一种基于深度学习的多组学智能诊断系统的制作方法
本发明涉及深度学习和生物医学的技术领域,尤其是指一种基于深度学习的多组学智能诊断系统。
背景技术:
随着新一代基因测序技术的发展,各类组学测序成本急剧下降,大量组学数据得以生成。传统疾病诊断系统中常对单个组学数据进行统计分析,但单个组学无法全方位描述疾病,因而同时考虑多种组学数据是疾病诊断的新趋势。但不同组学间存在异构且互补的信息,因此如何基于多组学数据进行疾病诊断是亟待解决的一个课题。
目前多组学诊断系统主要分为三类:1)基于统计方法的系统:计算cdds值,将cdds值与预存的cdds基准值进行对比,从而进行疾病预测。2)基于传统机器学习方法的系统:利用贝叶斯模型或贝叶斯网络,集成学习、多核学习、多视图矩阵分解等方法对多组学数据建模并做出预测。3)基于深度学习方法的系统:将多组学视为多个模态,各组学分别设计神经网络进行初步处理,随后设计整合网络融合多组学并做出预测。总体而言,现有的多组学疾病诊断系统大都将各组学视为独立部分进行前序处理,后续多组学融合部分也只考虑简单融合。这意味着目前的系统无法充分捕获组学间的异构与互补特性,从而导致疾病诊断准确率低的问题。另一方面,目前的系统大都不具备结果可解释性,无法对模型所作决策提供证据支持。
技术实现要素:
本发明的目的在于克服现有技术的缺点与不足,提出了一种基于深度学习的多组学智能诊断系统,突破传统单组学系统信息源单一的局限性,充分捕获多组学间的异构与互补特性,从而增强疾病的智能诊断能力,同时,为诊断结果提供了一定程度的可解释性说明,增强了诊断结果的可信度。
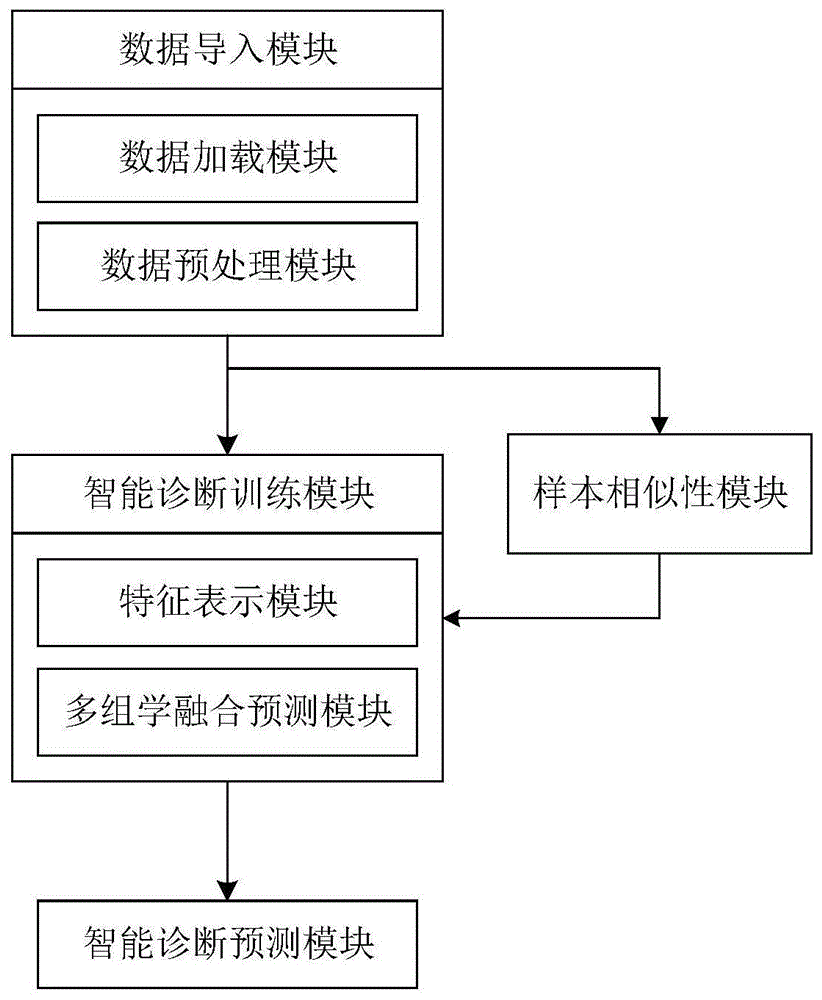
为实现上述目的,本发明所提供的技术方案为:一种基于深度学习的多组学智能诊断系统,包括:
数据导入模块,用于加载多组学数据和临床数据,并对数据进行预处理;
样本相似性模块,用于构建多组学样本相似性矩阵,利用相似性网络融合技术融合样本在不同组学下的相似性;
智能诊断训练模块,利用自动编码器将样本在不同组学下的表示转换为相应的向量形式,利用多视图注意力机制神经网络进行多组学特征融合,并将样本相似性模块结果整合到智能诊断训练过程中,最终得到最优诊断模型;
智能诊断预测模块,用于依据多组学数据进行疾病智能诊断,并提供诊断结果的解释。
所述数据导入模块包括数据加载模块、数据预处理模块,其中:
所述数据加载模块从本地读取多组学和临床数据,包括csv、rdata、xlsx格式,其中,多组学数据包括基因组学、转录组学、蛋白质组学、表观基因组学以及影像组学,临床数据包括生存时间、复发时间;
所述数据预处理模块用于提取多组学共享的样本、针对非影像组学去除空缺特征数超过20%的样本、去除空缺样本数超过20%的特征、均值填补空缺值、去除低均值和低方差特征、去除离群特征、数据归一化,针对影像数据则使用pyradiomics工具包提取特征。
所述样本相似性模块,用于构建多组学样本相似性矩阵,利用相似性网络融合技术融合样本在不同组学下的相似性,包括以下步骤:
1)基于核函数分别构建各组学的单组学样本相似性矩阵:
其中,f(v)为组学v的单组学样本相似性矩阵,s(v)为组学v的样本距离矩阵,ρ(v)(·)为组学v中的样本距离度量方法;
2)基于相似性网络融合方法融合各组学的单组学样本相似性矩阵,构建多组学样本相似性矩阵:
首先,通过k最近邻方法计算各组学的局部单组学样本相似性矩阵:
其中,l(v)为组学v的局部单组学样本相似性矩阵,
然后,进行迭代融合:
其中,f为多组学样本相似性矩阵,w为总的多组学类型数量。
所述智能诊断训练模块包括特征表示模块、多组学融合预测模块,其中:
所述特征表示模块,用于对样本进行特征表示,利用自动编码器将样本在不同组学下的表示转换为相应的向量形式,包括以下步骤:
1)对每个组学分别使用编码器encoder(·)进行编码:
h(v)=encoder(m(v),θ(v))
其中,
2)对每个组学分别使用解码器decoder(·)进行解码:
其中,
所述多组学融合预测模块,利用多视图注意力机制神经网络进行多组学特征融合,并将样本相似性模块结果整合到智能诊断训练过程中,最终训练得到最优诊断模型,包括以下步骤:
1)每个组学对应一个独立的神经网络输入层,且以特征表示模块中步骤1)生成的组学的表征作为具体的输入向量;
2)使用注意力机制计算组学v相对于组学i的权重a(v)(i):
其中,h(v)为组学v的表征,h(i)为组学i的表征,h(j)为组学j的表征,w为组学总数;
3)依据上述权重计算加权后的组学v的表征
4)通过求平均融合各组学表征,得到多组学融合表示o:
5)依据上述所得多组学融合表示o进行预测;
6)模型损失函数l为:
其中,y为真实诊断结果,
7)使用反向传播训练模型,得到最优智能诊断模型。
所述智能诊断预测模块,用于依据多组学数据进行疾病智能诊断,并提供诊断结果的解释,包含以下步骤:
1)使用数据导入模块中的数据加载模块加载多组学数据,使用数据预处理模块处理多组学数据;
2)经过步骤1)处理后的多组学数据输入由智能诊断训练模块训练出的最优诊断模型中进行疾病诊断;
3)提取上述步骤2)中最优诊断模型的注意力机制求出的组学v相对于组学i的权重a(v)(i),然后求和
本发明与现有技术相比,具有如下优点与有益效果:
1、通过深度学习方法挖掘各组学互补特性,增强了智能诊断系统的准确性。
2、对疾病预测结果赋予一定程度的可解释性,有利于医疗人员了解结果的形成原理,也有助于结果的进一步验证工作。
3、将分子水平多种基因数据与影像组学相结合,有利于进一步揭示疾病病理。
附图说明
图1为本发明系统各个模块的关系示意图。
图2为本发明系统训练和预测的流程图。
图3为本发明系统所使用自动编码器的结构示意图。
图4为本发明系统所使用多视图注意力机制神经网络结构示意图。
具体实施方式
下面结合具体实施例对本发明作进一步说明。
本实施例所提供的基于深度学习的多组学智能诊断系统,是使用python语言开发的可在windows设备上运行的多组学智能诊断系统,系统各模块间关系如图1所示,系统训练和预测的流程图如图2所示。它包括有:
数据导入模块,用于加载多组学数据和临床数据,并对数据进行预处理;
样本相似性模块,用于构建多组学样本相似性矩阵,利用样本相似性融合技术融合样本在不同组学下的相似性;
智能诊断训练模块,利用自动编码器将样本在不同组学下的表示转换为相应的向量形式,利用多视图注意力机制神经网络进行多组学特征融合,并将样本相似性模块结果整合到智能诊断过程中,最终训练得到最优的智能诊断模型;
智能诊断预测模块,用于依据多组学数据进行疾病智能诊断,并提供诊断结果的解释。
所述数据导入模块包括数据加载模块、数据预处理模块,其中:
所述数据加载模块从本地读取csv、rdata、xlsx等格式的肝癌数据,即肝癌多组学数据和肝癌临床数据,其中肝癌的多组学数据包括五个组学:基因组学、转录组学、蛋白质组学、表观基因组学以及影像组学,肝癌临床数据包括生存时间等;
所述数据预处理模块用于提取肝癌中多组学共享的样本、针对肝癌数据中的非影像组学去除空缺特征数超过20%的样本、去除空缺样本数超过20%的特征、均值填补空缺值、去除低均值和低方差特征、去除离群特征、数据归一化,针对肝癌的影像数据则使用pyradiomics工具包提取特征。
所述样本相似性模块,用于构建肝癌中多组学样本相似性矩阵,利用样本相似性融合技术融合样本在不同组学下的相似性,包括以下步骤:
1)基于核函数分别构建各组学的单组学样本相似性矩阵:
其中f(v)为组学v的单组学样本相似性矩阵,s(v)为组学v的样本距离矩阵,ρ(v)(·)为组学v中的样本距离度量方法,例如欧式距离,
2)基于相似性网络融合(snf)方法融合各组学的单组学样本相似性矩阵,构建肝癌的多组学样本相似性矩阵:
首先,通过k最近邻方法计算各组学的局部单组学样本相似性矩阵:
其中l(v)为组学v的局部单组学样本相似性矩阵,
然后,进行迭代融合:
其中f为肝癌的多组学样本相似性矩阵,w为总的多组学类型数量,在肝癌中w=5。
所述智能诊断训练模块包括特征表示模块、多组学融合预测模块,其中:
所述特征表示模块,用于对肝癌样本进行特征表示,如图3所示,利用自动编码器模型将肝癌样本在不同组学下的表示转换为相应的向量形式,包括以下步骤:
1)对每个组学分别使用编码器encoder(·)进行编码:
h(v)=encoder(m(v),θ(v))
其中
2)对每个组学分别使用解码器decoder(·)进行解码:
其中
所述多组学融合预测模块,如图4所示,利用多视图注意力机制神经网络进行多组学特征融合,并将肝癌的样本相似性模块结果整合到智能诊断训练过程中,最终训练得到肝癌生存的最优诊断模型,包括以下步骤:
1)每个组学对应一个独立的神经网络输入层且以特征表示模块中步骤1)生成的组学的表征作为具体的输入向量;
2)使用注意力机制计算组学v相对于组学i的权重a(v)(i)
其中h(v)为组学v的表征,h(i)为组学i的表征,h(j)为组学j的表征,w为组学总数;
3)依据上述权重计算加权后的组学v的表征
4)通过求平均融合各组学表征,得到多组学融合表示:
5)依据上述所得多组学融合表示o进行疾病预测。
6)模型损失函数l为:
y为真实的肝癌患者生存时间,y为系统诊断肝癌患者生存时间,loss(·)为分类损失函数,m(v)为组学v的样本-特征矩阵,
7)使用反向传播训练模型,得到关于肝癌患者生存时间的最优智能诊断模型。
所述智能诊断预测模块,用于依据肝癌的多组学数据进行疾病智能诊断,并提供诊断结果的解释,包含以下步骤:
1)使用数据导入模块中的数据加载模块加载新的肝癌多组学数据,使用数据预处理模块处理新的肝癌多组学数据;
2)经过步骤1)处理后的多组学数据将输入由智能诊断训练模块训练出的关于肝癌患者生存时间的最优智能诊断模型中进行疾病诊断,即预测新的肝癌患者的生存时间;
3)提取上述步骤2)中最优诊断模型的注意力机制求出的组学v相对于组学i的权重a(v)(i),然后求和
以上所述实施例只为本发明之较佳实施例,并非以此限制本发明的实施范围,故凡依本发明之形状、原理所作的变化,均应涵盖在本发明的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!