一种基于多重PCR的基因组DNA完整性检测的方法与流程
一种基于多重pcr的基因组dna完整性检测的方法
技术领域
1.本发明属于生物信息学和生物技术领域,具体地,本发明涉及一种基于多重pcr的基因组dna完整性检测的方法。
背景技术:
2.在分子生物学实验中,基因组dna完整性是极重要的质控指标之一,譬如全基因组测序,全外显子组测序,目标区域扩增子测序等,都要求基因组dna完整,否则实验会面临结果变差甚至失败的风险,但轻微的降解不会显著影响实验结果,得到的结果虽不是最优但亦可解决相应问题。当材料受限(比如ffpe,法医样本,本身高度降解)但必须进行样本检测时,为了准确地预估实验成功率,显然需要一种准确且高灵敏度的dna完整性量化检测方法。
3.目前,dna完整性的检测主要有琼脂糖电泳,安捷伦2200生物分析仪,以及基于qpcr的dna完整性检测方法,虽都能在不同程度上实现对基因组dna完整性的检测,但也存在着各种短板。
4.1)琼脂糖电泳无疑是dna完整性检测最常见的方法。常规抽提出的完整基因组dna长度通常超过20k bp,琼脂糖电泳对这一长度的dna分子几无分辨力,因此完整基因组电泳结果呈现为清晰单一的条带。但基因组若发生降解,断裂形成小片段,在胶图上会呈现典型的弥散条带。琼脂糖电泳的方法虽简单高效,但是对dna完整性的判断依据非常主观,不同人对相同的电泳图可能有不同的结果判读,仅能够较明确地判断dna是否完整,无法将dna完整性这一指标量化。此外,某些类型的dna损伤,比如单链断裂,或碱基发生化学修饰也会抑制下游的实验,但这些损伤因基因组dna未完全降解为小片段,而无法在琼脂糖电泳中体现,这些dna损伤甚至会造成dna迁徙率改变,呈现出完全偏离事实的结果。另一方面,dna需求量也较高,弥散条带通常需要至少50ng才可检测。
5.2)安捷伦2200生物分析仪,本质和琼脂糖电泳类似,但通过高精度的仪器实现较低的样本用量。并且仪器配套的软件也可以根据分析结果提供较准确的din值衡量dna完整程度。但需要投入较高的仪器与耗材的投入,实验成本偏高。
6.3)基于qpcr的dna完整性检测,以kapa公司的kapa human genomic dna quantification and qc kit为代表,使用扩增产物长度呈梯度的3对引物(41bp,129bp,305bp),基于dna发生降解时,长片段较短片段扩增效率更容易降低的特点。通过3对引物间的ct值来衡量dna的降解程度,ct越大的降解越严重。但该方案需要的反应数较高,每个样本需要9个反应孔检测,样品需求量较高且检测通量偏低。
7.因此,本领域迫切需要开发一种可以指导dna测序文库构建的,仅需微量核酸样本就可以实施的,且低成本、高效率的基因组dna完整性检测方法。
技术实现要素:
8.本发明的目的是提供一种可以指导dna测序文库构建的,仅需微量核酸样本就可
以实施的,且低成本、高效率的基因组dna完整性检测方法。
9.本发明的第一方面,提供了一种评估核酸样本的基因组完整性的方法,包括:
10.(a)提供一待测的核酸样本;
11.(b)提供第一反应体系和第二反应体系,其中所述第一反应体系含有待测dna的待测的核酸样本,并且所述第一反应体系中含有完整性评估检测的引物组,所述引物组包括n对引物,其中n为≥3的正整数,所述引物组扩增所得的扩增产物的长度呈梯度增加;
12.所述第二反应体系与所述第一反应体系的条件相同,其中,所述第二反应体系含有完整的标准基因组dna的标准品但不含待测dna的待测样本;
13.(c)对所述第一反应体系和所述第二反应体系分别进行多重pcr扩增,从而获得对应于待测dna的第一扩增产物和对应于标准基因组dna的第二扩增产物;
14.(d)分别检测步骤(c)获得的对应于待测dna的第一扩增产物的峰高f1和对应于标准基因组dna的第二扩增产物的峰高f2,并基于所述峰高f1和f2获得核酸样本的相对扩增度k
rel
;
15.(e)基于所述核酸样本的相对扩增度k
rel
,评估所述核酸样本的基因组完整性。
16.在另一优选例中,将第一反应体系中所述待测dna的待测样本用完整的标准基因组dna的标准品加以替换,就构成所述第二反应体系。
17.在另一优选例中,在步骤(e)中,所述的评估为定量评估。
18.在另一优选例中,相对扩增度k
rel
越高,则提示所述核酸样本的基因组完整性越高。
19.在另一优选例中,步骤(c)中,所述第一扩增产物和第二扩增产物的各自扩增条带(也称为“子扩增产物”)的数量为m,较佳地m为3-8个,更佳地,4-6个。
20.在另一优选例中,所述第一扩增产物的扩增条带的数量和第二扩增产物的扩增条带的数量是相同的,并且第一扩增产物的扩增条带和第二扩增产物的扩增条带是一一对应的。
21.在另一优选例中,所述第一扩增产物和第二扩增产物中的各扩增条带长度各自独立地≥50bp,较佳地,50-500bp,更佳地,50-300bp。
22.在另一优选例中,所述待测的核酸样本选自下组:组织抽提的dna、细胞抽提的dna、石蜡包埋组织抽提的dna、或其组合。
23.在另一优选例中,待测的核酸样本来源于石蜡包埋样本抽提dna。
24.在另一优选例中,步骤(b)中,n为3-6,较佳地,3-5,更佳地,4。
25.在另一优选例中,所述步骤(d)中,“基于所述峰高f1和f2获得核酸样本的相对扩增度k
rel”包括进行归一化处理,获得各扩增条带的相对峰高,并基于相对峰高获得核酸样本的相对扩增度k
rel
。
26.在另一优选例中,所述的归一化为对同一反应体系内各扩增条带的峰高进行归一化处理,从而获得归一化的峰高(即同一反应体系内的归一化处理)。
27.在另一优选例中,所述归一化处理包括:对于第一反应体系或待测样本而言,某一扩增条带的相对扩增度为该扩增条带的峰高与该第一反应体系中最短扩增产物峰高的比值;以及
28.对于第二反应体系或标准基因组dna而言,某一扩增条带的相对扩增度为该扩增
条带的峰高与该第二反应体系中最短扩增产物峰高的比值。
29.在另一优选例中,“基于相对峰高获得核酸样本的相对扩增度k
rel”包括用公式q进行结算,从而获得核酸样本的相对扩增度k
rel
:
30.k
rel
={w2(f2/f1*z1/z2)+w3(f3/f1*z1/z3)+
…
+wj(fj/f1*z1/zj)}
1/2
ꢀꢀꢀꢀ
(q);
31.式中,
32.f1为长度最短的待测dna的第一扩增条带的峰高,
33.z1为与长度最短的标准基因组dna的第一扩增条带的峰高;
34.f2为待测dna的第二扩增条带的峰高,
35.z2为标准基因组dna的第二扩增条带的峰高;
36.f3为待测dna的第三扩增条带的峰高,
37.z3为标准基因组dna的第三扩增条带的峰高;
38.fj为待测dna的第j扩增条带的峰高,
39.zj为标准基因组dna的第j扩增条带的峰高;
40.w2为第二扩增条带的权重值;
41.w3为第三扩增条带的权重值,
42.wj为第j扩增条带的权重值;
43.其中,j为≥3的正整数。
44.在另一优选例中,j为3,4,5,6,7,8,9,或10。
45.在另一优选例中,所述的第二至第j个扩增条带的权重值之和(即w2+w3+
…
+wj的总和)s为100、或10000。
46.在另一优选例中,所述的第一个扩增条带的权重值w1为10。
47.在另一优选例中,所述的第j个扩增条带的权重值wj与第j对引物在引物组中的权重或摩尔百分比是相对应的(或成正比)。
48.在另一优选例中,所述每个引物对上设有一个或多个相同或不同的荧光基团。
49.在另一优选例中,所述引物为4对引物,分别为第一引物对:seq id no:1和seq id no:2;第二引物对:seq id no:3和seq id no:4;第三引物对:seq id no:5和seq id no:6;第四引物对:seq id no:7和seq id no:8。
50.在另一优选例中,所述4对引物(8个引物)的比例为(0.5-2):(0.5-2):(1-4):(1-4):(1.5-6):(1.5-6):(2-8):(2-8),较佳地,(0.5-1):(0.5-1):(1-2):(1-2):(1.5-3):(1.5-3):(2-4):(2-4)。
51.在另一优选例中,所述荧光基团选自下组:fam、hex、rox、temra、cy5、texas red、hex、vic、tet、joe、tamra、lc red610、lc red640、lccyan500、yakima yel low、或其组合。
52.在另一优选例中,所述的标准基因组dna为人的全基因组dna标准品(如thermofisher提供的taqman
tm
control genomic dna(human))。
53.在另一优选例中,在反应体系中,所述标准基因组dna的浓度为0.1-1ng/μl,较佳地,0.2-0.8ng/μl,更佳地,0.4-0.6ng/μl。
54.在另一优选例中,所述待测的核酸样本中的待测dna的浓度为1-10ng/μl,较佳地,2-8ng/μl,更佳地,4-6ng/μl。
55.本发明第二方面提供了一种试剂盒,包括:
56.(a)完整性评估检测的引物组,所述引物组包括n对引物,其中n为≥3的正整数,所述引物组扩增所得的扩增产物的长度呈梯度增加;和
57.(b)任选的完整的标准基因组dna的标准品。
58.本发明第三方面提供了一种评估核酸样本的基因组完整性的设备,包括:
59.(a)反应单元,所述反应单元用于进行多重pcr扩增反应,其中所述反应单元包括第一反应体系和第二反应体系,所述第一反应体系含有待测dna的待测的核酸样本,并且所述第一反应体系中含有完整性评估检测的引物组,所述引物组包括n对引物,其中n为≥3的正整数,所述引物组扩增所得的扩增产物的长度呈梯度增加;
60.所述第二反应体系与所述第一反应体系的条件相同,其中,所述第二反应体系含有完整的标准基因组dna的标准品但不含待测dna的待测样本;
61.(b)长度区分检测单元,所述长度区分检测单元用于对反应单元所获得的对应于待测dna的第一扩增产物和对应于标准基因组dna的第二扩增产物分别进行电泳检测,从而获得所述第一扩增产物的峰高f1和所述第二扩增产物的峰高f2;
62.(c)计算单元;所述计算单元用于基于所述第一扩增产物的峰高f1和所述第二扩增产物的峰高f2,从而计算获得核酸样本的相对扩增度k
rel
,并基于核酸样本的相对扩增度k
rel
,从而评估所述核酸样本的基因组完整性;
63.(d)输出单元,所述输出单元用于输出所述计算单元的评估结果。
64.应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。
附图说明
65.图1显示了实施例中随机挑选的5份石蜡dna样本的毛细管电泳结果与琼脂糖电泳结果示意图,a)中4个蓝色峰分别对应各长度片段扩增产物,峰高代表扩增产物得率。b)中为对应的5个降解样本的琼脂糖电泳结果,marker:100,250,500,750,1000,2000bp。可见dna降解越严重,较长片段越难以扩增,得率越低,在多重pcr中更容易被小片段引物竞争。
66.图2显示了实施例中石蜡样品dna,两种计算方法得到的d值与文库浓度的相关性散点图。横坐标表示本发明方法计算出的d值,其中d1为使用4对引物数据计算、d2为使用3对引物数据计算;纵坐标为文库浓度:所有样品均采用相同起始量,相同实验步骤进行文库构建,因此可简单地使用文库浓度表示样品中原始的能够转化到最终文库的分子数。红线为预设的样品等级的判定边界,设定b级(d>7)为完整性较好,建库无问题;c级(5<d<7)设定为完整性可接受,建库有风险;d级(d<5)设定为完整度较差,不可建库。结果可见判定等级在b级(d>7)以上的样本,文库浓度普遍在20ng/ul以上,c级(5<d<7)样本构建得到的文库,随着d值的提升,文库得率波动很大,而d级样本(d<5),文库普遍得率偏低(<10ng/ul),且易出现建库失败样本。两种计算方法得到的d1和d2与文库浓度间关系的趋势也相近,按照相同的分级标准,仅有少数处于临界点的样本的分级在两种计算方式下发生变化。
具体实施方式
67.本发明人经过广泛而深入的研究,首次意外地发现,使用多对扩增产物长度呈梯
度的引物对,对待测dna和标准基因组dna进行多重pcr扩增,分别检测对应于待测dna的第一扩增产物的峰高f1和对应于标准基因组dna的第二扩增产物的峰高f2,基于所述峰高f1和f2,从而获得核酸样本的相对扩增度k
rel
,并基于所述核酸样本的相对扩增度k
rel
,从而评估核酸样本的基因组完整性。
68.具体地,本发明基于长片段扩增效率更易受dna降解抑制的原理,使用多对扩增产物长度呈梯度的、标记了荧光的引物对,对待测dna进行多重pcr扩增,并对扩增产物进行毛细管电泳检测;借助毛细管电泳可以实现多重扩增产物的分离与定量的特点,使用计算公式将扩增产物各峰高与对照样本(完整基因组)峰高的比值转化为衡量dna完整程度的d值,实现对dna完整性的定量检测,并有效的控制反应数与样本需求量。在此基础上,本发明人完成了本发明。
69.术语说明
70.除非另外定义,否则本文中所用的全部技术与科学术语均具有如本发明所属领域的普通技术人员通常理解的相同含义。
71.如本文所用,在提到具体列举的数值中使用时,术语“约”意指该值可以从列举的值变动不多于1%。例如,如本文所用,表述“约100”包括99和101和之间的全部值(例如,99.1、99.2、99.3、99.4等)。
72.如本文所用,术语“含有”或“包括(包含)”可以是开放式、半封闭式和封闭式的。换言之,所述术语也包括“基本上由
…
构成”、或“由
…
构成”。
73.如本文所用,“同一性”、“序列同一性”可互换使用,通过沿着预定的比较窗(其可以是参考核苷酸序列或蛋白的长度的50%、60%、70%、80%、90%、95%或100%)比较两个对齐的序列,并且确定出现相同的残基的位置的数目来确定。通常地,这表示为百分比。核苷酸序列的序列同一性的测量是本领域技术人员熟知的方法。
74.标准基因组dna的标准品
75.标准基因组dna泛指完整度能够完全满足相应实验要求的dna样本。通常意义上,从正常人新鲜二倍体细胞或组织中提取的,溶解在te或类似缓冲液中,无蛋白、rna、多糖等污染,片段长度在20kb以上,且经过严格的qubit或类似基于荧光染料精确定量的dna可认为是标准基因组dna。市场上有很多商品化试剂也可作为本发明中的标准基因组dna使用,如thermofisher提供的taqman
tm
control genomic dna(human)。
76.相对扩增度k
rel
77.在本发明中,相对扩增度k
rel
包括用公式q进行结算,从而获得核酸样本的相对扩增度k
rel
:
78.k
rel
={w2(f2/f1*z1/z2)+w3(f3/f1*z1/z3)+
…
+wj(fj/f1*z1/zj)}
1/2
ꢀꢀꢀꢀ
(q);
79.式中,
80.f1为长度最短的待测dna的第一扩增条带的峰高,
81.z1为与长度最短的标准基因组dna的第一扩增条带的峰高;
82.f2为待测dna的第二扩增条带的峰高,
83.z2为标准基因组dna的第二扩增条带的峰高;
84.f3为待测dna的第三扩增条带的峰高,
85.z3为标准基因组dna的第三扩增条带的峰高;
86.fj为待测dna的第j扩增条带的峰高,
87.zj为标准基因组dna的第j扩增条带的峰高;
88.w2为第二扩增条带的权重值;
89.w3为第三扩增条带的权重值,
90.wj为第j扩增条带的权重值;
91.其中,j为≥3的正整数。
92.在另一优选例中,j为3,4,5,6,7,8,9,或10。
93.在另一优选例中,所述的第二至第j个扩增条带的权重值之和(即w2+w3+
…
+wj的总和)s为100、或10000。
94.在另一优选例中,所述的第一个扩增条带的权重值w1为10。
95.在另一优选例中,所述的第j个扩增条带的权重值wj与第j对引物在引物组中的权重或摩尔百分比是相对应的(或成正比)。
96.评估方法
97.在本发明中,本发明的评估方法可通过公式q进行计算,从而获得核酸样本的相对扩增度k
rel
:
98.k
rel
={w2(f2/f1*z1/z2)+w3(f3/f1*z1/z3)+
…
+wj(fj/f1*z1/zj)}
1/2
ꢀꢀꢀꢀ
(q);
99.式中,
100.f1为长度最短的待测dna的第一扩增条带的峰高,
101.z1为与长度最短的标准基因组dna的第一扩增条带的峰高;
102.f2为待测dna的第二扩增条带的峰高,
103.z2为标准基因组dna的第二扩增条带的峰高;
104.f3为待测dna的第三扩增条带的峰高,
105.z3为标准基因组dna的第三扩增条带的峰高;
106.fj为待测dna的第j扩增条带的峰高,
107.zj为标准基因组dna的第j扩增条带的峰高;
108.w2为第二扩增条带的权重值;
109.w3为第三扩增条带的权重值,
110.wj为第j扩增条带的权重值;
111.其中,j为≥3的正整数。
112.并基于核酸样本的相对扩增度k
rel
,从而评估待测样本的基因组的完整度。
113.在一优选实施方式中,当待测样本的相对扩增度k
rel
>7,表明待测样本的基因组完整度好。
114.在一优选实施方式中,当待测样本的相对扩增度k
rel
<5,表明待测样本的基因组完整度较差。
115.本发明的实验结果表明,本发明的方法可极大降低错误率,提高基因组完整性评估的准确率。
116.评估核酸样本的基因组完整性的方法
117.本发明提供了一种评估核酸样本的基因组完整性的方法,包括:
118.(a)提供一待测的核酸样本;
119.(b)提供第一反应体系和第二反应体系,其中所述第一反应体系含有待测dna的待测的核酸样本,并且所述第一反应体系中含有完整性评估检测的引物组,所述引物组包括n对引物,其中n为≥3的正整数,所述引物组扩增所得的扩增产物的长度呈梯度增加;
120.所述第二反应体系与所述第一反应体系的条件相同,其中,所述第二反应体系含有完整的标准基因组dna的标准品但不含待测dna的待测样本;
121.(c)对所述第一反应体系和所述第二反应体系分别进行多重pcr扩增,从而获得对应于待测dna的第一扩增产物和对应于标准基因组dna的第二扩增产物;
122.(d)分别检测步骤(c)获得的对应于待测dna的第一扩增产物的峰高f1和对应于标准基因组dna的第二扩增产物的峰高f2,并基于所述峰高f1和f2获得核酸样本的相对扩增度k
rel
;
123.(e)基于所述核酸样本的相对扩增度k
rel
,评估所述核酸样本的基因组完整性。
124.在一具体实施方式中,该方法包括以下步骤:
125.1)在基因组上选择拷贝数稳定、且不存在与基因组其余位置同源性问题的区域,设计扩增产物长度不少于50bp,且不超过500bp(以50-300bp间为佳);且扩增产物间存在长度差异的多对引物(3-5,以4为佳),要求扩增产物长度呈均匀的梯度关系。所有引物对的正向或反向引物需标记荧光基团。
126.2)上述引物按一定比例混合后,以一定量的完整的人全基因组dna标准品为模板(1-10ng,5ng为佳)进行多重pcr扩增,扩增产物通过3730基因分析仪毛细管电泳分析后,得到扩增产物的荧光峰图,要求所有扩增产物的峰高比例接近,两两之间差异不超过50%;如差异符合要求,该引物浓度配比则确定为检测体系的引物配比;如差异过大,需重新调整引物之间用量的比例,原则为提高峰高较矮的引物浓度,降低峰高较高的引物浓度,重新实施本步骤。
127.3)使用上述确定的引物配比,以一定量的待测降解样本dna为模板,进行多重pcr扩增;同时需设置完整的人全基因组dna标准品模板为对照。待测样本与对照样本需在同一96孔板进行检测;且需设置一定量的重复(2-6重复,以3重复为佳)。
128.4)根据各降解样品中与完整的人基因组dna样品对应扩增产物峰高的比值,计算dna完整度分值,用于评估dna的完整性。
129.5)d值的计算采用双重校正的原则:第一次校正以多重体系中最短扩增片段的峰高为基准,计算剩余扩增片段峰高相较于最短扩增片段峰高的比值;第二次校正计算待测样品与完整基因组样品间对应扩增产物峰高的比值比;最终将多个峰高的比值比加权平均后转化为分值。本发明基于的原理是长片段扩增效率更易受dna降解抑制的原理,因此扩增产物片段越长,其对应权重也越大。
130.6)本发明所得到的d值,是待测样本相较于完整基因组的评分,其分值大小并不具有实际意义,仅可作为样品之间完整度差异的衡量。当该值被用于预测实验成功率时,需要首先选择一定数量的样本作为训练集,得出d值与该类型实验结果的对应关系,该检测条件下得出的d值可用于后续同一类型实验的指导。如石蜡样本用于dna测序文库的构建:石蜡样本dna降解严重,且伴随着多种dna损伤,反映dna降解程度的d值和最终是否能够成功构建出足够的dna测序文库存在关联。为了确定d值与文库得率间的规律,首先对降解程度不一的各类样本进行d值检测及dna文库构建,并根据构建出的文库得率与相应d值设定样本
的分级标准,该标准即可用于后续该类型实验,预测dna文库的得率。
131.7)可用于标记的荧光基团可至少有4种(fam,hex,rox,temra),对应4组检测体系,在大样本量情况下,使用不同检测体系的扩增产物可在单根毛细管中进行分离检测,进一步降低实验成本。
132.试剂盒
133.本发明提供了一种试剂盒,包括:
134.(a)完整性评估检测的引物组,所述引物组包括n对引物,其中n为≥3的正整数,所述引物组扩增所得的扩增产物的长度呈梯度增加;和
135.(b)任选的完整的标准基因组dna的标准品。
136.评估核酸样本的基因组完整性的设备
137.本发明提供了一种评估核酸样本的基因组完整性的设备,包括:
138.(a)反应单元,所述反应单元用于进行多重pcr扩增反应,其中所述反应单元包括第一反应体系和第二反应体系,所述第一反应体系含有待测dna的待测的核酸样本,并且所述第一反应体系中含有完整性评估检测的引物组,所述引物组包括n对引物,其中n为≥3的正整数,所述引物组扩增所得的扩增产物的长度呈梯度增加;
139.所述第二反应体系与所述第一反应体系的条件相同,其中,所述第二反应体系含有完整的标准基因组dna的标准品但不含待测dna的待测样本;
140.(b)长度区分检测单元,所述长度区分检测单元用于对反应单元所获得的对应于待测dna的第一扩增产物和对应于标准基因组dna的第二扩增产物分别进行电泳检测,从而获得所述第一扩增产物的峰高f1和所述第二扩增产物的峰高f2;
141.(c)计算单元;所述计算单元用于基于所述第一扩增产物的峰高f1和所述第二扩增产物的峰高f2,从而计算获得核酸样本的相对扩增度k
rel
,并基于核酸样本的相对扩增度k
rel
,从而评估所述核酸样本的基因组完整性;
142.(d)输出单元,所述输出单元用于输出所述计算单元的评估结果。
143.本发明的主要优点包括:
144.(1)本发明的进样量仅需5ng dna(可低至1ng),检测时间不超过4小时,且手动时间仅需要约15分钟,成本低,通量高,仅pcr成本与3730上机成本,并且检测结果能够直接用于预估降解样本的实验成功率,起到指导实验的作用。
145.(2)本发明首次使用多对扩增产物长度呈梯度的引物对,对待测dna和标准基因组dna进行多重pcr扩增,分别检测对应于待测dna的第一扩增产物的峰高f1和对应于标准基因组dna的第二扩增产物的峰高f2,基于所述峰高f1和f2,从而获得核酸样本的相对扩增度k
rel
,并基于所述核酸样本的相对扩增度k
rel
,从而评估核酸样本的基因组完整性。
146.下面结合具体实施,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,例如sambrook等人,分子克隆:实验室手册(new york:cold spring harbor laboratory press,1989)中所述的条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数按重量计算。
147.如无特别说明,本发明实施例中所用的试剂和材料均为市售产品。
148.实施例1
149.评估核酸样本的基因组完整性
150.1.在拷贝数稳定且无同源序列干扰的区域设计4对引物,反向引物5'端标记fam荧光修饰,扩增产物长度梯度分别为75bp、145bp、222bp以及298bp。引物序列为:
151.75bp扩增引物:上游引物序列(seq id no:1):tgagccaaaaattcagaatacaagga(26bp);下游引物序列(seq id 2):[fam]ttgcttggaaggcaggcaaac(21bp);
[0152]
145bp扩增引物:上游引物序列(seq id no:3):cactgagccccagagacctgac(22bp);下游引物序列(seq id 4):[fam]aatgcacacctccagggaaaac(22bp);
[0153]
222bp扩增引物:上游引物序列(seq id no:5):agggtgctgggatcagagagag(22bp);下游引物序列(seq id 6):[fam]gctactggagggtggcaaaatg(22bp);
[0154]
298bp扩增引物:上游引物序列(seq id no:7):tcctccaccaagctgatgtgtt(22bp);下游引物序列(seq id 8):[fam]ttcaggcctgtccccgaaatag(22bp)。
[0155]
2.上述引物干粉配制为100mm的引物母液,按seq id no.1:seq id no.2:seq id no.3:seq id no.4:seq id no.5:seq id no.6:seq id no.7:seq id no.8为1:1:2:2:3:3:4:4的比例混合,并加水稀释为总终浓度20μm。
[0156]
3.石蜡包埋样本中的dna,使用qiagen公司的generead dna ffpe kit进行抽提,浓度经qubit测定后稀释为5ng/μl参照上述方法测定基因组dna完整度;
[0157]
4.使用qubit测定样品浓度与完整基因组对照浓度,并稀释浓度至5ng/μl;
[0158]
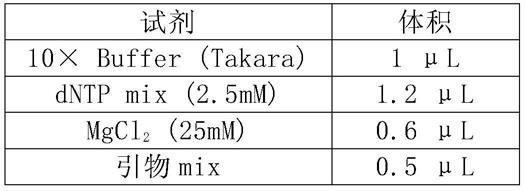
5.使用takara taq hs(5u/μl)及配套10x pcr buffer,takara dntp(2.5mm)进行pcr扩增,每个样本2个平行重复;扩增体系如表1所示:
[0159]
表1 pcr反应体系
[0160][0161][0162]
6.按表2所示的pcr条件,在abi 2720仪器上进行pcr反应
[0163]
表2 pcr反应条件
[0164][0165]
7.扩增产物稀释10倍后,配制表3所示的体系;
[0166]
表3 3730基因分析仪上机体系
[0167]
试剂体积hi-di9μlliz5000.1μl稀释后扩增产物1μl总体积10μl
[0168]
表4 3730基因分析仪上机预反应条件
[0169]
步骤反应温度反应时间循环数变性95℃5min1冷藏4℃forever1
[0170]
8.abi 3730基因分析仪上机(表4),snapshot/str程序;
[0171]
9.随机挑选5个样本,分别进行的毛细管电泳结果与琼脂糖电泳结果明确表明,dna降解越严重,较长片段越难以扩增,得率越低,在多重pcr中更容易被小片段引物竞争(图1,a,b);
[0172]
10.数据分析:样本完整度分值d计算:
[0173]
a)abi 3730基因分析仪原始数据导入genemapper软件(abi),计算每个样本与完整基因组对照的4个扩增产物(75bp、145bp、222bp以及298bp)的峰高;
[0174]
b)将每个反应的4个峰高值基于同一反应体系中75bp的峰高进行归一化(使用峰高值除以75bp的峰高值),记作b、c、d;类似地,完整基因组对照同样进行归一化处理,并计算技术重复反应孔的平均值,记作b0、c0、d0;
[0175]
c)计算使用到所有4个峰高值,则综合d值(即核酸样本的总体的相对扩增度k
rel
)为:
[0176]
计算也可只使用到3个峰高值,则综合d值为:
[0177]
原始数据和结果见表6和表7;
[0178]
d)将同一样品2个技术重复反应的d取平均后,记作该样品的d值,同时可以计算相应的标准差与cv值。
[0179]
11.判定等级(表5所示)
[0180]
表5判定等级
[0181][0182]
9|所有样本统一取100ng基因组dna,使用neb公司的ultr ii dna library prep kit for illumina构建dna测序文库,所有文库使用相同的片段筛选标准与pcr扩增循环数。
[0183]
结果如表6和表7所示。
[0184]
表6完整基因组对照样本的峰高原始数据与计算结果
[0185][0186][0187]
从表6可以看出,实验共分3个独立批次进行,分别标记为板号1,2,3;每次独立实
验亦进行了不同的完整基因组检测的重复次数(4或6次,见编号列)。经3730基因分析仪检测分析,体系中的4对引物(75bp、145bp、222bp与298bp)均可成功扩增出目标片段,并成功计算出峰高(数据见相应列),及相应的相对扩增度b0,c0,d0(数据见相应列)。在完整基因组dna样本的每次独立实验内,4种长度的扩增产物峰高相近。体现了实验良好的稳定性与一致性。但不同批次间标准dna样品的相对扩增度存在较大差异,这也表明使用经校正的相对扩增度进行dna完整性评估会获得更好的准确性(表7)。
[0188]
表7 63个石蜡降解样本原始数据与计算结果
[0189]
[0190]
[0191]
[0192]
[0193][0194]
[0195]
从表7可以看出,实验共分3个独立批次进行,分别标记为板号1,2,3。每个待测样本设置一次重复检测。经3730基因分析仪检测,体系中的4对引物(75bp、145bp、222bp与298bp)均计算出峰高(数据见相应列),及相应的相对扩增度b,c,d(数据见相应列)。使用对应板号的完整基因组对照的相对扩增度b0,c0,d0(表六)计算出经校正的相对扩增度(b/b0,c/c0,d/d0,数据见对应列)。计算使用到所有4个峰高值的d值,计算只使用到3个峰高值的d值,并计算待测样本间两重复的均值记作完整度评判指标。可见两种方案计算出的d值虽在数值上存在些微差异,但所有样本整体的d值分布并无太大区别。所有待测样本统一取100ng基因组dna,使用neb公司的ultr ii dna library prep kit for illumina构建dna测序文库,所有文库使用相同的片段筛选标准与pcr扩增循环数。得到的文库使用qubit测定浓度(数值见glib浓度列),文库浓度分别与d1,d2的散点图表明,d1,d2值均与文库浓度呈显著的正相关(图2),均可用于样本构建文库成功率的衡量指标,体现了本发明良好的灵活性以及准确性。
[0196]
讨论
[0197]
虽然dna的进样量相同,但因降解程度不同,样本中能够转化到最终文库中的有效分子数存在差异,有效分子数越多,构建出的文库质量越高,可简单的体现为文库浓度较高,反之,有效分子数越少,文库质量越差,文库浓度较低甚至无浓度。我们的数据充分表明,在相同建库流程下,判定等级在b级(d>7)以上的样本,文库浓度普遍在20ng/ul以上,c级(5<d<7)样本构建得到的文库,随着d值的提升,文库得率波动很大,而d级样本(d<5),文库普遍得率偏低(<10ng/ul),且易出现建库失败样本(结果见附图2)。以上可明确证明,本方法开发的d值检测能够用于降解石蜡样本dna文库构建成功率的预测,起到指导实验的作用。
[0198]
d值的两种计算方法:横坐标表示本发明方法计算出的d值,其中d1为使用4对引物数据计算、d2为使用3对引物数据计算,虽计算公式不尽相同,但基本原则都是采用双重校正的原则:第一次以多重体系中最短扩增片段的峰高为基准,计算剩余扩增片段峰高相较于最短扩增片段峰高的比值;第二次计算待测样品与完整基因组样品间对应扩增产物峰高的比值比;最终将多个峰高的比值比加权平均后转化为分值。并且扩增产物片段越长,其对应权重也越大,在本实施例中,d1的各片段权重(扩增产物长度从短至长)分别为,20、30、50;d2的各片断权重分别为30、70。两种计算方法得到的d1和d2与文库浓度间关系的趋势也相近,按照相同的分级标准(b级(d>7)、c级(5<d<7)、d级(d<5)),仅有少数处于临界点的样本的分级在两种计算方式下发生变化:44(d1:7.38;d2:6.99),54(d1:7.15;d2:6.55),56号(d1:7.21;d2:6.70)样本。本发明的使用原则在于以固定的检测体系和计算方法,结合预实验结果建立预测模型,并以该模型预测样本的实验结果。本实例也表明了实验体系及计算方法并不要求固定,可根据实际情况灵活设置。
[0199]
在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1