一种基于深度神经网络的乳腺癌生存期预测方法与流程
本发明属于生物医学工程领域,涉及一种基于深度神经网络的乳腺癌生存期预测方法。
背景技术:
乳腺癌是女性最常见的恶性肿瘤之一,根据资料显示,全球每年约有120万新增的女性乳腺癌患者,并且每年约有50万女性死于乳腺癌。经研究,乳腺癌是一种容易引起转移的疾病,经手术治疗后约50%的病例可以治愈,其余50%的病例则可能发生复发或转移。随着乳腺癌的发病率逐渐升高,对癌症患者进行精准预后预测是当前所面临癌症问题的关键。预后指的是预测疾病的可能病程和结局,不仅包括预测某段时间内发生某种结局的可能性,而且包括判断疾病的特定后果。生存期预测作为癌症预后预测的重要研究内容之一,具有十分重要的意义。
目前,对于乳腺癌生存期预测主要包括以下两类方法:生存期临床预测方法和生存期计算预测方法。
生存期临床预测方法是指临床医生利用临床数据并结合一些非正式的主观方法对患者的生存期做出判断的过程,生存期临床预测评估较为灵活,但因受制于认知偏差的影响而不可避免的降低了预测准确性,而且癌症早期阶段的一些生存期预测因素在晚期即失去了预测价值。现有研究表明,生存期临床预测结果往往高于实际的生存时间,与实际生存期相比,生存期临床预测方法往往会高估45%,预测误差在一周内的病例仅占25%,即使是富有经验的临床肿瘤专家,预测生存期的准确率也只有20%左右。
生存期计算预测方法利用预测算法对生存期相关因素进行分析,并建立生存期预测模型。随着机器学习的进步与发展,各种相关技术已被应用于癌症研究,进行预测模型的开发,准确决策的提供。然而目前机器学习在癌症预测诊断的主流方向是早期病症辅助筛查,癌症术后的治疗主要靠医生定期随访,病人定期到院复查,这大大降低了术后治疗方案的实时性和方便性,若利用机器学习对乳腺癌患者的术后生存期进行预测,有助于筛选出可能需要进行预防性治疗的高风险患者并针对性进行书后预防性治疗,提高患者的术后生存率。
技术实现要素:
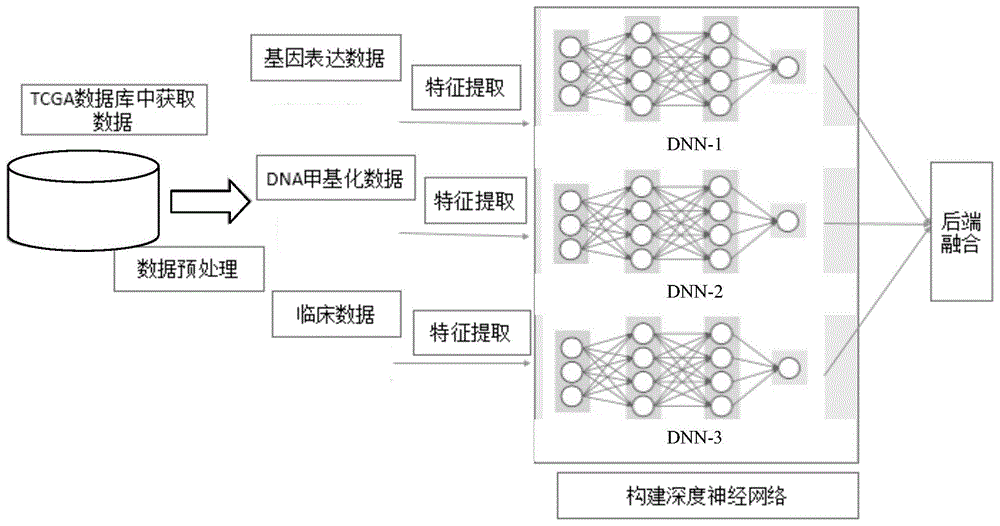
为解决上述背景技术中存在的问题,本发明提出一种基于深度神经网络的乳腺癌生存期预测方法,其采用深度神经网络融合乳腺癌的多组学数据进行生存期预测,从tcga数据库中获取乳腺癌临床数据、基因表达数据、dna甲基化数据,提取数据特征,分别构建深度神经网络模型,然后进行后端融合,提升乳腺癌生存期预测性能,获得生存期预测模型。
本发明解决上述问题的技术方案是:一种基于深度神经网络的乳腺癌生存期预测方法,其特殊之处在于,包括以下步骤:
1)获取数据;所述数据为临床数据和组学数据,组学数据包括基因表达数据和dna甲基化数据;
2)对数据进行预处理;
3)利用最大相关最小冗余算法对数据集特征进行提取;
4)针对临床数据、基因表达数据和dna甲基化数据分别构建深度神经网络,然后进行后端融合。
优先地,所述步骤2)中对数据进行预处理,具体为:
对于基因表达数据,首先将超过10%缺失值样本的基因剔除,然后采用基于权值近邻填充算法填充剩余的缺失值,最后用标准分数将表达数据归一化,并设定相应的阀值,将每个表达值离散化;
对dna甲基化数据做标准分数归一化处理,从而确保两个组学数据的范围在同一尺度内。
优先地,所述步骤3)中数据集特征提取,具体为:
利用最大相关最小冗余算法(mrmr)计算样本标签和特征之间的相关性来给特征集排序。
对于每一个样本si,假设它的特征集合为f,那么特征fi和样本si之间的相关性rel可以表示为:
其中p(fi,si)表示两个变量之间的联合概率分布函数。而特征fi和其它所有被选择出的特征之间的冗余度red可表示为:
根据上述两个条件,拥有与类别最大相关性和与己选特征最小冗余度的特征应当满足下列条件:
mrmr=max(rel(fi,s)-red(fi,f)),
通过这种标准,每次选取一个新的特征并在下一个循环中作为己排序特征,在所有的特征都被计算过后,该样本的所有相关特征完成排序;
在排序步骤中,将基因表达和dna甲基化两种特征分别输入到mrmr算法中,然后用网格搜索法分别选出基因表达和dna甲基化在不同特征数下的auc值,从而选择出最有特征子集。
优先地,所述步骤4)包括以下步骤:
4.1)对基因表达、dna甲基化和临床信息三种模态数据进行预处理,对具有缺失值的样本进行填充,对所有模态数据进行z-score标准化;
4.2)为了有效提取三种不同模态数据的完整信息,分别构建三个深度神经网络dnn-1、dnn-2和dnn-3;
4.3)将dnn-1、dnn-2和dnn-3三个深度神经网络进行后端决策水平融合,公式为:
odnn=α*odnn-1+β*odnn-2+γ*odnn-3,
α+β+γ=1,α≥0,β≥0,γ≥0,
其中α、β、γ是三个权重系数,用于平衡每个dnn子模型的贡献大小。
本发明的优点:
本发明采用深度神经网络融合乳腺癌的多组学数据进行生存期预测,从tcga数据库中获取乳腺癌临床数据、基因表达数据、dna甲基化数据,提取数据特征,分别构建深度神经网络模型,然后进行后端融合,提升乳腺癌生存期预测性能,获得生存期预测模型;本发明在乳腺癌生存期预测中作用显著。
附图说明
图1示出一种基于深度神经网络的乳腺癌生存期预测方法的流程图。
具体实施方式
为使本发明实施方式的目的、技术方案和优点更加清楚,下面将结合本发明实施方式中的附图,对本发明实施方式中的技术方案进行清楚、完整地描述,显然,所描述的实施方式是本发明一部分实施方式,而不是全部的实施方式。基于本发明中的实施方式,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施方式,都属于本发明保护的范围。因此,以下对在附图中提供的本发明的实施方式的详细描述并非旨在限制要求保护的本发明的范围,而是仅仅表示本发明的选定实施方式。
参见图1,一种基于深度神经网络的乳腺癌生存期预测方法,包括以下步骤:
1)获取数据。具体为从tcga数据库中获取临床数据和组学数据,组学数据包括基因表达数据和dna甲基化数据。
2)对数据进行预处理,具体为:
对于基因表达数据,首先将超过10%缺失值样本的基因剔除,然后采用基于权值近邻填充算法填充剩余的缺失值,最后用标准分数将表达数据归一化,并设定相应的阀值,将每个表达值离散化;
对dna甲基化数据做标准分数归一化处理,从而确保两个组学数据的范围在同一尺度内。
3)利用最大相关最小冗余算法对数据集特征进行提取;其具体为:
利用最大相关最小冗余算法(mrmr)计算样本标签和特征之间的相关性来给特征集排序。
对于每一个样本si,假设它的特征集合为f,那么特征fi和样本si之间的相关性rel可以表示为:
其中p(fi,si)表示两个变量之间的联合概率分布函数。而特征fi和其它所有被选择出的特征之间的冗余度red可表示为:
根据上述两个条件,拥有与类别最大相关性和与己选特征最小冗余度的特征应当满足下列条件:
mrmr=max(rel(fi,s)-red(fi,f)),
通过这种标准,每次选取一个新的特征并在下一个循环中作为己排序特征,在所有的特征都被计算过后,该样本的所有相关特征完成排序;
在排序步骤中,将基因表达和dna甲基化两种特征分别输入到mrmr算法中,然后用网格搜索法分别选出基因表达和dna甲基化在不同特征数下的auc值,从而选择出最有特征子集。
4)针对临床数据、基因表达数据和dna甲基化数据分别构建深度神经网络,然后进行后端融合。所述步骤4)包括以下步骤:
4.1)对基因表达、dna甲基化和临床信息三种模态数据进行预处理,对具有缺失值的样本进行填充,对所有模态数据进行z-score标准化;
4.2)为了有效提取三种不同模态数据的完整信息,分别构建三个深度神经网络dnn-1、dnn-2和dnn-3;
4.3)将dnn-1、dnn-2和dnn-3三个深度神经网络进行后端决策水平融合,公式为:
odnn=α*odnn-1+β*odnn-2+γ*odnn-3
α+β+γ=1,α≥0,β≥0,γ≥0,
其中α、β、γ是三个权重系数,用于平衡每个dnn子模型的贡献大小。
本发明设计一种深度神经网络模型融合多模态多组学数据,用来预测乳腺癌生存期,从实验结果来看,性能和效果优于其他癌症生存期预测方法。经过试验,本发明与其它单模态深度神经网络模型比较,基于深度神经网络的多模态数据融合技术,在乳腺癌生存期预测中作用显著。相较于其他机器学习算法如回归算法、支持向量机、随机森林等,本发明设计的模型预测效果更优。
以上所述仅为本发明的实施例,并非以此限制本发明的保护范围,凡是利用本发明说明书及附图内容所作的等效结构或等效流程变换,或直接或间接运用在其他相关的系统领域,均同理包括在本发明的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!