在时,间隔体单元也可以将药物单元连接于抗体单元。间隔体单元为两种主要类型:非自切割的和自切割的。非自切割间隔体单元是其中间隔体单元的部分或全部在氨基酸单元(或葡萄糖苷酸单元)从抗体-药物偶联物切割(特别是酶促切割)后保持与药物部分结合的间隔体单元。非自切割间隔体单元的实例包括,但不限于(甘氨酸-甘氨酸)间隔体单元和甘氨酸间隔体单元(两者均描绘于以下方案1中(也参见u.s.8,309,093,其通过引用并入本文))。方案1当包含甘氨酸-甘氨酸间隔体单元或甘氨酸间隔体单元的偶联物通过酶(例如,肿瘤细胞相关蛋白酶、癌细胞相关蛋白酶或淋巴细胞相关蛋白酶)发生酶促切割时,甘氨酸-甘氨酸-药物部分或甘氨酸-药物部分从l-aa-ww-切割。在一个实施方式中,独立的水解反应在靶细胞内发生,从而切割甘氨酸-药物部分键并释放药物。在一些实施方式中,非自切割间隔体单元(-y-)是-gly-。在一些实施方式中,非自切割间隔体单元(-y-)是-gly-gly-。在一个实施方式中,提供其中间隔体单元不存在(y=0)的药物-接头偶联物或其药学上可接受的盐或溶剂合物。或者,包含自切割间隔体单元的偶联物可以允许释放药物部分。自切割间隔体单元自发地与第二化学部分分离(如果其与第一部分的键被切割)。在一些实施方式中,-yy-是p-氨基苯甲醇(pab)单元,其亚苯基部分被qm取代,其中q是-c1-c8烷基、-c1-c8烯基、-c1-c8炔基、-o-(c1-c8烷基)、-o-(c1-c8烯基)、-o-(c1-c8炔基)、-卤素、-硝基或-氰基,且m是0-4的整数。烷基、烯基和炔基(无论是单独的或作为另一基团的部分)可以是任选取代的。在一些实施方式中,-y-是通过pab基团的氨基氮原子连接于-ww-,且通过碳酸酯、氨基甲酸酯或醚基团直接连接于-d的pab基团。非受任何特定理论或机制的限制,以下方案2(也参见u.s.8,309,093)描绘了通过氨基甲酸酯或碳酸酯基团直接连接于-d的pab基团的药物释放的可能机制,如toki等人,2002,j.org.chem.67:1866-1872所述的。方案2在方案2中,q是-c1-c8烷基、-c1-c8烯基、-c1-c8炔基、-o-(c1-c8烷基)、-o-(c1-c8烯基)、-o-(c1-c8炔基)、-卤素、-硝基或-氰基;m是0-4的整数,且p的范围为1-约20。烷基、烯基和炔基(无论是单独的或作为另一基团的部分)可以是任选取代的。自切割间隔体的其它实例包括,但不限于与pab基团电子类似的芳香化合物,如2-氨基咪唑-5-甲醇衍生物(hay等人,1999,bioorg.med.chem.lett.9:2237)和邻位-或对位-氨基苯甲基缩醛类。可以使用在酰胺键水解时发生环化的间隔体,如取代的和未取代的4-氨基丁酰胺类(rodrigues等人,1995,chemistrybiology2:223)、适当取代的双环[2.2.1]和双环[2.2.2]环系统(storm等人,1972,j.amer.chem.soc.94:5815)及2-氨基苯基丙酰胺类(amsberry等人,1990,j.org.chem.55:5867)。在甘氨酸的α位被取代的含胺药物的消去反应(kingsbury等人,1984,j.med.chem.27:1447)也是自切割间隔体的实例。在一个方面,间隔体单元(-yy-)由式(x)-(xii)表示(参见以下)(也参见u.s.8,309,093),其中q是-c1-c8烷基、-c1-c8烯基、-c1-c8炔基、-o-(c1-c8烷基)、-o-(c1-c8烯基)、-o-(c1-c8炔基)、-卤素、-硝基或-氰基,且m是0-4的整数。自切割间隔体的其它实例包括,但不限于与pab基团电子类似的芳香化合物,如2-氨基咪唑-5-甲醇衍生物(参见,例如,hay等人,1999,bioorg.med.chem.lett.9:2237)和邻位或对位-氨基苯甲基缩醛类。可以使用在酰胺键水解时发生环化的间隔体,如取代的和未取代的4-氨基丁酰胺类(参见,例如,rodrigues等人,1995,chemistrybiology2:223)、适当取代的双环[2.2.1]和双环[2.2.2]环系统(参见,例如,storm等人,1972,j.amer.chem.soc.94:5815)及2-氨基苯基丙酰胺类(参见,例如,amsberry等人,1990,j.org.chem.55:5867)。在甘氨酸的α位被取代的含胺药物的消去反应(参见,例如,kingsbury等人,1984,j.med.chem.27:1447)也是自切割间隔体的实例。其它合适的间隔体单元公开于公布的美国专利申请no.2005-0238649中,其公开内容通过引用并入本文。用于生成adc的另一途径包括使用异双功能交联剂,其将抗-egfr抗体连接于药物部分。可以使用的交联剂的实例包括n-琥珀酰亚胺基4-(5-硝基-2-吡啶二硫代)-戊酸酯或高度水溶性的类似物n-磺基琥珀酰亚胺基4-(5-硝基-2-吡啶二硫代)-戊酸酯、n-琥珀酰亚胺基-4-(2-吡啶二硫代)丁酸酯(spdb)、n-琥珀酰亚胺基-4-(5-硝基-2-吡啶二硫代)丁酸酯(snpb)和n-磺基琥珀酰亚胺基-4-(5-硝基-2-吡啶二硫代)丁酸酯(ssnpb)、n-琥珀酰亚胺基-4-甲基-4-(5-硝基-2-吡啶二硫代)戊酸酯(smnp)、n-琥珀酰亚胺基-4-(5-n,n-二甲基甲酰胺基-2-吡啶二硫代)丁酸酯(scpb)或n-磺基琥珀酰亚胺基4-(5-n,n-二甲基甲酰胺基-2-吡啶二硫代)丁酸酯(sscpb))。本发明的抗体可以用交联剂n-琥珀酰亚胺基4-(5-硝基-2-吡啶二硫代)-戊酸酯、n-磺基琥珀酰亚胺基4-(5-硝基-2-吡啶二硫代)-戊酸酯、spdb、snpb、ssnpb、smnp、scpb或sscpb进行修饰,然后可以与稍过量的包含硫醇部分的特定药物反应以获得adc的优良产率。优选地,交联剂是如下所示式的化合物(也参见美国专利no.6,913,748,其通过引用并入本文):其中r、r1、r2和r3相同或不同,且是h、甲基、乙基或者具有3-6个碳原子的直链、支链或环状烷基,n是0或1-4的整数,x和y相同或不同,且是h、conr4r5或no2,条件是x和y不同时是h,r4和r5相同或不同且各自是h、甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基或叔丁基,且z是so3-m+或h,其中m+代表金属离子或四烷基铵离子,条件是当x和/或y是no2时,z不是h。另外的异双功能交联剂及使用其制备adc的方法描述于美国专利no.6,913,748中,其明确地通过引用并入本文。在一个实施方式中,带电接头(也称为前带电接头(pro-chargedlinker))用于将抗-egfr抗体与药物偶联以形成adc。带电接头包括在细胞加工后变成带电的接头。细胞加工后特定adc的接头中或药物上带电基团的存在提供了几种优势,例如(i)adc的更高水溶性,(ii)在水性溶液中以更高浓度操作的能力,(iii)每抗体连接更多数目的药物分子的能力,潜在地导致更高效力,(iv)带电偶联物物质保留在靶细胞内的潜能,导致更高效力,和(v)多药抗性细胞的更高敏感性,该细胞不能将带电药物物质从细胞排出。一些合适的带电或前带电交联剂的实例及其合成显示于美国专利no.8,236,319的图1-10中,且通过引用并入本文。优选地,带电或前带电交联剂是包含显著提高adc(特别是具有2-20个偶联的药物的adc)的溶解性的磺酸酯、磷酸酯、羧基或季铵取代基的那些交联剂。从包含前带电部分的接头制备的偶联物在偶联物于细胞中代谢后将产生一个或多个带电部分。在进一步的实施方式中,本发明的adc包含具有如下所示式的接头(也参见美国专利no.8,236,319,其通过引用并入本文):其中y′表示使得与抗体的反应成为可能的官能团;q表示使得药物能够通过二硫键、硫醚、硫酯、肽、腙、酯、醚、氨基甲酸酯或酰胺键连接的官能团;r1、r2、r3、r4、r5、r6、r7、r8、r9和r10相同或不同且是h、具有1-6个碳原子的直链烷基、具有3-6个碳原子的支链或环状烷基、具有2-6个碳原子的直链、支链或环状烯基或炔基、阴离子(例如,但不限于so3-、x-so3-、opo32-、x-opo32-、po32-、x-po32-、co2-)、阳离子(例如,但不限于含氮杂环、n+r11r12r13或x-n+r11r12r13)或苯基,其中r11、r12和r13相同或不同且是h、具有1-6个碳原子的直链烷基或者具有3-6个碳原子的支链或环状烷基,且x表示苯基或具有1-6个碳原子的直链烷基或者具有3-6个碳原子的支链或环状烷基;l、m和n是0或1-4的整数;a是苯基或取代的苯基,其中取代基是具有1-6个碳原子的直链烷基或者具有3-6个碳原子的支链或环状烷基,或者选自阴离子(例如,但不限于so3-、x-so3-、opo32-、x-opo32-、po32-、x-po32-、co2-)和阳离子(例如,但不限于含氮杂环、n+r11r12r13或x-n+r11r12r13)的带电取代基,其中x具有以上定义的相同含义,且其中g是0或1;z是任选的式(och2ch2)p的聚乙烯氧单元(其中p是0或2-约1000的整数)或者f1-e1-p-e2-f2单元(其中e1和e2相同或不同且是c=o、o或nr14,其中r14是h、具有1-6个碳原子的直链烷基、具有3-6个碳原子的支链或环状烷基、具有2-6个碳原子的直链、支链或环状烯基或炔基;p是长度2-20个氨基酸的肽单元,其中e1或e2可以通过末端氮、末端碳或者通过肽的一个氨基酸的侧链连接于肽,并且f1和f2相同或不同且是任选的式(och2ch2)p的聚乙烯氧单元(其中p是0或2-约1000的整数),条件是当z不是f1-e1-p-e2-f2时,r1、r2、r3、r4、r5、r6、r7、r8、r9和r10中至少一个是带电的取代基,或者当g是1时,a、r1、r2、r3、r4、r5、r6、r7、r8、r9和r10中至少一个是带电的取代基。可以用于该组合物和方法的接头的其它实例包括缬氨酸-瓜氨酸、马来酰亚胺基己酰基、氨基苯甲酸类、p-氨基苯甲基氨基甲酰基(pab)、溶酶体酶可切割的接头、马来酰亚胺基己酰基-聚乙二醇(mc(peg)6-oh)、n-甲基-缬氨酸瓜氨酸、n-琥珀酰亚胺基4-(n-马来酰亚胺基甲基)环己烷-1-羧酸酯(smcc)、n-琥珀酰亚胺基4-(2-吡啶二硫代)丁酸酯(spdb)和n-琥珀酰亚胺基4-(2-吡啶基硫代)戊酸酯(spp)(也参见us2011/0076232)。用于本发明中的另一接头包括抗生物素蛋白-生物素连接以提供含抗生物素蛋白-生物素的adc(也参见美国专利no.4,676,980,pct公开no.wo1992/022332a2,wo1994/016729a1,wo1995/015770a1,wo1997/031655a2,wo1998/035704a1,wo1999/019500a1,wo2001/09785a2,wo2001/090198a1,wo2003/093793a2,wo2004/050016a2,wo2005/081898a2,wo2006/083562a2,wo2006/089668a1,wo2007/150020a1,wo2008/135237a1,wo2010/111198a1,wo2011/057216a1,wo201i/058321a1,wo2012/027494a1和ep77671b1),其中一些这样的接头抗生物素酶切割。可以用于本发明中的另外的接头包括粘连蛋白/dockerin对以提供含粘连蛋白-dockerin的adc(参见pct公开no.wo2008/097866a2,wo2008/097870a2,wo2008/103947a2和wo2008/103953a2)。用于本发明中的另外的接头可以包含非肽聚合物(其实例包括,但不限于聚乙二醇、聚丙二醇、聚氧乙烯化多元醇、聚乙烯醇、多糖、葡聚糖、聚乙烯基乙醚、pla(聚(乳酸))、plga(聚(乳酸-乙醇酸))及其组合,其中优选的聚合物是聚乙二醇)(也参见pct公开no.wo2011/000370)。另外的接头也描述于wo2004-010957、u.s.公开no.20060074008、u.s.公开no.20050238649和u.s.公开no.20060024317中,其各自通过全文引用并入本文中)。对于包含类美登素的adc,类美登素上的许多位置可以用作化学连接连接部分的位置。在一个实施方式中,类美登素包含含有反应性化学基团的连接部分,其为美登醇及其类似物的c-3酯,其中该连接部分包含二硫键和该化学反应性基团包含n-琥珀酰亚胺基或n-磺基琥珀酰亚胺基酯。例如,具有羟基的c-3位置、用羟甲基修饰的c-14位置、用羟基修饰的c-15位置和具有羟基的c-20位置均是可用的。连接部分最优选连接于美登醇的c-3位置。药物通过接头与抗体的偶联可以通过本领域中已知的任何技术完成。多种不同的反应可用于将药物和接头共价连接于抗体。这可以通过抗体的氨基酸残基(包括赖氨酸的氨基、谷氨酸和天冬氨酸的游离羧基、半胱氨酸的巯基及芳族氨基酸的各个部分)的反应完成。最常使用的非特异性共价连接方法之一是将化合物的羧基(或氨基)与抗体的氨基(或羧基)连接的碳二亚胺反应。另外,双功能试剂如二醛或亚氨酸酯已用于将化合物的氨基连接于抗体的氨基。还可用于将药物连接于抗体的是希夫碱反应。这一方法包括含二醇或羟基的药物的高碘酸盐氧化,因此形成随后与结合剂反应的醛。连接通过与抗体的氨基形成希夫碱而发生。异硫氰酸酯也可以用作偶联剂以将药物与抗体共价连接。其它技术是本领域技术人员已知的且在本发明的范围内。在某些实施方式中,中间体(其为接头的前体)在适宜条件下与药物反应。在某些实施方式中,使用在药物或中间体上的反应性基团。药物和中间体之间的反应产物或衍生化的药物随后在适宜条件下与抗-egfr抗体反应。示例性接头、延伸体单元、氨基酸单元、自切割间隔体单元的合成和结构描述于美国专利申请公开no.20030083263、20050238649和20050009751中,其各自通过引用并入本文。adc的稳定性可以通过标准分析技术如质谱、hplc和分离/分析技术lc/ms来测量。iv.抗-egfradc的纯化adc的纯化可以按照收集具有特定dar的adc的方式进行。例如,hic树脂可以用于将高药物负载的adc与具有最佳药物-抗体比率(dar)(例如,4或更低的dar)的adc分离。在一个实施方式中,疏水性树脂加入adc混合物中以使得不需要的adc,即,高药物负载的adc,结合树脂且可以选择性地从混合物移除。在某些实施方式中,adc的分离可以通过将adc混合物(例如,包含4或更低的adc载药种类和6或更高的adc载药种类的混合物)与疏水性树脂接触来实现,其中树脂的量足以允许从adc混合物移除的载药种类的结合。树脂和adc混合物混合在一起,使得待移除的adc种类(例如,6或更高的载药种类)结合于树脂且可以与adc混合物中的其它adc种类分离。该方法中使用的树脂的量是基于待除去种类与树脂之间的重量比,其中所使用的树脂的量不允许所需的载药种类大量结合。因此,该方法可以用于将平均dar5.5降低到小于4。进一步地,本文中描述的纯化方法可以用于分离具有任何所需范围的载药种类的adc,例如4或更低的载药种类、3或更低的载药种类、2或更低的载药种类、1或更低的载药种类。特定分子种类基于该种类和疏水性树脂之间的疏水相互作用而与表面结合。在一个实施方式中,本发明的方法是指依赖于疏水性树脂与adc混合物的互混的纯化过程,其中添加到混合物中的树脂的量决定哪些种类(例如具有6或更高的dar的adc)将结合。在抗体从表达系统(例如,哺乳动物表达系统)产生和纯化后,抗体被还原并通过偶联反应与药物偶联。所得的adc混合物通常包含具有一定范围的dar(例如,1-8)的adc。在一个实施方式中,adc混合物包含4或更低的载药种类和6或更高的载药种类。根据本发明的方法,adc混合物可以使用例如,但不限于批处理的方法纯化,使得选择具有4或更低的载药种类的adc并与具有较高药物载量的adc(例如,具有6或更高的载药种类的adc)分离。值得注意的是,本文描述的纯化方法可以用于分离具有任何所需范围的dar(例如,4或更低的dar、3或更低的dar、2或更低的dar)的adc。因此,在一个实施方式中,包含4或更低的载药种类和6或更高的载药种类的adc混合物可以与疏水性树脂接触以形成树脂混合物,其中与adc混合物接触的疏水性树脂的量足以允许6或更高的载药种类与树脂结合,但不允许4或更低的载药种类的显著结合,并从adc混合物除去疏水性树脂,使得获得包含adc的组合物,其中组合物包含少于15%的6或更高的载药种类,且其中adc包含与阿里他汀偶联的抗体。在单独的实施方式中,本发明的方法包括使包含4或更低的载药种类和6或更高的载药种类的adc混合物与疏水性树脂以形成树脂混合物,其中与adc混合物接触的疏水性树脂的量足以允许6或更高的载药种类与树脂结合,但不允许4或更低的载药种类的显著结合,并从adc混合物除去疏水性树脂,使得获得包含adc的组合物,其中组合物包含少于15%的6或更高的载药种类,且其中adc包含与阿里他汀偶联的抗体,其中疏水性树脂重量是adc混合物中6或更高的载药种类重量的3-12倍。本文所述的adc分离方法可以使用批纯化方法进行。批纯化处理一般包括将adc混合物添加到容器中的疏水性树脂中,混合和随后将树脂与上清液分离。例如,在批纯化的情况中,疏水性树脂可以在所需的平衡缓冲液中制备或相对于所需的平衡缓冲液进行平衡。因此可以获得疏水性树脂的浆料。adc混合物然后可以与浆料接触以吸附待通过疏水性树脂分离的特定种类的adc。包含不与疏水性树脂材料结合的所需adc的溶液然后可以例如通过过滤或通过允许浆料沉降和除去上清液而与浆料分离。所得的浆料可以进行一个或多个洗涤步骤。为了洗脱结合的adc,盐浓度可以降低。在一个实施方式中,本发明中使用的方法包括不超过50g的疏水性树脂。因此,批方法可以用于使包含4或更低的载药种类和6或更高的载药种类的adc混合物与疏水性树脂接触以形成树脂混合物,其中与adc混合物接触的疏水性树脂的量足以允许6或更高的载药种类与树脂结合但不允许4或更低的载药种类的显著结合;并从adc混合物除去疏水性树脂,使得获得包含adc的组合物,其中该组合物包含少于15%的6或更高的载药种类,且其中adc包含与阿里他汀偶联的抗体。在单独的实施方式中,批方法用于使包含4或更低的载药种类和6或更高的载药种类的adc混合物与疏水性树脂接触以形成树脂混合物,其中与adc混合物接触的疏水性树脂的量足以允许6或更高的载药种类与树脂结合但不允许4或更低的载药种类的显著结合;并从adc混合物除去疏水性树脂,使得获得包含adc的组合物,其中该组合物包含少于15%的6或更高的载药种类,且其中adc包含与阿里他汀偶联的抗体,其中疏水性树脂的重量是adc混合物中6或更高的载药种类的重量的3-12倍。或者,在单独的实施方式中,纯化可以使用循环处理进行,从而树脂填装在容器中且adc混合物经过疏水性树脂床直到待分离的特定种类的adc被除去。上清液(含有所需的adc种类)然后从容器泵送且树脂床可以进行洗涤步骤。循环处理可以用于使包含4或更低的载药种类和6或更高的载药种类的adc混合物与疏水性树脂接触以形成树脂混合物,其中与adc混合物接触的疏水性树脂的量足以允许6或更高的载药种类与树脂结合但不允许4或更低的载药种类的显著结合;并从adc混合物除去疏水性树脂,使得获得包含adc的组合物,其中该组合物包含少于15%的6或更高的载药种类,且其中adc包含与阿里他汀偶联的抗体。在单独的实施方式中,循环处理用于使包含4或更低的载药种类和6或更高的载药种类的adc混合物与疏水性树脂接触以形成树脂混合物,其中与adc混合物接触的疏水性树脂的量足以允许6或更高的载药种类与树脂结合但不允许4或更低的载药种类的显著结合;并从adc混合物除去疏水性树脂,使得获得包含adc的组合物,其中该组合物包含少于15%的6或更高的载药种类,且其中adc包含与阿里他汀偶联的抗体,其中疏水性树脂的重量是adc混合物中6或更高的载药种类的重量的3-12倍。或者,流过处理(flowthroughprocess)可以用于纯化adc混合物以获得包含具有某些所需dar的大部分adc的组合物。在流过处理中,树脂填装在容器(例如,柱)中,且adc混合物经过填装的树脂以使得所需的adc种类基本上不与树脂结合而流过树脂,且不希望的adc种类结合于树脂。流过处理可以以单轮模式(其中目标adc种类由于单次通过容器的树脂而获得)进行或以多轮模式(其中目标adc种类由于多次通过容器的树脂而获得)进行。进行流过处理以使得所选择的树脂重量结合不希望的adc群体,而所需的adc(例如,dar2-4)流过树脂并在一轮或多轮后收集在流通液中。流过处理可以用于使包含4或更低的载药种类和6或更高的载药种类的adc混合物与疏水性树脂接触,其中与adc混合物接触的疏水性树脂的量足以允许6或更高的载药种类与树脂结合但不允许4或更低的载药种类的显著结合,其中4或更低的载药种类经过树脂并随后在一轮或多轮后收集,使得获得包含所需adc(例如,dar2-4)的组合物,其中该组合物包含少于15%的6或更高的载药种类,且其中adc包含与阿里他汀偶联的抗体。在单独的实施方式中,流过处理用于通过使包含4或更低的载药种类和6或更高的载药种类的adc混合物经过疏水性树脂而使adc混合物与树脂接触,其中与adc混合物接触的疏水性树脂的量足以允许6或更高的载药种类与树脂结合但不允许4或更低的载药种类的显著结合,其中4或更低的载药种类经过树脂并随后收集,使得获得包含adc的组合物,其中该组合物包含少于15%的6或更高的载药种类,且其中adc包含与阿里他汀偶联的抗体,其中疏水性树脂重量的量是adc混合物中6或更高的载药种类的重量的3-12倍。在流过处理后,树脂可以用一个或多个洗液进行洗涤以进一步回收具有所需dar范围的adc(在洗涤滤液中发现)。例如,具有降低的电导率的多个洗液可以用于进一步回收具有目标dar的adc。从树脂的洗涤获得的洗脱材料可以随后与从流过处理获得的滤液组合以获得具有目标dar的adc的更高回收率。前述批、循环和流过处理纯化方法是基于使用疏水性树脂来分离高载药种类的adc与低载药种类的adc。疏水性树脂包含与adc的疏水性质相互作用的疏水基团。adc上的疏水基团与疏水性树脂内的疏水基团相互作用。蛋白质疏水性越高,其与疏水性树脂的相互作用越强。疏水性树脂通常包含疏水性配体(例如烷基或芳基)与其偶联的基础基质(例如交联的琼脂糖或合成的共聚物材料)。许多疏水性树脂可商购获得。实例包括但不限于具有低或高取代的phenylsepharosetm6fastflow(pharmacialkbbiotechnology,ab,sweden);phenylsepharosetmhighperformance(pharmacialkbbiotechnology,ab,sweden);octylsepharosetmhighperformance(pharmacialkbbiotechnology,ab,sweden);fractogeltmemdpropyl或fractogeltmemdphenyl柱(e.merck,germany);macro-preptmmethyl或macro-preptm.t-butylsupports(bio-rad,california);wphi-propyl(c3)tm(j.t.baker,newjersey)及toyopearltm醚、己基、苯基或丁基(tosohaas,pa)。在一个实施方式中,疏水性树脂是丁基疏水性树脂。在另一实施方式中,疏水性树脂是苯基疏水性树脂。在另一实施方式中,疏水性树脂是己基疏水性树脂、辛基疏水性树脂或癸基疏水性树脂。在一个实施方式中,疏水性树脂是具有正丁基配体的甲基丙烯酸聚合物(例如,butyl-600m)。用于纯化adc混合物以获得具有所需dar的组合物的进一步方法描述于美国申请no.14/210,602(美国专利申请公开no.us2014/0286968)中,其通过引入全文并入。v.抗-egfr抗体和抗-egfradc的用途本发明的抗体和抗体部分(及adc)优选能够在体内和在体外中和人egfr活性。因此,本发明的这些抗体和抗体部分可以用于抑制hegfr活性,例如,在包含hegfr的细胞培养物中、在人类受试者中或在具有本发明抗体与之交叉反应的egfr的其它哺乳动物受试者中。在一个实施方式中,本发明提供用于抑制hegfr活性的方法,包括使hegfr与本发明的抗体或抗体部分接触以使得hegfr活性被抑制。例如,在包含或疑似包含hegfr的细胞培养物中,本发明的抗体或抗体部分可以添加到培养基中以抑制培养物中的hegfr活性。在另一实施方式中,本发明的方法用于降低受试者中的hegfr活性,有利地来自患有其中egfr活性有害的疾病或障碍的受试者。本发明提供用于降低患有这种疾病或障碍的受试者中的egfr活性的方法,该方法包括向受试者施用本发明的抗体或抗体部分以使得受试者中的egfr活性降低。优选地,egfr是人egfr,且受试者是人类受试者。或者,受试者可以是表达本发明的抗体能够与其结合的egfr的哺乳动物。再进一步的受试者可以是egfr已经被引入其中的哺乳动物(例如,通过施用egfr或通过egfr转基因的表达)。本发明的抗体可以施用于人类受试者用于治疗目的。而且,本发明的抗体可以施用于表达抗体能够与其结合的egfr的非人哺乳动物用于兽医学目的或者作为人类疾病的动物模型。关于后者,这样的动物模型可用于评估本发明的抗体的治疗效力(例如,施用剂量和时程的测试)。如本文中使用的,术语“其中egfr活性有害的障碍”意图包括其中患有该障碍的受试者中egfr的存在已经表明为或怀疑是造成该障碍的病理生理或是造成障碍恶化的因素的疾病和其他障碍。因此,其中egfr活性有害的障碍是其中egfr活性的降低预期缓解障碍的症状和/或进展的障碍。例如,此类障碍可以由例如可以用例如如上所述的抗-egfr抗体检测的来自患有该障碍的受试者的生物流体中egfr浓度的提高(例如,受试者的肿瘤、血清、血浆、滑膜液等中egfr浓度的提高)来证明。可以用本发明的抗体(例如,aba)或其抗原结合片段治疗的障碍的非限制性实例包括以下讨论的那些障碍。例如,合适的障碍包括,但不限于多种癌症,包括但不限于乳腺癌、肺癌、神经胶质瘤、前列腺癌、胰腺癌、结肠癌、头颈癌和肾癌。可用使用本文公开的组合物和方法治疗的癌症的其它实例包括鳞状细胞癌(例如,肺鳞癌或头颈鳞状细胞癌)、三阴性乳腺癌、非小细胞肺癌、结肠直肠癌和间皮瘤。在一个实施方式中,本文公开的抗体和adc用于治疗实体肿瘤,例如,抑制过表达egfr或本身为egfr阳性的实体肿瘤的生长或减小其尺寸。在一个实施方式中,本发明涉及egfr扩增的肺鳞癌的治疗。在一个实施方式中,本文公开的抗体和adc用于治疗egfr扩增的头颈鳞状细胞癌。在另一实施方式中,本文公开的抗体和adc用于治疗三阴性乳腺癌(tnbc)。本文所述的疾病和障碍可以通过本发明的抗-egfr抗体或adc以及包含这种抗-egfr抗体或adc的药物组合物治疗。在某些实施方式中,本文公开的抗体和adc施用于需要的受试者以治疗很可能表现出升高水平的表皮生长因子受体(egfr)的晚期实体肿瘤类型。这样的肿瘤的实例包括,但不限于头颈鳞状细胞癌、非小细胞肺癌、三阴性乳腺癌、结肠直肠癌和多形性成胶质细胞瘤。在某些实施方式中,本发明包括用于抑制或降低患有实体肿瘤的受试者中的实体肿瘤生长的方法,所述方法包括向患有实体肿瘤的受试者施用本文所述的抗-egfr抗体或adc,使得实体肿瘤生长被抑制或降低。在某些实施方式中,实体肿瘤是非小细胞肺癌或成胶质细胞瘤。在进一步的实施方式中,实体肿瘤是egfrviii阳性肿瘤或egfr-表达实体肿瘤。在进一步的实施方式中,实体肿瘤是egfr扩增的实体肿瘤或egfr过表达的实体肿瘤。在某些实施方式中,本文所述的抗-egfr抗体或adc单独地或与另外的药剂(例如,放射和/或替莫唑胺)结合施用于患有多形性成胶质细胞瘤(glioblastimamultiforme)的受试者。在某些实施方式中,本发明包括用于抑制或降低患有鉴别为egfr表达或egfr过表达肿瘤(或egfrviii表达肿瘤)的实体肿瘤的受试者中的实体肿瘤生长的方法,所述方法包括向患有实体肿瘤的受试者施用本文所述的抗-egfr抗体或adc,使得实体肿瘤生长被抑制或降低。用于鉴别egfr表达肿瘤(例如,egfr过表达肿瘤)的方法是本领域已知的,且包括fda-批准的测试和验证分析法。例如,egfrpharmdxtm分析(dakonorthamerica,inc.)是用于鉴定经常规固定用于组织学评价的正常或肿瘤组织中的egfr表达的定性免疫组织化学(ihc)试剂盒系统。egfrpharmdx特别地检测egfr-表达细胞中的egfr(her1)蛋白。另外,基于pcr的分析也可以用于鉴别egfr过表达肿瘤。例如,这些分析可以使用对于变体egfr基因(例如,seqidno:33)和/或cdna特异性的引物并导致egfr基因/cdna或其部分的扩增。扩增的pcr产物随后可以例如使用本领域中已知的标准方法通过凝胶电泳进行分析以测定pcr产物的大小。这样的测试可以用于鉴别可用本文所述的方法和组合物治疗的肿瘤。本领域中可得的用于基因疗法的任何方法可根据本发明使用。对于基因疗法的方法的一般综述参见goldspiel等人,1993,clinicalpharmacy12:488-505;wu和wu,1991,biotherapy3:87-95;tolstoshev,1993,ann.rev.pharmacol.toxicol.32:573-596;mulligan,science260:926-932(1993)及morganandanderson,1993,ann.rev.biochem.62:191-217;may,1993,tibtech11(5):155-215。可以使用的重组dna
技术领域:
中通常已知的方法描述于ausubel等人(eds.),currentprotocolsinmolecularbiology,johnwiley&sons,ny(1993)及kriegler,genetransferandexpression,alaboratorymanual,stocktonpress,ny(1990)中。基因治疗的各种方法的详细描述提供在us20050042664a1中,其通过引用并入本文。在另一方面,本申请特征在于治疗(例如,治愈、抑制、缓解、延迟或防止其发作、或者预防其重现或复发)或预防受试者的egfr-相关障碍的方法。该方法包括:以足以治疗或预防egfr-相关障碍的量向受试者施用egfr结合剂(特别是拮抗剂),例如,本文所述的抗-egfr抗体或其片段。egfr拮抗剂,例如,抗-egfr抗体或其片段,可以如本文所述单独地或与其它治疗形式结合施用于受试者。本发明的抗体或adc或其抗原结合部分可以单独或组合地使用以治疗这类疾病。应理解,本发明的抗体或其抗原结合部分可以单独地或与另外的药剂(例如,治疗剂)结合使用,所述另外的药剂由本领域技术人员根据预期的目的进行选择。例如,另外的药剂可以是本领域公认为可用于治疗本发明的抗体所治疗的疾病或病症的治疗剂。另外的药剂也可以是赋予治疗组合物有益特性的试剂,例如,影响组合物的粘度的试剂。应进一步理解,包括在本发明内的组合是可用于其预期目的的那些组合。以下给出的药剂是用于说明目的而非旨在用于限制。作为本发明的部分的组合可以是本发明的抗体和至少一种选自以下列表的另外的药剂。组合也可以包括超过一种另外的药剂,例如,两种或三种另外的药剂,如果该组合使得所形成的组合物可以执行其预期功能。组合疗法可以包括与一种或多种另外的治疗剂(例如,一种或多种细胞因子和生长因子抑制剂、免疫抑制剂、抗炎剂(例如,系统抗炎剂)、抗纤维化剂、代谢抑制剂、酶抑制剂和/或细胞毒性或细胞生长抑制剂、有丝分裂抑制剂、抗肿瘤抗生素、免疫调节剂、用于基因疗法的载体、烷化剂、抗血管生成剂、抗代谢物、含硼剂、化疗保护剂、激素、抗激素剂、皮质类固醇、光敏治疗剂、寡核苷酸、放射性核素剂、拓扑异构酶抑制剂、酪氨酸激酶抑制或放射致敏剂)一起配制和/或共同施用的一个或多个egfr拮抗剂,例如,抗-egfr抗体或其片段,如本文中进一步描述的。在特定实施方式中,本文所述的抗-egfr结合蛋白,例如,抗-egfr抗体,与抗癌剂或抗肿瘤剂组合使用。术语“抗癌剂”和“抗肿瘤剂”是指用于治疗恶性肿瘤如癌性生长的药物。药物治疗可以单独地或与其它治疗如手术或放疗结合使用。根据牵涉的器官的性质,几类药物可以用于癌症治疗中。例如,乳腺癌通常受到雌激素的刺激,且可以用使性激素失活的药物治疗。类似地,前列腺癌可以用使雄激素(男性性激素)灭活的药物进行治疗。可以与本发明的抗-egfr抗体或adc结合使用的抗癌剂特别地包括以下药剂:除了上述抗癌剂外,本文所述的抗-egfr抗体和adc可以与第ii节中描述的药剂组合施用。进一步地,前述抗癌剂也可以用于本发明的adc中。在特定实施方式中,抗-egfr抗体或adc可以单独地或与另一抗癌剂组合施用,该抗癌剂与抗体结合或协同地发挥作用以治疗与egfr活性相关的疾病。这样的抗癌剂包括,例如,本领域公知的药剂(例如,细胞毒素、化疗剂、小分子和放射)。抗癌剂的实例包括,但不限于panorex(glaxo-welcome)、美罗华(idec/genentech/hoffmanlaroche)、米罗他(wyeth)、坎帕斯(millennium)、泽娃灵(idecandscheringag)、百克沙(corixa/gsk)、爱必妥(imclone/bms)、阿瓦斯丁(genentech)和赫塞汀(genentech/hoffmanlaroche)。其它抗癌剂包括,但不限于美国专利no.7,598,028和国际公开no.wo2008/100624中公开的那些,其内容通过引用并入本文。一种或多种抗癌剂可以与本发明的抗体或其抗原结合部分的施用同时施用,或在其之前或之后施用。在本发明的特定实施方式中,本文所述的抗-egfr抗体或adc可以用于与凋亡剂(如bcl-xl抑制剂或bcl-2(b-细胞淋巴瘤2)抑制剂(例如,abt-199(venetoclax))的组合疗法中来治疗受试者的癌症如白血病。在一个实施方式中,本文所述的抗-egfr抗体或adc可以用于与bcl-xl抑制剂的组合疗法中用于治疗癌症。在一个实施方式中,本文所述的抗-egfr抗体或adc可以用于与venetoclax的组合疗法中用于治疗癌症。在本发明的特定实施方式中,本文所述的抗-egfr抗体或adc可以用于与nampt的抑制剂(参见us2013/0303509中的抑制剂实例;abbvie,inc.,其通过引用并入本文)的组合疗法中来治疗需要的受试者。nampt(也称为前b细胞集落增强因子(pbef)和内脂素)是催化烟酰胺的磷酸核糖基化的酶且是挽救(salvage)nad的两个途径之一中的限速酶。在本发明的一个实施方式中,本文所述的抗-egfr抗体或adc与nampt抑制剂组合施用以治疗受试者的癌症。在本发明的特定实施方式中,本文所述的抗-egfr抗体或adc可以用于与sn-38(其是拓扑异构酶抑制剂依立替康的活性代谢物)的组合疗法中。在本发明的其它实施方式中,本文所述的抗-egfr抗体或adc可以用于与parp(聚adp核糖聚合酶)抑制剂(例如,veliparib)的组合疗法中以治疗癌症,包括乳腺癌、卵巢癌和非小细胞肺癌。可以与本文所述的抗-egfr抗体或抗-egfradc共同施用和/或一起配制的另外的治疗剂的进一步实例包括,但不限于以下一种或多种:吸入性类固醇、β-激动剂,例如,短效或长效β-激动剂、白三烯或白三烯受体的拮抗剂、组合药物如advair、ige抑制剂,例如抗-ige抗体(例如,奥马珠单抗)、磷酸二酯酶抑制剂(例如,pde4抑制剂)、黄嘌呤类、抗胆碱能药物、肥大细胞稳定剂如色甘酸、il-4抑制剂、il-5抑制剂、嗜酸细胞活化趋化因子/ccr3抑制剂、组胺或其受体(包括h1、h2、h3和h4)的拮抗剂,及前列腺素d或其受体(dp1和crth2)的拮抗剂。这样的组合可以用于治疗例如,哮喘和其它呼吸系统障碍。可以与本文所述的抗-egfr抗体或抗-egfradc共同施用和/或一起配制的另外的治疗剂的其它实例包括,但不限于替莫唑胺、依鲁替尼、duvelisib和idelalisib中的一种或多种。可以与一个或多个抗-egfr抗体或其片段共同施用和/或一起配制的治疗剂的其它实例包括以下一种或多种:tnf拮抗剂(例如,tnf受体的可溶性片段,例如,p55或p75人tnf受体或其衍生物,例如,75kdtnfr-igg(75kdtnf受体-igg融合蛋白,enbrel));tnf酶拮抗剂,例如,tnf转化酶(tace)抑制剂;毒蕈碱受体拮抗剂;tgf-β拮抗剂;干扰素γ;吡非尼酮;化疗剂,例如,甲氨蝶呤、来氟米特或西罗莫司(雷帕霉素)或其类似物,例如,cci-779;cox2和cpla2抑制剂;nsaid;免疫调节剂;p38抑制剂、tpl-2、mk-2和nfkb抑制剂,及其它。其它优选组合是细胞因子抑制性抗炎药物(csaid);其它人类细胞因子或生长因子(例如,il-1、il-2、il-3、il-4、il-5、il-6、il-7、il-8、il-15、il-16、il-18、il-21、il-31、干扰素、emap-ii、gm-csf、fgf、egf、pdgf和内皮素-1)以及这些细胞因子和生长因子的受体的抗体或拮抗剂。本发明的抗体或其抗原结合部分可以与细胞表面分子如cd2、cd3、cd4、cd8、cd25、cd28、cd30、cd40、cd45、cd69、cd80(b7.1)、cd86(b7.2)、cd90、ctla、ctla-4、pd-1或它们的配体包括cd154(gp39或cd40l)的抗体组合。优选的治疗剂组合可以在炎症级联的不同点介入;优选的实例包括tnf拮抗剂如嵌合的、人源化的或人tnf抗体、阿达木单抗(humira;d2e7;pct公开no.wo97/29131和美国专利no.6,090,382,其通过引用并入本文)、ca2(remicadetm)、cdp571及可溶性p55或p75tnf受体,其衍生物(p75tnfr1gg(enbreltm)或p55tnfr1gg(lenercept),以及tnf转化酶(tace)抑制剂;类似地,il-1抑制剂(白介素-1-转化酶抑制剂,il-1ra等)出于相同原因可以有效地使用。其它优选组合包括白介素4。本发明的药物组合物可以包括“治疗有效量”或“预防有效量”的本发明的抗体或抗体部分。“治疗有效量”是指在必要的剂量和持续时间下有效地实现所需治疗结果的量。治疗有效量的抗体或抗体部分可以由本领域技术人员确定和可以按照如疾病状态及个体的年龄、性别和体重及抗体或抗体部分在个体中引起所需反应的能力的因素变化。治疗有效量也是其中抗体或抗体部分的任何毒性或有害效应远不如治疗有益效果的量。“预防有效量”是指在必要的剂量和持续时间下有效地实现所需预防结果的量。通常,由于预防剂量在疾病之前或早期阶段中使用于受试者,预防有效量低于治疗有效量。剂量方案可以进行调整以提供最佳的所需反应(例如,治疗或预防反应)。例如,可以施用单次大剂量(bolus),可以随时间施用几个分剂量或者可以按照治疗情况所指示的成比例地降低或提高剂量。特别有利的是配制单位剂量形式的肠胃外施用组合物以为了施用方便和剂量均一性。如本文中使用的单位剂量形式是指适合作为用于待治疗的哺乳动物受试者的单一剂量的物理离散单元,各单元包含与所需药用载体结合的计算为产生所述治疗效果的预定量的活性化合物。本发明的单位剂量形式的规格由(a)活性化合物的独特性质和需实现的特定治疗或预防效应决定,和(b)配制这种活性化合物以在个体中实现敏感的治疗的领域中固有的限定,并与其直接相关。本发明的adc、抗体或抗体部分的治疗或预防有效量的示例性非限制性范围是0.1-20mg/kg,更优选1-10mg/kg。在一个实施方式中,本文所述的抗体和adc的剂量是1-6mg/kg,包括本文所述的个体剂量,例如,1mg/kg、2mg/kg、3mg/kg、4mg/kg、5mg/kg和6mg/kg。在另一实施方式中,本文所述的抗体和adc的剂量是1-200μg/kg,包括本文所述的个体剂量,例如,1μg/kg、2μg/kg、3μg/kg、4μg/kg、5μg/kg、10μg/kg、20μg/kg、30μg/kg、40μg/kg、50μg/kg、60μg/kg、80μg/kg、100μg/kg、120μg/kg、140μg/kg、160μg/kg、180μg/kg和200μg/kg。应当注意,剂量值可随待缓解的病症的类型和严重性而变化。还应理解,对于任何特定受试者,具体剂量方案应按照个体需要和施用组合物或监控组合物的施用的人员的专业判断随时间进行调整,且本文中给出的剂量范围仅是示例性的而不旨在限制本发明组合物的范围和实施。在一个实施方式中,本文所述的抗-egfr抗体,例如,aba,或其抗原结合部分作为adc以0.1-30mg/kg的剂量施用于需要的受试者,例如,患有癌症的受试者。在另一实施方式中,抗-egfr抗体,例如,aba,或其抗原结合部分作为adc以1-15mg/kg的剂量施用于需要的受试者,例如,患有癌症的受试者。在另一实施方式中,抗-egfr抗体,例如,aba,或其抗原结合部分作为adc以1-10mg/kg的剂量施用于需要的受试者,例如,患有癌症的受试者。在另一实施方式中,抗-egfr抗体,例如,aba,或其抗原结合部分作为adc以2-3mg/kg的剂量施用于需要的受试者,例如,患有癌症的受试者。在另一实施方式中,抗-egfr抗体,例如,aba,或其抗原结合部分作为adc以1-4mg/kg的剂量施用于需要的受试者,例如,患有癌症的受试者。在一个实施方式中,本文所述的抗-egfr抗体,例如,aba,或其抗原结合部分作为adc以1-200μg/kg的剂量施用于需要的受试者,例如,患有癌症的受试者。在另一实施方式中,抗-egfr抗体,例如,aba,或其抗原结合部分作为adc以5-150μg/kg的剂量施用于需要的受试者,例如,患有癌症的受试者。在另一实施方式中,抗-egfr抗体,例如,aba,或其抗原结合部分作为adc以5-100μg/kg的剂量施用于需要的受试者,例如,患有癌症的受试者。在另一实施方式中,抗-egfr抗体,例如,aba,或其抗原结合部分作为adc以5-90μg/kg的剂量施用于需要的受试者,例如,患有癌症的受试者。在另一实施方式中,抗-egfr抗体,例如,aba,或其抗原结合部分作为adc以5-80μg/kg的剂量施用于需要的受试者,例如,患有癌症的受试者。在另一实施方式中,抗-egfr抗体,例如,aba,或其抗原结合部分作为adc以5-70μg/kg的剂量施用于需要的受试者,例如,患有癌症的受试者。在另一实施方式中,抗-egfr抗体,例如,aba,或其抗原结合部分作为adc以5-60μg/kg的剂量施用于需要的受试者,例如,患有癌症的受试者。在另一实施方式中,抗-egfr抗体,例如,aba,或其抗原结合部分作为adc以10-80μg/kg的剂量施用于需要的受试者,例如,患有癌症的受试者。在一个实施方式中,本文所述的抗-egfradc,例如,aba-vc-mmae,以0.1-6mg/kg的剂量施用于需要的受试者,例如,患有癌症的受试者。在另一实施方式中,本文所述的抗-egfradc,例如,aba-vc-mmae,以0.5-4mg/kg的剂量施用于需要的受试者,例如,患有癌症的受试者。在另一实施方式中,本文所述的抗-egfradc,例如,aba-vc-mmae,以1.8-2.4mg/kg的剂量施用于需要的受试者,例如,患有癌症的受试者。在另一实施方式中,本文所述的抗-egfradc,例如,aba-vc-mmae,以1-4mg/kg的剂量施用于需要的受试者,例如,患有癌症的受试者。在另一实施方式中,本文所述的抗-egfradc,例如,aba-vc-mmae,以约1mg/kg的剂量施用于需要的受试者,例如,患有癌症的受试者。在另一实施方式中,本文所述的抗-egfradc,例如,aba-vc-mmae,以3-6mg/kg的剂量施用于需要的受试者,例如,患有癌症的受试者。在另一实施方式中,本文所述的抗-egfradc,例如,aba-vc-mmae,以3mg/kg的剂量施用于需要的受试者,例如,患有癌症的受试者。在另一实施方式中,本文所述的抗-egfradc,例如,aba-vc-mmae,以2-3mg/kg的剂量施用于需要的受试者,例如,患有癌症的受试者。在另一实施方式中,本文所述的抗-egfradc,例如,aba-vc-mmae,以6mg/kg的剂量施用于需要的受试者,例如,患有癌症的受试者。在另一实施方式中,与药物(例如,pbd)偶联的本文所述的抗-egfr抗体(adc)以1-200μg/kg的剂量施用于需要的受试者,例如,患有癌症的受试者。在另一实施方式中,本文所述的抗-egfradc以5-100μg/kg的剂量施用于需要的受试者,例如,患有癌症的受试者。在另一实施方式中,本文所述的抗-egfradc以5-90μg/kg的剂量施用于需要的受试者,例如,患有癌症的受试者。在另一实施方式中,本文所述的抗-egfradc以5-80μg/kg的剂量施用于需要的受试者,例如,患有癌症的受试者。在另一实施方式中,本文所述的抗-egfradc以5-70μg/kg的剂量施用于需要的受试者,例如,患有癌症的受试者。在另一实施方式中,本文所述的抗-egfradc以5-60μg/kg的剂量施用于需要的受试者,例如,患有癌症的受试者。如上所述的剂量可能可用于本文公开的抗-egfradc或抗体的施用。在另一方面,本申请提供用于检测体外样品(例如,生物样品,如血清、血浆、组织、活检样品)中egfr的存在的方法。所述方法可以用于诊断疾病,例如,癌症。该方法包括:(i)使样品或对照样品与本文所述的抗-egfr抗体或其片段接触,及(ii)检测抗-egfr抗体或其片段与样品或对照样品之间复合物的形成,其中样品中复合物的形成相对于对照样品的统计学显著的改变指示样品中egfr的存在。考虑到其与人egfr结合的能力,本发明的抗-人egfr抗体或其部分(以及其adc)可以用于使用常规免疫分析法如酶联免疫吸附分析(elisa)、放射免疫分析(ria)或组织免疫组织化学检测人egfr(例如,在生物样品中,如血清或血浆)。在一个方面,本发明提供用于检测生物样品中的人egfr的方法,包括使生物样品与本发明的抗体或抗体部分接触和检测与人egfr结合的抗体(或抗体部分)或者未结合的抗体(或抗体部分),由此检测生物样品中的人egfr。抗体用可检测物质直接或间接地标记以利于结合的或未结合的抗体的检测。合适的可检测物质包括各种酶、辅基、荧光材料、发光材料和放射性材料。合适的酶的实例包括辣根过氧化物酶、碱性磷酸酶、β-半乳糖苷酶或乙酰胆碱酯酶;合适的辅基的实例包括链霉亲和素/生物素和抗生物素蛋白/生物素;合适的荧光材料的实例包括伞形酮、荧光素、异硫氰酸荧光素、罗丹明、二氯三嗪基胺荧光素、丹黄酰氯或藻红蛋白;发光材料的实例包括鲁米诺,且合适的放射性材料的实例包括3h、14c、35s、90y、99tc、111in、125i、131i、177lu、166ho或153sm。作为标记抗体的替代方案,人egfr可以利用以可检测物质标记的rhegfr标准品和未标记的抗-人egfr抗体通过竞争免疫分析法在生物流体中测定。在这一分析中,生物样品、标记的rhegfr标准品和抗-人egfr抗体合并在一起且测定与未标记的抗体结合的标记rhegfr标准品的量。生物样品中人egfr的量与结合于抗-egfr抗体的标记rhegfr标准品的量成反比。类似地,人egfr也可以利用以可检测物质标记的rhegfr标准品和未标记的抗-人egfr抗体通过竞争免疫分析在生物流体中测定。在再另一方面,本申请提供用于体内检测egfr的存在的方法(例如,受试者的体内成像)。所述方法可以用于诊断疾病,例如,egfr-相关障碍。该方法包括:(i)在允许抗体或片段与egfr结合的条件下向受试者或对照受试者施用本文所述的抗-egfr抗体或其片段,且(ii)检测抗体或片段与egfr之间复合物的形成,其中受试者中复合物的形成相对于对照受试者的统计学显著的改变指示egfr的存在。vi.药物组合物本发明还提供包含本发明的抗体或其抗原结合部分或者adc及药学上可接受的载体的药物组合物。包含本发明的抗体或adc的药物组合物用于(但不限于)诊断、检测或监测疾病,用于预防、治疗、控制或缓解疾病或其一种或多种症状和/或用于研究。在特定的实施方式中,组合物包含本发明的一个或多个抗体。在另一实施方式中,药物组合物包含本发明的一个或多个抗体或adc及本发明抗体或adc以外的一种或多种用于治疗其中egfr活性有害的障碍的预防或治疗剂。优选地,已知可用于或已经用于或当前正用于预防、治疗、控制或缓解疾病或其一种或多种症状的预防或治疗剂。根据这些实施方式,组合物可以进一步包含载体、稀释剂或赋形剂。本发明的抗体和抗体部分或adc可以并入适合施用于受试者的药物组合物中。通常,药物组合物包含本发明的抗体或抗体部分和药学上可接受的载体。如本文中使用的,“药学上可接受的载体”包括生理上相容的任何或所有溶剂、分散介质、包衣、抗细菌剂和抗真菌剂、等渗剂和吸收延迟剂等。药学上可接受的载体的实例包括水、盐水、磷酸盐缓冲盐水、右旋糖、甘油、乙醇等等以及其组合中的一种或多种。在许多情况中,优选在组合物中包括等渗剂,例如,糖类、多元醇如甘露醇、山梨醇或氯化钠。药学上可接受的载体可以进一步包含少量的辅助物质如湿润剂或乳化剂、防腐剂或缓冲剂,其增强抗体或抗体部分或adc的储存期或有效性。在一个实施方式中,本发明特征在于包含抗-egfr抗体药物偶联物、蔗糖、聚山梨醇酯80和组氨酸的冻干制剂,其中该制剂具有约5-7的ph,其中抗-egfr抗体药物偶联物包含与单甲基阿里他汀e(mmae)偶联的抗-egfr抗体或其抗原结合部分。在一个实施方式中,本发明进一步提供包含含有阿里他汀(例如,mmae)偶联的本文所述的抗-egfr抗体或其抗原结合部分的抗-egfradc、糖(例如,蔗糖)、表面活性剂(例如,聚山梨醇酯,如聚山梨醇酯80)和组氨酸的冻干制剂。在一个实施方式中,冻干制剂包含1-20mg的组氨酸、约320-410mg的糖、约0.1-0.9mg的表面活性剂和约1-150mg的含有与阿里他汀(例如,mmae)偶联的本文所述的抗-egfr抗体或其抗原结合部分的抗-egfradc。本发明还提供包含约1-100mg/ml的含有与阿里他汀(例如,mmae)偶联的本文所述的抗-egfr抗体或其抗原结合部分的抗-egfradc、约1-10mg/ml的组氨酸、约50-90mg/ml的糖(例如,蔗糖)和约0.01-0.2mg/ml的表面活性剂(例如,聚山梨醇酯80)的水性制剂。各种递送系统是已知的且可以用于施用一个或多个本发明的抗体或adc或者一个或多个本发明的抗体与可用于预防、控制、治疗或缓解疾病或其一种或多种症状的预防剂或治疗剂的组合,例如,包封在脂质体中、微粒、微胶囊、能够表达抗体或抗体片段的重组细胞、受体介导的内吞作用(参见,例如,wu和wu,j.biol.chem.262:4429-4432(1987))、核酸构建为逆转录病毒或其它载体的部分,等等。施用本发明的预防剂或治疗剂的方法包括,但不限于肠胃外施用(例如,皮内、肌肉内、腹膜内、静脉内和皮下)、硬膜外施用、肿瘤内施用和粘膜施用(例如,鼻内和口腔途径)。另外,肺部施用可以通过,例如,使用吸入器或喷雾器及具有雾化剂的制剂来采用。参见,例如,美国专利no.6,019,968、5,985,320、5,985,309、5,934,272、5,874,064、5,855,913、5,290,540和4,880,078及pct公开no.wo92/19244、wo97/32572、wo97/44013、wo98/31346和wo99/66903,其各自通过引用全文并入本文中。在一个实施方式中,本发明的抗体、组合疗法或本发明的组合物使用alkermes肺部药物递送技术(alkermes,inc.,cambridge,mass.)施用。在特定的实施方式中,本发明的预防剂或治疗剂肌肉内、静脉内、肿瘤内、口服、鼻内、肺部或皮下施用。预防剂或治疗剂可以通过任何方便的途径施用,例如通过输注或浓注、通过经上皮或皮肤粘膜内衬层(例如,口腔粘膜、直肠和肠粘膜等)的吸收,且可以与其它生物活性剂一起施用。施用可以是系统的或局部的。在特定的实施方式中,可能希望的是局部地施用本发明的预防剂或治疗剂至需要治疗的区域;这可以通过,例如,且不限于局部输注、通过注射或借助于植入物施用,所述植入物是多孔或非多孔材料的,包括膜和基质,如sialastic膜、聚合物、纤维基质(例如,)或胶原基质。在一个实施方式中,有效量的一个或多个本发明的抗体拮抗剂局部施用至受试者的影响区域以预防、治疗、控制和/或缓解障碍或其症状。在另一实施方式中,有效量的一个或多个本发明的抗体与本发明的抗体之外的有效量的一个或多个疗法(例如,一种或多种预防剂或治疗剂)组合局部施用至受试者的影响区域以预防、治疗、控制和/或缓解障碍或其一种或多种症状。在另一实施方式中,本发明的预防剂或治疗剂可以在受控释放或持续释放系统中递送。在一个实施方式中,泵可以用于实现受控释放或持续释放(参见langer,同上;seffon,1987,crccrit.ref.biomed.eng.14:20;buchwald等人,1980,surgery88:507;saudek等人,1989,n.engl.j.med.321:574)。在另一实施方式中,聚合材料可以用于本发明疗法的受控释放或持续释放(参见例如,medicalapplicationsofcontrolledrelease,langer和wise(eds.),crcpres.,bocaraton,fla.(1974);controlleddrugbioavailability,drugproductdesignandperformance,smolen和ball(eds.),wiley,newyork(1984);ranger和peppas,1983,j.,macromol.sci.rev.macromol.chem.23:61;也参见levy等人,1985,science228:190;during等人,1989,ann.neurol.25:351;howard等人,1989,j.neurosurg.71:105;美国专利no.5,679,377;美国专利no.5,916,597;u.s.pat.no.5,912,015;美国专利no.5,989,463;美国专利no.5,128,326;pct公开no.wo99/15154,及pct公开no.wo99/20253)。用于持续释放制剂中的聚合物的实例包括,但不限于聚(2-羟乙基甲基丙烯酸酯)、聚(甲基甲基丙烯酸酯)、聚(丙烯酸)、聚(乙烯-共-乙酸乙烯酯)、聚(甲基丙烯酸)、聚乙醇酸类(plg)、聚酸酐类、聚(n-乙烯基吡咯烷酮)、聚(乙烯醇)、聚丙烯酰胺、聚(乙二醇)、聚交酯类(pla)、聚(丙交酯-共-乙交酯类)(plga)和聚原酸酯。在优选的实施方式中,持续释放制剂中使用的聚合物是惰性的、无可渗滤杂质、储存稳定的、无菌的和可生物降解的。在又一实施方式中,受控释放或持续释放系统可以接近于预防或治疗靶标布置,因此仅需要全身剂量的一部分(参见,例如,goodson,medicalapplicationsofcontrolledrelease,同上,vol.2,pp.115-138(1984))。受控释放系统在langer(1990,science249:1527-1533)的综述中讨论。本领域技术人员已知的任何技术可以用于产生包含一种或多种本发明的治疗剂的持续释放制剂。参见,例如,u.s.专利no.4,526,938,pct公开wo91/05548,pct公开wo96/20698,ning等人,1996,“intratumoralradioimmunotheraphyofahumancoloncancerxenograftusingasustained-releasegel”,radiotherapy&oncology39:179-189,song等人,1995,“antibodymediatedlungtargetingoflong-circulatingemulsions”,pdajournalofpharmaceuticalscience&technology50:372-397,cleek等人,1997,“biodegradablepolymericcarriersforabfgfantibodyforcardiovascularapplication”,pro.int′l.symp.control.rel.bioact.mater.24:853-854和lam等人,1997,“microencapsulationofrecombinanthumanizedmonoclonalantbodyforlocaldelivery,”proc.int′l.symp.controlrel.bioact.mater.24:759-760,其各自通过引用全文并入本文。在其中本发明的组合物是编码预防剂或治疗剂的核酸的特定实施方式中,核酸可以通过将其构建为适宜核酸表达载体的部分并施用以使得其变成细胞内的(例如,通过使用逆转录病毒载体(参见美国专利no.4980,286),或者通过直接注射,或者通过使用微粒轰击(例如,基因枪;biolistic,dupont),或者用脂质或细胞表面受体或转移剂涂布或通过将其与已知进入细胞核的同源框样肽(参见,例如,joliot等人,1991,proc.natl.acad.sci.usa88:1864-1868)连接而施用)来体内施用以促进其编码的预防剂或治疗剂的表达。或者,核酸可以细胞内引入并通过同源重组整合到宿主细胞dna内用于表达。本发明的药物组合物配制为与其预定施用途径相容。施用途径的实例包括,但不限于肠胃外施用,例如,静脉内、皮内、皮下、口服、鼻内(例如,吸入)、透皮(例如,局部)、经粘膜和直肠施用。在特定的实施方式中,组合物按照适应于静脉内、皮下、肌肉内、口服、鼻内或局部施用于人类的药物组合物的常规程序配制。通常,用于静脉内施用的组合物是无菌等渗水性缓冲液中的溶液。必要的情况下,组合物也可以包括增溶剂和局部麻醉剂如利诺卡因以减轻注射部位的疼痛。如果本发明的方法包括鼻内施用组合物,该组合物可以配制成气溶胶形式、喷雾、薄雾或滴剂的形式。特别地,根据本发明使用的预防剂或治疗剂可以方便地以来自加压的包装或喷雾器的气溶胶喷雾呈现形式利用合适的推进剂(例如,二氯二氟甲烷、三氯氟甲烷、二氯四氟乙烷、二氧化碳或其它合适的气体)递送。在加压气溶胶的情况中,剂量单位可以通过提供阀以递送测定的量来确定。可以配制用于吸入器或吹入器中的包含化合物和合适的粉末基质如乳糖或淀粉的粉末混合物的胶囊和药盒(由例如明胶构成)。如果本发明的方法包括口服施用,组合物可以以片剂、胶囊、扁囊剂、胶帽、溶液、悬浮液等等的形式配制用于口服。片剂或胶囊可以用药学上可接受的赋形剂如结合剂(例如,预胶化玉米淀粉、聚乙烯吡咯烷酮或羟丙基甲基纤维素)、填充剂(例如,乳糖、微晶纤维素或磷酸氢钙)、润滑剂(例如,硬脂酸镁、滑石或二氧化硅)、崩解剂(例如,土豆淀粉或羟基乙酸淀粉钠)或湿润剂(例如,月桂基硫酸钠)通过常规方式制备。片剂可以通过本领域公知的方法包衣。用于口服施用的液体制剂可以采用溶液、糖浆或悬浮液的形式(但不限于此),或者它们可以表现为在使用前用水或其它合适的媒介复原的干燥产物。这样的液体制剂可以通过常规方式用药学上可接受的添加剂如悬浮剂(例如,山梨醇糖浆、纤维素衍生物或氢化食用脂肪)、乳化剂(例如,卵磷脂或阿拉伯树胶)、非水性媒介(例如,杏仁油、油酯、乙醇或分馏植物油)和防腐剂(例如,甲基-或丙基-p-羟基苯甲酸酯或山梨酸)制备。适宜情况下,制剂也可以包含缓冲盐、调味剂、着色剂和甜味剂。用于口服施用的制剂可以适当地配制用于预防剂或治疗剂的缓释、控缓或持续释放。本发明的方法可以包括肺部施用用雾化剂配制的组合物,例如,通过使用吸入器或喷雾器。参见,例如,美国专利no.6,019,968、5,985,320、5,985,309、5,934,272、5,874,064、5,855,913、5,290,540和4,880,078,及pct公开no.wo92/19244、wo97/32572、wo97/44013、wo98/31346和wo99/66903,其各自通过引用全文并入本文。在特定的实施方式中,本发明的抗体、组合疗法和/或本发明的组合物使用alkermes肺部药物递送技术(alkermes,inc.,cambridge,mass.)施用。本发明的方法可以包括施用配制用于通过注射(例如,通过浓注或持续输注)肠胃外施用的组合物。用于注射的制剂可以以具有添加的防腐剂的单位剂型(例如,在安瓿中或在多剂量容器中)存在。组合物可以采取如油性或水性媒介中的悬浮液、溶液或乳液的这类形式,且可以包含配制试剂如悬浮剂、稳定剂和/或分散剂。或者,活性成分可以是用于在使用前以合适的媒介复原(例如,无菌的无热原水)的粉末形式。本发明的方法可以另外地包括施用配制为储库制剂的组合物。这样的长效制剂可以通过植入(例如,皮下或肌肉内)或通过肌肉内注射施用。因此,例如,组合物可以用合适的聚合物或疏水性材料(例如,作为可接受油中的乳液)或离子交换树脂,或者作为难溶衍生物(例如,作为难溶盐)来配制。本发明的方法包括施用作为中性或盐形式配制的组合物。药学上可接受的盐包括与阴离子(如源自盐酸、磷酸、乙酸、草酸、酒石酸等的那些阴离子)形成的那些盐及与阳离子(如源自钠、钾、铵、钙、铁氢氧化物、异丙胺、三乙胺、2-乙基氨基乙醇、组氨酸、普鲁卡因等的那些阳离子)形成的那些盐。一般地,组合物的成分单独地或在单位剂型中混合在一起提供,例如,作为标明活性剂的量的气密容器(如安瓿或小囊)中的干燥的冻干粉末或无水浓缩物。在施用模式是输注的情况中,组合物可以用包含无菌药用级水或盐水的输液瓶分配。在施用模式是通过注射的情况中,可以提供无菌注射用水或盐水的安瓿,以使得成分可以在施用前混合。特别地,本发明也提供其中一种或多种预防剂或治疗剂或本发明的药物组合物包装在标明药剂的量的气密容器如安瓿或小囊中。在一个实施方式中,一种或多种预防剂或治疗剂或本发明的药物组合物作为气密容器中干燥的冻干粉末或无水浓缩物供应且可以(例如,用水或盐水)重构至适宜的浓度用于施用于受试者。优选地,一种或多种预防剂或治疗剂或本发明的药物组合物以至少5mg、至少10mg、至少15mg、至少25mg、至少35mg、至少45mg、至少50mg、至少75mg或至少100mg的单位剂量作为气密容器中干燥的冻干粉末供应。冻干的预防剂或治疗剂或本发明的药物组合物应当在2℃-8℃下储存在其原始容器中且预防剂或治疗剂或本发明的药物组合物应当在重构后1周内、5天内、72小时内、48小时内、24小时内、12小时内、6小时内、5小时内、3小时内或1小时内施用。在替代的实施方式中,一种或多种预防剂或治疗剂或本发明的药物组合物以液体形式在标明药剂的量和浓度的气密容器中供应。优选地,液体形式的所施用组合物在气密容器中以至少0.25mg/ml、至少0.5mg/ml、至少1mg/ml、至少2.5mg/ml、至少5mg/ml、至少8mg/ml、至少10mg/ml、至少15mg/kg、至少25mg/ml、至少50mg/ml、至少75mg/ml或至少100mg/ml供应。液体形式应当在2℃-8℃下储存在其原始容器中。本发明的抗体和抗体部分可以并入适合于肠胃外施用的药物组合物中。优选地,抗体或抗体部分制备为包含0.1-250mg/ml抗体的注射溶液。注射溶液可以由燧石或琥珀瓶、安瓿或预装注射器中的液体或冻干剂型构成。缓冲剂可以是ph5.0-7.0(最佳ph6.0)的l-组氨酸(1-50mm),最佳5-10mm。其它合适的缓冲剂包括,但不限于琥珀酸钠、柠檬酸钠、磷酸钠或磷酸钾。氯化钠可以用于以0-300mm的浓度(对于液体剂型最佳150mm)改变溶液的毒性。对于冻干剂型可以包括冻干保护剂,主要0-10%蔗糖(最佳0.5-1.0%)。其它合适的冻干保护剂包括海藻糖和乳糖。对于冻干剂型可以包括增容剂,主要1-10%甘露醇(最佳2-4%)。在液体和冻干剂型中可以使用稳定剂,主要1-50mml-甲硫氨酸(最佳5-10mm)。其它合适的增容剂包括甘氨酸、精氨酸,表面活性剂可以包括为0-0.05%聚山梨醇酯-80(最佳0.005-0.01%)。另外的表面活性剂包括,但不限于聚山梨醇酯20和brij表面活性剂。制备为用于肠胃外施用的注射溶液的包含本发明的抗体和抗体部分的药物组合物可以进一步包含可用作辅剂的试剂,如用于提高治疗蛋白质(例如,抗体)的吸收或扩散的那些。特别有用的辅剂是透明质酸酶如(重组人透明质酸酶)。在注射溶液中添加透明质酸酶改善肠胃外施用(特别是皮下施用)后的人体生物利用度。还允许以较低的疼痛和不适及最小的注射部位反应发生率获得更大的注射部位体积(即,大于1ml)(参见wo2004078140,us2006104968,通过引用并入本文)。本发明的组合物可以为多种形式。这些包括,例如,液体、半固体和固体剂型,如液体溶液(例如,注射和输注溶液)、分散体或悬浮液、片剂、丸剂、粉末剂、脂质体和栓剂。优选的形式取决于预期的施用模式和治疗应用。典型的优选组合物为注射或输注溶液的形式,如类似于用于以其它抗体对人进行被动免疫的那些的组合物。优选的施用模式是肠胃外施用(例如,静脉内、皮下、腹膜内、肌肉内)。在优选的实施方式中,抗体通过静脉内输注或注射施用。在另一优选实施方式中,抗体通过肌肉内或皮下注射施用。治疗组合物通常在制造和储存条件下必须是无菌的和稳定的。组合物可以配制为溶液、微乳液、分散体、脂质体或适合于高药物浓度的其它有序结构。无菌注射溶液可以通过将活性化合物(即,抗体或抗体部分)以需要的量合并到具有以上列举的成分之一或组合的适宜溶剂中,且随后在需要时过滤除菌来制备。一般地,分散体通过将活性化合物合并到含有基础分散介质和来自以上列举的那些的所需的其它成分的无菌媒介中制备。在用于制备无菌注射溶液的无菌的冻干粉末的情况中,优选的制备方法是从预先无菌过滤的活性成分加任何另外的所需成分的溶液产生其粉末的真空干燥和喷雾干燥。溶液的适当流动性,例如,通过使用如卵磷脂的涂层、在分散体的情况下通过维持所需的颗粒大小和通过使用表面活性剂来保持。注射组合物的延长吸收可以通过在组合物中包括延迟吸收的试剂(例如,单硬脂酯盐和明胶)来产生。本发明的抗体和抗体部分或adc可以通过本领域中已知的多种方法施用,虽然对于许多治疗应用,优选的施用途径/模式是皮下注射、静脉内注射或输注。如本领域技术人员应理解的,施用途径和/或模式根据所需的结果变化。在某些实施方式中,活性化合物可以用保护化合物免于快速释放的载体制备,如控释制剂,包括植入物、透皮贴片和微包封递送系统。可以使用可生物降解的、生物相容的聚合物,如乙烯乙酸乙烯酯、聚酸酐、聚乙醇酸、胶原蛋白、聚原酸酯和聚乳酸。用于制备这样的制剂的许多方法是获得专利的或总体上本领域技术人员已知的。参见,例如,sustainedandcontrolledreleasedrugdeliverysystems,j.r.robinson,ed.,marceldekker,inc.,newyork,1978。在某些实施方式中,本发明的抗体或抗体部分或adc可以,例如,与惰性稀释剂或可吸收的食用载体口服施用。化合物(及其它成分,如果需要)也可以包封在硬壳或软壳明胶胶囊中、压制成片剂或直接加入受试者的饮食中。对于口服治疗施用,化合物可以与赋形剂结合并以可摄食片剂、口含片、糖锭、胶囊、酏剂、悬浮液、糖浆、干胶片等等的形式使用。为通过肠胃外施用以外的方式施用本发明的化合物,可能有必要用防止其灭活的材料包装化合物或者与防止其灭活的材料共同施用该化合物。在其它实施方式中,本发明的抗体或抗体部分或adc可以与基于聚合物的物质偶联以使得所述基于聚合物的物质可以赋予本发明的所述抗体或抗体部分足够的大小,从而本发明的所述抗体或抗体部分受益于增强的渗透性和保留效应(epr效应)(也参见pct公开no.wo2006/042146a2及u.s.公开no.2004/0028687a1、2009/0285757a1和2011/0217363a1,以及美国专利no.7,695,719(其各自通过引用并出于所有目的全文并入本文))。补充的活性化合物也可以并入组合物中。在某些实施方式中,本发明的抗体或抗体部分或adc与一种或多种可用于治疗其中egfr活性有害的障碍的另外的治疗剂一起配制和/或共同施用。例如,本发明的抗-hegfr抗体或抗体部分或adc可以与一种或多种结合其它靶标的另外的抗体(例如,结合细胞因子的抗体或结合细胞表面分子的抗体)一起配制和/或共同施用。此外,一个或多个本发明的抗体可以与两种或更多种前述治疗剂组合使用。这样的组合疗法可以有利地利用较低剂量的施用治疗剂,因此避免与各种单一疗法相关的可能的毒性或并发症。在某些实施方式中,egfr或其片段的抗体或adc连接于本领域中已知的半衰期延长媒介。这样的媒介包括,但不限于fc域、聚乙二醇和葡聚糖。这样的媒介描述于,例如,u.s.申请序列no.09/428,082和公开的pct申请no.wo99/25044,其通过引用为任何目的在此并入。本领域技术人员很容易明白,本文所述的本发明方法的其它合适的改进和适应是显而易见的且可以使用合适等同物进行而不脱离本发明或本文公开的实施方式的范围。尽管现在已经详细地描述了本发明,本发明通过参照以下实施例会更清楚地理解,该实施例仅用于说明的目的而不旨在限制。仅出于举例的目的,本发明包括但不限于如下的技术方案:技术方案1.一种抗-人表皮生长因子受体(抗-hegfr)抗体或其抗原结合部分,其a)结合氨基酸序列cgadsyemeedgvrkc(seqidno:45)内的表位或在竞争结合分析中与第二抗-hegfr抗体竞争结合表皮生长因子受体变体iii(egfrviii)(seqidno:33),其中所述第二抗-egfr抗体包含含有seqidno:1中所示的氨基酸序列的重链可变域和含有seqidno:5中所示的氨基酸序列的轻链可变域;b)如通过表面等离子体共振测定的,以约1x10-6m或更低的解离常数(kd)结合egfr(1-525)(seqidno:47);和c)在体内人非小细胞肺癌(nsclc)异种移植分析中相对于非egfr特异性的人igg抗体以至少约50%的肿瘤生长抑制%(tgi%)抑制肿瘤生长,其中所述人igg抗体在所述nsclc异种移植分析中以与所述抗-hegfr抗体或其抗原结合部分相同的剂量和频率施用。技术方案2.技术方案1所述的抗体或其抗原结合部分,其以约1x10-6m和约1x10-10m之间的kd结合egfr(1-525)(seqidno:47),如通过表面等离子体共振测定的。技术方案3.技术方案1所述的抗体或其抗原结合部分,其以约1x10-6m和约1x10-7m之间的kd结合egfr(1-525)(seqidno:47),如通过表面等离子体共振测定的。技术方案4.技术方案1-3任一项所述的抗体或其抗原结合部分,其以约8.2x10-9m或更低的kd结合egfrviii(seqidno:33),如通过表面等离子体共振测定的。技术方案5.技术方案1-3任一项所述的抗体或其抗原结合部分,其以约8.2x10-9m和约6.3x10-10m之间的kd结合egfrviii(seqidno:33),如通过表面等离子体共振测定的。技术方案6.技术方案1-3任一项所述的抗体或其抗原结合部分,其以约8.2x10-9m和约2.0x10-9m之间的kd结合egfrviii(seqidno:33),如通过表面等离子体共振测定的。技术方案7.技术方案1-6任一项所述的抗体或其抗原结合部分,其在体内人非小细胞肺癌(nsclc)异种移植分析中相对于非egfr特异性的人igg抗体抑制肿瘤生长至少约60%。技术方案8.技术方案1-6任一项所述的抗体或其抗原结合部分,其在体内人非小细胞肺癌(nsclc)异种移植分析中相对于非egfr特异性的人igg抗体抑制肿瘤生长至少约70%。技术方案9.技术方案1-6任一项所述的抗体或其抗原结合部分,其在体内人非小细胞肺癌(nsclc)异种移植分析中相对于非egfr特异性的人igg抗体抑制肿瘤生长至少约80%。技术方案10.技术方案1-9任一项所述的抗体或其抗原结合部分,其中所述抗体或其抗原结合部分包含含有seqidno:12中所示的氨基酸序列的重链cdr3域、含有seqidno:11中所示的氨基酸序列的重链cdr2域和含有seqidno:10中所示的氨基酸序列的重链cdr1域;及含有seqidno:8中所示的氨基酸序列的轻链cdr3域、含有seqidno:7中所示的氨基酸序列的轻链cdr2域和含有seqidno:6中所示的氨基酸序列的轻链cdr1域。技术方案11.技术方案1-9任一项所述的抗体或其抗原结合部分,其中所述抗体或其抗原结合部分包含含有seqidno:9中所示的氨基酸序列的重链可变区和含有seqidno:5中所示的氨基酸序列的轻链可变区。技术方案12.技术方案1-9任一项所述的抗体或其抗原结合部分,包含含有seqidno:15中所示的氨基酸序列的重链和含有seqidno:13中所示的氨基酸序列的轻链。技术方案13.一种抗-hegfr抗体或其抗原结合部分,包含含有seqidno:40中所示的氨基酸序列的轻链cdr3域、含有seqidno:39中所示的氨基酸序列的轻链cdr2域和含有seqidno:38中所示的氨基酸序列的轻链cdr1域;和含有seqidno:37中所示的氨基酸序列的重链cdr3域、含有seqidno:36中所示的氨基酸序列的重链cdr2域和含有seqidno:35中所示的氨基酸序列的重链cdr1域。技术方案14.一种抗-hegfr抗体或其抗原结合部分,包含含有选自50、52、54、56、58、60、62、64、66、68、70、72、74、76和78的氨基酸序列的重链可变区及含有选自51、53、55、57、59、61、63、65、67、69、71、73、75、77和79的氨基酸序列的轻链可变区。技术方案15.一种抗-hegfr抗体或其抗原结合部分,包含选自seqidno:10、11和12,seqidno:16、17和18,seqidno:10、11和19,seqidno:20、11和12,seqidno:21、3和22,seqidno:16、17和19,seqidno:2、3和4,seqidno:10、3和12,seqidno:80、11和18,seqidno:80、3和18,seqidno:20、3和12,seqidno:80、11和12及seqidno:81、11和22的重链cdr组(cdr1、cdr2和cdr3);和选自seqidno:6、7和8,seqidno:23、24和25,seqidno:26、27和28,seqidno:29、30和31,seqidno:6、7和84,seqidno:82、83和31及seqidno:82、27和85的轻链cdr组(cdr1、cdr2和cdr3),其中所述抗体或其抗原结合部分不同时包含seqidno:2、3和4的重链cdr组及seqidno:6、7和8的轻链cdr组。技术方案16.技术方案1-15任一项所述的抗体或其抗原结合部分,包含含有seqidno:41中所示的氨基酸序列的重链恒定区和/或含有seqidno:43中所示的氨基酸序列的轻链恒定区。技术方案17.技术方案1-15任一项所述的抗体或其抗原结合部分,其中所述抗体或其抗原结合部分包含选自人igg恒定域、人igm恒定域、人ige恒定域和人iga恒定域的重链免疫球蛋白恒定域。技术方案18.技术方案17所述的抗体或其抗原结合部分,其中所述igg恒定域选自igg1恒定域、igg2恒定域、igg3恒定域和igg4恒定域。技术方案19.技术方案1-18任一项所述的抗体或其抗原结合部分,其中所述抗体是多特异性抗体。技术方案20.技术方案1-19任一项所述的抗体或其抗原结合部分,其中所述其抗原结合部分选自fab、fab’、f(ab’)2、fv、二硫键连接的fv、scfv、单域抗体和双抗体。技术方案21.技术方案1-20任一项所述的抗体或其抗原结合部分,其中所述抗体或其抗原结合部分与成像剂偶联。技术方案22.技术方案21所述的抗体或其抗原结合部分,其中所述成像剂选自放射标记、酶、荧光标记、发光标记、生物发光标记、磁性标记和生物素。技术方案23.技术方案22所述的抗体或其抗原结合部分,其中所述放射标记是铟。技术方案24.一种药物组合物,其包含技术方案1-23任一项所述的抗体或其抗原结合部分和药学上可接受的载体。技术方案25.一种抗体药物偶联物(adc),其包含与至少一种药物偶联的技术方案1-23任一项所述的抗体或其抗原结合部分。技术方案26.技术方案25所述的adc,其中所述至少一种药物选自抗-细胞凋亡剂、有丝分裂抑制剂、抗-肿瘤抗生素、免疫调节剂、用于基因疗法的核酸、烷化剂、抗-血管生成剂、抗-代谢物、含硼剂、化疗保护剂、激素剂、抗-激素剂、皮质类固醇、光敏治疗剂、寡核苷酸、放射性核素剂、放射致敏剂、拓扑异构酶抑制剂和酪氨酸激酶抑制剂。技术方案27.技术方案26所述的adc,其中所述有丝分裂抑制剂选自多拉司他汀、阿里他汀、类美登素和植物生物碱。技术方案28.技术方案27所述的adc,其中所述阿里他汀是单甲基阿里他汀f(mmaf)或单甲基阿里他汀e(mmae)。技术方案29.技术方案27所述的adc,其中所述类美登素选自dm1、dm2、dm3和dm4。技术方案30.技术方案26所述的adc,其中所述抗-肿瘤抗生素选自放射菌素、蒽环类、卡奇霉素和倍癌霉素。技术方案31.一种包含adc混合物和药学上可接受的载体的药物组合物,该adc混合物包含多种技术方案25-30任一项所述的adc。技术方案32.技术方案31所述的药物组合物,其中所述adc混合物具有2-4的平均药物-抗体比率(dar)。技术方案33.技术方案31所述的药物组合物,其中所述adc混合物包含各自具有2-8的dar的adc。技术方案34.一种用于治疗患有癌症的受试者的方法,包括将技术方案24或31-33任一项所述的药物组合物施用于所述受试者,使得所述患有癌症的受试者得到治疗。技术方案35.技术方案34所述的方法,其中所述癌症选自乳腺癌、肺癌、成胶质细胞瘤、前列腺癌、胰腺癌、结肠癌、头颈癌和肾癌。技术方案36.技术方案34所述的方法,其中所述癌症是鳞状细胞癌。技术方案37.技术方案36所述的方法,其中所述鳞状细胞癌是肺鳞癌或头颈鳞状细胞癌。技术方案38.技术方案34所述的方法,其中所述癌症是三阴性乳腺癌。技术方案39.技术方案34所述的方法,其中所述癌症是非小细胞肺癌。技术方案40.技术方案34-39中任一项所述的方法,其中所述癌症特征为具有egfr过表达。技术方案41.一种用于抑制或降低患有实体肿瘤的受试者中的实体肿瘤生长的方法,所述方法包括对所述患有实体肿瘤的受试者施用技术方案24或31-33中任一项所述的药物组合物,以使得所述实体肿瘤生长被抑制或降低。技术方案42.一种用于抑制或降低患有实体肿瘤的受试者中的实体肿瘤生长的方法,所述方法包括对所述患有实体肿瘤的受试者施用有效量的技术方案1-23或25-30中任一项所述的抗体或adc,以使得所述实体肿瘤生长被抑制或降低。技术方案43.技术方案41或42所述的方法,其中所述实体肿瘤是表达egfr的实体肿瘤或是egfrviii阳性实体肿瘤。技术方案44.技术方案41或42所述的方法,其中所述实体肿瘤是egfr过表达的实体肿瘤。技术方案45.技术方案41-44中任一项所述的方法,其中所述实体肿瘤是非小细胞肺癌或成胶质细胞瘤。技术方案46.技术方案34-45中任一项所述的方法,其中所述抗体、adc或药物组合物与另外的药剂或另外的疗法结合施用。技术方案47.技术方案46所述的方法,其中所述另外的药剂选自抗-pd1抗体、抗-ctla-4抗体、替莫唑胺、bcl-xl抑制剂、依鲁替尼、duvelisib、idelalisib、venetoclax和烟酰胺磷酸核糖基转移酶(nampt)抑制剂。技术方案48.技术方案46所述的方法,其中所述另外的疗法是放疗。技术方案49.技术方案46所述的方法,其中所述另外的药剂是化疗剂。技术方案50.一种分离的核酸,其编码技术方案1-23中任一项所述的抗体或其抗原结合部分。技术方案51.一种载体,包含技术方案50所述的核酸。技术方案52.一种宿主细胞,包含技术方案51所述的载体。技术方案53.技术方案52所述的宿主细胞,其是原核细胞或真核细胞。技术方案54.技术方案53所述的宿主细胞,其中所述真核细胞选自动物细胞、protest细胞、植物细胞和真菌细胞。技术方案55.技术方案54所述的宿主细胞,其中所述动物细胞选自哺乳动物细胞、昆虫细胞和鸟类细胞。技术方案56.技术方案55所述的宿主细胞,其中所述哺乳动物细胞选自cho细胞、cos细胞和sp2/0细胞。技术方案57.一种抗-hegfr抗体药物偶联物(adc),包含与阿里他汀偶联的抗-hegfr抗体,其中所述抗体包含含有seqidno:12的氨基酸序列的重链cdr3域,含有seqidno:11的氨基酸序列的重链cdr2域和含有seqidno:10的氨基酸序列的重链cdr1域,及含有seqidno:8的氨基酸序列的轻链cdr3域,含有seqidno:7的氨基酸序列的轻链cdr2域和含有seqidno:6的氨基酸序列的轻链cdr1域。技术方案58.技术方案57所述的adc,其中所述抗体包含含有seqidno:9所示的氨基酸序列的重链可变区和含有seqidno:5的氨基酸序列的轻链可变区。技术方案59.技术方案57或58所述的adc,其中所述抗体包含igg重链免疫球蛋白恒定域。技术方案60.技术方案59所述的adc,其中所述igg是igg1或igg4重链免疫球蛋白恒定域。技术方案61.技术方案57-60中任一项所述的adc,其中所述阿里他汀是单甲基阿里他汀f(mmaf)或单甲基阿里他汀e(mmae)。技术方案62.技术方案57或58所述的adc,其中所述抗体包含含有seqidno:15的氨基酸序列的重链和含有seqidno:13的氨基酸序列的轻链。技术方案63.技术方案61所述的adc,其中所述抗体通过包含马来酰亚胺基己酰基,缬氨酸-瓜氨酸,p-氨基苯甲醇的接头(mc-vc-paba)与阿里他汀共价连接。技术方案64.技术方案57-63中任一项所述的adc,其中所述adc包含放射标记。技术方案65.技术方案64所述的adc,其中所述放射标记是铟。技术方案66.一种药物组合物,包含技术方案57-65中任一项所述的adc和药学上可接受的载体。技术方案67.一种药物组合物,包含含有技术方案57-65中任一项所述的adc的adc混合物,其中所述adc混合物中平均药物-抗体比率(dar)范围为2-4。技术方案68.一种药物组合物,包含含有抗-hegfr抗体药物偶联物(adc)的adc混合物和药学上可接受的载体,其中所述adc混合物的平均药物-抗体比率(dar)为2-4,且其中所述adc包含与抗-hegfr抗体偶联的单甲基阿里他汀e(mmae),所述抗-hegfr抗体包含含有seqidno:12的氨基酸序列的重链cdr3域,含有seqidno:11的氨基酸序列的重链cdr2域和含有seqidno:10的氨基酸序列的重链cdr1域,及含有seqidno:8的氨基酸序列的轻链cdr3域,含有seqidno:7的氨基酸序列的轻链cdr2域和含有seqidno:6的氨基酸序列的轻链cdr1域。技术方案69.技术方案68所述的药物组合物,其中所述抗体的重链可变区包含seqidno:9所示的氨基酸序列,和所述抗-egfr抗体的轻链可变区包含seqidno:5所示的氨基酸序列。技术方案70.技术方案68或69所述的药物组合物,其中所述抗体包含igg重链免疫球蛋白恒定域。技术方案71.技术方案70所述的药物组合物,其中所述igg是igg1或igg4重链免疫球蛋白恒定域。技术方案72.技术方案68或69所述的药物组合物,其中所述抗体包含含有seqidno:15所示的氨基酸序列的重链和含有seqidno:13的氨基酸序列的轻链。技术方案73.技术方案68-72中任一项所述的药物组合物,其中所述mmae通过包含马来酰亚胺基己酰基、val-cit、paba的接头与所述抗体偶联。技术方案74.一种治疗患有癌症的受试者的方法,包括对所述受试者施用技术方案68-73中任一项所述的药物组合物,使得所述患有癌症的受试者得到治疗。技术方案75.技术方案74所述的方法,其中所述癌症选自乳腺癌、肺癌、成胶质细胞瘤、前列腺癌、胰腺癌、结肠癌、头颈癌和肾癌。技术方案76.技术方案74所述的方法,其中所述癌症是鳞状细胞癌。技术方案77.技术方案76所述的方法,其中所述鳞状细胞癌是肺鳞癌或头颈鳞状细胞癌。技术方案78.技术方案74所述的方法,其中所述癌症是三阴性乳腺癌。技术方案79.技术方案74所述的方法,其中所述癌症是非小细胞肺癌。技术方案80.技术方案74-79中任一项所述的方法,其中所述特征为具有egfr过表达。技术方案81.一种用于抑制或降低患有实体肿瘤的受试者中的实体肿瘤生长的方法,所述方法包括对所述患有实体肿瘤的受试者施用技术方案66-73中任一项所述的药物组合物,使得所述实体肿瘤生长被抑制或降低。技术方案82.技术方案81所述的方法,其中所述实体肿瘤是非小细胞肺癌或成胶质细胞瘤。技术方案83.技术方案81所述的方法,其中所述实体肿瘤是鳞状细胞癌。技术方案84.技术方案81-83中任一项所述的方法,其中所述实体肿瘤是egfrviii阳性实体肿瘤或是表达egfr的实体肿瘤。技术方案85.技术方案81-83中任一项所述的方法,其中所述实体肿瘤过表达egfr。技术方案86.技术方案74-85中任一项所述的方法,其中所述药物组合物与另外的药剂或另外的疗法结合施用。87.技术方案86所述的方法,其中所述另外的药剂选自抗-pd1抗体、抗-ctla-4抗体、替莫唑胺、bcl-xl抑制剂、依鲁替尼、duvelisib、idelalisib、venetoclax和烟酰胺磷酸核糖基转移酶(nampt)抑制剂。技术方案88.技术方案86所述的方法,其中所述另外的疗法是放疗。技术方案89.技术方案86所述的方法,其中所述另外的药剂是化疗剂。实施例实施例1:改进的抗-egfr抗体的鉴别抗体1抗体1(ab1)是人源化抗-egfr抗体。ab1的重链可变区(vh)氨基酸序列以下提供为seqidno:1。ab1的vhcdr氨基酸序列下面加下划线且如下:gysissdfawn(vhcdr1;seqidno:2);yisysgntryqpslks(vhcdr2;seqidno:3),及agrgfpy(vhcdr3;seqidno:4)。ab1vh序列ab1的轻链可变区(vl)氨基酸序列以下提供为seqidno5。ab1的vlcdr氨基酸序列下面加下划线且如下:hssqdinsnig(vlcdr1;seqidno:6);hgtnldd(vlcdr2;seqidno:7)和vqyaqfpwt(vlcdr3;seqidno:8)。ab1vl序列进行筛选以鉴别具有优于ab1的改善性能的抗-egfr抗体。ab1变体鉴别的详细信息提供如下。ab1变体vl和vh文库的制备ab1变体抗体通过筛选三个包含ab1的变体重链或轻链可变区的单链(scfv)文库来鉴别。scfv文库之一包含ab1变体可变重链(即,包含ab1变体可变重链和ab1可变轻链的scfv),而第二scfv文库包含ab1变体可变轻链(即,包含ab1变体可变轻链和ab1可变重链的scfv)。ab1变体vh和vl区也组合成第三scfv文库,其随后经过多轮筛选以选择具有比ab1更高的结合亲和力的vh和vlab1变体的组合。ab1变体vh和vl文库的设计显示于图16中。为产生变体ab1vh区,至少部分地基于ab1的vh区与vh4-4(seqidno:93)和vh4-b种系序列的比对选择十二个氨基酸残基用于突变(参见图16a,其中突变的氨基酸残基指明为“x”)。p101d改变(图16a中指明为“z”)也基于人种系和ab1序列产生。使用如上所述的密码子选择,通过靶向十二个残基(图16a中的“x”残基)计算出,109文库将提供大多数的vhab1变体,每克隆具有四个或更少的突变残基。如图16a中描述的,一个框架残基(q1e)也发生改变以防止n-末端焦谷氨酸形成。为产生变体ab1vl区,至少部分地基于ab1的vl区与igkv1-12(l5)种系序列的比对选择十一个氨基酸残基用于突变(参见图16b中标记为“x”的残基)(igkv1-12描述于seqidno:94中)。33和52位的残基(图16b中分别标记为“1”和“2”)设计为具有有限的多样性。33位的残基限制于l、v、i或f,且52位的残基限制于s、a、t和p。基于前述的酵母文库使用benatuil等人,(2010)proteineng,design,andselection,23(4):155-159中描述的转化方法产生。scfvab1变体文库的筛选包含ab1变体vh或vl区的单链可变片段(scfv)从相应的文库表达,并基于scfv结合截短的野生型(wt)人egfr1-525和突变体egfr(ca)的能力进行筛选。egfr(ca)是包含295和307位的半胱氨酸至丙氨酸突变的egfr变体(参见garrett等人,(2009)pnasusa106(13):5082-5087)。筛选了包含约1x109个克隆的文库。既然ab1结合egfr(ca)但基本上不结合wtegfr,初始轮的筛选使用egfr(ca)来鉴别相对于ab1具有对于egfr(ca)的改善的结合亲和力(kon、koff或两种速率)的ab1变体重链和轻链。但是,较后轮的筛选使用egfr(1-525)来鉴别相对于ab1对于egfr(1-525)具有改善的结合亲和力(kon、koff或两种速率)的ab1变体。文库筛选使用两种方法进行,包括磁珠分选(macs(磁性细胞分离技术))和基于facs的分析(对于macs和facs参见,例如,chao等人,(2006)natureprotocols1:755-765;feldhaus等人,(2003)naturebiotech21:163-170,及vanantwerp(2000)biotechnol.prog.16:31-37)。进行基于磁珠富集的至少两轮筛选,接着至少三轮基于流式细胞分选的筛选(feldhaus和siegel,flowcytometryprotocols,第2版的第17章,ed.hawleyandhawley,vol.263)。使用平衡和koff选择来鉴别相对于ab1具有改善的结合的scfv。总的说来,三个文库针对相对于ab1具有改善的结合的ab1变体vh和vl区进行筛选。使用ab1变体vh和vl文库进行四至五轮筛选,且对于组合ab1变体vh/vl文库进行五至六轮筛选。ab1变体vl文库、ab1变体vh文库及组合ab1变体vh和vl文库的筛选导致鉴别包含相对于ab1具有对于egfr(egfr(1-525)和egfr(ca))的更高结合亲和力的ab1变体vh和vl区的scfv。鉴别改进的抗体鉴别为具有改善的结合特征(包括与egfr(1-525)的特异性结合)的十五个ab1变体scfv选择用于转化到igg蛋白(特别地igg1抗体)中。十五个ab1变体抗体在本文中描述,且包括抗体a(全文中称为“aba”)(参见vhseqidno:9;vlseqidno:5)、抗体b(在本文中称为“abb”)(参见vhseqidno:64;vlseqidno:65)、抗体c(在本文中称为“abc”)(参见vhseqidno:66;vlseqidno:67)和抗体d(在本文中称为“abd”)(参见vhseqidno:68;vlseqidno:69)、抗体e(在本文中称为“abe”)(参见vhseqidno:50;vlseqidno:51)、抗体f(在本文中称为“abf”)(参见vhseqidno:52;vlseqidno:53)、抗体g(在本文中称为“abg”)(参见vhseqidno:72;vlseqidno:73)、抗体h(在本文中称为“abh”)(参见vhseqidno:54;vlseqidno:55)、抗体j(在本文中称为“abj”)(参见vhseqidno:56;vlseqidno:57)、抗体k(在本文中称为“abk”)、抗体l(在本文中称为“abl”)(参见vhseqidno:58;vlseqidno:59)、抗体m(在本文中称为“abm”)(参见vhseqidno:76;vlseqidno:77)、抗体n(在本文中称为“abn”)(参见vhseqidno:60;vlseqidno:61)、抗体o(在本文中称为“abo”)(参见vhseqidno:62;vlseqidno:63)和抗体p(在本文中称为“abp”)(参见vhseqidno:78;vlseqidno:79)。来自vh和vl文库的选择的克隆分别与ab1vl或vh区配对(参见aba、abb、abc、abd、abe和abf)。ab1变体抗体vh区的氨基酸序列在以下提供。cdr加下划线,且相对于ab1的氨基酸变化以黑体突出显示。abavhabbvhabcvhabdvhabevhabfvhabgvhabhvhabjvhabkvhablvhabmvhabnvhabovhabqvhabpvhab1变体抗体vl区的氨基酸序列在以下提供。cdr加下划线,且相对于ab1的氨基酸变化以黑体突出显示。abavlabbvlabcvlabdvlabevlabfvlabgvlabhvlabjvlabkvlablvlabmvlabnvlabovlabqvlabpvl十五个ab1变体vh和/或vl域的核酸序列亚克隆到表达载体是用于表达全长igg抗体(参见美国专利no.8,187,836和美国专利no.8,455,219中公开的载体和方法,这两者通过引用并入本文)。抗体表达载体按照标准方法瞬时转染到hek293细胞中(参见durocher等人,(2002)nucleicacidres.30(2;e9))。用于表达各ab1变体的重链的前导序列的氨基酸序列是mefglswlflvailkgvqc(seqidno:88),而用于表达各ab1变体的轻链的前导序列的氨基酸序列为mrvpaqllgllllwfpgsrc(seqidno:89)。ab1抗体变体随后通过蛋白a色谱从培养基纯化用于亲和力和功能评估。抗体aba鉴别的ab1变体抗体之一是aba。aba具有与ab1相同的可变轻链序列(seqidno:5),包括相同的cdr1、cdr2和cdr3氨基酸序列(分别描述于seqidno:6、7和8中)。aba的vh氨基酸序列以下提供在seqidno:9中。aba的vhcdr氨基酸序列如下:gysisrdfawn(cdr1;seqidno:10);yisyngntryqpslks(cdr2;seqidno:11)和asrgfpy(cdr3;seqidno:12),且在下面加下划线。aba的重链可变区中相对于ab1不同的残基以下以黑体显示。abavh氨基酸序列图1和2提供ab1和aba的vh和vl区(图1)及完整重链和轻链(图2)的氨基酸序列的比对。ab1和aba的轻链氨基酸序列相同(seqidno:13)。但是,ab1和aba的重链氨基酸序列在两个序列之间具有六个氨基酸差异,其中三个是在cdr中。ab1vh氨基酸序列和abavh氨基酸序列之间的差异在图1中加轮廓并在各vhcdr中存在。aba的可变重链的cdr1域包括丝氨酸(ab1)至精氨酸的氨基酸变化。可变重链的cdr2域包括从ab1中的丝氨酸至aba中的天冬酰胺的氨基酸变化。最后,可变重链的cdr3域包括从ab1中的甘氨酸至aba中的丝氨酸的氨基酸变化。aba内氨基酸变化中的两个是在重链的恒定区中(d354e和l356m)。aba中的fc区氨基酸突变代表从z,a异型至z,非-a异型的人igg异型变化。除了其它变化外,第一氨基酸从谷氨酰胺(q)变成谷氨酸(e),如例如在图1中所述的。ab1变体抗体序列与ab1序列的比较表1提供ab1变体抗体aba、abg、abk、abm和abp与ab1相比的重链和轻链cdr氨基酸序列的比对。表2提供ab1、aba、abg、abk、abm和abp的抗-egfr抗体cdr共有序列的比较。表1和3中的空白区表示该残基与ab1相同。表2:来自表1的ab1变体的cdr共有序列如表2中所述,ab1变体抗体aba、abg、abk、abm、abp各自在cdr3的可变重链中具有替代甘氨酸的丝氨酸残基(表2中以黑体/加下划线显示)。ab1的vh和vlcdr序列相对于抗体abb、abc、abd、abe、abf、abh、abj、abl、abn、abo和abq的比较描述于表3中。除了表3中描述的cdr改变外,abg具有vh框架2区内的氨基酸残基改变。ab1变体抗体的表征描述于以下实施例2-8中。实施例2:抗-egfr抗体的结合分析biacore分析进行biacore分析以比较ab1、ab2(具有与西妥昔单抗相同的六个cdr氨基酸序列的抗体)和实施例1中鉴别的抗-egfr抗体对三种形式的重组egfr的亲和力,具体地野生型egfr细胞外域(ecd)(egfr1-645)(seqidno:34)、egfrviii(egfr(1-29)-g-(298-645)(seqidno:46))和截短的野生型egfr1-525(egfr1(1-525)(seqidno:47))。ab2包括分别如seqidno:48和49中提供的ab2的重链和轻链氨基酸序列,且按照标准方法制备。尽管ab1和ab2都是抗-egfr抗体,它们具有明显不同的性质且结合独特的表位。ab2结合egfr的l2结构域(gan等人,(2012)cancerres72(12)1-7;li等人,(2005)cancercell7:301),而ab1结合egfr的cr1域(结构域ii)的氨基酸残基287-302(gan等人,(2012)72(12)1-7;johns等人,(2004)jbiolchem279:30375-30384)。egfr的结构域ii在egfr的伸展构象中暴露(li等人,(2005)cancercell7:301)。图17提供egfr的总体结构域组织的示意图并一般地指明ab2和ab1的表位位于何处。与ab2不同,ab1不结合egfrecd(seqidno:34)(或与其具有非常弱的结合)。虽然ab1可以结合激活的野生型egfr,但在体内对于egfrviii具有相对于ab2较高的结合亲和力。因此,ab2作为结合与ab1不同的表位且具有不同结合亲和力特征的第二抗-egfr抗体在本文所述的实验中用作对照。biacore分析使用具有cm5传感芯片的biacoret100进行,且测试抗体通过抗-人fc抗体(其使用标准胺偶联试剂盒按照制造商的说明(gehealthcare)氨基偶联于芯片)捕获。简言之,cm5芯片表面用edc/nhs激活。山羊抗-人fc特异性多克隆抗体(thermofisherscientificinc.,rockford,il)在10mm乙酸钠(ph4.5)中稀释至25μg/ml并在活化的表面上注射以实现固定。生物传感表面上的未反应部分用乙醇胺封闭。biacoret100是用于检测、表征和定量生物分子相互作用(包括抗体与其抗原之间的相互作用)的基于表面等离子体共振的生物传感器。抗原以80μl/分钟注射3分钟且随后解离15分钟。测试的egfrecd包括与myc和组氨酸标签融合的egfr的氨基酸1-645(egfr(1-645)-lesrgpf-myc-nmhtg-6his(“lesrgpf”(seqidno:90);“6his”(seqidno:91))。egfrviii变体也与myc和组氨酸标签融合(egfr(1-29)-g-(298-645)-lesrgpf-myc-nmhtg-6his),ecdegfr1-525(egfr1(1-525)-srgpf-myc-nmhtg-6his(“srgpf”(seqidno:92))也是如此。biacore分析中使用的运行缓冲液是hbs-ep+:10mmhepes,ph7.5,150mmnacl,3mmedta,0.05%tween20。来自biacore结合分析的结果显示于图3中。测试的ab1变体抗体中,abk对于截短的egfr(1-525)具有最高的结合亲和力,kd为1.7x10-9m,这比ab1的kd提高超过1300倍。如图3中所述,abe对于截短的egfr(1-525)显示最低的可测定亲和力,kd为5.9x10-7m,相对于ab1的kd仅提高约4倍。aba对于截短的egfr(1-525)显示10倍的kd提高(2.2x10-7m的kd(aba)vs2.3x10-6m的kd(ab1))。abb、abc、abd、abf、abg、abh、abj、abl、abm、abn、abo和abp相对于ab1的kd比率对于截短的egfr1-525也描述于图3中。也测试了ab1变体抗体、ab1和ab2对egfrviii的结合亲和力。如图3中所示,所有ab1变体与ab1相比显示出对于egfrviii的更高结合亲和力。例如,aba以2.3x10-9m的kd与egfrviii解离,而ab1具有9.4x10-9m的kd的解离常数。因此,aba相对于ab1具有四倍的亲和力提高(通过解离常数测定)。相对于ab1显示出对于截短的egfr(1-525)的更高解离常数的ab1变体抗体不必然显示对于egfrviii的亲和力的相同提高。例如,尽管abo相对于ab1显示出对于egfrviii的亲和力的最高倍数提高(63倍),但abk相对于ab1显示出对于截短的egfr(1-525)的最高倍数提高(超过1300倍的提高),如图3中所述。在另一实例中,abg在ab1变体抗体中显示出与egfrviii结合的第二高的倍数提高(具有超过47倍的提高),但相对于ab1在对于截短的egfr(1-525)的亲和力提高中列第六位(具有263倍的提高),如图3中所述。许多ab1变体在biacore测试中确定为对于egfr1-525和egfrviii两者具有比ab2低的亲和力,ab2对于截短的egfr(1-525)具有,例如,4.0x10-9m的kd。biacore分析揭示,ab1和ab1变体抗体均不结合egfr的全长ecd(egfr氨基酸1-645)。相反,ab2结合egfr的ecd。因此,尽管观察到ab1变体与野生型egfrecd的结合不存在或可忽略,但如也对于ab1所观察到的,对于截短的受体egfr(1-525)提高的结合亲和力随ab1变体提高。facs(荧光激活细胞分选)分析进行facs分析以测定aba与ab1和ab2相比对于肿瘤细胞(a431细胞)的结合特性。使用facs分析,测定荧光强度和细胞计数,其中与细胞结合的抗体的量反映在通过使用流式细胞分析软件的分析获得的荧光强度(即,几何平均数的值)中。具体地,由结合抗体的量代表的抗体结合活性通过测定几何平均数的值来评估。a431细胞使用细胞解离缓冲剂从大约80%汇合的烧瓶收获。收获的a431细胞在pbs/1%fbs(胎牛血清)(facs缓冲液)中洗涤一次,然后在facs缓冲液中以2.5x106细胞/ml重悬。100μl细胞/孔添加到圆底96-孔板。使用10μl的10x浓度抗体(最终浓度显示在图4中)。孔用facs缓冲液洗涤两次并重悬在facs缓冲液中稀释的50μl的次级抗体(alexafluor488)中。板在4℃下孵育一小时并用facs缓冲液洗涤两次。细胞重悬在100μl的pbs/1%甲醛中并在bectondickinsonlsrii流式细胞仪上进行分析。数据使用winlist流式细胞分析软件进行分析。来自ab1、ab2和aba与a431肿瘤细胞结合的facs分析的结果提供在图4中,其显示相对于与细胞一起孵育的抗体的浓度的几何平均数。如图4中所述,aba与ab1相比对于a431肿瘤细胞上的egfr具有更高的结合,但与ab2相比aba具有较低的结合亲和力。图4中的数据代表各抗体与其抗原的直接结合。如图3和4中所述,aba对于egfr具有提高的结合亲和力并相对于ab1显示对于具有高水平egfr的细胞(a431肿瘤细胞)提高的结合。也确定,相对于与a431细胞的结合,aba表现出与具有较低水平egfr的细胞的结合的更温和的提高(数据未显示)。实施例3:抗-egfr抗体的表位分析进行测试以确定实施例1中鉴别的抗-egfr抗体是否具有与抗体ab1相同的表位或者ab1变体抗体aba、abg、abk、abm和abp内的氨基酸变化是否影响表位结合。竞争试验分析竞争结合facs分析用于测定改善的抗-egfr抗体相比于ab1、ab2和对照1抗体(用作阴性对照的抗-cd20抗体(利妥昔单抗(roche))的表位特异性。使用表达egfrviii的u87mg细胞(人成胶质细胞瘤细胞系(获自a.scott,ludwiginstituteforcancerresearch)(u87mg细胞可作为atcchtb-14tm得自attc;也参见,例如,u-87mgcelllinehuman,sigma-aldrich)。u87mg细胞使用细胞解离缓冲剂从大约80%汇合的烧瓶收获。细胞在pbs/1%fbs(胎牛血清)(facs缓冲液)中洗涤一次,然后以2.5x106细胞/ml重悬在facs缓冲液中。100μl细胞/孔添加到圆底96-孔板。对于竞争facs,ab1通过添加最终浓度100nm的异硫氰酸荧光素(fitc)(具有或不具有竞争抗体)进行荧光偶联,且然后孔在facs缓冲液中洗涤两次,重悬在100μl的pbs/1%甲醛中,并在bectondickinsonlsrii流式细胞仪上进行分析。荧光在488nm下测量。来自竞争试验的结果描述于图5中并表明测试的ab1变体抗体(即,aba、abg、abk、abm和abp)识别与ab1相同的表位(egfr的结构域ii,其在“伸展的”egfr构象中暴露),假定荧光的几何平均计算随未标记ab1变体抗体浓度的提高而降低。结果也显示ab1/aba/abg/abk/abm/abp表位不同于ab2表位,因为没有观察到ab2和ab1之间或ab2和ab1变体抗体之间的竞争。对照1抗体在竞争试验中也未显示与ab1表位的可检测的结合,因为对照1抗体未能与fitc标记的ab1竞争结合细胞。成像分析为评估egfr表达肿瘤的抗体摄取的效率,使用111in标记的aba进行spect成像分析(参见,例如,khalil等人,internationaljournalofmolecularimaging,vol.2011,articleid796025,p.1-15)。两种肿瘤模型,sw48细胞(结肠肿瘤细胞;atccno.ccl-231tm)和ebc-1细胞(ebc-1细胞来自于人肺鳞癌细胞系,其是来自japaneseresearchresourcesbank(idno.rcb1965)(tokyo,japan))基于免疫组织化学分析由于其适度的egfr表达水平而选择。在注射肿瘤细胞到小鼠中和随后施用标记的aba抗体、标记的ab1或标记的阴性对照(非egfr结合igg)后,在注射后4、12、24、48、72、120和168小时使用nanospect/ct获取spect/ct图像。所有标记的抗体通过尾静脉注射给药。考虑到抗体的血液清除率的差异,数据报告为肿瘤与血液的比率。图20a和20b两者中提供的结果证明,与ab1和igg对照相比,更多的aba摄取到小鼠中的sw48(图20a)和ebc-1(图20b)细胞中。在ebc1模型中(图20b)(其是具有较低egfr表达水平的模型)中,ab1摄取类似于igg对照而aba摄取高于ab1或阴性对照。图20中提供的结果证明aba能够靶向特定的egfr-表达肿瘤。对于aba的类似成像结果也使用a431肿瘤细胞(源自已知具有高egfr表达水平的表皮样癌实体肿瘤(atccno.crl-1555))观察到。实施例4:肿瘤细胞系中抗-egfr抗体活性的体外分析scc-15和h292肿瘤细胞系中egfr信号传导的体外分析ab1、ab2、aba、abb、abc、abd、abg、abk、abm和abp在体外抑制肿瘤细胞系中egf-介导的egfr酪氨酸磷酸化的能力使用鳞状细胞癌细胞(scc)-15(crl-1623tm)和h292细胞(肺癌细胞系;crl-1848tm)通过western印迹分析进行评估。scc-15和h292细胞两者表达野生型egfr。表达野生型egfr的肿瘤细胞中通过抗体处理诱导的pegfr的下调表明那些抗体至少部分地通过抑制经由该受体的信号传导发挥功能。scc-15细胞(人舌鳞状细胞癌)是转化的角质细胞且在体内对于ab1是敏感的。尽管scc-15细胞在体内对于ab1是敏感的,h292(人非小细胞肺癌(nsclc))细胞在体外或体内对于egf-介导的egfr酪氨酸磷酸化的ab1抑制是抗性的。因此,两种细胞系用于测试aba、abb、abc、abd、abg、abk、abm和abp在体外抑制肿瘤细胞中的egfr信号传导的能力。细胞以每孔60,000-80,000接种在24-孔板中,或以每孔100,000-200,000接种在6-孔组织培养板中。细胞在生长培养基中孵育过夜。在适当时37℃下血清饥饿24小时后,细胞在37℃下与抗体孵育一小时,且然后在37℃下用重组人egf(r&dsystems)刺激10分钟。细胞然后用冰冷的pbs洗涤两次,且用补充completemini蛋白酶抑制剂混合物(roche)和0.1%np40(tergitol-np-40型;壬基苯氧基聚乙氧基乙醇)的100-200μl/孔的细胞裂解缓冲液(cellsignalingtechnolgy)进行裂解。在-80℃下速冻至少20分钟后,细胞裂解物通过在4℃下以14,000rpm离心10分钟进行澄清。澄清样品裂解物的蛋白质浓度通过bcaproteinassay(pierce_thermoscientific)测定。细胞裂解物(10μg)使用4-12%bis-tris,midi凝胶(lifetechnologies)通过sds-page解析并使用iblot干转移系统(lifetechnologies)转移到硝基纤维素膜上。印迹用5%乳/tween-tris缓冲盐水(ttbs)在室温下封闭一小时,用ttbs洗涤三次,且然后在4℃下与适宜的一级抗体(抗-磷酸酪氨酸(4g10)生物素偶联物,millipore,1∶1000稀释以检测磷酸化egfr;兔抗-egfr,lifespanbiosciences,1∶2000稀释以检测总egfr;兔抗-pan肌动蛋白,cellsignalingtechnology,1∶1000稀释以检测内部对照肌动蛋白)孵育过夜。在与一级抗体孵育过夜后,印迹用ttbs洗涤三次,持续五分钟,且然后在室温下与驴抗-兔抗体(jacksonlaboratories,1:2000dilution)孵育一小时以检测总egfr和肌动蛋白,或者与链霉亲和素-hrp偶联物(kpl,1∶1000稀释)孵育以检测磷酸化egfr。印迹然后用ttbs洗涤三次,且用westdurachemiluminescent底物(thermoscientific)处理。印迹通过使用las-4000扫描仪(fuji)进行扫描而可视化。ab2用作阳性对照(即,egfr磷酸化的抑制剂)且对照1抗体用作阴性对照(即,不结合egfr且因此对磷酸化egfr没有影响)。来自使用scc-15细胞的体外研究的western印迹分析结果提供在图7a中并表明ab1及抗体aba、abb、abc和abd不能够显著抑制scc-15细胞中的egfr信号传导,如通过没有显著的磷酸化egfr的下调所表明的。相反,如图7a中显示的磷酸化egfr的降低表明抗体abg、abk、abp和abm在体外降低scc-15细胞中的egfr活性,已知磷酸化egfr的水平相对于阴性对照和ab1较低且与阳性对照(ab2)水平相当。因此,多种ab1变体相对于ab1获得体外活性的提高。来自使用h292细胞的体外研究的western印迹分析结果提供在图7b中。图7b中的结果表明ab1及抗体aba、abb、abc和abd在h292细胞中体外显示对egfr信号传导的很少抑制,与ab2及抗体abg、abk、abm和abp相比没有显著的磷酸化egfr的降低。抗体abg、abk、abm和abp更有效地抑制h292细胞中egfr的酪氨酸磷酸化(参见图7b),优于ab1。ab2(阳性对照)也抑制egfr磷酸化,如图7b中所述,而阴性对照(对照1抗体)未显示可检测的磷酸化抑制,给出与ab2相当的结果。如下所述,尽管aba发现在野生型egfr阳性细胞(h292)中在体外抑制egfr磷酸化方面相对不活跃,但aba能够在体内抑制这些细胞的磷酸化,与在相同条件下测试的抗体ab1相比。因此,图7a和7b中描述的体外结果表明,尽管aba、abb、abc、abd、abg、abk、abm和abp与egfr的结合提高(相对于ab1),仅一些ab1变体抗体(即,abg、abk、abm和abp)能够在测试的肿瘤细胞系中在体外显著地抑制或降低egf-介导的信号传导。一般地,测试的ab1变体抗体在scc-15和h292细胞中体外抑制egf-介导的egfr酪氨酸磷酸化的能力与对于egfrviii和截短的egfr(1-525)两者的亲和力提高(如通过表面等离子体共振测定的)相关。图6提供了ab1、ab2和、ab1变体抗体与egfr(1-525)的结合亲和力的总结。图6中两个圆圈轮廓反映了相对于抗体在h292细胞或scc-15细胞中抑制egfr磷酸化(表明egfr活性的抑制)的能力的如上所述的体外结果(或来自相似测试的结果)。如图6中所描述的,ab1变体抗体可以分类为两个组(组1和2):在测试的肿瘤细胞系中基于图7中提供的结果不体外抑制egfr信号传导的那些变体抗体(包括ab1、aba、abb、abd、abe和abf;图6中指示为组1),及如图7中所述在测试的肿瘤细胞系中确实体外抑制egfr信号传导的那些变体抗体(包括abg、abh、abl、abk、abj、abm、abn、abo和abp;图6中指示为组2)。图6中提供的比较表明所有ab1变体抗体与ab1相比具有对于egfr(1-525)的更高亲和力,及具有低于1x10-4s-1的kd的那些变体抗体(组2)能够在体外显著地抑制egfr信号传导,如图7中呈现的结果所描述的。如图6中所述,ab1的成熟过程主要导致具有增强的解离速率的ab1变体。a431肿瘤细胞系的体外分析ab1和ab1变体抗体抑制egf-介导的egfr磷酸化的能力也使用a431人上皮瘤细胞通过磷酸-egfrelisa分析进行测试。a431细胞表达野生型egfr。细胞以每孔20,000在生长培养基中接种于胶原蛋白涂覆的96-孔盘中。24小时后,细胞在无血清培养基中洗涤并血清饥饿4小时。在适宜时,细胞用单克隆抗体预处理一小时,且随后在37℃下用重组egf刺激10分钟。在egf刺激后,细胞用冰冷的pbs洗涤两次,且用补充蛋白酶抑制剂的100μl/孔的细胞裂解缓冲液裂解和在-80℃下速冻至少20分钟。捕获板通过用50μl的抗-egfr抗体(r&dsystems,部件号841402,0.8μg/ml)预涂覆孔,随后用pbs/1%bsa处理1小时进行封闭,且在tween-tris缓冲盐水(ttbs)中洗涤三次而产生。细胞裂解物添加到捕获板并在4℃下孵育过夜。板在ttbs中洗涤五次,且与ptry-马辣根过氧化物酶(r&dsystems,dyc1095)孵育1小时。板在ttbs中洗涤五次,并向各孔添加100μl的3,3,5,5-四甲基联苯胺(tmb)和在室温下孵育直到显色。反应通过添加1nhcl终止,且od在450nm下读数。来自a431抑制研究的结果描述于图8a和8b中且表明ab1变体抗体在a431细胞中显示一定范围的抑制egfr活性的能力。如图8a中所示,ab1及变体aba、abb、abc、abd、abe和abf在a431细胞中不能有效地抑制egfr信号传导(通过egf-介导的egfr磷酸化测定),甚至以1333nm的抗体浓度。与ab1相反,变体抗体abg、abh、abj、abk、abl、abm、abn、abo、abp和abq比ab1更有效地阻断egfr磷酸化,即使以比ab2高的浓度。不结合egfr的对照1抗体对于a431细胞的egfr抑制没有作用。图8b对于抗体ab1、abp和ab2扩展了图8a中提供的数据。如图8b中描述的,ab1与ab2或abp相比显示低的抑制水平,即,平均10%或更低。尽管ab1测定为在抑制a431细胞中的egfr信号传导方面具有大于1333nm的ic50值,但abp具有大于40nm的ic50值,其相对于ab1提高。ab2具有大于2.3nm的ic50值。实施例5:抗-egfr抗体在体外角质细胞结合分析中的facs分析进行角质细胞facs结合分析以测定ab1变体抗体对正常人表皮角质细胞(nhek)细胞的结合亲和力。nhek细胞表达野生型egfr。细胞在大约80%汇合时使用胰蛋白酶收获、中和并在pbs/1%fbs(facs缓冲液)中洗涤一次,然后以2.5x106细胞/ml重悬在facs缓冲液中。每孔100μl细胞添加到圆底96-孔板。添加10μl的10x浓度的ab(最终浓度列举于图中)且板在4℃下孵育一时间。孔用facs缓冲液洗涤两次,然后重悬在facs缓冲液中稀释的50μl的二级ab(alexafluor488)中。板在4℃下孵育一小时,然后用facs缓冲液洗涤两次。细胞然后重悬在100μl的pbs/1%甲醛中并在bectondickinsonlsrii流式细胞仪上进行分析。数据使用winlist流式细胞分析软件进行分析。来自体外角质细胞结合分析的结果描述于图9中,其表明随着标记的aba、abg、abk、abm和abp抗体的浓度提高,测量的荧光由于角质细胞上egfr的结合而增加。ab2用作阳性对照并导致随着抗体添加到nhek细胞增加的荧光。ab1显示低得多的结合水平,甚至在高抗体浓度下。图9中呈现的结果表明测试的ab1变体抗体结合角质细胞上的野生型egfr并具有高于ab1(和阴性对照,对照1抗体)的对于正常人表皮角质细胞的亲和力。图9中的结果也显示抗体aba、abg、abk、abm和abp与ab2相比具有较低的与正常人表皮角质细胞的结合。这些结果表明ab1变体抗体能够比ab1更好地结合角质细胞上的野生型egfr。实施例6:肿瘤上抗-egfr抗体的体内分析ab1变体抗体对体内肿瘤生长的影响使用小鼠异种移植分析进行评估。scid和无胸腺cd-1裸鼠从charlesriver(wilmington,ma)获得。每笼十只小鼠。到达时的小鼠体重为18-20g。所有试验按照实验室动物护理评估和鉴定协会(associationfortheassessmentandaccreditationoflaboratoryanimalcare)认可的美国国立卫生研究院实验动物护理和使用指南(nationalinstitutesofhealthguideforcareanduseoflaboratoryanimals)进行。对于每项皮下研究,活细胞在第0天皮下接种到小鼠侧腹中。注射体积是0.2ml,由s-mem和matrigel(bd,franklinlakes,nj)的1∶1混合物组成。肿瘤以200-250mm3进行大小匹配。治疗开始于肿瘤大小匹配之日或之后24小时。人igg混合物对照用作阴性对照(类似于人血清的纯化的人igg;innovativeresearch)。小鼠在治疗开始时称重为大约25g。肿瘤体积每周评估两到三次。肿瘤的长度(l)和宽度(w)的测量通过电子卡尺进行且体积按照以下公开计算:v=(lxw2)/2。小鼠在肿瘤体积达到3,000mm3时或当发生皮肤溃疡时处死。适当量的抗体原液在施用前在磷酸盐缓冲盐水中稀释。药物如图中所示腹膜内施用。h292(人非小细胞肺癌(nsclc))细胞用于异种移植研究中。来自体内实验的结果显示ab1变体抗体aba、abg、abk、abm和abp相对于ab1能够显著地抑制肿瘤生长(参见图10)。ab1变体抗体与ab1相比也能够增加反应持续时间的持久性,例如,aba保持小于500mm3的肿瘤体积29天,与此相比ab1在肿瘤细胞注射后保持小于500mm3的肿瘤体积约18天(按照相同的剂量和施用时间表)。如图10中所示,在第20天,注射aba的小鼠显示大致300mm3的肿瘤体积,而在第20天注射ab1的小鼠显示具有大致700mm3的体积的肿瘤。至第29天,aba注射小鼠显示具有与第20天的大小(即,大约300mm3)相比相似的体积的肿瘤,而ab1注射小鼠具有增大到约1000mm3的肿瘤体积。计算百分肿瘤生长抑制(%tgi)以量化图10中描述的结果。图10中描述的抗体的%tgi如下:aba=74abg=88abk=90abm=84abp=86ab1=31ab2=73%tgi值是相对于用人igg对照处理的小鼠的肿瘤体积。如上所述,ab1相对于人igg对照导致31%tgi,而aba具有计算的74%的tgi。值得注意的是,ab1变体抗体相对于ab2显示相同或更大的%tgi。aba能够在h292异种移植肿瘤模型中体内提高反应的持久性和减小肿瘤体积(如图10中所述),但不能在相同细胞系中体外抑制egfr的磷酸化,如实施例4和图7中所述的。抗体aba体内活性也与抗体abg、abk、abm和abp相当,尽管对于egfr(1-525)和egfrviii具有较低结合亲和力(参见图3)。因此,尽管存在着aba在体外显示很少细胞信号传导抑制或没有细胞信号传导抑制的事实,aba在体内以类似于具有更强亲和力值的其它抗-egfr变体抗体(例如,abk、abm和abp)的方式降低或抑制h292肿瘤细胞生长。实施例7:使用abaadc的体内肿瘤生长抑制分析aba-vcmmaeadc抑制肿瘤生长的能力使用体内小鼠异种移植分析测定。本实施例中使用的abaadc按照实施例8中描述的方法偶联,但未按照本文所述的批纯化方法纯化。本实施例中使用的abaadc组合物的平均dar为3.7。小鼠异种移植分析(类似于实施例6中描述的那些)使用两种不同的nsclc细胞系nci-h1703和ebc1进行。来自两种不同细胞系的结果提供在图14中。如图14a中所示,aba-vcmmae(以1mg/kg体重的剂量)在减小肿瘤体积和提高nci-h1703细胞中的反应持久性方面优于ab1(以10mg/kg体重的剂量)且与包含ab1和mmaf及mc接头的ab1adc相比。单独的ab1(以10mg/kg的剂量)与不抑制肿瘤生长(以10mg/kg的剂量)的阴性对照,抗体对照2(抗-破伤风毒素抗体),相似地导致肿瘤体积抑制的总体缺乏。aba-vcmmaeadc的效力也使用ebc1细胞在异种移植小鼠模型中研究。来自该研究的结果显示于图14b中并表明aba-mcmmafadc(以3mg/kg体重的剂量)在减小肿瘤体积和提高反应的持久性方面与ab1(以3mg/kg体重的剂量)相比且与包含ab1和mmaf及me接头的ab1adc(以3mg/kg体重的剂量)相比更有效。总之,图14中的结果显示两种不同的aba阿里他汀adc(aba-vcmmae和aba-mcmmaf)相对于单独的ab1或ab1-mmafadc在体内有效地减小肿瘤体和提高反应的持久性。实施例8:纯化的抗-egfr抗体药物偶联物(adc)制备包含通过缬氨酸-瓜氨酸(vc)接头与单甲基阿里他汀e(mmae)连接的aba抗体的抗体药物偶联物(adc)。这种adc(称为aba-vcmmae)的示意图描述于图11中。aba抗体与vcmmae的偶联开始于aba的部分还原,接着与val-cit-mmae(vcmmae)反应。aba抗体(20mg/ml)通过添加tcep(tcep:mab的摩尔当量是2.1)部分还原,接着在0℃下孵育过夜。还原反应然后升温到20℃。为偶联所有硫醇,添加vcmmae至最终vcmmae:还原cys摩尔比为1.15。偶联反应在10%v/v的dmso存在下进行并允许在20℃下进行60分钟。在偶联反应后,添加过量的游离n(乙酰基)-半胱氨酸(2当量vs.vcmmae装载量)以淬灭未反应的vcmmae而产生cys-val-cit-mmae加合物。cys淬灭反应允许在20℃下进行大约30分钟。cys-淬灭的反应混合物按照以下进行纯化。以上偶联方法也可以用于将mcmmaf与抗体偶联。批纯化abaadc使用批纯化方法进行纯化。反应混合物用适宜量的水洗bu-hic树脂(toyopearl;tosohbiosciences)处理,即,七倍重量的树脂加入混合物中。树脂/反应混合物搅拌适当的时间,且通过分析型疏水相互作用色谱监测药物偶联物产物的除去、通过聚丙烯粗过滤器过滤和通过两倍柱床体积的缓冲液(0.28m氯化钠,7mm磷酸钾,ph7)洗涤。组合的滤液和清洗液合并,并通过hichplc分析产物谱。组合的滤液和清洗液用10倍透析体积(diavolumes)的15nm组氨酸缓冲液,ph6通过超滤/渗滤(uf/df)缓冲液交换到15mm组氨酸。在偶联和纯化后,进行所得adc混合物的分析性分析。采集样品并使用疏水相互作用色谱-高效液相色谱(hic-hplc)进行分析。使用的柱是tsk凝胶butyl-npr柱(4.6mmidx3.5cm,2.5μm,30℃;tosohbiosciencellc,japan)并以0.8ml/min的流速使用。流动相包括a:25mmna2hpo4,ph7,1.5m(nh4)2so和b:25mmna2hpo4,ph7(75%)至ipa(25%)。使用的梯度是0%相b2分钟,0-100%b12分钟,和保持1分钟。蛋白质含量使用uv分析进行分析。hic痕量分析表明对于aba-vcmmae的所得平均药物-抗体比率(dar)为3.1,如以下表4和图12中所述。平均dar通过合计0、1、2、3、4、5、6、7和8adc产物,乘以pa%(pa%是通过按照必要药物载量在a280的峰下测量的面积确定的峰面积百分比),和除以100来确定。表4:使用批纯化的aba-vcmmae的hic分析的结果(pa%)如可以从表4和图12中看到的,aba-vcmmae的批纯化产生2-4的dar。初始平均dar为4.1,其中纯化后的最终平均dar为3.1。aba-vcmmaeadc混合物也通过尺寸排阻色谱(sec)进行分析。sechplc使用tosohtsk凝胶gs3000swxl柱(7.58x30cm,5μm)进行。使用0.3ml/min的流速。流动相包括92.5%的25mmna2po4,ph6.8,350mmnacl和7.5%异丙醇(ipa)。稀释剂是流动相,且分析在214nm的uv下进行。secpa%@214nm结果提供在以下表5中。sec结果显示于图13中。表5:aba-vcmmae的sec结果(pa%)高分子量单体低分子量宽3.296.8纯化的2.297.8dar20.499.6dar41.096.22.8mababa95.9214nm下的secpa%结果因此,aba-vcmmae的批纯化导致2-4之间的dar,平均3.1。实施例9:使用aba-mmaeadc的体内肿瘤生长抑制分析如实施例8中所述的,制备纯化的aba-vcmmae组合物以使得组合物内adc的平均dar为3.1。纯化的adc随后进行测试以使用肺癌异种移植模型确定是否纯化的abaadc组合物在体内有效地抑制肿瘤生长。更具体地,进行异种移植肿瘤生长抑制分析以评估纯化的aba-vcmmae(ab1-vcmmaep)对于nci-h292细胞(人nsclc癌细胞系)的作用。图15a中提供的结果证明aba-vcmmaep(以3mg/kg体重的剂量)与未纯化的aba-vcmmae相比更有效地在nci-h292细胞中降低肿瘤体积和延长反应持续时间(提高反应的持久性)。类似地,图15b证明纯化的aba-vcmmaep(以6mg/kg体重的剂量)与aba-vcmmae的未纯化形式相比也更有效地抑制肿瘤生长。如图15a和15b中所描述的,纯化的aba-vcmmae比阴性对照2抗体(单独地或与mmae偶联)、未纯化或纯化形式的与mmae或mmaf偶联的ab1也更有效地降低肿瘤体积和延长反应持续时间。对照2抗体是不结合egfr的抗-破伤风毒素抗体。在评估纯化的aba-vcmmae(ab1-vcmmaep)对nci-h292细胞(肺癌细胞系;crl-1848tm)的作用的进一步研究中,纯化的aba-vcmmae以3mg/kg和6mg/kg的剂量相对于相似剂量的ab1-mcmmaf进行测试。在nsclc肿瘤模型中的这一第二研究的结果提供在图18中,且证明纯化的aba-vcmmaep与aba-vcmmae相比更有效地抑制肿瘤生长。实施例10:aba-vcmmae的流通工艺纯化包含具有降低的每抗体vc-mmae分子药物载量的aba-vcmmaeadc的组合物使用流通方法制备。aba-vcmmaeadc的制备描述于上述实施例7中。用于纯化具有dar范围(1-8)的adc的aba-vcmmae组合物的流通方法按照以下进行。5ml柱的bu-hic树脂首先用大约28mmnacl,7mm磷酸钾,ph7平衡。反应混合物随后使用1.95mnacl,50mm磷酸钾,ph7以1/6体积稀释,且以大约100mg蛋白质/ml树脂的比率以1ml/min的流速(大约5min保留时间或36cm/hr线性流)加载到树脂中。由1份异丙醇和10份(按体积)28mmnacl,7mm磷酸钾,ph7组成的清洗液作为约12倍柱体积的清洗液施用。产物在大约1倍柱体积后开始收集,直到uv信号平衡(其也可以在级分中收集)。级分通过hichplc分析且所需的等分试样汇合在一起并通过tff(切向流过滤)浓缩和用10倍透析体积的组氨酸缓冲液交换到15mm组氨酸,ph6中。以下表6提供纯化之前和之后反应混合物的纯度结果。表6:使用流通纯化的aba-vcmmae的hic分析的结果(pa%)与实施例8中描述的批纯化相比,蛋白质相对于树脂的负载对于各纯化模式(批纯化和流通纯化)是相当的。在批纯化方法中,在效能调节的基础上相对于adc使用2.26倍重量的树脂。树脂的密度为大约0.23g/ml。因此,例如,如果使用10gadc,则使用98.2ml树脂。使用的负载为(10gx1000ml/l)/98.2ml=102g/l负载。流通纯化试验目标为100g/l的负载。在各种情况中,负载溶液和清洗溶液是相同的,除了流通纯化清洗溶液为10%ipav/v。使用实施例8中描述的批纯化方法获得的纯化aba-vcmmae组合物和使用上述的流通方法获得的纯化aba-vcmmae组合物之间的比较提供在表7中。表7:aba-vcmmae批纯化相对于流通纯化的hic分析的结果(pa%)总之,流通和批纯化方法两者成功地获得包含约80%的adc具有2-4的dar的组合物,其中dar为0-1或5-8的adc的量限制于小于组合物中总adc群体的约20%,如例如表7中所示。实施例11:使用abammaeadc的体内肿瘤生长抑制分析aba-vcmmaeadc抑制成胶质细胞瘤肿瘤生长的能力使用体内小鼠异种移植分析测定。小鼠异种移植分析(类似于实施例6中进行的那些)使用u87mgde2-7细胞(其表达egfrviii)进行。u87细胞源自人恶性神经胶质瘤(atccno.htb-14tm)。对于该研究,肿瘤细胞与50%matrigel(bdbiosciences,franklinlakes,nj)混合并在第0天皮下接种3x106个细胞到6-8周龄nu/nu雌性小鼠的侧腹中(nu/nu雌性小鼠从charlesriver(wilmington,ma)获得)。肿瘤长度(l)和宽度(w)的测量通过电子卡尺获取且体积按照以下公式计算:v=(lxw2)/2。小鼠在肿瘤体积达到3,000mm3时或当发生皮肤溃疡时处死。图19中提供的结果证明aba-vcmmaep比阴性对照(对照2抗体;抗破伤风抗体)在抑制成胶质细胞瘤细胞生长方面更有效。序列总结通过引用并入本申请全文中提及的所有参考文献、专利、未决专利申请和公布的专利的内容在此通过引用明确并入。等同本领域技术人员将认识到或能够仅使用常规试验确定本文所述的本发明特定实施方式的许多等同。这样的等同旨在被以下权利要求涵盖。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1