用于CART细胞疗法的顺序方法与流程
相关申请的交叉引用
本申请要求2018年2月23日提交的美国临时专利申请第62/634,573号,2018年4月11日提交的美国临时专利申请第62/656,265号,2018年8月29日提交的美国临时专利申请第62/724,345号,和2018年9月26日提交的美国临时专利申请第62/736,730号的优先权,所有上述的公开内容均在此以其整体通过引用明确并入本文。
本发明涉及一种治疗癌症患者的方法,其通过向该患者施用包含cart细胞的组合物且向该患者施用通过接头连接于靶向部分的小分子。本发明还涉及一种用于此类方法的组合物。
背景技术:
基于淋巴细胞(例如,t细胞)的过继转移至患者中的免疫疗法是一种有价值的治疗癌症及其他疾病的疗法。基于淋巴细胞的过继转移的免疫疗法的发展已取得重大进步。在许多不同类型的免疫治疗剂中,所研发的最有前景的免疫治疗剂之一为表达嵌合抗原受体的t细胞(cart细胞)。嵌合抗原受体(car)是一种经设计以靶向特定抗原,例如肿瘤抗原的基因工程化受体。此靶向可引起针对肿瘤的细胞毒性,例如使得表达car的cart细胞可经由特定肿瘤抗原靶向及杀死肿瘤。
第一代car由识别区及活化信号传导结构域构成,识别区例如用于识别和结合由肿瘤表达的抗原的衍生自抗体的单链片段可变(scfv)区,活化信号传导结构域例如t细胞cd3ζ链可在car中充当t细胞活化信号。虽然cart细胞已在体外显示阳性结果,但其在临床试验中消除疾病(例如癌症)的成果有限。一个问题为无法在体内延长活化并扩增cart细胞群体。
为了解决此问题,共刺激结构域(例如,cd137、cd28或cd134)已包括于第二代car中,以达成在体内延长t细胞的活化。共刺激结构域的添加增强含car的t细胞的体内增殖及存活,且初始临床数据已显示此类构建体为有前景的治疗诸如癌症的疾病的治疗剂。
虽然cart细胞疗法已得到改善,但仍有若干问题。首先,可由于表达被cart细胞靶向的抗原(例如,肿瘤相关抗原)的正常细胞而出现“脱靶”毒性。其次,可发现不受调控的cart细胞活化,其中通过cart细胞对患病细胞(例如,癌细胞)进行的快速且不受控制的消除诱导一系列代谢紊乱,称为肿瘤溶解综合征或细胞因子释放综合征(crs),其对患者可为致命的。肿瘤溶解综合征及crs可由于施用不能容易调控且不受控制地活化的cart细胞而引起。因此,虽然cart细胞显示出作为治疗诸如癌症的疾病的工具的极大前景,但仍需要提供减少脱靶毒性及对cart细胞活化的更精确控制的额外cart细胞治疗方法。
技术实现要素:
在本文所描述的各种实施方案中,通过接头连接于靶向部分的小分子配体用作癌症与cart细胞之间的桥,引导cart细胞至癌症以改善癌症。在一个实施方案中,“小分子配体”可为例如叶酸化合物、dupa、nk-1r配体、caix配体、γ谷氨酰基转肽酶的配体、nkg2d配体或cck2r配体,其中每一种为特异性结合于癌细胞的小分子配体(即,与正常组织相比,这些配体的受体在癌症上过表达)。
在一个实施方案中,“小分子配体”连接于与由cart细胞表达的car结合的“靶向部分”。在各种实施方案中,“靶向部分”可选自例如2,4-二硝基苯酚(dnp)、2,4,6-三硝基苯酚(tnp)、生物素、地高辛(digoxigenin)、荧光素、荧光素异硫氰酸酯(fitc)、nhs-荧光素、五氟苯基酯(pfp)、四氟苯基酯(tfp)、打结素(knottin)、赛丁素(centyrin)及darpin。
“靶向部分”结合于由cart细胞表达的基因工程化的car的识别区。因此,car(例如,抗体、fab、fv、fc、(fab')2片段及其类似者的单链片段可变区(scfv))的识别区指向“靶向部分”。因此,通过接头连接于靶向部分的小分子配体充当癌症与cart细胞之间的桥,引导cart细胞至癌症以改善癌症。
在一个实施方案中,提供一种治疗癌症的方法。该方法包括i)向患者施用至少一个剂量的包含cart细胞的cart细胞组合物,其中cart细胞包含指向靶向部分的car;ii)向患者施用化合物或其药学上可接受的盐,其中该化合物包含由接头连接于靶向部分的小分子配体,且其中该化合物或其药学上可接受的盐以至少第一剂量递增顺序及第二剂量递增顺序施用。
在另一实施方案中,提供一种治疗癌症的方法。该方法包括i)向患者施用至少一个剂量的包含cart细胞的cart细胞组合物,其中cart细胞包含指向靶向部分的car;ii)向患者施用化合物或其药学上可接受的盐,其中该化合物包含通过接头连接于靶向部分的小分子配体,且其中该化合物或其药学上可接受的盐以第一剂量递增顺序施用,其中如果在第一剂量递增顺序中出现严重crs,则使用较低剂量递增顺序施用该化合物或其药学上可接受的盐,其中较低剂量递增顺序中的化合物或其药学上可接受的盐的第一剂量低于以第一剂量递增顺序施用的化合物或其药学上可接受的盐的第一剂量。在一个实施方案中,在较低剂量递增顺序中,可在分开的三天以全剂量的化合物或其药学上可接受的盐的约0.5%、约5%及约50%施用化合物或其药学上可接受的盐。
额外实施方案亦由以下列举的条款描述。亦涵盖与本专利申请的发明内容部分、说明性实施方案的详细描述部分、实施例部分或权利要求书所述的任何适用实施方案组合的任何以下实施方案。
1.一种治疗癌症的方法,该方法包括:
i)向患者施用至少一个剂量的包含cart细胞的cart细胞组合物,其中所述cart细胞包含指向靶向部分的car;
ii)向该患者施用化合物或其药学上可接受的盐,其中该化合物包含通过接头连接于靶向部分的小分子配体,且其中该化合物或其药学上可接受的盐以至少第一剂量递增顺序及第二剂量递增顺序施用。
2.如条款1的方法,其中该化合物或其药学上可接受的盐以至少第一剂量递增顺序、第二剂量递增顺序及第三剂量递增顺序施用。
3.如条款1的方法,其中该化合物或其药学上可接受的盐以至少第一剂量递增顺序、第二剂量递增顺序、第三剂量递增顺序及第四剂量递增顺序施用。
4.如条款1的方法,其中该化合物或其药学上可接受的盐以至少第一剂量递增顺序、第二剂量递增顺序、第三剂量递增顺序、第四剂量递增顺序及第五剂量递增顺序施用。
5.如条款1的方法,其中该化合物或其药学上可接受的盐以至少第一剂量递增顺序、第二剂量递增顺序、第三剂量递增顺序、第四剂量递增顺序、第五剂量递增顺序及第六剂量递增顺序施用。
6.如条款1至5中任一项的方法,其中向该患者施用第一剂量的所述cart细胞及第二剂量的所述cart细胞。
7.如条款6的方法,其中所述cart细胞的该第一剂量为监测该患者对所述cart细胞的耐受性的测试剂量。
8.如条款6的方法,其中所述cart细胞的该第二剂量包含比该第一剂量的所述cart细胞更高剂量的所述cart细胞。
9.如条款6至8中任一项的方法,其中所述cart细胞的该第一剂量包含约0.5×105个所述cart细胞/公斤患者体重至约1.5×106个所述cart细胞/公斤患者体重。
10.如条款6至9中任一项的方法,其中所述cart细胞的该第二剂量包含约0.8×106个所述cart细胞/公斤患者体重至约2×107个所述cart细胞/公斤患者体重。
11.如条款1至10中任一项的方法,其中该第一剂量递增顺序之后为期间不施用该化合物或其药学上可接受的盐的时间段。
12.如条款1至11中任一项的方法,其中该第二剂量递增顺序之后为期间不施用该化合物或其药学上可接受的盐的时间段。
13.如条款2至12中任一项的方法,其中该第三剂量递增顺序之后为期间不施用该化合物或其药学上可接受的盐的时间段。
14.如条款3至13中任一项的方法,其中该第四剂量递增顺序之后为期间不施用该化合物或其药学上可接受的盐的时间段。
15.如条款4至14中任一项的方法,其中该第五剂量递增顺序之后为期间不施用该化合物或其药学上可接受的盐的时间段。
16.如条款5至15中任一项的方法,其中该第六剂量递增顺序之后为期间不施用该化合物或其药学上可接受的盐的时间段。
17.如条款11至16中任一项的方法,其中该时间段为约7天。
18.如条款1至17中任一项的方法,其中该第一剂量递增顺序包含在分开的三天向该患者施用约1%、约10%及约100%的全剂量的该化合物或其药学上可接受的盐。
19.如条款1至18中任一项的方法,其中该第二剂量递增顺序包含在分开的三天向该患者施用约1%、约30%及约300%的全剂量的该化合物或其药学上可接受的盐。
20.如条款2至19中任一项的方法,其中该第三剂量递增顺序包含在分开的三天向该患者施用约1%、约50%及约500%的全剂量的该化合物或其药学上可接受的盐。
21.如条款3至20中任一项的方法,其中该第四剂量递增顺序包含在分开的三天向该患者施用约1%、约10%及约100%的全剂量的该化合物或其药学上可接受的盐。
22.如条款4至21中任一项的方法,其中该第五剂量递增顺序包含在分开的三天向该患者施用约1%、约30%及约300%的全剂量的该化合物或其药学上可接受的盐。
23.如条款5至22中任一项的方法,其中该第六剂量递增顺序包含在分开的三天向该患者施用约1%、约50%及约500%的全剂量的该化合物或其药学上可接受的盐。
24.如条款18至23中任一项的方法,其中该全剂量的该化合物或其药学上可接受的盐为约10µg/kg至约50µg/kg的该化合物或其药学上可接受的盐。
25.如条款18至24中任一项的方法,其中该全剂量的该化合物或其药学上可接受的盐为约20µg/kg至约40µg/kg的该化合物或其药学上可接受的盐。
26.如条款18至25中任一项的方法,其中该全剂量的该化合物或其药学上可接受的盐为约25µg/kg至约35µg/kg的该化合物或其药学上可接受的盐。
27.如条款18至26中任一项的方法,其中该全剂量的该化合物或其药学上可接受的盐为约30µg/kg的该化合物或其药学上可接受的盐。
28.如条款6至27中任一项的方法,其中在第1周期间向该患者施用该第一剂量的所述cart细胞及该第二剂量的所述cart细胞。
29.如条款6至28中任一项的方法,其中在第1周的星期一及星期四向该患者施用该第一剂量的所述cart细胞及该第二剂量的所述cart细胞。
30.如条款1至29中任一项的方法,其中用于该化合物或其药学上可接受的盐的该第一剂量递增顺序出现在第2周及第3周期间。
31.如条款1至30中任一项的方法,其中用于该化合物或其药学上可接受的盐的该第二剂量递增顺序出现在第4周及第5周期间。
32.如条款2至31中任一项的方法,其中用于该化合物或其药学上可接受的盐的该第三剂量递增顺序出现在第6周及第7周期间。
33.如条款30的方法,其中在分开的三天施用该化合物或其药学上可接受的盐,且该分开的三天为第2周的星期一及星期四及第3周的星期一。
34.如条款31的方法,其中在分开的三天施用该化合物或其药学上可接受的盐,且该分开的三天为第4周的星期一及星期四及第5周的星期一。
35.如条款32的方法,其中在分开的三天施用该化合物或其药学上可接受的盐,且该分开的三天为第6周的星期一及星期四及第7周的星期一。
36.如条款1至35中任一项的方法,其中在向该患者施用该cart细胞组合物之前耗尽该患者中的淋巴细胞。
37.如条款1至36中任一项的方法,其进一步包含向该患者施用血小板,向该患者施用浓缩红细胞,向该患者施用冷沉淀物,向该患者施用静脉内免疫球蛋白及/或向该患者提供抗菌剂疗法。
38.如条款1至37中任一项的方法,其中如果在该第一剂量递增顺序期间在患者中未观测到crs或神经毒性,则该方法前进至该第二剂量递增顺序。
39.如条款2至38中任一项的方法,其中如果在该第二剂量递增顺序期间在该患者中未观测到crs或神经毒性,则该方法前进至该第三剂量递增顺序。
40.如条款3至39中任一项的方法,其中如果在该第三剂量递增顺序期间在该患者中未观测到crs或神经毒性,则该方法前进至该第四剂量递增顺序。
41.如条款4至40中任一项的方法,其中如果在该第四剂量递增顺序期间在该患者中未观测到crs或神经毒性,则该方法前进至该第五剂量递增顺序。
42.如条款5至41中任一项的方法,其中如果在该第五剂量递增顺序期间在该患者中未观测到crs或神经毒性,则该方法前进至该第六剂量递增顺序。
43.如条款1至42中任一项的方法,其中如果在所述剂量递增顺序中的任一者期间在该患者中观测到发热而无低血压且未观测到神经毒性,则以导致该发热而无低血压的该剂量递增顺序水平向该患者施用所有后续剂量的该化合物或其药学上可接受的盐。
44.如条款1至43中任一项的方法,其中如果在任何剂量递增顺序中在该患者中出现严重crs或神经毒性,则以低于导致该患者中的严重crs或神经毒性的该剂量递增顺序水平的剂量递增顺序水平向该患者施用所有后续剂量的该化合物或其药学上可接受的盐。
45.一种治疗癌症的方法,该方法包括:
i)向患者施用至少一个剂量的包含cart细胞的cart细胞组合物,其中所述cart细胞包含指向靶向部分的car;
ii)向该患者施用化合物或其药学上可接受的盐,其中该化合物包含通过接头连接于靶向部分的小分子配体,且其中该化合物或其药学上可接受的盐以第一剂量递增顺序施用,其中如果在该第一剂量递增顺序中出现严重crs,则使用较低剂量递增顺序施用该化合物或其药学上可接受的盐,其中该较低剂量递增顺序中的该化合物或其药学上可接受的盐的该第一剂量低于以该第一剂量递增顺序施用的该化合物或其药学上可接受的盐的该第一剂量。
46.如条款45的方法,其中在该较低剂量递增顺序中,在分开的三天以全剂量的该化合物或其药学上可接受的盐的约0.5%、约5%及约50%施用该化合物或其药学上可接受的盐。
47.如条款46的方法,其中该化合物或其药学上可接受的盐的该全剂量是该化合物或其药学上可接受的盐的大约全剂量,是约10µg/kg至约50µg/kg的该化合物或其药学上可接受的盐。
48.如条款46至47中任一项的方法,其中该全剂量的该化合物或其药学上可接受的盐为约25µg/kg至约35µg/kg的该化合物或其药学上可接受的盐。
49.如条款46至48中任一项的方法,其中该全剂量的该化合物或其药学上可接受的盐为约30µg/kg的该化合物或其药学上可接受的盐。
50.如条款1至49中任一项的方法,其中该配体选自:叶酸化合物、dupa、nk-1r配体、caix配体、γ谷氨酰基转肽酶配体、nkg2d配体及cck2r配体。
51.如条款1至50中任一项的方法,其中该配体为叶酸化合物。
52.如条款1至50中任一项的方法,其中该配体为nk-1r配体。
53.如条款1至50中任一项的方法,其中该配体为dupa。
54.如条款1至50中任一项的方法,其中该配体为cck2r配体。
55.如条款1至50中任一项的方法,其中该配体为γ谷氨酰基转肽酶配体。
56.如条款1至55中任一项的方法,其中该靶向部分选自:2,4-二硝基苯酚(dnp)、2,4,6-三硝基苯酚(tnp)、生物素、地高辛、荧光素、荧光素异硫氰酸酯(fitc)、nhs-荧光素、五氟苯基酯、四氟苯基酯、打结素、赛丁素及darpin。
57.如条款1至56中任一项的方法,其中该靶向部分为fitc。
58.如条款1至56中任一项的方法,其中该靶向部分为dnp。
59.如条款1至56中任一项的方法,其中该靶向部分为tnp。
60.如条款1至59中任一项的方法,其中该接头包含聚乙二醇(peg)、聚脯氨酸、亲水性氨基酸、糖、非天然肽聚糖、聚乙烯吡咯啶酮、普洛尼克(pluronic)f-127或其组合。
61.如条款1至60中任一项的方法,其中该接头包含peg。
62.如条款1至61中任一项的方法,其中该化合物或其药学上可接受的盐具有下式:
其中b表示该小分子配体,l表示该接头,且t表示该靶向部分,且其中l包含具有下式的结构:
其中n为0至200的整数。
63.如条款62的方法,其中n为0至150的整数。
64.如条款62的方法,其中n为0至110的整数。
65.如条款62的方法,其中n为0至20的整数。
66.如条款62的方法,其中n为15至20的整数。
67.如条款62的方法,其中n为15至110的整数。
68.如条款1至67中任一项的方法,其中该癌症选自:肺癌、骨癌、胰脏癌、皮肤癌、头部癌、颈部癌、皮肤黑素瘤、眼内黑色素瘤、子宫癌、卵巢癌、子宫内膜癌(endometrialcancer)、直肠癌、胃癌、结肠癌、乳腺癌、三阴性乳腺癌、输卵管癌、子宫内膜癌(carcinomaoftheendometrium)、子宫颈癌、阴道癌、外阴癌、霍奇金氏病(hodgkin'sdisease)、食道癌、小肠癌、内分泌系统癌、甲状腺癌、副甲状腺癌、非小细胞肺癌、肾上腺癌、软组织肉瘤、骨肉瘤(包括儿科或非儿科骨肉瘤)、尿道癌、前列腺癌、慢性白血病、急性白血病、急性骨髓细胞性白血病、淋巴细胞性淋巴瘤、骨髓白血病、骨髓单核球性白血病、毛细胞白血病、胸膜间皮瘤、膀胱癌、伯基特氏淋巴瘤(burkitt'slymphoma)、尿管癌、肾脏癌、肾细胞癌、肾盂癌、中枢神经系统(cns)赘生物、原发性cns淋巴瘤、脊轴肿瘤、脑干神经胶质瘤、垂体腺瘤、及胃食道接合点的腺癌。
69.如条款1至51或56至68中任一项的方法,其中该癌症为表达叶酸受体的癌症。
70.如条项1至69中任一项的方法,其中该癌症为骨肉瘤。
71.如条款1至70中任一项的方法,其中该car具有识别区且该识别区为抗体的单链片段可变(scfv)区。
72.如条款1至57或60至71中任一项的方法,其中该car具有识别区且该car的该识别区为抗fitc抗体的单链片段可变(scfv)区。
73.如条款1至72中任一项的方法,其中该car具有共刺激结构域且该共刺激结构域选自:cd28、cd137(4-1bb)、cd134(ox40)及cd278(icos)。
74.如条款1至73中任一项的方法,其中该car具有活化信号传导结构域且该活化信号传导结构域为t细胞cd3ζ链或fc受体γ。
75.如条款1至57或60至74中任一项的方法,其中该car具有识别区且该识别区为抗fitc抗体的单链片段可变(scfv)区,其中该car具有共刺激结构域且该共刺激结构域为cd137(4-1bb),且其中该car具有活化信号传导结构域且该活化信号传导结构域为t细胞cd3ζ链。
76.如条款1至75中任一项的方法,其中在施用该化合物或其药学上可接受的盐之前,或在施用该cart细胞组合物之前,对该患者成像。
77.如条款1至76中任一项的方法,其中该化合物或其药学上可接受的盐不为抗体且不包含抗体的片段。
78.如条款1至77中任一项的方法,其中该靶向部分不包含肽表位。
79.如条款1至78中任一项的方法,其中不出现在患者中导致脱靶毒性的细胞因子释放,且其中出现对该癌症的cart细胞毒性。
80.如条款1至78中任一项的方法,其中不出现在患者中的脱靶组织毒性,且其中出现对该癌症的cart细胞毒性。
81.如条款1至78中任一项的方法,其中该癌症包含肿瘤,其中肿瘤大小在该患者体内减小,且其中不出现脱靶毒性。
82.如条款1至81中任一项的方法,其中所述cart细胞包含含有seqidno:1的核酸。
83.如条款1至82中任一项的方法,其中所述cart细胞包含含有seqidno:2的多肽。
84.如条款82的方法,其中该核酸编码嵌合抗原受体。
85.如条款1至81中任一项的方法,其中所述cart细胞包含含有seqidno:4的核酸。
86.如条款1至81或85中任一项的方法,其中所述cart细胞包含含有seqidno:5的多肽。
87.如条款85的方法,其中该核酸编码嵌合抗原受体。
88.如条款1至87中任一项的方法,其中该car包含人源化氨基酸序列。
89.如条款1至87中任一项的方法,其中该car由人源化氨基酸序列组成。
90.如条款1至89中任一项的方法,其进一步包含向该患者施用叶酸化合物;包含叶酸化合物的缀合物,其中包含叶酸化合物的该缀合物不包含靶向部分;或抑制所述cart细胞活化的药剂。
91.如条款90的方法,其中施用叶酸化合物。
92.如条款90的方法,其中施用叶酸或甲酰四氢叶酸。
93.如条款90的方法,其中施用包含叶酸化合物的该缀合物。
94.如条款93的方法,其中包含叶酸化合物的该缀合物包含连接于一或多个氨基酸的叶酸化合物。
95.如条款93的方法,其中包含叶酸化合物的该缀合物具有下式:
96.如条款91的方法,其中该叶酸化合物具有下式:
其中x1及y1各自独立地选自:卤素、r2、or2、sr3及nr4r5;
u、v及w表示各自独立地选自以下的二价部分:-(r6a)c=、-n=、-(r6a)c(r7a)-及-n(r4a)-;q选自:c及ch;t选自:s、o、n及-c=c-;
x2及x3各自独立地选自:氧、硫、-c(z)-、-c(z)o-、-oc(z)-、-n(r4b)-、-c(z)n(r4b)-、-n(r4b)c(z)-、-oc(z)n(r4b)-、-n(r4b)c(z)o-、-n(r4b)c(z)n(r5b)-、-s(o)-、-s(o)2-、-n(r4a)s(o)2-、-c(r6b)(r7b)-、-n(c≡ch)-、-n(ch2c≡ch)-、c1-c12亚烷基及c1-c12亚烷氧基,其中z为氧或硫;
r1选自:氢、卤素、c1-c12烷基及c1-c12烷氧基;
r2、r3、r4、r4a、r4b、r5、r5b、r6b及r7b各自独立地选自:氢、卤素、c1-c12烷基、c1-c12烷氧基、c1-c12烷酰基、c1-c12烯基、c1-c12炔基、(c1-c12烷氧基)羰基及(c1-c12烷基氨基)羰基;
r6及r7各自独立地选自:氢、卤素、c1-c12烷基及c1-c12烷氧基;或r6及r7一起形成羰基;
r6a及r7a各自独立地选自:氢、卤素、c1-c12烷基及c1-c12烷氧基;或r6a与r7a一起形成羰基;
p、r、s及t各自独立地为0或1;且
如果任何其他化学部分为该叶酸化合物的部分,则*表示与该缀合物的其余部分的任选的共价键。
97.如条款90的方法,其中抑制所述cart细胞活化的该药剂选自:淋巴细胞特异性蛋白质酪氨酸激酶抑制剂、pi3激酶抑制剂、il-2诱导性t细胞激酶抑制剂、jak抑制剂、btk抑制剂、ec2319及阻断cart细胞与该化合物或其药学上可接受的盐结合但不与该癌症结合的药剂。
98.如条款90的方法,其中施用抑制所述cart细胞活化的该药剂且该药剂为淋巴细胞特异性蛋白质酪氨酸激酶抑制剂。
99.如条款98的方法,其中该淋巴细胞特异性蛋白质酪氨酸激酶抑制剂为达沙替尼(dasatinib)。
100.如条款90的方法,其中施用抑制所述cart细胞活化的该药剂且该药剂为pi3激酶抑制剂。
101.如条款100的方法,其中该pi3激酶抑制剂为gdc0980。
102.如条款90的方法,其中施用抑制所述cart细胞活化的该药剂且该药剂为il-2诱导性t细胞激酶抑制剂。
103.如条款102的方法,其中该il-2诱导性t细胞激酶抑制剂为bms-509744。
104.如条款90的方法,其中抑制所述cart细胞的活化的该药剂经施用,且为阻断cart细胞与该化合物或其药学上可接受的盐结合但不与该癌症结合的药剂。
105.如条款104的方法,其中该药剂为荧光素胺、fitc或荧光素钠。
106.如条款105的方法,其中该药剂为荧光素钠。
107.如条款90至106中任一项的方法,其中施用该叶酸化合物、包含叶酸化合物的该缀合物(其中包含叶酸化合物的该缀合物不包含靶向部分)或抑制所述cart细胞活化的该药剂使得该患者中的细胞因子水平降低。
108.如条款107的方法,其中该细胞因子水平降低为降低至约未经治疗的患者中的细胞因子水平。
109.如条款90至108中任一项的方法,其中该癌症包含肿瘤且在向该患者施用该叶酸化合物、包含叶酸化合物的该缀合物(其中包含叶酸化合物的该缀合物不包含靶向部分)或抑制所述cart细胞活化的该药剂时,该患者中的肿瘤大小未增加。
110.如条款104至106中任一项的方法,当该crs级别达到1、2、3或4时,向该患者施用抑制所述cart细胞活化的该药剂。
111.如条款110的方法,其中当该crs级别达到3或4时,向该患者施用抑制所述cart细胞活化的该药剂。
112.如条款104至106中任一项的方法,其中以约0.01至约300微摩尔/公斤该患者体重的剂量施用抑制所述cart细胞活化的该药剂。
113.如条款1至112中任一项的方法,其中在该患者体内减轻或预防crs且该方法引起该患者中的肿瘤体积减小。
114.如条款1至113中任一项的方法,其中减轻或预防由crs所致的体重损失。
115.如条款90至114中任一项的方法,其进一步包含向该患者再施用该化合物或其药学上可接受的盐。
116.如条款115的方法,其中该化合物或其药学上可接受的盐的该后续施用致使cart细胞活化及该患者中的细胞因子水平增加。
117.如条款1至116中任一项的方法,其中在该化合物或其药学上可接受的盐前施用该cart细胞组合物。
118.如条款1至117中任一项的方法,其中所述cart细胞为自体性的。
119.如条款2至17中任一项的方法,其中该第一剂量递增顺序包含在分开的三天向该患者施用约1%、约2%及约20%的全剂量的该化合物或其药学上可接受的盐,其中该第二剂量递增顺序包含在分开的三天向该患者施用约1%、约6%及约60%的全剂量的该化合物或其药学上可接受的盐,且其中该第三剂量递增顺序包含在分开的三天向该患者施用约1%、约10%及约100%的全剂量的该化合物或其药学上可接受的盐。
120.如条款2至17中任一项的方法,其中该第一剂量递增顺序包含在分开的三天向该患者施用约1%、约10%及约100%的全剂量的该化合物或其药学上可接受的盐,且其中该第二剂量递增顺序包含在分开的三天向该患者施用约1%、约20%及约200%的全剂量的该化合物或其药学上可接受的盐。
121.如条款2至17中任一项的方法,其中该第一剂量递增顺序包含在分开的三天向该患者施用约1%、约10%及约100%的全剂量的该化合物或其药学上可接受的盐,且其中该第二剂量递增顺序包含在分开的三天向该患者施用约1%、约10%及约100%的全剂量的该化合物或其药学上可接受的盐。
122.如条款2至17中任一项的方法,其中该第一剂量递增顺序包含在分开的三天向该患者施用约1%、约20%及约200%的全剂量的该化合物或其药学上可接受的盐,且其中该第二剂量递增顺序包含在分开的三天向该患者施用约1%、约20%及约200%的全剂量的该化合物或其药学上可接受的盐。
123.如条款119至122中任一项的方法,其中该化合物或其药学上可接受的盐的该全剂量为约500纳摩尔/公斤。
附图说明
图1显示根据请求保护的方法的示例性给药方案的图示。在此示例性方案中,在于第2周开始施用通过接头连接于靶向部分的小分子配体(例如,ec17)之前,e2cart细胞(表达seqidno:4的cart细胞)施用出现在第1周的星期一及星期四。
在第2周,使用剂量递增顺序(即,顺序1),在星期一及星期四且接着在第3周的星期一施用通过接头连接于靶向部分的小分子配体(例如,ec17)。在第3周的星期二至星期日不施用通过接头连接于靶向部分的小分子配体(例如,ec17)。第2周至第3周称为“周期1”。
在第4周,使用剂量递增顺序(即,顺序2),在星期一及星期四且接着在第5周的星期一施用通过接头连接于靶向部分的小分子配体(例如,ec17)。在第5周的星期二至星期日不施用通过接头连接于靶向部分的小分子配体(例如,ec17)。第4周至第5周称为“周期2”。
在第6周,使用剂量递增顺序(即,顺序3),在星期一及星期四且接着在第7周的星期一施用通过接头连接于靶向部分的小分子配体(例如,ec17)。在第7周的星期二至星期日不施用通过接头连接于靶向部分的小分子配体(例如,ec17)。第6周至第7周称为“周期3”。第2周至第7周称为“疗程1”。
在顺序1期间,在第2周的星期一及星期四且接着在第3周的星期一施用通过接头连接于靶向部分的小分子配体(例如,ec17),其中所述通过接头连接于靶向部分的小分子配体(例如,ec17)的剂量分别为1%、10%及100%的全剂量(例如,31µg/kg)的连接于靶向部分的小分子配体(例如,ec17)。
在顺序2期间,在第4周的星期一及星期四且接着在第5周的星期一施用通过接头连接于靶向部分的小分子配体(例如,ec17),其中所述通过接头连接于靶向部分的小分子配体(例如,ec17)的剂量分别为1%、30%及300%的全剂量(例如,31µg/kg)的连接于靶向部分的小分子配体(例如,ec17)。
在顺序3期间,在第6周的星期一及星期四且接着在第7周的星期一施用通过接头连接于靶向部分的小分子配体(例如,ec17),其中所述通过接头连接于靶向部分的小分子配体(例如,ec17)的剂量分别为1%、50%及500%的全剂量(例如,31µg/kg)的连接于靶向部分的小分子配体(例如,ec17)。接着重复疗程1,呈现临床益处及可耐受毒性。
图2显示用于cart转导的构建体的总图。
图3概略地显示e2构建体相对于4m5.3构建体且显示e2构建体的图。
图4(上部图)为显示在用仅e2car-t细胞(●)及e2car-t细胞+ec-17(○)治疗时,hos-frα肿瘤的肿瘤体积的图表。图4(下部图)为显示在用仅e2car-t细胞(●)及e2car-t细胞+ec-17(○)治疗时携带hos-frα肿瘤的小鼠的体重变化的图表。
图5为显示在不用car-t(●);不用ec-17(■);用ec-17siw500nmol/kg(▲);用ec-17tiw(5、50、500nmol/kg开/关);用(♦)ec-17剂量递增(m/th/m开/关)治疗时,携带thp-1-frβaml的小鼠的体重变化的图表。
图6(左侧图)为显示肝脏转移性肿瘤负荷的图表。图6(右侧图)为显示非肝脏转移性肿瘤负荷的图表。
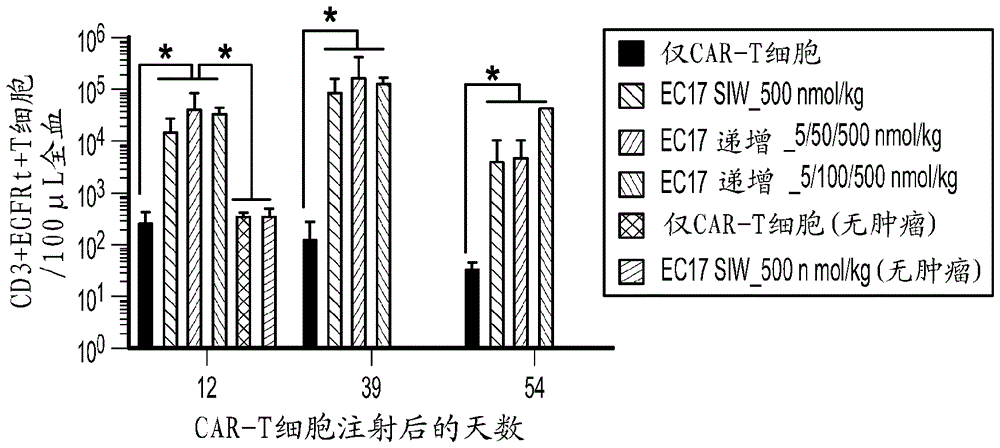
图7为以对数标度显示在第39天每100µl血液的循环thp1-frβ细胞计数的图表。
图8为显示在不同ec-17给药方案下自实体肝脏肿瘤分离的总e2cart细胞的百分比(y轴)的图表。对于各给药方案,最左侧条形为pd1+lag3+tim3+,左侧第二个条形为pd1+lag3+tim3-,中间条形为pd1+lag3-tim3+;右侧第二个条形为pd1+lag3-tim3-;最右侧条形为pd1-lag3-tim3-。
图9显示完全人类car构建体,其包括抗fitcscfv(克隆e2)、全长igg4间隔子(fc衍生的铰链-ch2(l235d,n297q)-ch3)、cd28tm跨膜结构域、4-1bb/cd3ζ细胞质活化结构域及表皮生长因子受体的非功能性截短细胞表面多肽(egfrt)。底部:通过流式细胞术对egfrt分选的(左侧饼图图表)car-t细胞制品及未分选“临床传真(clinicalfacsimile)”(右侧饼图图表)执行的cd4/cd8t细胞表型分析的实例。颜色键如所展示。
图10,a图:在37℃下在2小时培育之后,fr+靶细胞的3h-ec17摄取的kd值(自每细胞所结合的分子数目计算)。b图:在室温下在2小时培育之后,e2-car-t细胞(~24%egfrt+,~95:5cd8/cd4比率)的3h-ec17摄取的kd值(自总的细胞相关的放射活性dpm计算)。
图11显示通过基于3h-fa的结合测定(100nm,1小时,在37℃下),测量的肿瘤细胞上的功能性fr水平。
图12显示相较于无肿瘤小鼠,肿瘤中的ec17剂量研究结果及crs评估。a图:显示在无或有预先建立的mda-mb-231异种移植物的情况下,car-t细胞(约1千万“临床传真”)加上ec17在nsg小鼠中给药的3种不同方式的剂量安排的示意图。无肿瘤小鼠接受ec17siw500nmol/kg(在第2天及第10天2次剂量)。携带肿瘤的小鼠接受ec17如下:ec17siw500nmol/kg(在第2、10、17、24及31天5次剂量)、ec17m/th/m_递增-1(在星期一/星期四/星期一,随后中断1周,即在第2、6、10、17、20、24及31天重复5/50/500nmol/kg)或ec17m/th/m_递增-2(在星期一/星期四/星期一,随后中断1周,即在第2、6、10、17、20、24及31天重复5/100/1000nmol/kg)。b图:在将car-t细胞注射于携带肿瘤与无肿瘤小鼠中之后的第11及12天(即,在所有治疗群组中进行先前ec17给药之后约20及42小时)在小鼠血浆中检测到的以log2标度计的人类ifnγ的全身水平。c图:通过流式细胞术识别为人类cd3ε+egfrt+事件且每100µl全血所计数的小鼠血液中的循环car-t细胞。d图:接受仅car-t细胞,或以3种不同方式给药的car-t细胞加上ec17的携带肿瘤的小鼠的体重及肿瘤体积变化的测量结果(虚线指示各ec17剂量)。n=5只小鼠/组。所有数据均表示平均值±s.e.m。*p<0.05,通过单向anova测试。
图13显示ec17剂量递增的体内安全性及抗白血病活性。a图:显示经静脉内thp1-frβ异种移植的1天龄nsg小鼠中ec17加上未分选egfrtcar-t细胞(约6百万,第0天)的剂量安排的示意图。在car-t细胞注射之后3天起,ec17以3种不同方式给药:ec17siw500nmol/kg(在第3、10、17、24、31及38天5次剂量)、ec17tiw5/50/500nmol/kg(在星期一/星期三/星期五,随后中断9天,即在第3/5/7、17/19/21及31/33/35天重复3次5/50/500nmol/kg)或在星期一/星期四/星期一的ec17m/th/m_剂量递增(3个递增周期,第1周期为5/10/100nmol/kg,第2周期为5/30/300nmol/kg,且第3周期为5/50/500nmol/kg,即在第3/6/10、17/20/24及31/34/38天)。b图:体重变化的测量结果(n=5)。c图:在第31天每100µl小鼠全血发现的作为人类cd3ε+egfrt+事件的循环car-t细胞,以对数标度计。d图:左侧条形图:在研究结束时(第39天)所测量的所有群组中每100µl全血的循环肿瘤细胞(gfp+);中间条形图:表示肝脏转移性负荷的thp1-frβ浸润的肝脏重量;右侧条形图:所有非肝脏巨大转移的总肿瘤重量。e图:在预输注car-t细胞产物(三阴性)及自肝脏转移分离的肿瘤浸润car-t细胞上,对t细胞耗竭标记物pd1、lag3、tim3进行的流式细胞术分析。完全耗竭性t细胞的主要特征为共表达多个抑制性受体标记(即三阳性)。
图14显示儿科骨肉瘤的侵蚀性模型中的抗肿瘤活性及crs挽救。a图:显示在皮下hos-frα异种移植物的3天龄nsg小鼠(n=5)中,car-t细胞(约6百万,第0天)加上ec17的剂量安排的示意图。在car-t细胞注射之后3天(在肿瘤植入之后6天)起,3个周期为:在第1周期中以5/10/100nmol/kg,在第2周期中5/30/300nmol/kg,及在第3周期中5/50/500nmol/kg,在星期一/星期四/星期一,即在第3/6/10、17/20/24及31/34/38天进行ec17m/th/m“患者内”剂量递增。b图:肿瘤体积及体重变化的测量结果。由于肿瘤进展,在第23-31天将仅接受car-t细胞(无ec17)的五只小鼠安乐死,且在第33天(2只小鼠)及第47天(3只小鼠)分别将五只经ec17治疗的小鼠安乐死。c图:以对数标度标绘的每100µl全血的car-t细胞(人类cd3ε+egfrt+)(左侧)及在安乐死时的肿瘤浸润性car-t细胞(右侧)的流式细胞术分析。
定义
如本文所用,“一种(a或an)”可意指一或多种。如本文所用,关于数值,包括(例如)整数、分数及百分比,“约”通常是指本领域普通技术人员将考虑等同于所列举值(例如,具有相同功能或结果)的一系列数值(例如,所列举值的+/-5%至10%)。
如本文所用,术语“治疗(treat/treating/treated/treatment)”是指治疗性治疗及防治性或预防性治疗两者。
如本文所用,关于癌症,术语“改善(ameliorate/ameliorating/amelioration/ameliorated)”可意指减轻癌症的症状,减小肿瘤的大小,完全地或部分地移除肿瘤(例如,完全或部分反应),产生稳定的疾病,阻止癌症的进展(例如,无进展存活)或将由医师考虑为癌症的治疗性、防治性或预防性治疗的对于癌症的任何其他作用。
如本文所用,术语“施用(administer/administering/administered)”意指所有将化合物或其药学上可接受的盐或本文所述的cart细胞组合物引入患者的方式,包括(但不限于)口服、静脉内、肌肉内、皮下及经皮。
如本文所用,术语“脱靶毒性”意指治疗患者的医师不可接受的器官破坏或患者体重降低,或治疗患者的医师不可接受的对患者的任何其他作用,例如b细胞发育不全、发热、血压下降或肺水肿。
如本文所用,术语“转导”及“转染”等同地使用且所述术语意指通过任何人工方法,包括病毒及非病毒方法将核酸引入细胞中。
如本文所用,术语“剂量递增顺序”意指随时间施用增大剂量的化合物或其药学上可接受的盐。如本文所用,与“第二剂量递增顺序”、“第三剂量递增顺序”、“第四剂量递增顺序”、“第五剂量递增顺序”及“第六剂量递增顺序”等组合使用的“第一剂量递增顺序”的提及,意指出现多个剂量递增顺序,且对于在第一剂量递增顺序后的每一单独剂量递增顺序,后续剂量递增顺序中的化合物或其药学上可接受的盐的第一剂量低于先前剂量递增顺序中的化合物或其药学上可接受的盐的上次剂量(参见例如图1及附图说明中图1的解释)。
说明性实施方案的详细描述
在本文所描述的各种实施方案中,通过接头连接于靶向部分的小分子配体用作癌症与cart细胞(即,表达嵌合抗原受体的t细胞)之间的桥。该桥将cart细胞指向癌症以改善癌症。在一个实施方案中,“小分子配体”可为叶酸化合物、caix配体、dupa、nk-1r配体、γ谷氨酰基转肽酶的配体、nkg2d配体或cck2r配体,其中每一种为特异性结合于癌细胞类型的小分子配体(即,与正常组织相比,这些配体的每一种的受体在癌症上过表达)。
连接于小分子配体的“靶向部分”结合于由cart细胞表达的基因工程改造的car的识别区。因此,car的识别区(例如,抗体的单链片段可变区(scfv)、fab、fv、fc或(fab')2片段及类似者)指向“靶向部分”。因此,通过接头连接于靶向部分的小分子配体充当癌症与cart细胞之间的桥,将cart细胞指向癌症以改善癌症。在各种实施方案中癌症与cart细胞之间的桥可为实施例中显示的任何可应用的缀合物。
该桥为小的有机分子,因此可迅速地实现自血流清除(例如,约20分钟或更少)。在一个方面,cart细胞反应可仅仅靶向表达“桥”的小分子配体部分的受体的那些癌细胞,由此减小对正常组织的脱靶毒性。另外,此系统可为“通用”的,因为一种类型的cart细胞构建体可使用不同“桥”用于靶向多种癌症。说明性地,由cart细胞识别的靶向部分可保持恒定使得可使用一种类型的cart细胞构建体,同时可改变结合于癌症的小分子配体以允许靶向多种癌症。
在一个实施方案中,提供一种治疗癌症的方法。该方法包括i)向患者施用至少一个剂量的包含cart细胞的cart细胞组合物,其中cart细胞包含指向靶向部分的car;ii)向患者施用化合物或其药学上可接受的盐,其中该化合物包含由接头连接于靶向部分的小分子配体,且其中所述化合物或其药学上可接受的盐以至少第一剂量递增顺序及第二剂量递增顺序施用。
在另一实施方案中,提供一种治疗癌症的方法。该方法包括i)向患者施用至少一个剂量的包含cart细胞的cart细胞组合物,其中cart细胞包含指向靶向部分的car;ii)向患者施用化合物或其药学上可接受的盐,其中该化合物包含通过接头连接于靶向部分的小分子配体且其中所述化合物或其药学上可接受的盐以第一剂量递增顺序施用,其中如果在所述第一剂量递增顺序中出现严重crs,则使用较低剂量递增顺序施用所述化合物或其药学上可接受的盐,其中所述较低剂量递增顺序中的所述化合物或其药学上可接受的盐的所述第一剂量低于以所述第一剂量递增顺序施用的所述化合物或其药学上可接受的盐的所述第一剂量。在另一实施方案中,在较低剂量递增顺序中,可在分开的三天以全剂量的化合物或其药学上可接受的盐的约0.5%、约5%及约50%施用化合物或其药学上可接受的盐。
在以下条款清单及权利要求书和整个申请中描述的各种实施方案中,通过接头连接于靶向部分的小分子配体称为“化合物”。
若干实施方案由以下列举的条款描述。亦涵盖与本专利申请的发明内容部分、说明性实施方案的详细描述部分、实施例部分或本专利申请的权利要求书所述的任何适用实施方案组合的任何以下实施方案。
1.一种治疗癌症的方法,该方法包括:
i)向患者施用至少一个剂量的包含cart细胞的cart细胞组合物,其中所述cart细胞包含指向靶向部分的car;
ii)向该患者施用化合物或其药学上可接受的盐,其中该化合物包含通过接头连接于靶向部分的小分子配体,且其中该化合物或其药学上可接受的盐以至少第一剂量递增顺序及第二剂量递增顺序施用。
2.如条款1的方法,其中该化合物或其药学上可接受的盐以至少第一剂量递增顺序、第二剂量递增顺序及第三剂量递增顺序施用。
3.如条款1的方法,其中该化合物或其药学上可接受的盐以至少第一剂量递增顺序、第二剂量递增顺序、第三剂量递增顺序及第四剂量递增顺序施用。
4.如条款1的方法,其中该化合物或其药学上可接受的盐以至少第一剂量递增顺序、第二剂量递增顺序、第三剂量递增顺序、第四剂量递增顺序及第五剂量递增顺序施用。
5.如条款1的方法,其中该化合物或其药学上可接受的盐以至少第一剂量递增顺序、第二剂量递增顺序、第三剂量递增顺序、第四剂量递增顺序、第五剂量递增顺序及第六剂量递增顺序施用。
6.如条款1至5中任一项的方法,其中向该患者施用第一剂量的所述cart细胞及第二剂量的所述cart细胞。
7.如条款6的方法,其中所述cart细胞的该第一剂量为监测该患者对所述cart细胞的耐受性的测试剂量。
8.如条款6的方法,其中所述cart细胞的该第二剂量包含比该第一剂量的所述cart细胞更高剂量的所述cart细胞。
9.如条款6至8中任一项的方法,其中所述cart细胞的该第一剂量包含约0.5×105个所述cart细胞/公斤患者体重至约1.5×106个所述cart细胞/公斤患者体重。
10.如条款6至9中任一项的方法,其中所述cart细胞的该第二剂量包含约0.8×106个所述cart细胞/公斤患者体重至约2×107个所述cart细胞/公斤患者体重。
11.如条款1至10中任一项的方法,其中该第一剂量递增顺序之后为期间不施用该化合物或其药学上可接受的盐的时间段。
12.如条款1至11中任一项的方法,其中该第二剂量递增顺序之后为期间不施用该化合物或其药学上可接受的盐的时间段。
13.如条款2至12中任一项的方法,其中该第三剂量递增顺序之后为期间不施用该化合物或其药学上可接受的盐的时间段。
14.如条款3至13中任一项的方法,其中该第四剂量递增顺序之后为期间不施用该化合物或其药学上可接受的盐的时间段。
15.如条款4至14中任一项的方法,其中该第五剂量递增顺序之后为期间不施用该化合物或其药学上可接受的盐的时间段。
16.如条款5至15中任一项的方法,其中该第六剂量递增顺序之后为期间不施用该化合物或其药学上可接受的盐的时间段。
17.如条款11至16中任一项的方法,其中该时间段为约7天。
18.如条款1至17中任一项的方法,其中该第一剂量递增顺序包含在分开的三天向该患者施用约1%、约10%及约100%的全剂量的该化合物或其药学上可接受的盐。
19.如条款1至18中任一项的方法,其中该第二剂量递增顺序包含在分开的三天向该患者施用约1%、约30%及约300%的全剂量的该化合物或其药学上可接受的盐。
20.如条款2至19中任一项的方法,其中该第三剂量递增顺序包含在分开的三天向该患者施用约1%、约50%及约500%的全剂量的该化合物或其药学上可接受的盐。
21.如条款3至20中任一项的方法,其中该第四剂量递增顺序包含在分开的三天向该患者施用约1%、约10%及约100%的全剂量的该化合物或其药学上可接受的盐。
22.如条款4至21中任一项的方法,其中该第五剂量递增顺序包含在分开的三天向该患者施用约1%、约30%及约300%的全剂量的该化合物或其药学上可接受的盐。
23.如条款5至22中任一项的方法,其中该第六剂量递增顺序包含在分开的三天向该患者施用约1%、约50%及约500%的全剂量的该化合物或其药学上可接受的盐。
24.如条款18至23中任一项的方法,其中该全剂量的该化合物或其药学上可接受的盐为约10µg/kg至约50µg/kg的该化合物或其药学上可接受的盐。
25.如条款18至24中任一项的方法,其中该全剂量的该化合物或其药学上可接受的盐为约20µg/kg至约40µg/kg的该化合物或其药学上可接受的盐。
26.如条款18至25中任一项的方法,其中该全剂量的该化合物或其药学上可接受的盐为约25µg/kg至约35µg/kg的该化合物或其药学上可接受的盐。
27.如条款18至26中任一项的方法,其中该全剂量的该化合物或其药学上可接受的盐为约30µg/kg的该化合物或其药学上可接受的盐。
28.如条款6至27中任一项的方法,其中在第1周期间向该患者施用该第一剂量的所述cart细胞及该第二剂量的所述cart细胞。
29.如条款6至28中任一项的方法,其中在第1周的星期一及星期四向该患者施用该第一剂量的所述cart细胞及该第二剂量的所述cart细胞。
30.如条款1至29中任一项的方法,其中用于该化合物或其药学上可接受的盐的该第一剂量递增顺序出现在第2周及第3周期间。
31.如条款1至30中任一项的方法,其中用于该化合物或其药学上可接受的盐的该第二剂量递增顺序出现在第4周及第5周期间。
32.如条款2至31中任一项的方法,其中用于该化合物或其药学上可接受的盐的该第三剂量递增顺序出现在第6周及第7周期间。
33.如条款30的方法,其中在分开的三天施用该化合物或其药学上可接受的盐,且该分开的三天为第2周的星期一及星期四及第3周的星期一。
34.如条款31的方法,其中在分开的三天施用该化合物或其药学上可接受的盐,且该分开的三天为第4周的星期一及星期四及第5周的星期一。
35.如条款32的方法,其中在分开的三天施用该化合物或其药学上可接受的盐,且该分开的三天为第6周的星期一及星期四及第7周的星期一。
36.如条款1至35中任一项的方法,其中在向该患者施用该cart细胞组合物之前耗尽该患者中的淋巴细胞。
37.如条款1至36中任一项的方法,其进一步包含向该患者施用血小板,向该患者施用浓缩红细胞,向该患者施用冷沉淀物,向该患者施用静脉内免疫球蛋白及/或向该患者提供抗菌剂疗法。
38.如条款1至37中任一项的方法,其中如果在该第一剂量递增顺序期间在患者中未观测到crs或神经毒性,则该方法前进至该第二剂量递增顺序。
39.如条款2至38中任一项的方法,其中如果在该第二剂量递增顺序期间在该患者中未观测到crs或神经毒性,则该方法前进至该第三剂量递增顺序。
40.如条款3至39中任一项的方法,其中如果在该第三剂量递增顺序期间在该患者中未观测到crs或神经毒性,则该方法前进至该第四剂量递增顺序。
41.如条款4至40中任一项的方法,其中如果在该第四剂量递增顺序期间在该患者中未观测到crs或神经毒性,则该方法前进至该第五剂量递增顺序。
42.如条款5至41中任一项的方法,其中如果在该第五剂量递增顺序期间在该患者中未观测到crs或神经毒性,则该方法前进至该第六剂量递增顺序。
43.如条款1至42中任一项的方法,其中如果在所述剂量递增顺序中的任一者期间在该患者中观测到发热而无低血压且未观测到神经毒性,则以导致该发热而无低血压的该剂量递增顺序水平向该患者施用所有后续剂量的该化合物或其药学上可接受的盐。
44.如条款1至43中任一项的方法,其中如果在任何剂量递增顺序中在该患者中出现严重crs或神经毒性,则以低于导致该患者中的严重crs或神经毒性的该剂量递增顺序水平的剂量递增顺序水平向该患者施用所有后续剂量的该化合物或其药学上可接受的盐。
45.一种治疗癌症的方法,该方法包括:
i)向患者施用至少一个剂量的包含cart细胞的cart细胞组合物,其中所述cart细胞包含指向靶向部分的car;
ii)向该患者施用化合物或其药学上可接受的盐,其中该化合物包含通过接头连接于靶向部分的小分子配体,且其中该化合物或其药学上可接受的盐以第一剂量递增顺序施用,其中如果在该第一剂量递增顺序中出现严重crs,则使用较低剂量递增顺序施用该化合物或其药学上可接受的盐,其中该较低剂量递增顺序中的该化合物或其药学上可接受的盐的该第一剂量低于以该第一剂量递增顺序施用的该化合物或其药学上可接受的盐的该第一剂量。
46.如条款45的方法,其中在该较低剂量递增顺序中,在分开的三天以全剂量的该化合物或其药学上可接受的盐的约0.5%、约5%及约50%施用该化合物或其药学上可接受的盐。
47.如条款46的方法,其中该化合物或其药学上可接受的盐的该全剂量是该化合物或其药学上可接受的盐的大约全剂量,是约10µg/kg至约50µg/kg的该化合物或其药学上可接受的盐。
48.如条款46至47中任一项的方法,其中该全剂量的该化合物或其药学上可接受的盐为约25µg/kg至约35µg/kg的该化合物或其药学上可接受的盐。
49.如条款46至48中任一项的方法,其中该全剂量的该化合物或其药学上可接受的盐为约30µg/kg的该化合物或其药学上可接受的盐。
50.如条款1至49中任一项的方法,其中该配体选自:叶酸化合物、dupa、nk-1r配体、caix配体、γ谷氨酰基转肽酶配体、nkg2d配体及cck2r配体。
51.如条款1至50中任一项的方法,其中该配体为叶酸化合物。
52.如条款1至50中任一项的方法,其中该配体为nk-1r配体。
53.如条款1至50中任一项的方法,其中该配体为dupa。
54.如条款1至50中任一项的方法,其中该配体为cck2r配体。
55.如条款1至50中任一项的方法,其中该配体为γ谷氨酰基转肽酶配体。
56.如条款1至55中任一项的方法,其中该靶向部分选自:2,4-二硝基苯酚(dnp)、2,4,6-三硝基苯酚(tnp)、生物素、地高辛、荧光素、荧光素异硫氰酸酯(fitc)、nhs-荧光素、五氟苯基酯、四氟苯基酯、打结素、赛丁素及darpin。
57.如条款1至56中任一项的方法,其中该靶向部分为fitc。
58.如条款1至56中任一项的方法,其中该靶向部分为dnp。
59.如条款1至56中任一项的方法,其中该靶向部分为tnp。
60.如条款1至59中任一项的方法,其中该接头包含聚乙二醇(peg)、聚脯氨酸、亲水性氨基酸、糖、非天然肽聚糖、聚乙烯吡咯啶酮、普洛尼克(pluronic)f-127或其组合。
61.如条款1至60中任一项的方法,其中该接头包含peg。
62.如条款1至61中任一项的方法,其中该化合物或其药学上可接受的盐具有下式:
其中b表示该小分子配体,l表示该接头,且t表示该靶向部分,且其中l包含具有下式的结构:
其中n为0至200的整数。
63.如条款62的方法,其中n为0至150的整数。
64.如条款62的方法,其中n为0至110的整数。
65.如条款62的方法,其中n为0至20的整数。
66.如条款62的方法,其中n为15至20的整数。
67.如条款62的方法,其中n为15至110的整数。
68.如条款1至67中任一项的方法,其中该癌症选自:肺癌、骨癌、胰脏癌、皮肤癌、头部癌、颈部癌、皮肤黑素瘤、眼内黑色素瘤、子宫癌、卵巢癌、子宫内膜癌(endometrialcancer)、直肠癌、胃癌、结肠癌、乳腺癌、三阴性乳腺癌、输卵管癌、子宫内膜癌(carcinomaoftheendometrium)、子宫颈癌、阴道癌、外阴癌、霍奇金氏病(hodgkin'sdisease)、食道癌、小肠癌、内分泌系统癌、甲状腺癌、副甲状腺癌、非小细胞肺癌、肾上腺癌、软组织肉瘤、骨肉瘤(包括儿科或非儿科骨肉瘤)、尿道癌、前列腺癌、慢性白血病、急性白血病、急性骨髓细胞性白血病、淋巴细胞性淋巴瘤、骨髓白血病、骨髓单核球性白血病、毛细胞白血病、胸膜间皮瘤、膀胱癌、伯基特氏淋巴瘤(burkitt'slymphoma)、尿管癌、肾脏癌、肾细胞癌、肾盂癌、中枢神经系统(cns)赘生物、原发性cns淋巴瘤、脊轴肿瘤、脑干神经胶质瘤、垂体腺瘤、及胃食道接合点的腺癌。
69.如条款1至51或56至68中任一项的方法,其中该癌症为表达叶酸受体的癌症。
70.如条项1至69中任一项的方法,其中该癌症为骨肉瘤。
71.如条款1至70中任一项的方法,其中该car具有识别区且该识别区为抗体的单链片段可变(scfv)区。
72.如条款1至57或60至71中任一项的方法,其中该car具有识别区且该car的该识别区为抗fitc抗体的单链片段可变(scfv)区。
73.如条款1至72中任一项的方法,其中该car具有共刺激结构域且该共刺激结构域选自:cd28、cd137(4-1bb)、cd134(ox40)及cd278(icos)。
74.如条款1至73中任一项的方法,其中该car具有活化信号传导结构域且该活化信号传导结构域为t细胞cd3ζ链或fc受体γ。
75.如条款1至57或60至74中任一项的方法,其中该car具有识别区且该识别区为抗fitc抗体的单链片段可变(scfv)区,其中该car具有共刺激结构域且该共刺激结构域为cd137(4-1bb),且其中该car具有活化信号传导结构域且该活化信号传导结构域为t细胞cd3ζ链。
76.如条款1至75中任一项的方法,其中在施用该化合物或其药学上可接受的盐之前,或在施用该cart细胞组合物之前,对该患者成像。
77.如条款1至76中任一项的方法,其中该化合物或其药学上可接受的盐不为抗体且不包含抗体的片段。
78.如条款1至77中任一项的方法,其中该靶向部分不包含肽表位。
79.如条款1至78中任一项的方法,其中不出现在患者中导致脱靶毒性的细胞因子释放,且其中出现对该癌症的cart细胞毒性。
80.如条款1至78中任一项的方法,其中不出现在患者中的脱靶组织毒性,且其中出现对该癌症的cart细胞毒性。
81.如条款1至78中任一项的方法,其中该癌症包含肿瘤,其中肿瘤大小在该患者体内减小,且其中不出现脱靶毒性。
82.如条款1至81中任一项的方法,其中所述cart细胞包含含有seqidno:1的核酸。
83.如条款1至82中任一项的方法,其中所述cart细胞包含含有seqidno:2的多肽。
84.如条款82的方法,其中该核酸编码嵌合抗原受体。
85.如条款1至81中任一项的方法,其中所述cart细胞包含含有seqidno:4的核酸。
86.如条款1至81或85中任一项的方法,其中所述cart细胞包含含有seqidno:5的多肽。
87.如条款85的方法,其中该核酸编码嵌合抗原受体。
88.如条款1至87中任一项的方法,其中该car包含人源化氨基酸序列。
89.如条款1至87中任一项的方法,其中该car由人源化氨基酸序列组成。
90.如条款1至89中任一项的方法,其进一步包含向该患者施用叶酸化合物;包含叶酸化合物的缀合物,其中包含叶酸化合物的该缀合物不包含靶向部分;或抑制所述cart细胞活化的药剂。
91.如条款90的方法,其中施用叶酸化合物。
92.如条款90的方法,其中施用叶酸或甲酰四氢叶酸。
93.如条款90的方法,其中施用包含叶酸化合物的该缀合物。
94.如条款93的方法,其中包含叶酸化合物的该缀合物包含连接于一或多个氨基酸的叶酸。
95.如条款93的方法,其中包含叶酸化合物的该缀合物具有下式:
96.如条款91的方法,其中该叶酸化合物具有下式:
其中x1及y1各自独立地选自:卤素、r2、or2、sr3及nr4r5;
u、v及w表示各自独立地选自以下的二价部分:-(r6a)c=、-n=、-(r6a)c(r7a)-及-n(r4a)-;q选自:c及ch;t选自:s、o、n及-c=c-;
x2及x3各自独立地选自:氧、硫、-c(z)-、-c(z)o-、-oc(z)-、-n(r4b)-、-c(z)n(r4b)-、-n(r4b)c(z)-、-oc(z)n(r4b)-、-n(r4b)c(z)o-、-n(r4b)c(z)n(r5b)-、-s(o)-、-s(o)2-、-n(r4a)s(o)2-、-c(r6b)(r7b)-、-n(c≡ch)-、-n(ch2c≡ch)-、c1-c12亚烷基及c1-c12亚烷氧基,其中z为氧或硫;
r1选自:氢、卤素、c1-c12烷基及c1-c12烷氧基;
r2、r3、r4、r4a、r4b、r5、r5b、r6b及r7b各自独立地选自:氢、卤素、c1-c12烷基、c1-c12烷氧基、c1-c12烷酰基、c1-c12烯基、c1-c12炔基、(c1-c12烷氧基)羰基及(c1-c12烷基氨基)羰基;
r6及r7各自独立地选自:氢、卤素、c1-c12烷基及c1-c12烷氧基;或r6及r7一起形成羰基;
r6a及r7a各自独立地选自:氢、卤素、c1-c12烷基及c1-c12烷氧基;或r6a与r7a一起形成羰基;
p、r、s及t各自独立地为0或1;且
如果任何其他化学部分为该叶酸化合物的部分,则*表示与该缀合物的其余部分的任选的共价键。
97.如条款90的方法,其中抑制所述cart细胞活化的该药剂选自:淋巴细胞特异性蛋白质酪氨酸激酶抑制剂、pi3激酶抑制剂、il-2诱导性t细胞激酶抑制剂、jak抑制剂、btk抑制剂、ec2319及阻断cart细胞与该化合物或其药学上可接受的盐结合但不与该癌症结合的药剂。
98.如条款90的方法,其中施用抑制所述cart细胞活化的该药剂且该药剂为淋巴细胞特异性蛋白质酪氨酸激酶抑制剂。
99.如条款98的方法,其中该淋巴细胞特异性蛋白质酪氨酸激酶抑制剂为达沙替尼(dasatinib)。
100.如条款90的方法,其中施用抑制所述cart细胞活化的该药剂且该药剂为pi3激酶抑制剂。
101.如条款100的方法,其中该pi3激酶抑制剂为gdc0980。
102.如条款90的方法,其中施用抑制所述cart细胞活化的该药剂且该药剂为il-2诱导性t细胞激酶抑制剂。
103.如条款102的方法,其中该il-2诱导性t细胞激酶抑制剂为bms-509744。
104.如条款90的方法,其中抑制所述cart细胞的活化的该药剂经施用,且为阻断cart细胞与该化合物或其药学上可接受的盐结合但不与该癌症结合的药剂。
105.如条款104的方法,其中该药剂为荧光素胺、fitc或荧光素钠。
106.如条款105的方法,其中该药剂为荧光素钠。
107.如条款90至106中任一项的方法,其中施用该叶酸化合物、包含叶酸化合物的该缀合物(其中包含叶酸化合物的该缀合物不包含靶向部分)或抑制所述cart细胞活化的该药剂使得该患者中的细胞因子水平降低。
108.如条款107的方法,其中该细胞因子水平降低为降低至约未经治疗的患者中的细胞因子水平。
109.如条款90至108中任一项的方法,其中该癌症包含肿瘤且在向该患者施用该叶酸化合物、包含叶酸化合物的该缀合物(其中包含叶酸化合物的该缀合物不包含靶向部分)或抑制所述cart细胞活化的该药剂时,该患者中的肿瘤大小未增加。
110.如条款104至106中任一项的方法,当该crs级别达到1、2、3或4时,向该患者施用抑制所述cart细胞活化的该药剂。
111.如条款110的方法,其中当该crs级别达到3或4时,向该患者施用抑制所述cart细胞活化的该药剂。
112.如条款104至106中任一项的方法,其中以约0.01至约300微摩尔/公斤该患者体重的剂量施用抑制所述cart细胞活化的该药剂。
113.如条款1至112中任一项的方法,其中在该患者体内减轻或预防crs且该方法引起该患者中的肿瘤体积减小。
114.如条款1至113中任一项的方法,其中减轻或预防由crs所致的体重损失。
115.如条款90至114中任一项的方法,其进一步包含向该患者再施用该化合物或其药学上可接受的盐。
116.如条款115的方法,其中该化合物或其药学上可接受的盐的该后续施用致使cart细胞活化及该患者中的细胞因子水平增加。
117.如条款1至116中任一项的方法,其中在该化合物或其药学上可接受的盐前施用该cart细胞组合物。
118.如条款1至117中任一项的方法,其中所述cart细胞为自体性的。
119.如条款2至17中任一项的方法,其中该第一剂量递增顺序包含在分开的三天向该患者施用约1%、约2%及约20%的全剂量的该化合物或其药学上可接受的盐,其中该第二剂量递增顺序包含在分开的三天向该患者施用约1%、约6%及约60%的全剂量的该化合物或其药学上可接受的盐,且其中该第三剂量递增顺序包含在分开的三天向该患者施用约1%、约10%及约100%的全剂量的该化合物或其药学上可接受的盐。
120.如条款2至17中任一项的方法,其中该第一剂量递增顺序包含在分开的三天向该患者施用约1%、约10%及约100%的全剂量的该化合物或其药学上可接受的盐,且其中该第二剂量递增顺序包含在分开的三天向该患者施用约1%、约20%及约200%的全剂量的该化合物或其药学上可接受的盐。
121.如条款2至17中任一项的方法,其中该第一剂量递增顺序包含在分开的三天向该患者施用约1%、约10%及约100%的全剂量的该化合物或其药学上可接受的盐,且其中该第二剂量递增顺序包含在分开的三天向该患者施用约1%、约10%及约100%的全剂量的该化合物或其药学上可接受的盐。
122.如条款2至17中任一项的方法,其中该第一剂量递增顺序包含在分开的三天向该患者施用约1%、约20%及约200%的全剂量的该化合物或其药学上可接受的盐,且其中该第二剂量递增顺序包含在分开的三天向该患者施用约1%、约20%及约200%的全剂量的该化合物或其药学上可接受的盐。
123.如条款119至122中任一项的方法,其中该化合物或其药学上可接受的盐的该全剂量为约500纳摩尔/公斤。
因此,前述段落及以上条款清单中提供各种实施方案,且此“说明性实施方案的详细描述”、发明内容部分、实施例及权利要求书中描述的所有适用实施方案均适用于这些实施方案。
如本文所述,“患者”可为人类,或在兽医学应用的情况下,患者可为实验室动物、农畜、家畜或野生动物。在各个方面,患者可为实验室动物,诸如啮齿动物(例如小鼠、大鼠、仓鼠等)、兔、猴、黑猩猩、家畜(诸如犬、猫或兔)、农畜(诸如牛、马、猪、绵羊、山羊)或笼养野生动物,诸如熊、熊猫、狮子、虎、豹、大象、斑马、长颈鹿、大猩猩、海豚或鲸。
在各种实施方案中,待治疗的癌症可选自癌、肉瘤、淋巴瘤、黑色素瘤、间皮瘤、鼻咽癌、白血病、腺癌或骨髓瘤。在其他实施方案中,癌症可为肺癌、骨癌、胰腺癌、皮肤癌、头癌、颈癌、皮肤黑素瘤、眼内黑素瘤、子宫癌、卵巢癌、子宫内膜癌、直肠癌、胃癌、结肠癌、乳腺癌、三阴性乳腺癌、输卵管癌、子宫内膜癌、子宫颈癌、阴道癌、外阴癌、霍奇金氏病、食道癌、小肠癌、内分泌系统癌、甲状腺癌、副甲状腺癌、非小细胞肺癌、肾上腺癌、软组织肉瘤、骨肉瘤(包括儿科或非儿科骨肉瘤)、尿道癌、前列腺癌、慢性白血病、急性白血病(包括急性骨髓细胞性白血病)、淋巴细胞性淋巴瘤、骨髓白血病、骨髓单核细胞性白血病、毛细胞白血病、胸膜间皮瘤、膀胱癌、伯基特氏淋巴瘤、输尿管癌、肾癌、肾细胞癌、肾盂癌、中枢神经系统(cns)赘生物、原发性cns淋巴瘤、脊轴肿瘤、脑干神经胶质瘤、垂体腺瘤及胃食道结合部的腺癌。
在这些实施方案的一些方面,癌症为表达叶酸受体的癌症。在另一实施方案中,癌症为表达叶酸受体α的癌症。在另一实施方案中,癌症为表达叶酸受体β的癌症。在这些实施方案的一些方面,癌症为子宫内膜癌、非小细胞肺癌、卵巢癌、骨肉瘤(包括儿科或非儿科骨肉瘤)或三阴性乳腺癌。在另一实施方案中,所治疗的癌症为肿瘤。在另一实施方案中,癌症为恶性的。在另一实施方案中,癌症为骨肉瘤,包括儿科或非儿科骨肉瘤。
在一个实施方案中,“小分子配体”可为叶酸化合物、dupa(由psma阳性人类前列腺癌细胞及其他癌细胞类型结合的配体)、nk-1r配体(例如在结肠及胰脏的癌症上发现nk-1r配体的受体)、caix配体(例如在肾癌、卵巢癌、外阴癌及乳腺癌上发现caix配体的受体)、γ谷氨酰基转肽酶的配体(该转肽酶例如在卵巢癌、结肠癌、肝癌、星形胶质细胞瘤、黑色素瘤及白血病中过表达)、nkg2d配体(例如在肺癌、结肠癌、肾癌、前列腺癌上及在t细胞及b细胞淋巴瘤上发现nkg2d配体的受体)或cck2r配体(尤其在甲状腺、肺、胰脏、卵巢、脑、胃、胃肠道基质及结肠的癌症上发现cck2r配体的受体),其中每一种为特异性地结合于癌细胞类型的小分子配体(即与正常组织相比,这些配体的每一种的受体在癌症上过表达)。
在一个实施方案中,小分子配体可具有低于约10,000道尔顿、低于约9000道尔顿、低于约8,000道尔顿、低于约7000道尔顿、低于约6000道尔顿、低于约5000道尔顿、低于约4500道尔顿、低于约4000道尔顿、低于约3500道尔顿、低于约3000道尔顿、低于约2500道尔顿、低于约2000道尔顿、低于约1500道尔顿、低于约1000道尔顿或低于约500道尔顿的质量。在另一实施方案中,小分子配体可具有约1至约10,000道尔顿、约1至约9000道尔顿、约1至约8,000道尔顿、约1至约7000道尔顿、约1至约6000道尔顿、约1至约5000道尔顿、约1至约4500道尔顿、约1至约4000道尔顿、约1至约3500道尔顿、约1至约3000道尔顿、约1至约2500道尔顿、约1至约2000道尔顿、约1至约1500道尔顿、约1至约1000道尔顿或约1至约500道尔顿的质量。
在一个实施方案中,dupa衍生物可为连接于靶向部分的小分子配体的配体,且dupa衍生物描述于以引用的方式并入本文中的wo2015/057852中。
在一个实施方案中,在“连接于接头的小分子配体”的上下文中的小分子配体为叶酸化合物。在各种实施方案中,叶酸化合物可为叶酸、叶酸类似物或另一叶酸受体结合分子。在各种实施方案中,可使用的叶酸类似物包括亚叶酸(例如甲酰四氢叶酸)、喋酰聚谷氨酸及叶酸受体结合喋啶,诸如四氢喋呤、二氢叶酸、四氢叶酸及其脱氮及二脱氮类似物。术语“脱氮”及“二脱氮”类似物是指由碳原子取代天然存在的叶酸结构中的一或两个氮原子的本领域所知的类似物。例如,脱氮类似物包括1-脱氮、3-脱氮、5-脱氮、8-脱氮及10-脱氮类似物。二脱氮类似物包括例如1,5-二脱氮、5,10-二脱氮、8,10-二脱氮及5,8-二脱氮类似物。上述叶酸类似物一般称为“叶酸化合物”,反映其结合叶酸受体的能力。其他叶酸受体结合类似物包括氨基喋呤、氨甲喋呤(甲氨喋呤)、n10-甲基叶酸、2-脱氨基-羟基叶酸、脱氮类似物(诸如1-脱氮氨甲喋呤或3-脱氮氨甲喋呤)及3',5'-二氯-4-氨基-4-脱氧-n10-甲基喋酰谷氨酸(二氯甲氨喋呤)。
在另一实施方案中,在“连接于接头的小分子配体”的上下文中的小分子配体可具有下式:
其中x1及y1各自独立地选自:卤素、r2、or2、sr3及nr4r5;
u、v及w表示各独立地选自以下的二价部分:-(r6a)c=、-n=、-(r6a)c(r7a)-及-n(r4a)-,q选自:c及ch;t选自:s、o、n及-c=c-;
x2及x3各自独立地选自:氧、硫、-c(z)-、-c(z)o-、-oc(z)-、-n(r4b)-、-c(z)n(r4b)-、-n(r4b)c(z)-、-oc(z)n(r4b)-、-n(r4b)c(z)o-、-n(r4b)c(z)n(r5b)-、-s(o)-、-s(o)2-、-n(r4a)s(o)2-、-c(r6b)(r7b)-、-n(c≡ch)-、-n(ch2c≡ch)-、c1-c12亚烷基及c1-c12亚烷氧基,其中z为氧或硫;
r1选自:氢、卤素、c1-c12烷基及c1-c12烷氧基;
r2、r3、r4、r4a、r4b、r5、r5b、r6b及r7b各自独立地选自:氢、卤素、c1-c12烷基、c1-c12烷氧基、c1-c12烷酰基、c1-c12烯基、c1-c12炔基、(c1-c12烷氧基)羰基及(c1-c12烷基氨基)羰基;
r6及r7各自独立地选自:氢、卤素、c1-c12烷基及c1-c12烷氧基;或r6及r7一起形成羰基;
r6a及r7a各自独立地选自:氢、卤素、c1-c12烷基及c1-c12烷氧基;或r6a及r7a一起形成羰基;
p、r、s及t各自独立地为0或1;以及
如果任何其他化学部分为该叶酸化合物的一部分,则*表示与该缀合物的其余部分的任选的共价键。
在一个方面,结合于由cart细胞表达的car的“靶向部分”可选自例如2,4-二硝基苯酚(dnp)、2,4,6-三硝基苯酚(tnp)、生物素、地高辛、荧光素、荧光素异硫氰酸酯(fitc)、nhs-荧光素、五氟苯基酯、四氟苯基酯、打结素、赛丁素、darpin、亲和体(affibody)、affilin、抗运载蛋白(anticalin)、atrimer、avimer、双环肽、fn3骨架、cys-结、fynomer、kunitz结构域或obody。靶向部分的属性仅限于其应当由car识别且与car结合(较佳地特异性结合)且其具有相对较低分子量。在各个方面,示例性靶向部分为半抗原,包括小分子量有机分子。
在一个说明性实施方案中,靶向部分可具有以下说明性结构:
其中x为氧、氮或硫,且其中x附接至接头l;y为ora、nra2或nra3+;且y'为o、nra或nra2+;其中在各情况下各r独立地选自h、氟、磺酸、磺酸酯及其盐及其类似物;且ra为氢或烷基。
在一个说明性方面,接头可包含聚乙二醇(peg)、聚脯氨酸、亲水性氨基酸、糖、非天然肽聚糖、聚乙烯吡咯啶酮、普洛尼克f-127或其组合。
在另一说明性方面,本文所述的化合物或其药学上可接受的盐中的接头可包含直接连接(例如fitc的异硫氰酸酯基团与小分子配体的游离氨基之间的反应)或连接可经由中间接头。在一个实施方案中,如果存在,则中间接头可为本领域中已知的任何生物相容性接头,诸如二价接头。在一个说明性实施方案中,二价接头可包含约1至约30个碳原子。在另一说明性实施方案中,二价接头可包含约2至约20个碳原子。在其他实施方案中,采用较低分子量二价接头(即,具有约30至约300道尔顿的近似分子量的那些接头)。在另一实施方案中,适合的接头长度包括(但不限于)具有2个、3个、4个、5个、6个、7个、8个、9个、10个、11个、12个、13个、14个、15个、16个、17个、18个、19个、20个、21个、22个、23个、24个、25个、26个、27个、28个、29个、30个、31个、32个、33个、34个、35个、36个、37个、38个、39个或40个或更多个原子的接头。
在各种实施方案中,连接于靶向部分的小分子配体可具有下式:
其中b表示该小分子配体,l表示该接头,且t表示该靶向部分,且其中l包含具有下式的结构:
其中n为0至200的整数。在另一实施方案中,n可为0至150、0至110、0至100、0至90、0至80、0至70、0至60、0至50、0至40、0至30、0至20、0至15、0至14、0至13、0至12、0至11、0至10、0至9、0至8、0至7、0至6、0至5、0至4、0至3、0至2、0至1、15至16、15至17、15至18、15至19、15至20、15至21、15至22、15至23、15至24、15至25、15至26、15至27、15至28、15至29、15至30、15至31、15至32、15至33、15至34、15至35、15至36、15至37、15至38、15至39、15至40、15至50、15至60、15至70、15至80、15至90、15至100、15至110、15至120、15至130、15至140、15至150的整数,或n可为1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、50、60、70、80、90、100、108、110、120、130、140或150。
在另一实施方案中,接头可为可包括一或多个间隔子的二价接头。说明性间隔子显示于下表中。描述以下非限制性示例性间隔子,其中*指示与小分子配体或与靶向部分或与其他二价接头部分的连接点。
在其他实施方案中,连接于靶向部分的小分子配体(桥)可具有任何的以下结构。
在其他实施方案中,化合物或其药学上可接受的盐并非抗体,且不包含抗体片段。在又另一实施方案中,靶向部分不包含肽表位。
在一个说明性实施方案中,通过接头连接于靶向部分的小分子配体(桥)包含连接于小分子配体的荧光素异硫氰酸酯(fitc)。在一个方面,癌症可过表达小分子配体的受体。在另一方面,例如可转化细胞毒性t细胞或另一类型t细胞以表达包含抗fitcscfv的car。在此方面,作为小分子配体与癌症结合的结果,car可靶向以fitc分子装饰癌症的fitc。因此,可避免或降低对正常的非靶细胞的毒性。在此实施方案中,当表达抗fitccar的t细胞结合fitc时,cart细胞活化且改善癌症。
涵盖通过接头连接于靶向部分的小分子配体的“药学上可接受的盐”。如本文所用,术语“药学上可接受的盐”是指其抗衡离子可用于药物中的那些盐。在各种实施方案中,此类盐包括(但不限于):1)酸加成盐,其可由母体化合物的游离碱与无机酸的反应获得,所述无机酸为诸如盐酸、氢溴酸、硝酸、磷酸、硫酸及过氯酸及其类似物;或由母体化合物的游离碱与有机酸的反应形成,所述有机酸为诸如乙酸、乙二酸、(d)或(l)苹果酸、顺丁烯二酸、甲磺酸、乙磺酸、对甲苯磺酸、水杨酸、酒石酸、柠檬酸、丁二酸或丙二酸及其类似物;或(2)当母体化合物中存在的酸性质子由金属离子(例如碱金属离子、碱土金属离子或铝离子)置换时所形成的盐;或当母体化合物中存在的酸性质子与有机碱配位而形成的盐,该有机碱为诸如乙醇胺、二乙醇胺、三乙醇胺、三羟甲基氨基甲烷、n-甲基葡糖胺及其类似物。药学上可接受的盐为本领域技术人员所熟知,且涵盖与本文所述的实施方案有关的任何此类药学上可接受的盐。
在各种实施方案中,合适酸加成盐由形成无毒盐的酸形成。说明性实例包括乙酸盐、天冬氨酸盐、苯甲酸盐、苯磺酸盐、碳酸氢盐/碳酸盐、硫酸氢盐/硫酸盐、硼酸盐、樟脑磺酸盐、柠檬酸盐、乙二磺酸盐、乙磺酸盐、甲酸盐、反丁烯二酸盐、葡庚糖酸盐、葡糖酸盐、葡萄糖醛酸盐、六氟磷酸盐、海苯酸盐、盐酸盐/氯化物、氢溴酸盐/溴化物、氢碘酸盐/碘化物、羟乙磺酸盐、乳酸盐、苹果酸盐、顺丁烯二酸盐、丙二酸盐、甲磺酸盐、甲硫酸盐、萘酸盐、2-萘磺酸盐、烟酸盐、硝酸盐、乳清酸盐、草酸盐、棕榈酸盐、双羟萘酸盐、磷酸盐/磷酸氢盐/磷酸二氢盐、葡萄糖二酸盐、硬脂酸盐、丁二酸盐、酒石酸盐、甲苯磺酸盐及三氟乙酸盐。
在各种实施方案中,适合的碱盐由形成无毒盐的碱形成。说明性实例包括精氨酸、苄星青霉素、钙、胆碱、二乙胺、二乙醇胺、甘氨酸、赖氨酸、镁、葡甲胺、乙醇胺、钾、钠、三乙醇胺及锌的盐。亦可形成酸及碱的半盐,例如半硫酸盐及半钙盐。
在一个示例性方面,本文所述的化合物或其药学上的盐可含有一或多个手性中心,或可另外能够呈多个立体异构体存在。因此,各种实施方案可包括纯立体异构体以及立体异构体的混合物,诸如对映异构体、非对映异构体及对映异构性或非对映异构性富集的混合物。在一个方面,本文所述的化合物或其药学上可接受的盐能够呈几何异构体存在。因此,各种实施方案可包括纯几何异构体或几何异构体的混合物。
在一些方面,本文所述的化合物或其药学上可接受的盐可呈未溶剂化形式以及溶剂化形式,包括水合形式存在。一般而言,溶剂化形式等同于未溶剂化形式且涵盖于本发明的范围内。
本文所述的方法亦利用t淋巴细胞(例如细胞毒性t淋巴细胞),其经工程改造以表达识别且结合于桥的靶向部分(例如fitc、dnp或tnp)的嵌合抗原受体(car)。在一个实施方案中,本文中描述的car包含三个结构域,包括1)识别区(例如,抗体的单链片段可变(scfv)区、fab片段及其类似物),其识别且特异性结合于靶向部分,2)共刺激结构域,其增强t淋巴细胞的增殖及存活,3)活化信号传导结构域,其产生t淋巴细胞活化信号。
在各个方面,作为非限定性的实例,可使用结合以下的抗体的scfv区:2,4-二硝基苯酚(dnp)、2,4,6-三硝基苯酚(tnp)、生物素、地高辛、荧光素、荧光素异硫氰酸酯(fitc)、nhs-荧光素、五氟苯基酯、四氟苯基酯、打结素、赛丁素、darpin、亲和体、affilin、抗运载蛋白、atrimer、avimer、双环肽、fn3骨架、cys-结、fynomer、kunitz结构域或obody。在说明性非限制性实施方案中,scfv区可以下由制备:(i)结合靶向部分的本领域中已知的抗体,(ii)使用选定靶向部分(诸如半抗原)新制备的抗体及(iii)源自此类抗体的scfv区的序列变体,例如scfv区域具有与衍生出它们的scfv区域的氨基酸序列至少约80%、至少约90%、至少约91%、至少约92%、至少约93%、至少约94%、至少约95%、至少约96%、至少约97%、至少约98%、至少约99%或至少约99.5%序列同一性。
在一个方面,共刺激结构域用以在car结合于靶向部分后增强细胞毒性t淋巴细胞的增殖及存活。合适共刺激结构域包括(但不限于)cd28、cd137(4-1bb)、肿瘤坏死因子(tnf)受体家族的成员、cd134(ox40)、tnfr受体超家族的成员、cd27、cd30、cd150、dap10、nkg2d及cd278(icos)、在活化t细胞上表达的cd28超家族共刺激分子或其组合。本领域技术人员应了解可使用这些共刺激结构域的序列变体而没有不利地影响本发明,其中所述变体具有与改造出该变体的结构域相同或相似活性。在各种实施方案中,此类变体可与衍生出该变体的结构域的氨基酸序列具有至少约80%、至少约90%、至少约91%、至少约92%、至少约93%、至少约94%、至少约95%、至少约96%、至少约97%、至少约98%、至少约99%或至少约99.5%序列同一性。
在说明性实施方案中,活化信号传导结构域用以在car结合于靶向部分后活化t淋巴细胞(例如细胞毒性t淋巴细胞)。在各种实施方案中,适合的活化信号传导结构域包括t细胞cd3ζ链、cd3δ受体蛋白、mbl受体蛋白、b29受体蛋白及fc受体γ。技术人员应了解可使用这些活化信号传导结构域的序列变体,其中所述变体具有与改造出该变体的结构域相同或相似活性。在各种实施方案中,变体与衍生出该变体的结构域的氨基酸序列具有至少约80%、至少约90%、至少约91%、至少约92%、至少约93%、至少约94%、至少约95%、至少约96%、至少约97%、至少约98%、至少约99%或至少约99.5%序列同一性。
在一个方面,使用基因工程改造技术制备编码car的构建体。此类技术详细描述于以引用的方式并入本文中的sambrook等人,“molecularcloning:alaboratorymanual”,第3版,coldspringharborlaboratorypress,(2001)及以引用的方式并入本文中的greenandsambrook,“molecularcloning:alaboratorymanual”,第4版,coldspringharborlaboratorypress,(2012)中。
作为实例,可制备编码融合蛋白的质粒或病毒表达载体(例如,慢病毒载体、逆转录病毒载体、睡美人(sleepingbeauty)及附挂(piggyback)(包括非病毒介导的car基因递送系统的转位子/转位酶系统)),所述融合蛋白包含同框且在5'至3'方向上连接的识别区、一或多个共刺激结构域及活化信号传导结构域。在其他实施方案中,其他配置为可接受的,且包括识别区、活化信号传导结构域及一或多个共刺激结构域。在一个实施方案中,融合蛋白中的识别区的置放将通常使得实现该区域在cart细胞的外部展示。在一个实施方案中,car可包括其他组件,诸如确保融合蛋白适当输出至细胞表面的信号肽(例如cd8α信号肽);确保融合蛋白作为整合膜蛋白维持的跨膜结构域(例如cd8α跨膜结构域、cd28跨膜结构域或cd3ζ跨膜结构域)及赋予识别区的灵活性且允许强烈结合于靶向部分的铰链结构域(例如cd8α铰链)。
示例性car的图式显示于图2及3中。对于图2,可将融合蛋白序列并入至慢病毒表达载体中,其中“sp”为信号肽,car为抗fitccar,存在cd8α铰链及cd8α跨膜结构域,共刺激结构域为4-1bb且活化信号传导结构域为cd3ζ。car插入物的示例性核酸序列提供为seqidno:1及3且经编码氨基酸序列提供为seqidno:2。在又一实施方案中,seqidno:2可包含人源化或人类氨基酸序列或由人源化或人类氨基酸序列组成。
对于图3,展示示例性car构建体的图式,其中经表达car包含e2抗荧光素抗体片段,其中可将融合蛋白序列并入至表达载体中且其中car包含e2抗荧光素抗体片段、igg4铰链结构域、cd28跨膜结构域,且其中共刺激结构域为cd137(4-1bb),且活化信号传导结构域为cd3ζ。car可包含额外适合结构域。此car插入物的示例性核酸序列提供为seqidno:4且示例性经编码氨基酸序列提供为seqidno:5。如本文所用,“seqidno:4”意指开始于带下划线的“agc”密码子处且终止于带下划线的“ggc”密码子的序列。更长序列的此部分编码插入至t细胞膜中的car。较长序列的其他部分包括信号肽、egfrt域等的编码序列,其不是插入至膜中且充当嵌合抗原受体的car的部分。如本文中所使用,“seqidno:5”意指开始于带下划线的“s”处且终止于带下划线的“g”的序列。较长序列的此部分为插入至t细胞膜中的car的氨基酸序列。较长序列的其他部分包括信号肽、egfrt结构域等的氨基酸序列,其不是插入至膜中且充当嵌合抗原受体的car的部分。在又一实施方案中,seqidno:5可包含人源化或人类氨基酸序列或由人源化或人类氨基酸序列组成。seqidno:4及5如上文所描述且显示如下。较长核酸序列中的开始及终止密码子为带下划线的且较长序列为可用于转导用于如本文所描述的方法的t细胞的示例性序列。
seqidno:4(e2抗荧光素抗体片段car核酸序列(插入物))
seqidno:5(e2抗荧光素抗体片段car氨基酸序列(插入物))
另一示例性car构建体为概略地展示于图3中的4m5.3car。如本文中所使用,“seqidno:6”意指以下所示的开始于带下划线的“gac”密码子处且终止于带下划线的“ggc”密码子的序列。较长序列的此部分编码示例性4m5.3car。car插入至t细胞膜中。较长序列的其他部分包括信号肽、egfrt域等的编码序列,其不是插入至膜中且充当嵌合抗原受体的car的部分。如本文中所使用,“seqidno:7”意指开始于带下划线的“d”处且终止于带下划线的“g”的序列。较长序列的此部分为插入至t细胞膜中的car的氨基酸序列。较长序列的其他部分包括信号肽、egfrt域等的氨基酸序列,其不是插入至膜中且充当嵌合抗原受体的car的部分。在又另一实施方案中,seqidno:7可包含人源化或人类氨基酸序列或由人源化或人类氨基酸序列构成。seqidno:6及seqidno:7如上文所描述且其在下文展示。较长核酸序列中的起始密码子及终止密码子带下划线,且较长序列为可用于转导t细胞以制备4m5.3car的示例性序列。
seqidno:7[4m5.3-car氨基酸序列(插入物)]
seqidno:6[4m5.3-car核苷酸序列(插入序列)]
在一个实施方案中,car具有识别区且该识别区为抗fitc抗体的单链片段可变(scfv)区,具有共刺激结构域且该共刺激结构域为cd137(4-1bb),且具有活化信号传导结构域且该活化信号传导结构域为t细胞cd3ζ链。技术人员熟知抗fitcscfv与抗荧光素scfv为等同的术语。
在一个实施方案中,t淋巴细胞(例如细胞毒性t淋巴细胞)可通过用编码car构建体的表达载体转染t淋巴细胞群体而经基因工程改造以表达car构建体。适用于制备表达所选car构建体的t淋巴细胞的经转导群体的方法为技术人员所熟知,且描述于sambrook等人,“molecularcloning:alaboratorymanual”,第3版,coldspringharborlaboratorypress,(2001)(以引用的方式并入本文中)及green及sambrook,“molecularcloning:alaboratorymanual”,第4版,coldspringharborlaboratorypress,(2012)(以引用的方式并入本文中)中。
在一个实施方案中,提供包含seqidno:1、seqidno:3、seqidno:4或seqidno:6的核酸的cart细胞。在另一实施方案中,提供包含seqidno:2、seqidno:5或seqidno:7的多肽的cart细胞。在另一说明性方面,提供包含seqidno:1、seqidno:3、seqidno:4或seqidno:6且编码嵌合抗原受体的核酸(例如分离的核酸)。在又另一实施方案中,提供包含seqidno:2、seqidno:5或seqidno:7的嵌合抗原受体多肽。在另一实施方案中,提供包含seqidno:1、seqidno:3、seqidno:4或seqidno:6的载体。在另一方面,提供包含seqidno:1、seqidno:3、seqidno:4或seqidno:6的慢病毒载体。在又另一实施方案中,seqidno:2、seqidno:5或seqidno:7可包含人源化或人类氨基酸序列或由人源化或人类氨基酸序列构成。
在这些实施方案中的每一者中,涵盖与seqidno:1至seqidno:7具有至少约80%、至少约90%、至少约95%、至少约97%、至少约98%、至少约99%或至少约99.5%序列同一性的变体核酸序列或氨基酸序列。在另一实施方案中,核酸序列可为与seqidno:1、seqidno:3、seqidno:4或seqidno:6具有至少约80%、至少约90%、至少约95%、至少约97%、至少约98%、至少约99%或至少约99.5%序列同一性的变体核酸序列,只要该变体序列编码seqidno:2(针对seqidno:1及seqidno:3)、seqidno:5(针对seqidno:4)或seqidno:7(针对seqidno:6)的多肽即可。在另一实施方案中,核酸序列或氨基酸序列可以是沿200个核酸的一段与seqidno:1、seqidno:3、seqidno:4或seqidno:6或沿200个氨基酸的一段与seqidno:2、seqidno:5或seqidno:7具有至少约80%、至少约90%、至少约95%、至少约97%、至少约98%、至少约99%或至少约99.5%序列同一性的变体核酸或氨基酸序列。在一个实施方案中,可例如通过使用gap程序(geneticscomputergroup,软件;目前可在http://www.accelrys.com上经由accelrys获得)来实现对序列之间的同一性或相似性百分比的测定,且可使用(例如)clustalw算法(vnti软件,informax公司)来实现比对。可使用所关注的核酸或氨基酸序列来搜索序列数据库。数据库搜索算法典型地基于blast软件(altschul等人,1990)。在一些实施方案中,可沿核酸或氨基酸序列的全长来测定同一性百分比。
与由seqidno:1、seqidno:3、seqidno:4或seqidno:6表示的核酸互补的核酸及杂交至由seqidno:1、seqidno:3、seqidno:4或seqidno:6表示的核酸的那些核酸或在高度严格条件下杂交至其互补序列的那些核酸亦在本发明的范围内。根据本发明,“高度严格条件”意指在65℃下于5xsspe及50%甲酰胺中杂交,且在65℃下于0.5xsspe中洗涤。用于高度严格、低严格及中等严格杂交的条件描述于sambrook等人,“molecularcloning:alaboratorymanual”,第3版,coldspringharborlaboratorypress,(2001)(以引用的方式并入本文中)及green及sambrook,“molecularcloning:alaboratorymanual”,第4版,coldspringharborlaboratorypress,(2012)(以引用的方式并入本文中)。在一些说明性方面,杂交沿核酸的全长发生。
在一个实施方案中,尽管用于本文中所描述方法中的t淋巴细胞(例如用以制备cart细胞的细胞毒性t淋巴细胞)可为自体细胞,但亦可使用异源细胞,诸如当经治疗的患者已接受高剂量化疗或放射治疗而破坏患者的免疫系统时。在一个实施方案中,可使用同种异体细胞。
在一个方面,t淋巴细胞可通过本领域中熟知的方式自患者获得。例如,t细胞(例如细胞毒性t细胞)可通过以下获得:收集来自患者的外周血液,对血液进行菲科尔(ficoll)密度梯度离心,且随后使用阴性t细胞分离试剂盒(诸如easysep™t细胞分离试剂盒)以自外周血液分离t细胞群体。在一个说明性实施方案中,t淋巴细胞(例如细胞毒性t细胞)群体不必为纯净的,且可含有其他细胞,诸如其他类型的t细胞(在例如细胞毒性t细胞的情况下)、单核细胞、巨噬细胞、自然杀伤细胞及b细胞。在一个方面,所收集的群体可包含至少约90%的所选细胞类型、至少约91%、92%、93%、94%、95%、96%、97%、98%、99%或100%的所选细胞类型。
在一个实施方案中,在获得t淋巴细胞(例如用以制备cart细胞的细胞毒性t细胞)之后,在促进细胞活化的条件下培养所述细胞。在此实施方案中,培养条件可使得细胞可施用于患者,而不涉及针对培养基的组分的反应性。例如,培养条件可不包括牛血清产物,诸如牛血清白蛋白。在一个说明性方面,活化可通过在细胞毒性t细胞的情况下将诸如抗cd3抗体的已知活化剂引入至培养基中来达成。其他合适的活化剂包括抗cd28抗体。在一个方面,可在促进活化的条件下将淋巴细胞群体培养约1天至约4天。在一个实施方案中,适当活化水平可通过利用流式细胞术测定的细胞大小、增殖速率或活化标记物来测定。
在一个说明性实施方案中,在已于促进活化的条件下培养t淋巴细胞(例如用以制备cart细胞的细胞毒性t淋巴细胞)群体之后,所述细胞可经编码car的表达载体转染。上文描述用于各种实施方案中的合适的载体及转染方法。在一个方面,在转染之后,可立即向患者施用所述细胞,或可将细胞培养至少约1天、2天、3天、4天、5天、6天、7天、8天、9天、10天、11天、12天、13天、14天、15天、16天、17天、18天或更多天,或在约5天与约12天之间、在约6天与约13天之间、在约7天与约14天之间或在约8天与约15天之间,例如以为细胞自转染中恢复留出时间。在一个方面,合适的培养条件可类似于在具有或不具有用以促进活化的试剂下培养细胞以活化的条件。
因此,如上文所描述,在一个说明性方面,本文中所描述的治疗方法可进一步包含1)获得自体或异源t淋巴细胞(例如用以制备cart细胞的细胞毒性t淋巴细胞)的群体,2)在促进细胞活化的条件下培养t淋巴细胞,以及3)用编码car的表达载体转染淋巴细胞以形成cart细胞。
在一个实施方案中,可使用缺少任何动物产物(诸如牛血清)的培养基来培养cart细胞。在另一实施方案中,可使用技术人员典型地用以避免受细菌、真菌及支原体污染的组织培养条件。在一示例性实施方案中,在施用于患者之前,将细胞(例如cart细胞)沉淀,洗涤,且再悬浮于药学上可接受的载体或稀释剂中。包含car表达t淋巴细胞(例如细胞毒性t淋巴细胞)的示例性组合物包括在无菌290mosm生理盐水中、在可输注的冷冻介质(含有plasma-lytea、右旋糖、氯化钠注射剂、人类血清白蛋白及dmso)中、在具有2%人类血清白蛋白的0.9%nacl中或在任何其他无菌290mosm可输注物质中包含所述细胞的组合物。可替代地,在另一实施方案中,视培养基的属性而定,cart细胞可作为组合物在培养基中施用,或在施用之前经浓缩且再悬浮于培养基中。在各种实施方案中,可经由任何合适的手段来向患者施用cart细胞组合物,诸如胃肠外施用,例如皮内、皮下、肌肉内、腹膜内、静脉内或鞘内。
在一个方面,cart细胞的总数目及向患者施用的组合物中的细胞的浓度将取决于以下因素而变化,所述因素包括正在使用的t淋巴细胞(例如细胞毒性t淋巴细胞)的类型、car的结合特异性、靶向部分及小分子配体的属性、癌症的属性、癌症在患者中的部位、用以向患者施用组合物的手段以及正经治疗的患者的健康、年龄、体重。在各种实施方案中,包含经转导cart细胞的合适的组合物包括具有约0.1ml至约200ml与约0.1ml至约125ml的体积的那些。
在各种实施方案中,向患者施用的经转导cart细胞可包含约1×105至约1×1015或1×106至约1×1015个经转导cart细胞。在各种实施方案中,可向患者施用约1×105至约1×1010、约1×106至约1×1010、约1×106至约1×109、约1×106至约1×108、约1×106至约2×107、约1×106至约3×107、约1×106至约1.5×107、约1×106至约1×107、约1×106至约9×106、约1×106至约8×106、约1×106至约7×106、约1×106至约6×106、约1×106至约5×106、约1×106至约4×106、约1×106至约3×106、约1×106至约2×106、约2×106至约6×106、约2×106至约5×106、约3×106至约6×106、约4×106至约6×106、约4×106至约1×107、约1×106至约1×107、约1×106至约1.5×107、约1×106至约2×107、约0.2×106至约1×107、约0.2×106至约1.5×107、约0.2×106至约2×107、约0.2×106至约3×107、约0.2×106至约4×107、约0.2×106至约5×107、约0.2×105至约1.5×106、约0.5×105至约1.5×106、约0.2×105至约1.4×106、约0.2×105至约1.3×106、约0.5×105至约1.3×106、约0.8×106至约2×107、约0.8×106至约1.5×107、约0.9×106至约1.2×107、或约0.5×106、1×106、5×106、6×106、7×106、8×106、9×106、1×107、1.5×107、2×107、3×107、4×107或5×107个cart细胞。这些量可针对每kg患者体重。
在其他实施方案中,cart细胞组合物中向患者施用的cart细胞的剂量选自:约100万、约200万、约300万、约400万、约500万、约600万、约700万、约800万、约900万、约1000万、约1100万、约1200万、约1250万、约1300万、约1400万及约1500万的所述cart细胞。这些量可针对每kg患者体重。
在此段落中所描述的任何实施方案中,cart细胞剂量可为每kg患者体重的cart细胞的数目。在一个方面,在本文中所描述的任何实施方案中,可向患者施用单次剂量或多次剂量的cart细胞。在一个说明性实施方案中,可向患者施用第一剂量的cart细胞及第二剂量的cart细胞。在一个方面,cart细胞的第一剂量可为用以监测患者对cart细胞的耐受性的测试剂量,且cart细胞的第二剂量可包含高于cart细胞的第一剂量的cart细胞的剂量。在一个实施方案中,cart细胞的第一剂量可包含约0.5×105的cart细胞至约1.5×106的cart细胞。在另一实施方案中,cart细胞的第二剂量可包含约0.8×106的cart细胞至约2×107的cart细胞。在包括cart细胞的第一剂量及第二剂量的这些实施方案中,可施用本文中所描述的cart细胞的任何剂量。
在本文中所描述的任何实施方案中,cart细胞可在化合物或其药学上可接受的盐之前或之后施用。如将理解,除非另外陈述,否则针对本文所述的任何方法的步骤的名称i)、ii)及iii)等不指示次序。
可使用本领域中已知的任何合适的方法,将本文中所描述的化合物或其药学上可接受的盐或cart细胞组合物施用于患者。如本文所述,术语术语“施用(administering)”或“施用(administered)”包括向患者引入化合物或其药学上可接受的盐或cart细胞组合物的所有手段,包括(但不限于)口服、静脉内、肌肉内、皮下、经皮及其类似手段。在一个方面,本文中所描述的化合物或其药学上可接受的盐可以单位剂型及/或含有常规的无毒性药学上可接受的载体、佐剂及媒介物的制剂来施用。
在一个方面,如本文中所描述的化合物或其药学上可接受的盐或cart细胞组合物可直接施用至血流中、至肌肉中或至内部器官中。在各种实施方案中,适用于此类胃肠外施用的途径包括静脉内、动脉内、腹膜内、鞘内、硬膜外、脑室内、尿道内、胸骨内、颅内、瘤内、肌肉内及皮下递送。在一个实施方案中,用于胃肠外施用的手段包括针(包括微针)注射器、无针注射器及输注技术。
在一个说明性方面,胃肠外制剂典型地为可含有诸如盐、碳水化合物及缓冲剂(较佳地在3至9的ph值下)的载体或赋形剂的水溶液,但其可能更适合经配制为无菌非水性溶液,或配制为干燥形式以结合诸如无菌、无热原水或无菌盐水的合适媒介物的来使用。在其他实施方案中,本文中所描述的液体制剂中的任一者可经调适以用于如本文中所描述的胃肠外施用。可易于使用本领域技术人员所熟知的标准医药技术在无菌条件下制备,通过冻干来产生用于胃肠外制剂的无菌冻干粉末。在一个实施方案中,可通过使用诸如并入溶解度增强剂的适当配制技术来增加用于制备胃肠外制剂的化合物或其药学上可接受的盐的溶解度。
在一个实施方案中,待施用于患者的化合物或其药学上可接受的盐的量可视正经治疗的癌症、化合物或其药学上可接受的盐的施用途径及组织分布而显著变化。在一个方面,待施用于患者的量可基于体表面积、质量及医师评估。
在各种实施方案中,可按以下来施用化合物或其药学上可接受的盐:1)至少第一剂量递增顺序及第二剂量递增顺序,2)至少第一剂量递增顺序、第二剂量递增顺序及第三剂量递增顺序,3)至少第一剂量递增顺序、第二剂量递增顺序、第三剂量递增顺序及第四剂量递增顺序,4)至少第一剂量递增顺序、第二剂量递增顺序、第三剂量递增顺序、第四剂量递增顺序及第五剂量递增顺序,5)至少第一剂量递增顺序、第二剂量递增顺序、第三剂量递增顺序、第四剂量递增顺序、第五剂量递增顺序及第六剂量递增顺序,或可包括6)一或多个额外剂量递增顺序。
在各种实施方案中,对于第一、第二、第三、第四、第五或第六等剂量递增顺序,待施用于患者的化合物或其药学上可接受的盐的量可为约0.1µg/kg至约2000µg/kg、0.1µg/kg至约1500µg/kg、0.1µg/kg至约1000µg/kg、0.1µg/kg至约500µg/kg、约0.1µg/kg至约100µg/kg、约0.1µg/kg至约80µ/kg、约0.1µg/kg至约70µg/kg、约0.1µg/kg至约50µg/kg、约0.1µg/kg至约40µg/kg、约0.1µg/kg至约30µg/kg、约0.3µg/kg至约500µg/kg、约0.3µg/kg至约400µg/kg、约0.3µg/kg至约300µg/kg、约0.3µg/kg至约200µg/kg、约0.3µg/kg至约100µg/kg、约0.3µg/kg至约90µg/kg、约0.1µg/kg至约400µg/kg、约0.1µg/kg至约350µg/kg、约0.1µg/kg至约300µg/kg、约0.1µg/kg至约250µg/kg、约0.1µg/kg至约200µg/kg、约0.1µg/kg至约150µg/kg或约0.3µg/kg至约150µg/kg或5µg/kg至约2000µg/kg、5µg/kg至约1500µg/kg、5µg/kg至约1000µg/kg、5µg/kg至约500µg/kg、约5µg/kg至约100µg/kg、约5µg/kg至约80µ/kg、约5µg/kg至约70µg/kg、约5µg/kg至约50µg/kg、约5µg/kg至约40µg/kg或约5µg/kg至约30µg/kg。在另一实施方案中,对于第一剂量递增顺序及第四剂量递增顺序,待施用于患者的化合物或其药学上可接受的盐的量可为约0.1µg/kg至约100µg/kg、约0.1µg/kg至约80µg/kg、约0.1µg/kg至约70µg/kg、约0.1µg/kg至约50µg/kg、约0.1µg/kg至约40µg/kg、约0.1µg/kg至约30µg/kg或约0.3µg/kg至约30µg/kg、约5µg/kg至约100µg/kg、约5µg/kg至约80µg/kg、约5µg/kg至约70µg/kg、约5µg/kg至约50µg/kg、约5µg/kg至约40µg/kg、约5µg/kg至约30µg/kg或约5µg/kg至约2000µg/kg或约5µg/kg至约1500µg/kg或约5µg/kg至约1000µg/kg或约5µg/kg至约500µg/kg。在另一实施方案中,对于第二剂量递增顺序及第五剂量递增顺序,待施用于患者的化合物或其药学上可接受的盐的量可为约0.3µg/kg至约500µg/kg、约0.3µg/kg至约400µg/kg、约0.3µg/kg至约300µg/kg、约0.3µg/kg至约200µg/kg、约0.3µg/kg至约100µg/kg或约0.3µg/kg至约90µg/kg或约5µg/kg至约100µg/kg、约5µg/kg至约80µg/kg、约5µg/kg至约70µg/kg、约5µg/kg至约50µg/kg、约5µg/kg至约40µg/kg、约5µg/kg至约30µg/kg或约5µg/kg至约2000µg/kg或约5µg/kg至约1500µg/kg或约5µg/kg至约1000µg/kg或约5µg/kg至约500µg/kg。在又另一实施方案中,对于第三剂量递增顺序及第六剂量递增顺序,待施用于患者的化合物或其药学上可接受的盐的量可为0.1µg/kg至约400µg/kg、约0.1µg/kg至约350µg/kg、约0.1µg/kg至约300µg/kg、约0.1µg/kg至约250µg/kg、约0.1µg/kg至约200µg/kg、约0.1µg/kg至约150µg/kg或约0.3µg/kg至约150µg/kg或约5µg/kg至约100µg/kg、约5µg/kg至约80µg/kg、约5µg/kg至约70µg/kg、约5µg/kg至约50µg/kg、约5µg/kg至约40µg/kg、约5µg/kg至约30µg/kg或约5µg/kg至约2000µg/kg或约5µg/kg至约1500µg/kg或约5µg/kg至约1000µg/kg或约5µg/kg至约500µg/kg。在这些实施方案中,“kg”为患者体重的公斤数。
在这些实施方案中的任一者中,化合物或其药学上可接受的盐的量的范围可为基于化合物或其药学上可接受的盐的“全剂量”的计算百分比的范围,其中化合物或其药学上可接受的盐的“全剂量”可为约30µg/kg,且其中百分比为化合物或其药学上可接受的盐的“全剂量”的约百分之1至约百分之100(见图1中的顺序1)、约百分之1至约百分之300(见图1中的顺序2)及约百分之1至约百分之500(见图1中的顺序3)。在其他实施方案中,以剂量递增顺序施用的化合物或其药学上可接受的盐的“全剂量”的百分比可为约百分之1、约百分之2、约百分之3、约百分之4、约百分之5、约百分之6、约百分之7、约百分之8、约百分之9、约百分之10、约百分之20、约百分之30、约百分之40、约百分之50、约百分之60、约百分之70、约百分之80、约百分之90、约百分之100、约百分之200、约百分之300、约百分之400或约百分之500,且施用量可分别为约0.3µg/kg、约3µg/kg、约9µg/kg、约15µg/kg、约30µg/kg、约90µg/kg或约150µg/kg,或约百分之17、约百分之333、约百分之1666或约百分之3333,且施用量可分别为约5µg/kg、约100µg/kg、约500µg/kg或约1000µg/kg。在这些实施方案中,“kg”为患者体重的公斤数。
在另一实施方案中,以剂量递增顺序施用的化合物或其药学上可接受的盐的“全剂量”的百分比可为:对第一剂量递增顺序约百分之1、约百分之2及约百分之20,对第二剂量递增顺序约百分之1、约百分之6及约百分之60,且对第三剂量递增顺序约百分之1、约百分之10及约百分之100,且化合物或其药学上可接受的盐的“全剂量”可为500纳摩尔/公斤。在此实施方案中,“kg”为患者体重的公斤数。在此实施方案中,化合物或其药学上可接受的盐可在星期一、星期四及星期一施用,其中各剂量递增周期之间有约6天。
在另一实施方案中,以剂量递增顺序施用的化合物或其药学上可接受的盐的“全剂量”的百分比可为:对第一剂量递增顺序约百分之1、约百分之10及约百分之100,且对第二剂量递增顺序约百分之1、约百分之20及约百分之200,且化合物或其药学上可接受的盐的“全剂量”可为500纳摩尔/公斤。在此实施方案中,“kg”为患者体重的公斤数。在此实施方案中,化合物或其药学上可接受的盐可在星期一、星期四及星期一施用,其中各剂量递增周期之间有约6天。
在另一实施方案中,以剂量递增顺序施用的化合物或其药学上可接受的盐的“全剂量”的百分比可为:对第一剂量递增顺序约百分之1、约百分之10及约百分之100,且对第二剂量递增顺序约百分之1、约百分之10及约百分之100,且化合物或其药学上可接受的盐的“全剂量”可为500纳摩尔/公斤。在此实施方案中,“kg”为患者体重的公斤数。在此实施方案中,化合物或其药学上可接受的盐可在星期一、星期四及星期一施用,其中在各剂量递增周期之间有约6天。在另一实施方案中,化合物或其药学上可接受的盐可在星期一、星期四及星期一施用,其中在各剂量递增周期之间有9天。在其他实施方案中,剂量递增可重复一次、两次、三次、四次、五次、六次、七次、八次、九次或十次或任何适当次数。
在又另一实施方案中,以剂量递增顺序施用的化合物或其药学上可接受的盐的“全剂量”的百分比可为:对第一剂量递增顺序约百分之1、约百分之20及约百分之200,且对第二剂量递增顺序约百分之1、约百分之20及约百分之200,且化合物或其药学上可接受的盐的“全剂量”可为500纳摩尔/公斤。在此实施方案中,“kg”为患者体重的公斤数。在此实施方案中,化合物或其药学上可接受的盐可在星期一、星期四及星期一施用,其中各剂量递增周期之间有约6天。在其他实施方案中,剂量递增可重复一次、两次、三次、四次、五次、六次、七次、八次、九次或十次或任何适当次数。
在另一实施方案中,以第一剂量递增顺序或第四剂量递增顺序施用的化合物或其药学上可接受的盐的“全剂量”的百分比可为以递增量的化合物或其药学上可接受的盐的“全剂量”(约10µg/kg至约50µg/kg、约20µg/kg至约40µg/kg、约25µg/kg至约35µg/kg或约30µg/kg)的约百分之1、约百分之10及约百分之100,且所施用化合物或其药学上可接受的盐的量可分别为约0.3µg/kg、约3µg/kg及约30µg/kg。在又另一实施方案中,以第二剂量递增顺序或第五剂量递增顺序施用的化合物或其药学上可接受的盐的“全剂量”的百分比可为以递增量的化合物或其药学上可接受的盐的“全剂量”(约10µg/kg至约50µg/kg、约20µg/kg至约40µg/kg、约25µg/kg至约35µg/kg或约30µg/kg)的约百分之1、约百分之30及约百分之300,且所施用化合物或其药学上可接受的盐的量可分别为约0.3µg/kg、约9µg/kg及约90µg/kg。在再另一实施方案中,以第三剂量递增顺序或第六剂量递增顺序施用的化合物或其药学上可接受的盐的“全剂量”的百分比可为以递增量的化合物或其药学上可接受的盐的“全剂量”(约10µg/kg至约50µg/kg、约20µg/kg至约40µg/kg、约25µg/kg至约35µg/kg或约30µg/kg)的约百分之1、约百分之50及约百分之500,且所施用化合物或其药学上可接受的盐的量可分别为约0.3µg/kg、约15µg/kg及约150µg/kg。在这些实施方案中,“kg”为患者体重的公斤数。
在各种实施方案中,待施用于患者的量可例如在约0.05mg至约30mg、0.05mg至约25.0mg、约0.05mg至约20.0mg、约0.05mg至约15.0mg、约0.05mg至约10.0mg、约0.05mg至约9.0mg、约0.05mg至约8.0mg、约0.05mg至约7.0mg、约0.05mg至约6.0mg、约0.05mg至约5.0mg、约0.05mg至约4.0mg、约0.05mg至约3.0mg、约0.05mg至约2.0mg、约0.05mg至约1.0mg、约0.05mg至约0.5mg、约0.05mg至约0.4mg、约0.05mg至约0.3mg、约0.05mg至约0.2mg、约0.05mg至约0.1mg、约0.01mg至约2mg、约0.3mg至约10mg、约0.1mg至约20mg或约0.8至约3mg的范围内。
在其他实施方案中,化合物或其药学上可接受的盐的剂量可在例如约50纳摩尔/公斤患者体重至约3000纳摩尔/公斤患者体重、约50纳摩尔/公斤患者体重至约2000纳摩尔/公斤患者体重、约50纳摩尔/公斤患者体重至约1000纳摩尔/公斤患者体重、约50纳摩尔/公斤患者体重至约900纳摩尔/公斤患者体重、约50纳摩尔/公斤患者体重至约800纳摩尔/公斤患者体重、约50纳摩尔/公斤患者体重至约700纳摩尔/公斤患者体重、约50纳摩尔/公斤患者体重至约600纳摩尔/公斤患者体重、约50纳摩尔/公斤患者体重至约500纳摩尔/公斤患者体重、约50纳摩尔/公斤患者体重至约400纳摩尔/公斤患者体重、约50纳摩尔/公斤患者体重至约300纳摩尔/公斤患者体重、约50纳摩尔/公斤患者体重至约200纳摩尔/公斤患者体重、约50纳摩尔/公斤患者体重至约100纳摩尔/公斤患者体重、约100纳摩尔/公斤患者体重至约300纳摩尔/公斤患者体重、约100纳摩尔/公斤患者体重至约500纳摩尔/公斤患者体重、约100纳摩尔/公斤患者体重至约1000纳摩尔/公斤患者体重、约100纳摩尔/公斤患者体重至约2000纳摩尔/公斤患者体重的范围内。在其他实施方案中,该剂量可为约1纳摩尔/公斤患者体重、约5纳摩尔/公斤患者体重、约10纳摩尔/公斤患者体重、约20纳摩尔/公斤患者体重、约25纳摩尔/公斤患者体重、约30纳摩尔/公斤患者体重、约40纳摩尔/公斤患者体重、约50纳摩尔/公斤患者体重、约60纳摩尔/公斤患者体重、约70纳摩尔/公斤患者体重、约80纳摩尔/公斤患者体重、约90纳摩尔/公斤患者体重、约100纳摩尔/公斤患者体重、约150纳摩尔/公斤患者体重、约200纳摩尔/公斤患者体重、约250纳摩尔/公斤患者体重、约300纳摩尔/公斤患者体重、约350纳摩尔/公斤患者体重、约400纳摩尔/公斤患者体重、约450纳摩尔/公斤患者体重、约500纳摩尔/公斤患者体重、约600纳摩尔/公斤患者体重、约700纳摩尔/公斤患者体重、约800纳摩尔/公斤患者体重、约900纳摩尔/公斤患者体重、约1000纳摩尔/公斤患者体重、约2000纳摩尔/公斤患者体重、约2500纳摩尔/公斤患者体重或约3000纳摩尔/公斤患者体重。在又其他实施方案中,该剂量可为约0.1纳摩尔/公斤患者体重、约0.2纳摩尔/公斤患者体重、约0.3纳摩尔/公斤患者体重、约0.4纳摩尔/公斤患者体重或约0.5纳摩尔/公斤患者体重、约0.1纳摩尔/公斤患者体重至约1000纳摩尔/公斤患者体重、约0.1纳摩尔/公斤患者体重至约900纳摩尔/公斤患者体重、约0.1纳摩尔/公斤患者体重至约850纳摩尔/公斤患者体重、约0.1纳摩尔/公斤患者体重至约800纳摩尔/公斤患者体重、约0.1纳摩尔/公斤患者体重至约700纳摩尔/公斤患者体重、约0.1纳摩尔/公斤患者体重至约600纳摩尔/公斤患者体重、约0.1纳摩尔/公斤患者体重至约500纳摩尔/公斤患者体重、约0.1纳摩尔/公斤患者体重至约400纳摩尔/公斤患者体重、约0.1纳摩尔/公斤患者体重至约300纳摩尔/公斤患者体重、约0.1纳摩尔/公斤患者体重至约200纳摩尔/公斤患者体重、约0.1纳摩尔/公斤患者体重至约100纳摩尔/公斤患者体重、约0.1纳摩尔/公斤患者体重至约50纳摩尔/公斤患者体重、约0.1纳摩尔/公斤患者体重至约10纳摩尔/公斤患者体重或约0.1纳摩尔/公斤患者体重至约1纳摩尔/公斤患者体重。在其他实施方案中,该剂量可为约0.3纳摩尔/公斤患者体重至约1000纳摩尔/公斤患者体重、约0.3纳摩尔/公斤患者体重至约900纳摩尔/公斤患者体重、约0.3纳摩尔/公斤患者体重至约850纳摩尔/公斤患者体重、约0.3纳摩尔/公斤患者体重至约800纳摩尔/公斤患者体重、约0.3纳摩尔/公斤患者体重至约700纳摩尔/公斤患者体重、约0.3纳摩尔/公斤患者体重至约600纳摩尔/公斤患者体重、约0.3纳摩尔/公斤患者体重至约500纳摩尔/公斤患者体重、约0.3纳摩尔/公斤患者体重至约400纳摩尔/公斤患者体重、约0.3纳摩尔/公斤患者体重至约300纳摩尔/公斤患者体重、约0.3纳摩尔/公斤患者体重至约200纳摩尔/公斤患者体重、约0.3纳摩尔/公斤患者体重至约100纳摩尔/公斤患者体重、约0.3纳摩尔/公斤患者体重至约50纳摩尔/公斤患者体重、约0.3纳摩尔/公斤患者体重至约10纳摩尔/公斤患者体重或约0.3纳摩尔/公斤患者体重至约1纳摩尔/公斤患者体重。在这些实施方案中,“kg”为患者体重的公斤数。
在另一实施方案中,在cart细胞的施用(例如以本文中所描述的量中的任一者)之后,执行使用化合物(例如其中各剂量递增步骤为50nmol/kg且随后500nmol/kg)或其药学上可接受的盐的第一剂量递增步骤及第二剂量递增步骤。在此实施方案中,在例如第一周及第二周中的第一剂量递增步骤及第二剂量递增步骤之后,相对于第二周中所施用的最末剂量,化合物或其药学上可接受的盐的水平可在第三周保持恒定(例如在500nmol/kg处保持恒定)。在本文中所描述的任何实施方案中,相对于前一周中所施用的最末剂量,化合物或其药学上可接受的盐的水平可在后一周保持恒定。
在各种其他实施方案中,化合物或其药学上可接受的盐的剂量可介于例如约10纳摩尔/公斤患者体重至约10000纳摩尔/公斤患者体重、约10纳摩尔/公斤患者体重至约5000纳摩尔/公斤患者体重、约10纳摩尔/公斤患者体重至约3000纳摩尔/公斤患者体重、约10纳摩尔/公斤患者体重至约2500纳摩尔/公斤患者体重、约10纳摩尔/公斤患者体重至约2000纳摩尔/公斤患者体重、约10纳摩尔/公斤患者体重至约1000纳摩尔/公斤患者体重、约10纳摩尔/公斤患者体重至约900纳摩尔/公斤患者体重、约10纳摩尔/公斤患者体重至约800纳摩尔/公斤患者体重、约10纳摩尔/公斤患者体重至约700纳摩尔/公斤患者体重、约10纳摩尔/公斤患者体重至约600纳摩尔/公斤患者体重、约10纳摩尔/公斤患者体重至约500纳摩尔/公斤患者体重、约10纳摩尔/公斤患者体重至约400纳摩尔/公斤患者体重、约10纳摩尔/公斤患者体重至约300纳摩尔/公斤患者体重、约10纳摩尔/公斤患者体重至约200纳摩尔/公斤患者体重、约10纳摩尔/公斤患者体重至约150纳摩尔/公斤患者体重、约10纳摩尔/公斤患者体重至约100纳摩尔/公斤患者体重、约10纳摩尔/公斤患者体重至约90纳摩尔/公斤患者体重、约10纳摩尔/公斤患者体重至约80纳摩尔/公斤患者体重、约10纳摩尔/公斤患者体重至约70纳摩尔/公斤患者体重、约10纳摩尔/公斤患者体重至约60纳摩尔/公斤患者体重、约10纳摩尔/公斤患者体重至约50纳摩尔/公斤患者体重、约10纳摩尔/公斤患者体重至约40纳摩尔/公斤患者体重、约10纳摩尔/公斤患者体重至约30纳摩尔/公斤患者体重、约10纳摩尔/公斤患者体重至约20纳摩尔/公斤患者体重、约200纳摩尔/公斤患者体重至约900纳摩尔/公斤患者体重、约200纳摩尔/公斤患者体重至约800纳摩尔/公斤患者体重、约200纳摩尔/公斤患者体重至约700纳摩尔/公斤患者体重、约200纳摩尔/公斤患者体重至约600纳摩尔/公斤患者体重、约200纳摩尔/公斤患者体重至约500纳摩尔/公斤患者体重、约250纳摩尔/公斤患者体重至约600纳摩尔/公斤患者体重、约300纳摩尔/公斤患者体重至约600纳摩尔/公斤患者体重、约300纳摩尔/公斤患者体重至约500纳摩尔/公斤患者体重或约400纳摩尔/公斤患者体重至约600纳摩尔/公斤患者体重的范围内。在这些实施方案中,“kg”为患者体重的公斤数。
在上文所描述的所有剂量实施方案中,在任何步骤处以剂量递增顺序施用的化合物或其药学上可接受的盐的“全剂量”的百分比可为化合物或其药学上可接受的盐的“全剂量”的约百分之1、约百分之2、约百分之3、约百分之4、约百分之5、约百分之6、约百分之7、约百分之8、约百分之9、约百分之10、约百分之20、约百分之30、约百分之40、约百分之50、约百分之60、约百分之70、约百分之80、约百分之90、约百分之100、约百分之200、约百分之300、约百分之400或约百分之500。化合物或其药学上可接受的盐的“全剂量”可为在前文段落中描述为以剂量递增顺序施用的剂量的,化合物或其药学上可接受的盐的剂量中的任一者。
在各种其他实施方案中,化合物或其药学上可接受的盐的剂量可介于例如约1纳摩尔/公斤至约10000纳摩尔/公斤、约1纳摩尔/公斤至约5000纳摩尔/公斤、约1纳摩尔/公斤至约3000纳摩尔/公斤、约1纳摩尔/公斤至约2500纳摩尔/公斤、约1纳摩尔/公斤至约2000纳摩尔/公斤、约1纳摩尔/公斤至约1000纳摩尔/公斤、约1纳摩尔/公斤至约900纳摩尔/公斤、约1纳摩尔/公斤至约800纳摩尔/公斤、约1纳摩尔/公斤至约700纳摩尔/公斤、约1纳摩尔/公斤至约600纳摩尔/公斤、约1纳摩尔/公斤至约500纳摩尔/公斤、约1纳摩尔/公斤至约400纳摩尔/公斤、约1纳摩尔/公斤至约300纳摩尔/公斤、约1纳摩尔/公斤至约200纳摩尔/公斤、约1纳摩尔/公斤至约150纳摩尔/公斤、约1纳摩尔/公斤至约100纳摩尔/公斤、约1纳摩尔/公斤至约90纳摩尔/公斤、约1纳摩尔/公斤至约80纳摩尔/公斤、约1纳摩尔/公斤至约70纳摩尔/公斤、约1纳摩尔/公斤至约60纳摩尔/公斤、约1纳摩尔/公斤至约50纳摩尔/公斤、约1纳摩尔/公斤至约40纳摩尔/公斤、约1纳摩尔/公斤至约30纳摩尔/公斤或约1纳摩尔/公斤至约20纳摩尔/公斤的范围内。在这些实施方案中,“kg”为患者体重的公斤数。
在另一实施方案中,可向患者施用约20μg/kg患者体重至约3mg/kg患者体重的化合物或其药学上可接受的盐。在另一方面,量可为约0.2mg/kg患者体重至约0.4mg/kg患者体重。
在上述剂量实施方案中的任一者中,可向患者施用化合物或其药学上可接受的盐的单次剂量或多次剂量。
在一个实施方案中,连接于靶向部分的小分子配体可在cart细胞组合物之前施用于患者。在另一实施方案中,连接于靶向部分的小分子配体可与cart细胞组合物同时但在不同制剂中或在相同制剂中施用于患者。在又另一实施方案中,连接于靶向部分的小分子配体可在cart细胞组合物之后施用于患者。
在一个说明性方面,施用cart细胞与连接于靶向部分的小分子之间的时间安排可取决于以下因素而广泛变化,所述因素包括所使用cart细胞的类型、car的结合特异性、靶向部分及小分子配体的属性、癌症的属性、癌症在患者中的部位、用以向患者施用cart细胞及连接于靶向部分的小分子配体的手段以及患者的健康、年龄及体重。在一个方面,连接于靶向部分的小分子配体可在cart细胞之前或之后诸如约3小时、6小时、9小时、12小时、15小时、18小时、21小时、24小时、27小时、30小时、33小时、36小时、39小时、42小时、45小时、48小时或51小时内或约0.5天、1天、1.5天、2天、2.5天、3天、4天、5天、6天、7天、8天、9天、10天或更多天内施用。
在一个实施方案中,本领域中已知的任何适用给药时程可用于化合物或其药学上可接受的盐或cart细胞组合物的施用。在一个方面,为化合物或其药学上可接受的盐及cart细胞组合物选择的给药时程可考虑化合物或其药学上可接受的盐的浓度,及所施用cart细胞的数目,以调节cart细胞组合物的细胞毒性及控制crs。
在一个示例性实施方案中,第一剂量递增顺序、第二剂量递增顺序、第三剂量递增顺序、第四剂量递增顺序、第五剂量递增顺序、第六剂量递增顺序或任何额外剂量递增顺序可随后为其间不施用化合物或其药学上可接受的盐的一段时间。在各种说明性实施方案中,不施用化合物或其药学上可接受的盐的该时间段可为1天、2天、3天、4天、5天、6天、7天、8天、9天或10天。在另一实施方案中,不施用化合物或其药学上可接受的盐的该时间段可为6天、7天或8天。在又另一实施方案中,不施用化合物或其药学上可接受的盐的该时间段可为7天。
在一个方面,在第1周期间向患者施用cart细胞的第一剂量及cart细胞的第二剂量,例如在星期一及星期四施用。在此实施方案中,化合物或其药学上可接受的盐的第一剂量递增顺序可随后在第2周及第3周期间进行。例如,化合物或其药学上可接受的盐可在分开的三天施用,且分开的三天可为第2周的星期一及星期四以及第3周的星期一(见图1中的顺序1)。在此实施方案中,化合物或其药学上可接受的盐的第二剂量递增顺序可随后在第4周及第5周期间进行(见图1中的顺序2)。例如,化合物或其药学上可接受的盐可在分开的三天施用,且分开的三天可为第4周的星期一及星期四以及第5周的星期一。在此实施方案中,化合物或其药学上可接受的盐的第三剂量递增顺序可随后在第6周及第7周期间进行(见图1中的顺序3)。例如,化合物或其药学上可接受的盐可在分开的三天施用,且分开的三天可为第6周的星期一及星期四以及第7周的星期一。在其他实施方案中,后续剂量递增顺序可遵循类似顺序,或可以在顺序3结束之后约7天重复顺序3作为“疗程2”,且不进行使用化合物或其药学上可接受的盐的额外治疗直至“疗程3”开始为止(“疗程2”的时长基于图1中针对“疗程1”所示的时间长度)。在一个实施方案中,患者可接受四个治疗疗程。
在另一说明性实施方案中,提供癌症的治疗方法。该方法包括i)向患者施用至少一个剂量的包含cart细胞的cart细胞组合物,其中cart细胞包含指向靶向部位的car;ii)向患者施用化合物或其药学上可接受的盐,其中该化合物包含通过接头连接于靶向部位的小分子配体且其中以第一剂量递增顺序施用化合物或其药学上可接受的盐,其中如果在第一剂量递增顺序下出现严重crs,则使用较低剂量递增顺序来施用该化合物或其药学上可接受的盐,其中较低剂量递增顺序中的化合物或其药学上可接受的盐的第一剂量低于以第一剂量递增顺序施用的化合物或其药学上可接受的盐的第一剂量。在此实施方案中,在较低剂量递增顺序中,在分开的三天以化合物或其药学上可接受的盐的全剂量的约0.5%、约5%及约50%来施用化合物或其药学上可接受的盐。在此实施方案中,化合物或其药学上可接受的盐的全剂量可为约10µg/kg至约50µg/kg、约20µg/kg至约40µg/kg、约25µg/kg至约35µg/kg或约30µg/kg。在这些实施方案中,“kg”为患者体重的公斤数。
在各种相关实施方案中,在向患者施用cart细胞组合物之前,可使用例如环磷酰胺(cytoxan)、氟达拉宾(fludarabine)及/或依托泊苷(etopside)耗尽患者中的淋巴细胞,且方法可进一步包括额外步骤,包括但不限于;向患者施用血小板、向患者施用浓缩红细胞、向患者施用冷沉淀物、向患者施用静脉内免疫球蛋白、使用钙补充剂、酸-柠檬酸盐-右旋糖及/或肝素治疗及/或为患者提供抗微生物剂治疗。在一个实施方案中,淋巴耗尽发生在cart细胞施用之前至少约24小时。
在另一方面,方法可包括crs监测步骤。在一个说明性方面,如果在第一剂量递增顺序期间在患者中未观测到crs或神经毒性,则该方法可前进至第二剂量递增顺序。如果在第二剂量递增顺序期间在患者中未观测到crs或神经毒性,则该方法可前进至第三剂量递增顺序。如果在第三剂量递增顺序期间未观测到crs或神经毒性,则该方法可前进至第四剂量递增顺序。如果在第四剂量递增顺序期间在患者中未观测到crs或神经毒性,则该方法可前进至第五剂量递增顺序。如果在第五剂量递增顺序期间在患者中未观测到crs或神经毒性,则该方法可前进至第六剂量递增顺序,以此类推。
在另一说明性实施方案中,如果在剂量递增顺序中的任一者期间在患者中观测到发热而无低血压(即非严重crs)且在患者中未观测到神经毒性,则可按导致发热而无低血压的剂量递增顺序水平向患者施用化合物或其药学上可接受的盐的所有后续剂量。在另一方面,如果在任何剂量递增顺序下在患者中出现crs(即严重crs)或神经毒性,则可按低于导致患者中的严重crs或神经毒性的剂量递增顺序水平的剂量递增顺序水平来向患者施用化合物或其药学上可接受的盐的所有后续剂量。
在一个方面,除在治疗开始之后持续少于48小时的≤4级发热、在治疗开始之后持续少于24小时的≤3级发冷、在治疗开始之后持续少于24小时的≤3级咳嗽、在治疗开始之后持续少于7天的≤3级转氨酶、在治疗开始之后持续少于48小时的≤3级低血压、在治疗开始之后持续少于48小时的≤3级crs、与可通过苯海拉明(benadryl)及/或肾上腺素解决的dmso相关的≤3级过敏反应或通过口服或iv麻醉剂治疗控制的≤3级疼痛以外,或另外在cart细胞输注之后约3周出现的毒性(包括持续多达5天的≤3级发冷、持续多达2周的≤3级转氨酶、持续多达2周的≤3级crs、≤4级淋巴细胞减少症、≤4级白细胞减少症或通过口服或iv麻醉剂治疗控制的持续多达2周的≤3级疼痛)以外,严重crs可包括需要使用西妥昔单抗的任何毒性、任何≥3级自身免疫毒性、任何≥3级的可归因于cart细胞施用或化合物或其药学上可接受的盐的施用且在治疗开始之后28天内出现的毒性。
在一个实施方案中,为防止或抑制患者中的crs,方法可进一步包含向患者施用叶酸化合物、包含叶酸化合物的缀合物(其中包含叶酸化合物的缀合物不包含靶向部分)或抑制cart细胞活化的药物。在此实施方案中,任一叶酸化合物、包含叶酸化合物的缀合物(其中包含叶酸化合物的缀合物不包含靶向部分)或抑制cart细胞活化的药物在本文中可称为“救援药剂”。在一个实施方案中,可施用诸如叶酸的叶酸化合物以预防或抑制患者中的crs。在此实施方案中,叶酸化合物抑制桥(即通过接头连接于靶向部分的小分子配体)与肿瘤上桥的受体的相互作用,从而抑制肿瘤溶解及防止或抑制患者中的crs。
在一个实施方案中,作为桥与肿瘤结合的抑制剂施用的叶酸化合物可为例如叶酸、叶酸类似物或另一叶酸受体结合分子。在各种实施方案中,可使用的叶酸类似物包括亚叶酸、喋酰聚谷氨酸及叶酸受体结合喋啶,诸如四氢喋呤、二氢叶酸、四氢叶酸及其脱氮及二脱氮类似物。术语“脱氮”及“二脱氮”类似物是指由碳原子取代天然存在的叶酸化合物结构中的一个或两个氮原子的本领域公认的类似物。例如,脱氮类似物包括1-脱氮、3-脱氮、5-脱氮、8-脱氮及10-脱氮类似物。二脱氮类似物包括例如1,5-二脱氮、5,10-二脱氮、8,10-二脱氮及5,8-二脱氮类似物。前述叶酸类似物通常称为“叶酸化合物”,此反映其与叶酸受体结合的能力。其他叶酸受体结合类似物包括氨基喋呤、氨甲喋呤(甲氨喋呤)、n10-甲基叶酸、2-脱氨基-羟基叶酸、脱氮类似物(诸如1-脱氮氨甲喋呤或3-脱氮氨甲喋呤)及3',5'-二氯-4-氨基-4-脱氧-n10-甲基喋酰谷氨酸(二氯甲氨喋呤)。
在另一实施方案中,作为桥与肿瘤结合的抑制剂施用的叶酸化合物具有式
其中x1及y1各自独立地选自:卤素、r2、or2、sr3及nr4r5;
u、v及w表示各自独立地选自以下的二价部分:-(r6a)c=、-n=、-(r6a)c(r7a)-及-n(r4a)-;q选自:c及ch;t选自:s、o、n及-c=c-;
x2及x3各自独立地选自:氧、硫、-c(z)-、-c(z)o-、-oc(z)-、-n(r4b)-、-c(z)n(r4b)-、-n(r4b)c(z)-、-oc(z)n(r4b)-、-n(r4b)c(z)o-、-n(r4b)c(z)n(r5b)-、-s(o)-、-s(o)2-、-n(r4a)s(o)2-、-c(r6b)(r7b)-、-n(c≡ch)-、-n(ch2c≡ch)-、c1-c12亚烷基及c1-c12亚烷氧基,其中z为氧或硫;
r1选自:氢、卤素、c1-c12烷基及c1-c12烷氧基;
r2、r3、r4、r4a、r4b、r5、r5b、r6b及r7b各自独立地选自:氢、卤素、c1-c12烷基、c1-c12烷氧基、c1-c12烷酰基、c1-c12烯基、c1-c12炔基、(c1-c12烷氧基)羰基及(c1-c12烷基氨基)羰基;
r6及r7各自独立地选自:氢、卤素、c1-c12烷基及c1-c12烷氧基;或r6及r7一起形成羰基;
r6a及r7a各自独立地选自:氢、卤素、c1-c12烷基及c1-c12烷氧基;或r6a与r7a一起形成羰基;
p、r、s及t各自独立地为0或1;且
如果任何其他化学部分为该叶酸化合物的部分,则*表示与该缀合物的其余部分的任选的共价键。
在又另一实施方案中,可施用包含叶酸化合物的缀合物以预防或抑制患者中的细胞因子释放综合征(crs)。crs可对患者产生有害影响,包括但不限于体重损失、高热、肺水肿及血压危险地下降。
在此实施方案中,包含叶酸化合物的缀合物不包含靶向部分,且因此,缀合物抑制桥与肿瘤的相互作用以预防肿瘤溶解及减少患者中的crs。在此实施方案中,包含叶酸化合物的缀合物中的叶酸化合物部分可包含连接于不包含靶向部分的化学部分的前述段落中所描述的任一叶酸化合物。在一个方面,包含叶酸化合物的缀合物可包含连接于不包含靶向部分的一或多个氨基酸的叶酸化合物。说明性地,包含叶酸化合物的缀合物可具有式
此化合物亦可称为“ec923”。在这些实施方案中,可向患者施用叶酸化合物或包含叶酸化合物的缀合物,相对于桥(即通过接头连接于靶向部分的小分子配体)摩尔过量,诸如叶酸化合物或包含叶酸化合物的缀合物相对于通过接头连接于靶向部分的小分子配体过量10倍、过量100倍、过量200倍、过量300倍、过量400倍、过量500倍、过量600倍、过量700倍、过量800倍、过量900倍、过量1000倍或过量10,000倍。相对于通过接头连接于靶向部分的小分子配体的量,抑制桥与肿瘤的相互作用所需要的叶酸化合物或包含叶酸化合物的缀合物的量可由技术人员来确定。
在另一实施方案中,可向患者施用抑制cart细胞活化的药剂来抑制cart细胞活化及抑制或预防患者中的crs。在一个方面,药剂可选自:淋巴细胞特异性蛋白质酪氨酸激酶抑制剂(例如达沙替尼(dasatinib)、pi3激酶抑制剂(例如gdc0980)、托西路单抗(tociluzumab)、il-2诱导性t细胞激酶的抑制剂(例如bms-509744)、jak抑制剂、btk抑制剂、sip激动剂(例如西普尼莫德(siponimod)及奥扎尼莫(ozanimod))及阻断cart细胞结合至桥但不结合癌症的药剂(例如荧光素胺、fitc或荧光素钠)。技术人员应了解,在生理条件下或例如在缓冲液中在生理ph下,fitc(即荧光素)可呈盐(例如荧光素钠)形式或呈其非盐形式。因此,在一个实施方案中,在将荧光素施用于患者时,其可处于其盐形式(例如荧光素钠)与其非盐形式之间的平衡中。在另一实施方案中,抑制cart细胞活化的救援药剂可为下式化合物:
此化合物亦可称为“ec2319”。
在各种实施方案中,救援药剂可以约0.001nm至约100mm、约0.01nm至约100mm、约1nm至约100mm、约10nm至约100mm、约50nm至约100mm或约100nm至约100mm的浓度,以包括(例如)0.1ml、0.2ml、0.3ml、0.4ml、0.5ml、0.6ml、0.7ml、0.8ml、0.9ml、1ml、2ml、3ml、4ml、5ml、10ml、100ml或1000ml的任何适当体积施用。在其他实施方案中,救援药剂可以约0.01至约300微摩尔/公斤患者体重、约0.06至约100微摩尔/公斤患者体重、约0.06至约90微摩尔/公斤患者体重、约0.06至约80微摩尔/公斤患者体重、约0.06至约70微摩尔/公斤患者体重、约0.06至约60微摩尔/公斤患者体重、约0.06至约50微摩尔/公斤患者体重、约0.06至约40微摩尔/公斤患者体重、约0.06至约30微摩尔/公斤患者体重、约0.06至约20微摩尔/公斤患者体重、约0.06至约10微摩尔/公斤患者体重、约0.06至约8微摩尔/公斤患者体重或约0.06至约6微摩尔/公斤患者体重的剂量施用。
在这些实施方案中,可向患者施用救援药剂,相对于化合物或其药学上可接受的盐(即通过接头连接于靶向部分的小分子配体)摩尔过量,诸如救援药剂相对于通过接头连接于靶向部分的小分子配体约10倍过量、约20倍过量、约30倍过量、约40倍过量、约50倍过量、约60倍过量、约70倍过量、约80倍过量、约90倍过量、约100倍过量、约200倍过量、约300倍过量、约400倍过量、约500倍过量、约600倍过量、约700倍过量、约800倍过量、约900倍过量、约1000倍过量或约10,000倍过量。相对于通过接头连接于靶向部分的小分子配体的量,抑制化合物或其药学上可接受的盐与肿瘤及/或cart细胞的相互作用所需要的救援药剂的量可由技术人员来确定。
在另一实施方案中,可向患者施用超过一次剂量的叶酸化合物、包含叶酸化合物的缀合物(其中包含叶酸化合物的缀合物不包含靶向部分)或抑制cart细胞活化的药剂。
在本文中所描述的“救援药剂”实施方案中,叶酸化合物、包含叶酸化合物的缀合物(其中包含叶酸化合物的缀合物不包含靶向部分)或抑制cart细胞活化的药剂可在化合物或其药学上可接受的盐之前及/或之后施用于患者。在另一方面,可在施用叶酸化合物、包含叶酸化合物的缀合物(其中包含叶酸化合物的缀合物不包含靶向部分)或抑制cart细胞活化的药剂之前及之后施用化合物或其药学上可接受的盐。在此实施方案中,化合物或其药学上可接受的盐的后续施用可导致患者中的cart细胞活化及细胞因子水平增加。
在另一实施方案中,施用叶酸化合物、包含叶酸化合物的缀合物(其中包含叶酸化合物的缀合物不包含靶向部分)或抑制cart细胞活化的药剂可引起患者中细胞因子水平降低。在另一实施方案中,细胞因子水平降低为降低至约未经治疗的患者中的细胞因子水平。在其他实施方案中,当crs级别达到1、2、3或4时,或当crs级别达到3或4时,向患者施用抑制cart细胞活化的药剂。
在本文中描述的叶酸化合物为通过接头连接于靶向部分的配体的实施方案中的任一者中,在用本文中所描述的方法治疗之前可为患者规定叶酸化合物缺失型饮食,或患者可在饮食中施用叶酸化合物。在患者经施用叶酸化合物的实施方案中,剂量可例如介于约50nmol/kg患者体重至约3000nmol/kg患者体重、约50nmol/kg患者体重至约2000nmol/kg患者体重、约50nmol/kg患者体重至约1000nmol/kg患者体重、约50nmol/kg患者体重至约900nmol/kg患者体重、约50nmol/kg患者体重至约800nmol/kg患者体重、约50nmol/kg患者体重至约700nmol/kg患者体重、约50nmol/kg患者体重至约600nmol/kg患者体重、约50nmol/kg患者体重至约500nmol/kg患者体重、约50nmol/kg患者体重至约400nmol/kg患者体重、约50nmol/kg患者体重至约300nmol/kg患者体重、约50nmol/kg患者体重至约200nmol/kg患者体重、约50nmol/kg患者体重至约100nmol/kg患者体重、约100nmol/kg患者体重至约300nmol/kg患者体重、约100nmol/kg患者体重至约500nmol/kg患者体重、约100nmol/kg患者体重至约1000nmol/kg患者体重、约100nmol/kg患者体重至约2000nmol/kg患者体重的范围。在其他实施方案中,剂量可为约100nmol/kg、约150nmol/kg、约200nmol/kg、约250nmol/kg、约300nmol/kg、约350nmol/kg、约400nmol/kg、约450nmol/kg、约500nmol/kg、约600nmol/kg、约700nmol/kg、约800nmol/kg、约900nmol/kg、约1000nmol/kg、约2000nmol/kg或约3000nmol/kg患者体重。在这些实施方案中,“kg”为患者体重的公斤数。在一个方面,可例如每日、每周、两周一次、一周三次或使用叶酸化合物的施用的任何适合的方案施用叶酸化合物。
在本文中所描述的各种实施方案中,cart细胞可在cart细胞施用后长至约10天、长至约15天、长至约20天、长至约25天、长至约30天、长至约35天、长至约40天、长至约45天、长至约50天、长至约55天、长至约60天、长至约65天、长至约70天、长至约75天或长至约80天保持循环cart细胞的数量增加。
在本文中所描述的各种实施方案中,化合物或其药学上可接受的盐的半最大有效浓度(ec50)可为约1pm至约2nm、约1pm至约5nm、约1pm至约10nm、约1pm至约20nm、约1pm至约30nm、约1pm至约40nm、约1pm至约50nm、约1pm至约60nm、约1pm至约70nm、约1pm至约80nm、约1pm至约90nm、约1pm至约100nm、约1pm至约200nm、约1pm至约300nm、约1pm至约400nm、约1pm至约500nm、约1pm至约600nm、约1pm至约700nm、约1pm至约800nm、约1pm至约900nm、约1pm至约1nm、约1pm至约900pm、约1pm至约800pm、约1pm至约700pm、约1pm至约600pm、约1pm至约500pm、约1pm至约400pm、约1pm至约300pm、约1pm至约200pm、约1pm至约100pm、约1pm至约90pm、约1pm至约80pm、约1pm至约70pm、约1pm至约60pm、约1pm至约50pm、约1pm至约40pm、约1pm至约30pm、约1pm至约20pm、约1pm至约10pm或约1pm至约5pm。
在本文中所描述的各种实施方案中,化合物或其药学上可接受的盐与cart细胞的结合的kd可为约1nm至约100nm、约1nm至约200nm、约1nm至约300nm、约1nm至约400nm、约1nm至约500nm、约1nm至约600nm、约1nm至约700nm、约1nm至约800nm、约1nm至约900nm、约100nm至约500nm、约100nm至约400nm、约100nm至约300nm、约100nm至约200nm、约100nm至约150nm或约130nm。
在本文中所描述的各种实施方案中,可使用经egfrt分选或未分选的cart细胞。在另一实施方案中,可使用具有低分化特征的“临床传真”批次的cart细胞。在另一实施方案中,可使用“研究批次”的cart细胞。“临床传真”批次(约39%egfrt+)可包含呈约66%tscm及约32%tcm的cd4+亚组及呈约95%tscm及约3%tcm的cd8亚组。研究批次(约23%egfrt+)可包含呈约32%tscm、约53%tcm、约11%tem及约3.7%teff的cd4亚组及呈约44%tscm、约0.28%tcm、约3.4%tem及约52%teff的cd8亚组。
在本文中所描述的各种说明性实施方案中,可在cart细胞之前或之后约1、约2、约3、约4、约5、约6、约7、约8、约9或约10天或在cart细胞之前或之后的任一适当天时,首次向患者施用化合物或其药学上可接受的盐。
在本文中所描述的方法的一个实施方案中,在向患者施用化合物或其药学上可接受的盐之前或在向患者施用cart细胞组合物之前使癌症成像。在一个说明性实施方案中,成像通过pet成像进行。在其他说明性实施方案中,成像通过mri成像或spect/ct成像进行。成像方法可为本领域中已知的任何适合的成像方法。在一个实施方案中,成像方法可涉及使用本文中所描述的小分子配体,但其连接于适用于本文中所描述的成像类型的成像剂,以判定患者的叶酸受体表达是否为阳性。在另一实施方案中,可出于此目的使用免疫组织化学分析。
在本文中所描述的实施方案中的任一者中,即使出现针对癌症的cart细胞毒性,在患者中亦可能不会出现产生脱靶毒性的细胞因子释放。在本文中所描述的任何实施方案中,即使出现针对癌症的cart细胞毒性,在患者中亦可能不会出现脱靶组织毒性。在本文中所描述的任何实施方案中,癌症可包含肿瘤,且即使未出现脱靶毒性,患者中肿瘤大小亦可减小。在本文中所描述的实施方案中的任一者中,可减少或预防crs且方法可引起患者中肿瘤体积减小。在本文中所描述的任何实施方案中,可减小或预防归因于crs及cart细胞耗竭的体重损失。在本文中所描述的任何实施方案中,癌症可包含肿瘤且可获得肿瘤的完全反应。
实施例1
fitc-叶酸的合成
在无水二甲亚砜(dmf)中,在四甲基胍及二异丙胺存在的情况下,使叶酸-γ-乙二胺与荧光素异硫氰酸酯(fitc)异构体i(sigma-aldrich)偶联。将粗产物载入于xterrarp18制备型hplc柱(waters)上且用以99%5mm磷酸钠(流动相a,ph7.4)及1%乙腈(流动相b)开始且在20ml/min的流动速率下在10min内达至90%a及10%b的梯度条件洗脱。在这些条件下,fitc-叶酸主峰通常在27-50min洗脱。通过分析型反相hplc和uv检测器监测fitc-叶酸级分的质量。将纯度大于98.0%(lcms)的级分冻干以获得最终fitc-叶酸产物。如本领域中已知,具有此结构的化合物亦称为ec17。
实施例2
fitc-peg12-叶酸的合成
将universal聚乙二醇(peg)novatagtm树脂(0.2g)装入肽合成容器中且用异丙醇(i-proh)(3×10ml)及二甲基甲酰胺(dmf,3×10ml)洗涤。使用含20%哌啶的dmf(3×10ml)实施9-芴基甲氧基羰基(fmoc)去保护。进行kaiser测试以评估反应进展。随后向容器引入fmoc-l-谷氨酸5-叔丁酯(fmoc-glu-(o-t-bu)-oh)(23.5mg)的dmf溶液、n,n-二异丙基乙胺(i-pr2net)(4当量)及六氟磷酸苯并三唑-1-基-氧基三吡咯啶基鏻(pybop)(2当量)。使用含20%哌啶的dmf(3×10ml)实施fmoc去保护。随后向容器引入n10-tfa-pte-oh(22.5mg)、dmf、i-pr2net(4当量)及pybop(2当量)的溶液。氩气鼓泡2h,且用dmf(3×3ml)及i-proh(3×3ml)洗涤树脂。树脂在二氯甲烷(dcm)中溶胀之后,添加1m羟基苯并三唑(hobt)的dcm/三氟乙烷(tfe)(1:1)(2×3ml)溶液。氩气鼓泡1h,移除溶剂,且用dmf(3×3ml)及i-proh(3×3ml)洗涤树脂。树脂在dmf中溶胀之后,添加fmoc-nh-(peg)12-cooh(46.3mg)的dmf溶液、i-pr2net(4当量)及pybop(2当量)。氩气鼓泡2h,且用dmf(3×3ml)及i-proh(3×3ml)洗涤树脂。使用含20%哌啶的dmf(3×10ml)实施fmoc去保护。进行kaiser测试以评估反应进展。随后向容器引入fitc(lifetechnologies21.4mg)的dmf溶液及i-pr2net(4当量),随后氩气鼓泡2h,且用dmf(3×3ml)及i-proh(3×3ml)洗涤树脂。随后向容器添加含2%nh2nh2的dmf(2×2ml)。使用tfa:h2o:三异丙基硅烷(tis)(95:2.5:2.5)(裂解溶液)自树脂裂解最终化合物且在真空下浓缩。经浓缩的产物在et2o中沉淀,且在真空下干燥。使用制备型rp-hplc(流动相:a=10mm乙酸铵ph=7,b=acn;方法:在13ml/min下在30分钟内0%b至30%b)纯化粗产物。将纯净级分汇集且冻干,得到fitc-peg12-叶酸。
实施例3
fitc-peg20-叶酸的合成
将乙二胺、聚合物结合(200-400目)树脂(50mg)装入肽合成容器中且用dcm(3ml)、随后dmf(3ml)溶胀。随后向容器引入dmf中的fmoc-peg20-cooh溶液(131mg,1.0当量)、i-pr2net(6.0当量)及pybop(4.0当量)。氩气鼓泡6h,排出偶联溶液,且用dmf(3×10ml)及i-proh(3×10ml)洗涤树脂。进行kaiser测试以评估反应进展。在各氨基酸偶联之前,使用含20%哌啶的dmf(3×10ml)实施fmoc去保护。重复以上顺序,以完成与fmoc-glu-otbu(72mg,2.0当量)的反应及tfa.喋酸(41mg,1.2当量)偶联步骤。将树脂用含2%肼的dmf3×10ml(5min)洗涤以裂解喋酸上的三氟-乙酰基保护基,且先后用i-proh(3×10ml)及dmf(3×10ml)洗涤。树脂在氩气下干燥30min。使用裂解溶液使叶酸-肽自树脂裂解。引入10ml裂解混合物且氩气鼓泡1.5h。裂解混合物排出至干净烧瓶中。将树脂用更多裂解混合物洗涤3次。将合并的混合物减压浓缩成较小体积(约5ml)且在乙醚中沉淀。
沉淀物通过离心收集,用乙醚洗涤(3次)且在高真空下干燥。在室温下,将经干燥的叶酸-peg20-eda(1.0当量)用含fitc(50mg,1.5当量)的dmso及dipea处理。通过lcms监测反应的进展。在8h之后,起始物质耗尽,得到产物。通过制备型hplc(流动相a=10mm乙酸铵,ph=7;有机相b=乙腈;方法:在13ml/min下,35分钟内0%b至30%b)纯化粗反应混合物且得到fitc-peg20-叶酸,产率60%。
实施例4
fitc-peg108-叶酸的合成
将乙二胺、聚合物结合(200-400目)树脂(50mg)装于肽合成容器中且用dcm(3ml)、随后dmf(3ml)溶胀。随后向容器引入dmf中的fmoc-peg36-cooh溶液(161mg,1.0当量)、i-pr2net(6.0当量)及pybop(4.0当量)。氩气鼓泡6h,排出偶联溶液,且用dmf(3×10ml)及i-proh(3×10ml)洗涤树脂。进行kaiser测试以评估反应进展。在各氨基酸偶联之前,使用含20%哌啶的dmf(3×10ml)实施fmoc去保护。重复以上顺序,以完成具有2xfmoc-peg36-cooh(161mg,1.0当量)、fmoc-glu-otbu(72mg,2.0当量)的反应及tfa.喋酸(41.0mg,1.2当量)偶联步骤。结束时,将树脂用含2%肼的dmf3×10ml(5min)洗涤以裂解喋酸上的三氟-乙酰基保护基,且先后用i-proh(3×10ml)及dmf(3×10ml)洗涤。树脂在氩气下干燥30min。使用裂解溶液使叶酸-肽自树脂裂解。引入10ml裂解混合物且氩气鼓泡1.5h。裂解混合物排出至干净烧瓶中。将树脂用更多裂解溶液洗涤3次。将合并的混合物减压浓缩成较小体积(约5ml)且在乙醚中沉淀。
沉淀物通过离心收集,用乙醚洗涤(3次)且在高真空下干燥。在室温下将经干燥的叶酸-peg108-eda(1.0当量)用含fitc(50mg,1.5当量)的dmso及dipea处理。通过lcms监测反应进展。在10h之后,起始物质耗尽,得到产物。通过制备型hplc(流动相a=10mm乙酸铵,ph=7;有机相b=乙腈;方法:在13ml/min下,35分钟内0%b至30%b)纯化粗反应混合物且得到fitc-peg108-叶酸,产率64%。
实施例5
fitc-dupa的合成
通过如下固相方法合成dupa-fitc。universalnovatagtm树脂(50mg,0.53mm)用dcm(3ml)、随后dmf(3ml)溶胀。将20%哌啶于dmf(3×3ml)中的溶液添加至树脂,且氩气鼓泡5min。用dmf(3×3ml)及异丙醇(i-proh,3×3ml)洗涤树脂。树脂在dmf中溶胀之后,添加dupa-(otbu)-oh(1.5当量)、hatu(2.5当量)及i-pr2net(4.0当量)的dmf溶液。氩气鼓泡2h,且用dmf(3×3ml)及i-proh(3×3ml)洗涤树脂。树脂在dcm中溶胀之后,添加1mhobt于dcm/tfe(1:1)(2×3ml)中的溶液。氩气鼓泡1h,移除溶剂且用dmf(3×3ml)及i-proh(3×3ml)洗涤树脂。树脂在dmf中溶胀之后,添加fmoc-phe-oh(2.5当量)、hatu(2.5当量)及dipea(4.0当量)的dmf溶液。氩气鼓泡2h,且用dmf(3×3ml)及i-proh(3×3ml)洗涤树脂。重复以上顺序用于另外2个偶联步骤,以添加8-氨基辛酸及荧光素异硫氰酸酯或若丹明b异硫氰酸酯。使用裂解溶液使最终化合物自树脂裂解且在真空下浓缩。浓缩产物在乙醚中沉淀,且在真空下干燥。使用制备型rp-hplc[λ=488nm;溶剂梯度:25分钟内1%b至80%b,80%b洗涤30分钟操作;a=10mmnh4oac,ph=7;b=乙腈(acn)]纯化粗产物。在真空下移除acn,且冻干纯净级分,以产生呈淡褐色-橙色固体状的fitc-dupa。rp-hplc:tr=8.0min(a=10mmnh4oac,ph=7.0;b=acn,溶剂梯度:10分钟内1%b至50%b,80%b洗涤15分钟操作)。1hnmr(dmso-d6/d2o):δ0.98-1.27(ms,9h);1.45(b,3h);1.68-1.85(ms,11h);2.03(m,8h);2.6-3.44(ms,12h);3.82(b,2h);4.35(m,1h);6.53(d,j=8.1hz,2h),6.61(dd,j=5.3,3.5hz,2h);6.64(s,2h);7.05(d,j=8.2hz,2h),7.19(m,5h);7.76(d,j=8.0hz,1h);8.38(s,1h)。hrms(esi)(m/z):c51h59n7o15s的(m+h)+计算值,1040.3712,实验值,1040.3702。uv/vis:λ最大值=491nm。
实施例6
fitc-peg12-dupa的合成
将1,2-二氨基乙烷三苯甲基-树脂(0.025g)装入至肽合成容器且先后用i-proh(3×10ml)及dmf(3×10ml)洗涤。随后向容器引入fmoc-nh-(peg)12-cooh(42.8mg)的dmf溶液、i-pr2net(2.5当量)及pybop(2.5当量)。将所得溶液用ar鼓泡1h,排出偶联溶液,且用dmf(3×10ml)及i-proh(3×10ml)洗涤树脂。进行kaiser测试以评估反应进展。使用含20%哌啶的dmf(3×10ml)实施fmoc去保护。重复此程序以完成所有偶联步骤(2×1.5当量fmoc-phe-oh及1.5当量8-氨基辛酸及1.2当量dupa用于其对应偶联步骤中的每一者)。dupa偶联之后,将树脂用dmf(3×10ml)及i-proh(3×10ml)洗涤且在减压下干燥。在肽合成容器中,使用裂解溶液,使肽自树脂裂解。将15ml裂解溶液添加至肽合成容器中,且在ar下鼓泡反应15min。将树脂用两个额外10ml量的裂解溶液处理,各5min。将裂解混合物浓缩至约5ml且用乙醚沉淀。沉淀物通过离心收集,用乙醚洗涤(3次),且在高真空下干燥,产生粗物质的回收。在室温下向粗制dupa-(peg)12-eda(10mg)及fitc(5.6mg)于二甲亚砜(dmso,1ml)中的搅拌溶液中添加i-pr2net(5当量)且在氩气下搅拌6h。反应通过lcms监测且通过制备型hplc(流动相:a=10mm乙酸铵ph=7,b=acn;方法:在13ml/min下30分钟内0%b至50%b)纯化。将经纯净级分汇集且冻干,得到fitc-peg12-dupa。
实施例7
fitc-peg11-nk1的合成
在室温下在氩气下向nk-1(0.02g,0.0433mmol,1.0当量)、o-(2-氨基乙基)-o'-[2-(boc-氨基)乙基]癸乙二醇(bocnh-peg11-nh2)(sigma,0.0336g,0.0521mmol,1.2当量)、六氟磷酸苯并三唑-1-基-氧基三吡咯啶基鏻(pybop)(0.027g,0.0521mmol,1.2当量)于无水ch2cl2中的搅拌溶液中添加n,n-二异丙基乙胺(dipea)(0.076ml,0.4338mmol,10当量)。反应进展通过lcms监测且通过制备型rp-hplc(waters,xbridgetmprepc18,5μm;19×100mm柱,流动相a=20mm乙酸铵缓冲液,ph7,b=乙腈,梯度30分钟内10-100%b,13ml/min,λ=220nm,254nm)纯化。收集纯净级分,蒸发所有有机溶剂,且样品冻干48小时,得到nk1-peg11-nhboc。产率:40.13mg(97%)。向干燥dcm中的nk1-peg11-nhboc(0.0165g,0.015mmol)添加三氟乙酸(tfa,20当量),且在室温下搅拌反应混合物4h。移除过量tfa,且将剩余溶液用水稀释且使用ch2cl2(3×5ml)萃取。经合并的有机层用盐水洗涤、经(na2so4)干燥且浓缩。所得残余物在真空下干燥且未经进一步纯化即用于下一步骤。在室温下在氩气下将nk1-peg11-nh2(0.008g,0.0081mmol,1.0当量)、荧光素异硫氰酸酯(fitc)(sigma,0.0037g,0.0097mmol,1.2当量)于干燥二甲亚砜(dmso,0.3ml)中的搅拌溶液添加至二异丙基乙胺(0.0028ml,0.0162mmol,2.0当量)。反应进展通过lcms监测且产物通过制备型rp-hplc(waters,xbridgetmprepc18,5μm;19×100mm柱,流动相a=20mm乙酸铵缓冲液,ph7,b=乙腈,梯度30分钟内10-100%b,13ml/min,λ=280nm)纯化。收集纯净级分,蒸发所有有机溶剂,且样品冻干48h,得到fitc-peg11-nk1,产率为8.54mg(77%)。
*注意:通过自碱配体开始的二步程序合成nk-1化合物,该碱配体通过使用文献中的程序制备。(参考:designanddevelopmentofneurokinin-1receptor-bindingagentdeliveryconjugates,申请案号:pct/us2015/44229;以引用的方式并入本文中)。
实施例8
fitc-peg2-ca9的合成
在50ml圆底烧瓶中,使用teflon磁性搅拌棒,使ca9配体(53.6mg)溶解于dmf(2-3ml)中。使用真空移除环境空气且用氮气置换,此循环三次。圆底烧瓶保持在恒定氮气下。向烧瓶添加28.9mgn-(3-二甲基氨基丙基)-n'-乙基碳二亚胺盐酸盐(edc),随后添加21.6mg1-羟基苯并三唑水合物(hobt)及18.9μlboc-peg2-nh2(sigmaaldrich)。添加5.4μl三乙胺(tea)且反应搅拌过夜。反应混合物使用hplc纯化且用uhplc-ms(目标m/z为831)确认。使用高真空旋转蒸发移除乙腈且冻干产物。将化合物与1:1tfa:dcm混合30分钟。使用高真空旋转蒸发移除tfa/dcm,随后高真空30分钟。随后使化合物溶解于dmf中且与5摩尔当量的i-pr2net、16mg荧光素异硫氰酸酯(lifetechnologies)合并,且搅拌1小时。通过hplc纯化反应混合物且用uhplc-ms(目标m/z为1120)确认目标化合物。样品冻干且储存于-20℃下。
实施例9
t细胞制备
通过使用ficoll密度梯度离心(gehealthcarelifesciences),自健康供体的全血分离人类外周血液单核细胞(pbmc)。随后通过使用easysep™人类t细胞分离试剂盒(stemcelltechnologies)自pbmc分离t细胞。将t细胞在具有40-100iu/ml人类il-2(miltenyibiotech)、2%人类ab型血清及1%青霉素/链霉素硫酸盐的texmacs培养基(miltenyibiotechinc)中培养。将dynabeads人类t-活化剂cd3/cd28(thermofisherscientific)以1:1比率添加至t细胞以活化t细胞。活化后12-24小时,在8µg/ml聚凝胺(santacruizbiotech)存在的情况下,通过在22-32℃下在1,200g下离心感染90分钟,将t细胞用fitc-car慢病毒粒子转导。在移除活化珠粒之前,将含有具有car修饰的t细胞(car-t)及不具有car修饰的t细胞(未转化t)的t细胞混合物在活化珠粒存在的情况下培养6天。荧光活化的细胞分选术用于基于其gfp表达分选出car-t细胞(gfp阳性)及未转化t细胞(gfp阴性)。在注射至小鼠中之前将经分选的t细胞培养7-15天。当使用t细胞混合物时,在小鼠注射之前将car-t细胞与未转化t细胞以所需比率混合。
实施例10
编码car基因的慢病毒载体的产生
重叠pcr方法用于产生包含抗荧光素的scfv的car构建体。合成衍生自抗荧光素(4-4-20)抗体的抗荧光素的scfv,4m5.3(kd=270fm,762bp)。如图2中所示,通过重叠pcr使编码人类cd8α信号肽(sp,63bp)、铰链及跨膜区(249bp)、4-1bb的细胞质域(cd137,141bp)及cd3ζ链(336bp)的序列与抗荧光素scfv融合。将所得car构建体(1551bp)插入至ecori/noti裂解慢病毒表达载体pcdh-ef1-mcs-(pgk-gfp)(图2,systembiosciences)中。慢病毒载体中car构建体的序列通过dna测序来确认。
一种示例性car核酸编码序列可包含:
在上文所示的示例性核酸序列(seqidno:1)中,第一atg为起始密码子。一种示例性car氨基酸序列可包含:
一种示例性插入物可包含:
在上文所描述的示例性插入物(seqidno:3)中,第一gccacc序列可包含限制酶裂解位点,随后为atg起始密码子。经编码的氨基酸序列可包含:
实施例11
用于人类t细胞转导的含有car基因的慢病毒的产生
为制备含有抗荧光素(即,抗fitc)单链片段可变(scfv)car的慢病毒,将hek-293tn包装细胞系与编码抗荧光素scfvcar的慢病毒载体及第2代慢病毒包装质粒混合物(cellecta)或virapowerlentivrial包装混合物(thermofisher)共转染。转染24及48小时之后,收获含有具有car基因的慢病毒的上清液且通过标准聚乙二醇病毒浓缩法(clontech)浓缩病毒粒子,以供将来用人类t细胞转导。
实施例12
自人类pbmc分离人类t细胞
通过ficoll密度梯度离心(gehealthcarelifesciences)自人类外周血液单核细胞(pbmc)分离t细胞。在洗去剩余ficoll溶液后,通过使用easysep™人类t细胞分离试剂盒(stemcelltechnologies)分离t细胞。将经纯化的t细胞在具有1%青霉素及链霉素硫酸盐的texmacstm培养基(miltenyibiotechinc)中在人类il-2(100iu/ml,miltenyibiotechinc)存在的情况下培养。t细胞以1×106个细胞/毫升的密度在多孔板中培养。将t细胞分开且每2-3天重新投料。
实施例13
人类t细胞的转导
在人类il-2(100iu/ml)存在的情况下将经分离的t细胞用与抗cd3/cd28抗体(lifetechnologies)偶联的dynabeads活化12-24小时,随后用编码抗荧光素car基因的慢病毒转导。72小时后收获细胞且通过使用流式细胞术测量gfp荧光细胞来鉴别转导t细胞上car的表达。
实施例14
fitc-叶酸的合成
在无水二甲亚砜(dmf)中,在四甲基胍及二异丙胺存在的情况下,使叶酸-γ-乙二胺与荧光素异硫氰酸酯(fitc)异构体i(sigma-aldrich)偶联。将粗产物载入于xterrarp18制备型hplc柱(waters)上且用以99%5mm磷酸钠(流动相a,ph7.4)及1%乙腈(流动相b)开始且在20ml/min的流动速率下在10min内达至90%a及10%b的梯度条件洗脱。在这些条件下,fitc-叶酸主峰通常在27-50min洗脱。通过分析型反相hplc和uv检测器监测fitc-叶酸级分的质量。将纯度大于98.0%(lcms)的级分冻干以获得最终fitc-叶酸产物。
实施例15
fitc-dupa的合成
通过如下固相方法合成dupa-fitc。universalnovatag树脂(50mg,0.53mm)用二氯甲烷(dcm)(3ml)、随后二甲基甲酰胺(dmf,3ml)溶胀。将20%哌啶的dmf(3×3ml)溶液添加至树脂,且氩气鼓泡5min。用dmf(3×3ml)及异丙醇(i-proh,3×3ml)洗涤树脂。树脂在dmf中溶胀之后,添加dupa-(otbu)-oh(1.5当量)、hatu(2.5当量)及dipea(4.0当量)于dmf中的溶液。氩气鼓泡2h,且用dmf(3×3ml)及i-proh(3×3ml)洗涤树脂。树脂在dcm中溶胀之后,添加1mhobt于dcm/三氟乙烷(tfe)(1:1)(2×3ml)中的溶液。氩气鼓泡1h,移除溶剂且用dmf(3×3ml)及i-proh(3×3ml)洗涤树脂。树脂在dmf中溶胀之后,添加fmoc-phe-oh(2.5当量)、hatu(2.5当量)及dipea(4.0当量)于dmf中的溶液。氩气鼓泡2h,且用dmf(3×3ml)及i-proh(3×3ml)洗涤树脂。重复以上顺序进行另外2个偶联步骤,以添加8-氨基辛酸及荧光素异硫氰酸酯或若丹明b异硫氰酸酯。使用三氟乙酸(tfa):h2o:三异丙基硅烷混合物(95:2.5:2.5)自树脂裂解最终化合物且在真空下浓缩。浓缩产物在乙醚中沉淀,且在真空下干燥。使用制备型rp-hplc[λ=488nm;溶剂梯度:25分钟内1%b至80%b,80%b洗涤30分钟操作;a=10mmnh4oac,ph=7;b=乙腈(acn)]纯化粗产物。在真空下移除acn,且冻干纯化的级分,以产生呈淡褐色-橙色固体状的dupa-fitc。rp-hplc:tr=8.0min(a=10mmnh4oac,ph=7.0;b=acn,溶剂梯度:10分钟内1%b至50%b,80%b洗涤15分钟操作)。1hnmr(dmso-d6/d2o):δ0.98-1.27(ms,9h);1.45(b,3h);1.68-1.85(ms,11h);2.03(m,8h);2.6-3.44(ms,12h);3.82(b,2h);4.35(m,1h);6.53(d,j=8.1hz,2h),6.61(dd,j=5.3,3.5hz,2h);6.64(s,2h);7.05(d,j=8.2hz,2h),7.19(m,5h);7.76(d,j=8.0hz,1h);8.38(s,1h)。hrms(esi)(m/z):c51h59n7o15s的(m+h)+计算值,1040.3712,实验值,1040.3702。uv/vis:λ最大值=491nm。
实施例16
fitc-ca9的合成
在50ml圆底烧瓶中,使用teflon磁性搅拌棒使ca9配体(53.6mg,实验室中合成)溶解于所要量的n,n-二甲基甲酰胺(dmf)(2-3ml)中。使用真空移除环境空气且用氮气置换,此循环三次。随后将圆底烧瓶保持在恒定氮气下。向烧瓶先后添加28.9mgn-(3-二甲基氨基丙基)-n'-乙基碳二亚胺盐酸盐(edc)及21.6mg1-羟基苯并三唑水合物(hobt)及18.9μlboc-peg2-nh2(购自sigmaaldrich)。最后添加5.4μl三乙胺(tea)且使反应搅拌过夜。反应混合物使用hplc纯化且用uhplc-ms(目标m/z为831)确认。使用高真空旋转蒸发移除乙腈且置于冻干器上48小时。用1:1tfa:dcm进行boc去保护,历时30分钟。使用高真空旋转蒸发移除tfa/dcm,随后高真空30分钟。随后使化合物溶解于dmf中且与5摩尔当量的n,n-二异丙基乙胺(dipea)合并。将16mg荧光素异硫氰酸酯(购自lifetechnologies)添加至溶液且搅拌1小时。通过hplc纯化反应混合物且用uhplc-ms证实目标化合物(靶m/z为1120)。将样品置于冻干器上48小时且将化合物储存于-20℃下。
实施例17
合成fitc-nk1r
在室温下在氩气下向nk-1(0.02g,0.0433mmol,1.0当量)、o-(2-氨基乙基)-o'-[2-(boc-氨基)乙基]癸乙二醇(bocnh-peg11-nh2)(sigma,0.0336g,0.0521mmol,1.2当量)、六氟磷酸苯并三唑-1-基-氧基三吡咯啶基鏻(pybop)(0.027g,0.0521mmol,1.2当量)于干燥ch2cl2中的搅拌溶液中添加n,n-二异丙基乙胺(dipea)(0.076ml,0.4338mmol,10当量)。反应进展通过lcms监测且通过制备型rp-hplc(waters,xbridgetmprepc18,5μm;19×100mm柱,流动相a=20mm乙酸铵缓冲液,ph7,b=乙腈,梯度30分钟内10-100%b,13ml/min,λ=220nm,254nm)纯化。收集纯净级分,蒸发所有有机溶剂,且样品冻干48小时,得到nk1-peg11-nhboc。产率:40.13mg(97%)。向于干燥ch2cl2中的nk1-peg11-nhboc(0.0165g,0.015mmol)添加三氟乙酸(tfa,20当量),且反应混合物在室温下搅拌4h。移除过量tfa,用水稀释,且使用ch2cl2(3×5ml)萃取。经合并的有机层用盐水洗涤、经(na2so4)干燥且浓缩。所得残余物在真空下干燥且未经进一步纯化即用于下一步骤。在室温下在氩气下将nk1-peg11-nh2(0.008g,0.0081mmol,1.0当量)、荧光异硫氰酸盐(fitc)(sigma,0.0037g,0.0097mmol,1.2当量)于干燥二甲亚砜(dmso,0.3ml)中的搅拌溶液添加二异丙基乙胺(0.0028ml,0.0162mmol,2.0当量)。反应进展通过lcms监测且通过制备型rp-hplc(waters,xbridgetmprepc18,5μm;19×100mm柱,流动相a=20mm乙酸铵缓冲液,ph7,b=乙腈,梯度30分钟内10-100%b,13ml/min,λ=280nm)纯化。收集纯净级分,蒸发所有有机溶剂,且样品冻干48小时,得到nk1-peg11-fitc(5)。产率:8.54mg(77%)。
通过自碱配体开始的二步程序合成nk-1化合物,该碱配体通过使用文献程序制备。(参考:designanddevelopmentofneurokinin-1receptor-bindingagentdeliveryconjugates,申请案号:pct/us2015/44229,以全文引用的方式并入本文中)。
实施例18
hos-fr-α及aml研究概要
将根据以下规则施用连续递增剂量的ec17:
•如果无crs或神经毒性,则按计划通过ec17剂量递增推进。
•如果crs级别≤2,则以在相同周期中引起不严重crs的顺序水平施用所有后续ec17剂量。
•如果crs级别>2,则取消当前周期中其他ec17剂量,且按引起scrs的以下次高顺序,开始所有后续周期。
a.肿瘤植入
hos-frα(即hos-143b-lv-frα)为用人类frα稳定转染的hos-143b(atcccrl-8303)亚克隆。hos-143b最初衍生自患有骨肉瘤的13岁的高加索女性。肿瘤细胞在5%co2潮湿大气中在37℃下在具有5%fbs的叶酸缺失型rpmi1640中生长。针对体内研究,以1×106个细胞/只动物皮下接种肿瘤细胞。
b.car-t细胞施用
经egfrt分选的抗fitce2scfv-cart细胞在t细胞冷冻培养基中冷冻。在到达时,即刻将冷冻car-t细胞的小瓶储存于-80℃下。car-t细胞在37℃下快速解冻,用pbs洗涤两次,且以6百万有活性的egfrt+e2car-t细胞(cd4/cd8为约1:1)/只动物用于动物注射。在输注当天取出小的等分试样以用于e2-cart细胞表型的流式细胞术分析。
c.制备药物给药溶液
在给药开始时,通过以下制备ec17给药溶液:称取适当量的各化合物,在pbs(ph7.4)中重构,通过0.22µmpvdf针筒过滤器无菌过滤药物溶液,且在-20℃下冷冻等分试样用于每日给药。
d.化合物施用
至一天结束(3-4pm),给予所有ec17剂量,以允许过夜显现可能的crs(细胞因子释放综合征)。在第二天早晨,根据crs分级系统(参见下文)对动物评分。
e.评估
crs分级系统
肿瘤生长、体重及总体评定:肿瘤大小监测2-3次/周且体重测量2-3次/周。在紧接在任何ec17剂量之后的数天,每日进行体重测量且注意力集中于总体动物形态及行为。
安乐死:当小鼠的重量损失≥20%时,当肿瘤达至≥1500mm3时,进行安乐死。如果小鼠在短时间内(例如,归因于严重头部倾斜)损失大量重量,或当其按照crs分级系统接近濒死状态时,则亦进行安乐死。
ec17宿主内剂量递增治疗(3个周期)
抗肿瘤活性(n=5)。
如图4中所见:
群组1(仅e2-cart细胞)显示在无ec17治疗的情况下hos-frα强力生长。
群组2(ec175/100/1000nmol/kg,m/th/m)显示贯穿此研究,显著的肿瘤消退而没有体重损失或crs。
慢ec17剂量递增(m/th/m)方案为安全的。在携带hos-frα肿瘤的小鼠中未观测到体重损失或细胞因子释放综合征(crs)的症状。
此e2-cart细胞研究的一种目标为使用在car-t输注之后3天开始的多个给药方案,测试ec17/e2car-t细胞活性及相关毒性(例如scrs)。三个不同ec17给药方案包括(a)每周一次500nmol/kg(siw),(b)以9天的间隔,在星期一/星期三/星期五以5、50及500nmol/kg的“tiw样”,及(c)以7天的间隔,在星期一/星期四/星期一以5、100及1000nmol/kg的“tiw样”。
实验组(c=周期):
*组4-5:对于每个周期的第2周,跳过给药。
研究概要
第1组(未治疗):仅用于疗效比较。
第2组(仅car-t):
第3组(ec17siw):
第4组(ec17真实tiw):
第5组-ec17低剂量“tiw”(m/th/m,停止6天)具有/疗效:
a.肿瘤植入
thp-1-frβaml肿瘤细胞在5%co2潮湿大气中在37℃下在具有5-10%fbs的叶酸缺失型rpmi1640中生长。以5×106个细胞/只动物静脉内注射无血清叶酸缺失型rpmi1640培养基中的thp-1-frß肿瘤细胞。
b.car-t细胞施用
将抗fitce2scfv-cart细胞冷冻于t细胞冷冻培养基中。在到达时,即刻将冷冻car-t细胞的小瓶储存于-80℃下。car-t细胞在37℃下快速解冻,用car-t细胞培养基洗涤两次,且以6百万个有活性的egfrt+e2car-t细胞(cd4/cd8在约1:1)/只动物用于动物注射。在输注当天取出小的等分试样以用于e2-cart细胞表型的流式细胞术分析。
c.制备药物给药溶液
在给药开始时,通过以下制备ec17及双-eda-fitc给药溶液:称取适当量的各化合物,在pbs(ph7.4)中重构,通过0.22µmpvdf针筒过滤器无菌过滤药物溶液,且在-20℃下冷冻等分试样用于每日给药。
d.化合物施用
至一天结束(3-4pm),给予所有ec17剂量,以允许过夜显现可能的crs(细胞因子释放综合征)。在第二天早晨时,根据crs分级系统(参见下文)对动物评分。
ec17剂量计划表:
群组1-2:未给予ec17剂量
群组3:
群组4:
群组5:
监测/疗效:
需要在ec17剂量之后数天进行每日体重测量。注意力集中于总体动物形态及行为。如果小鼠在短时间内损失大量重量,或当小鼠按照crs分级系统或神经毒性接近濒死状态时,则进行安乐死。
crs分级系统
肿瘤生长、体重及总体评定:体重测量2-3次/周。在紧接在任何ec17剂量之后的数天,每日进行体重测量且注意力集中于总体动物形态及行为。
流式细胞术分析
全血细胞分析:自预定体积的经edta处理全血移除血浆,且用rbc裂解溶液裂解rbc。白细胞沉淀随后再悬浮于流式细胞术染色溶液(1%牛血清白蛋白,50mg/ml人类igg(equitechbio,cat#slh56-0001)、0.9%叠氮化钠于磷酸盐缓冲盐水中,ph=7.4)中,且白细胞表面标记物染色使用以下抗体进行:抗人类cd45(克隆hi30,ebioscience#47-0459-42,1:20(v/v)稀释),抗人类cd137(克隆4b4-1,bdbioscience#564092,1:20(v/v)稀释),抗人类cd8α[克隆rpa-t8,bdbioscience,目录号#557746,1:20(v/v)稀释],抗人类cd4[克隆sk3,ebioscience目录号#46-0047-42,1:20(v/v)稀释],抗人类egfr[r&dsystems,克隆hu1,目录号#fab9577b,@1:10(v/v)],抗人类pd1[bdbiosciences,克隆eh12.1,目录号#562511,@1:20(v/v)],抗人类lag3[bdbiosciences,克隆t47-530,目录号565616,@1:20(v/v)],抗人类tim3[bdbiosciences,克隆7d3,目录号565558,@1:20(v/v)],抗人类cd3ε[bdbiosciences,克隆sk7,目录号557832,@1:20(v/v)]。白细胞染色后,将细胞用pbs洗涤且再悬浮于含有53,000个countbrighttm珠粒[invitrogen目录号c36950]的冷pbs中且转移至流式细胞术的收集管。在gallios流式细胞仪(beckmancoulter,brea,ca)上收集流式细胞术数据。根据invitrogen的说明书,计算各血液样品中cart细胞的浓度的判定。cart细胞鉴别为人类cd3ε+egfrt+事件且容易地使用kaluzatm流式细胞术软件区分及计数。随后各输注小鼠的循环中的cart细胞数目以每50µl分析的全血的cart细胞总数目表示在图上。统计显著性通过利用未配对的双尾学生t测试来判定,其中显著性设定为p<0.05。
肿瘤及组织分析:收获实体肿瘤(100-1000mm3),称重,且切碎成小碎片,随后转移至含有20ml肿瘤消化混合物的50ml管中。酶促肿瘤消化混合物由补充有抗生素的无血清及叶酸缺失型rpmi1640培养基中0.5mg/mliv型胶原蛋白酶(sigma-aldrich,目录号#c5138)、0.5mg/ml透明质酸酶(sigma-aldrich,目录号#h3506)及0.1mg/mldnasei(sigma-aldrich,目录号#dn25)组成。肿瘤片段在水平震荡器上在37℃下在300rpm下消化一小时。随后,肿瘤消化物在400×g下离心5分钟,且肿瘤细胞沉淀进行红血球裂解步骤,随后用冷磷酸盐缓冲盐水(pbs,ph7.4)洗涤且最后经40µm尼龙细胞过滤器过滤。
结果及结论
a.ec17施用的三个不同剂量计划表显示不同体重损失模式及不同crs水平
如图5中所见,群组1(未治疗)及群组2(仅car-t)没有显示诸多体重损失且无crs。群组3(ec17siw)显示在各ec17剂量之后ec17剂量依赖型体重损失且贯穿研究展示级别1-2的crs。群组5(ec17剂量递增)显示在前两个周期期间很少的体重损失,但在第二次ec17剂量周期结束之后开始显示与群组2类似的体重损失。群组4(ec17tiw开/关)显示三个治疗组中最轻微的体重损失。群组4或5两者都没有显示任何大于1的crs。
群组3
群组4
群组5
ec17施用的三个不同剂量计划表显示不同抗肿瘤活性。
如图6中所见,所有群组中在第39天评定肝及非肝肿瘤负荷。群组2(仅car-t)与群组1(未治疗)相比没有显示任何差异。群组4显示三个不同ec17剂量计划表中最佳的抗肿瘤活性。
循环aml细胞系thp1-frβ的消除
在使用cd19特异性抗all疗法的研究中,已报告cart细胞在血液中的扩增及维持与导致对疗法的完全反应的循环cd19+all细胞的消除相关。在此实验中,我们在我们的小鼠aml模型中进行类似测定,以通过探索我们的经gfp标记的人类aml细胞系thp1-frβ响应于cart适配分子ec17的不同给药方案的消失,来测定我们的受适配子控制的e2cart细胞的抗肿瘤活性。图7显示循环thp1-frβ细胞,其中y轴展示在对数标度上每100µl全血的gfp+细胞总数目。不同条柱表示不同治疗群组。证实,在将aml细胞输注至没有接受cart细胞也没有接受ec17的小鼠中后40天,高aml细胞负荷存在于循环中。在接受aml细胞及e2cart细胞两者的输入但无ec17治疗的小鼠中,没有看到血液中aml细胞的减少。这证实,通过具有同种异体thp1细胞的人类t细胞的可能同种异体反应性的可忽略的抗肿瘤活性。有趣地,看见以不同给药方案用e2cart细胞加ec17输注的所有三个群组小鼠中的循环thp1细胞负荷的显著减少,由此展现在三个不同适配子给药方案下的cart疗法的有效性。
e2-car-t耗尽表型
在慢性抗原刺激下,例如在慢性病毒感染或癌症的情形下,t细胞经历耗尽过程,其中它们不再能够增殖、分泌炎性细胞因子或杀死呈现抗原的靶细胞,cart细胞在car的慢性刺激下通过持续存在的scfv刺激抗原亦具有耗尽的可能性。因为我们的e2cart细胞仅对结合至fr+肿瘤细胞的表面的我们的桥分子ec17的存在起反应,所以我们具有通过停止用ec17治疗且从而除去表面结合抗原的存在来预防e2cart细胞的慢性抗原刺激的能力。通过表面抗原的缺失表征的中止阶段预防e2cart细胞的慢性抗原暴露且预防可产生的所得耗尽。
为确认我们的ec17给药方案不引起e2cart细胞耗竭,对来自thp1-frβ肝癌转移的单细胞制剂及在经耗尽t细胞上特异性表达的表面标记物进行流式细胞术分析。随着t细胞接近耗尽,表面抑制受体pd1、lag3及tim3的共表达增加。图8展示柱状图,其中y轴说明自实体肝肿瘤分离的总e2cart细胞的百分比,其表达由不同条柱表示的三种表面标记物的各种组合。值得注意的是,同时表达所有三种标记物的经完全耗尽t细胞由各组中的左侧条柱表示。左侧组展示将e2cart细胞预输注至自三个不同ec17治疗群组分离的肝肿瘤cart细胞。抑制性受体表达在预输注cart细胞产物中几乎为零,这是由于t细胞中的大部分对于所有三种表面标记物为阴性。重要的是,所有三个经ec17治疗群组e2cart细胞具有经耗尽的三阳性细胞,且有趣的是,群组4(ec17tiw)表达最少数目的双及三阳性事件且进一步由显著数量的三阴性cart细胞组成。这些数据表明在此小鼠实验中所采用的所有三个ec17给药方案可用于临床,这是由于存在显著抗肿瘤活性且存在极少cart耗尽的诱导。
实施例19至25-参见图9至14。
实施例19
细胞系及试剂
除非另外指出,否则所有fr+及fr-阴性癌细胞系保持在补充有10%热灭活胎牛血清的rpmi-1640培养基(gibcobrl),该培养基不含2.4μm叶酸(fa)(ffrpmi)或含该叶酸(rpmi)。将kb(具有hela标记物的表达frα的人类子宫颈癌)及cho-β(经人类frβ转染的中国仓鼠卵巢细胞)分别用作frα及frβ的来源以用于放射性配体结合测定。mda-mb-231表示人类tnbc(三阴性乳腺癌)细胞系的frα亚克隆。对于aml研究,提供frβ阳性(thp1-frβ)及fr-阴性(thp1-fg12)细胞系的表达绿色荧光蛋白(gfp)的同基因对。两者均由thp-1(atcc,tib-202)构建,thp-1是用于研究儿科aml的常用细胞模型,其最初衍生自患有急性单核细胞性白血病的1岁男婴。对于骨肉瘤研究,通过用编码人类frα的folr1基因对fr阴性hos-143b(atcc,crl8303)的慢病毒转导来构建hos-frα。hos-143b最初自13岁高加索女性的原发肿瘤构建且在nsg小鼠中高度致瘤。用慢病毒萤火虫荧光素酶转导fr+hos-frαfluc及fr-阴性hos-143bfluc的表达gfp的生物发光对。
legendplextm人类细胞因子组购自biolegend(sandiego,ca)。基于乳酸脱氢酶(ldh)cytotox96®非放射性细胞毒性测定试剂盒购自promega(madison,wi)。用于多色流式细胞术的可商购的抗人类抗体为:来自thermofisherscientific(waltham,ma)的cd45ra(克隆hi100)、cd45ro(克隆uchl1)、cd4(克隆sk3)及cd69(克隆fn50);来自bdbioscience(sanjose,ca)的cd3ε(克隆sk7)、cd8α(克隆rpa-t8)、cd137/4-1bb(克隆4b4-1)、cd25(克隆m-a251)、pd1(克隆eh12.1)、lag3(克隆t47-530)及tim3(克隆7d3);来自r&dsystems(minneapolis,mn)的生物素化抗人类egfr(西妥昔单抗(cetuximab),克隆hu1);及来自biolegend(sandiego,ca)的frα(克隆lk26)。荧光团缀合的抗生物素亦购自biolegend。apc缀合的抗fitc小鼠igg2a/kappa抗体(克隆naweslee)、countbrighttm珠粒(invitrogen)、膜联蛋白v染色缓冲液及alexafluor-647缀合的膜联蛋白v购自thermofisherscientific。对于肿瘤组织的酶促消化,iv型胶原蛋白酶、透明质酸酶及dnasei皆购自sigma-aldrich(st.louis,mo)。
ec17或叶酸-fitc[fa-(γ)-乙二胺-fitc]在endocyte合成。3h-ec17购自moravekbiochemicals(brea,ca),呈约0.952ci/mmol的比活性,或在endocyte处通过使fitc与通过vitrax(placentia,ca)制得的3h-fa-(γ)-乙二胺缀合来制备,呈约1.2ci/mmol的比活性。3h-fa亦购自vitrax,呈59ci/mmol的比活性。对于crs救援,荧光素钠给药溶液由购自purduepharmacy(ndc17478-250-25)的ak-fluor®25%(荧光素注射液,usp)稀释。
实施例20
人源化car构建体及经car改性的t细胞
先前研究使用含有cd8α及4-1bb/cd3ζ信号传递域的铰链及跨膜序列(即,fitc-4m5.3-scfv-cd8α铰链-cd8αtm-4-1bb/cd3ζ)的gfp+第二代抗fitcscfv(克隆4m5.3)car。为了转译成第一次人体内测试,研发第二代完全人类fitc特异性(克隆e2)car构建体(本文中称为e2)(图9,顶部图)。经car改性的t细胞展示于图9,底部饼状图中。
本文中所描述的构建体为包括以下的fitc特异性car构建体:(1)完全人类抗fitcscfv(克隆e2,kd=0.75nm),(2)融合至cd28-跨膜结构域的igg4铰链-ch2(l235d、n297q)-ch3间隔子,(3)第二代4-1bb/cd3ζ-胞内域,及(4)细胞表面人类egfrt标记(图9,顶部图)(seqidno:4及5分别为核酸及氨基酸序列)。为产生经car改性的t细胞,在与car编码的ephiv7慢病毒载体共转染的293t细胞中产生慢病毒。供体cd4+及cd8+t细胞通过免疫磁性选择纯化且单独地或以约50:50比率转导。一般而言,实施仅一轮的cd3/cd28珠粒活化,随后为一或两轮的快速体外扩增。针对临床前评价,使用若干批次的经egfrt分选的cd4、cd8及未分选的cd4/cd8car-t细胞。在冷冻保存之前及解冻之后分析所有car-t细胞制品以通过流式细胞术判定egfrt表达及cd4/cd8比率。使用表面标记物的组合,在输注当天分析cd4+及cd8+car-t细胞亚组的分化状态且定义为tn,cd45ra+cd45ro-cd62l+cd95-初始t细胞;tscm,cd45ra+cd45ro-cd62l+cd95+干细胞记忆t细胞;tcm,cd45ra-cd45ro+cd62l+cd95+中枢记忆t细胞;tem,cd45ra-cd45ro+cd62l-cd95+效应记忆细胞;及teff,cd45ra+cd45ro-cd62l-cd95+效应t细胞。针对下文所描述的临床前测试,研究包括两个批次的经egfrt分选的纯cd4及cd8亚组(在以约1:1比率混合之后)及若干批次的包括具有低分化特征的“临床传真”制品的未分选的约1:1egfrt+cd4/cd8混合物。
在所合成及评价的一系列不同car构建体中,选择完全人类抗fitcscfv(fitc-e2)car用于临床前研发(图9)。此第二代完全人类car由抗fitcscfv(克隆e2)、ch2区域中具有双突变(l235d及n297q)以减少与fcγr结合的igg4-fc间隔子/铰链、cd28跨膜结构域及通过t2a核糖体跳跃序列附接至细胞表面egfrt标记的4-1bb/cd3ζ信号传导结构域组成(即,fitc-e2-scfv-igg4铰链-cd28tm-4-1bb/cd3ζ-t2a-egfrt)。针对临床前研究,经egfrt分选的及未分选的e2car-t细胞两者皆以约1:1cd4/cd8比率制备,且在各体内实验的cart细胞输注(第0天)时常规地通过流式细胞术进行t细胞亚型表型分析。a经egfrt分选的car-t细胞的典型表达模式包括均呈大致42%tscm、10%tcm、12%tem及34%teff的cd4及cd8亚组(图9,左侧上的饼图)。仅将经egfrt分选的car-t细胞用于共培养及药物动力学研究。针对肿瘤疗法,使用具有mda-mb-231的低分化特征(图9,右侧上的饼图)的“临床传真”批次,且研究批次用于thp1-frβ及hos-frα研究。“临床传真”批次(约39%egfrt+)包括呈约66%tscm及约32%tcm的cd4+亚组及呈约95%tscm及约3%tcm的cd8亚组。研究批次(约23%egfrt+)更经分化且包括呈约32%tscm、约53%tcm、约11%tem及约3.7%teff的cd4亚组及呈约44%tscm、约0.28%tcm、约3.4%tem及约52%teff的cd8亚组。
实施例21
ec17cam的双特异性亲和力
在基于细胞的放射性配体结合分析中使用3h-ec17评定ec17cam(cam在本申请中等同于“桥”或“化合物”)的双特异性亲和力。针对与fr+靶的结合,将kb及cho-β细胞在24孔组织培养板中预接种过夜且与0.1、0.5、1、5、10、20及40nm于ffrpmi中的3h-ec17在37℃下一起培育2h。随后,将细胞用磷酸盐缓冲盐水(pbs,ph7.4)冲洗且用1%十二烷基硫酸钠裂解。全细胞裂解物针对放射性等级及细胞蛋白质含量通过标准piercebca蛋白质测定来定量。计算每个细胞结合的3h-ec17分子的数目以分别判定frα(kb)及frβ(cho-β)的解离常数(kd)(图10)。
已在临床中出于免疫疗法及光学成像的目的测试ec17。为直接地定量其双特异性结合亲和力,合成3h-ec17且对分别表示frα+及frβ+靶细胞的kb及cho-β细胞系及对表示效应细胞的未分选egfrtcar-t细胞实施放射性配体结合测定。当结合至其靶标时,ec17展现对于frα及frβ两者类似的亲和力,分别具有1.7nm及0.8nm的低kd值(图10,a图)。在结合至未分选e2-car-t细胞(约24%egfrt+,约95:5cd8/cd4比率)时,估计kd值在约130nm(图10,b图)。
实施例22
肿瘤模型
所有动物护理及使用根据nih指南且遵循由purdueuniversityanimaluseandcarecommittee审批通过的方案进行。雌性4至5周龄的nod/scidγ(nsgtm)小鼠(物料编号:005557)购自thejacksonlaboratory(barharbor,me)。除非专门指示,否则所有动物在到达时及在整个研究中保持fa缺失型饮食(testdiet,st.louis,mo)。为建立皮下异种移植,分别将mda-mb-231及hos-frα以2.5×106及1×106个细胞/只动物植入于右侧腹区域中。针对静脉内异种移植,以5×106个细胞/只动物接种thp1-frβ细胞。皮下肿瘤用测径规测量每周2-3次且使用椭圆形公式(长度×宽度2)/2来计算。按照研究设计或在以下情况时进行安乐死:(i)动物的体重已损失≥20%或接近濒死状态,(ii)皮下肿瘤的大小达至≥1500mm3,或(iii)动物显示出肿胀的腹部及严重窘迫(即,thp1-frβ)。静脉内给予所有动物剂量(car-t细胞、ec17、荧光素钠)。
实施例23
肿瘤疗法
在治疗背景下,可在car-t细胞注射之前或之后给予ec17cam。如本文所述,在cart细胞2-3.5天后施用第一剂量的ec17以允许观察携带肿瘤的小鼠中的人类t细胞的时期。两个批次的未分选e2-cart细胞(23%或39%egfrt+,1:1cd4/cd8)用于体内研究。在用于各实验的输注日(第0天),将冷冻cart细胞在37℃下快速解冻,用达尔伯克(dulbecco's)1×pbs(ph7.4)洗涤2×并以所需egfrt+e2-car-t细胞剂量注射至尾部静脉中。此外,通过流式细胞术分析小的等分试样的cart细胞的cart细胞的cd4与cd8比率及分化状态。在ec17给药的第一天,根据携带肿瘤动物的肿瘤大小或静脉内移植(即,thp1-frβ)后相同天数将其随机分配为多组。
对于mda-mb-231研究,小鼠接受高剂量(约10百万)的具有低分化特征的“临床传真”的e2-cart细胞(约39%egfrt+)(图9)。两天后,在平均肿瘤大小为约293±39mm3下开始3种不同的ec17治疗方案(图12)。一周一次(siw)以500nmol/kg在星期一给予ec17给药,或以5、50或100及500或1000nmol/kg(即,5/50/500或5/100/1000)的递增剂量在星期一、星期四及星期一给予ec17给药,周期之间中断6天。对照小鼠维持不治疗的(接受cart细胞但无ec17)。对于比较,不含肿瘤同胎仔畜的群组也在没有或具有500nmol/kg的ec17siw的情况下接受相同数目cart细胞。
对于aml研究(图13),小鼠在接受低剂量的约6百万e2-cart细胞(约23%egfrt+)之前一天经静脉内注入thp1-frβ肿瘤细胞。在car-t细胞输注后约3.5天,以3种不同方式给药ec17,包括i)以500nmol/kgsiw,ii)以5/50/500nmol/kg在星期一/星期三/星期五三次,随后为周期之间9天的中断(tiw开/关),及iii)以第1周期5/10/100nmol/kg、第2周期5/30/300nmol/kg及第3周期的5/50/500nmol/kg的递增剂量,均在星期一/星期四/星期一,周期之间中断6天(m/th/m_开/关)。在第31天,采集动物外周组织用于将小鼠血液中识别的car阳性t细胞量化为人类cd3ε+egfrt+事件且经计算为每100µl全血的绝对数目。在安乐死之后或在第38天研究结束时,通过利用流式细胞术测量血液中的gfp+肿瘤细胞且收集体内发现的肝重量(具有转移性病变)及非肝巨型转移的总重量来评估携带thp1-frβ肿瘤的小鼠体内的总肿瘤负荷。将肝转移的肿瘤片段酶消化至单细胞悬浮液中且使用抗人类pd1、lag3及tim3(克隆eh12.1、t47-530及7d3,分别地)分析活细胞群的car-t细胞耗竭状态。
对于骨肉瘤研究(图14),两个群组的小鼠在接受用于thp1-frβ研究(图13)的约6百万的相同car-t细胞制剂前3天经皮下移植hos-frα肿瘤细胞。在car-t细胞输注后约3.5天,一个群组的小鼠经给予至多3个周期的ec17:第1周期以5/10/100nmol/kg,第2周期以5/30/300nmol/kg及第3周期以5/50/500nmol/kg,全部遵循星期一/星期四/星期一方案,周期之间中断6天。在研究结束时,计数每100μl小鼠血液的循环cd3ε+egfrt+cart细胞。亦采集hos-frα肿瘤(+/-ec17治疗)并消化用于肿瘤浸润性cart细胞的流式细胞术分析。
针对攻击性骨肉瘤模型的剂量递增试验
为了我们既定目的,hos-frα为具有约5.82±1.45pmol/mg蛋白质的功能性fr水平的低fr-表达但最具攻击性肿瘤模型。平行于上文所描述的thp1-frβ研究,两个群组的具有hos-frα肿瘤的3日龄小鼠经以相同剂量(约6百万)给予相同e2-cart细胞。一个群组经相同加速ec17剂量递增方案治疗:第1周期以5/10/100nmol/kg,第2周期以5/30/300nmol/kg及第3周期以5/50/500nmol/kg,按星期一/星期四/星期一排程,随后中断6天(图14,a图)。由于hos-frα肿瘤在无ec17治疗的情况下强力地生长,因此在此car-t细胞剂量下的加速ec17剂量递增为安全的(无crs或体重损失)且在前两个治疗周期内引起肿瘤生长的明显延迟(图14,b图)。在协议授权的仅因肿瘤大小(≥1500mm3)所致的安乐死之后,对全血执行的流式细胞术分析展示ec17依赖性car-t细胞扩增至第47天但在第33天时更高(图14,c图,左边直方图)。第33天的离体肿瘤分析指示经ec17治疗的动物中的低但显著的瘤内cd3ε+egfrt+car-t细胞群体,其占约1%总存活消化肿瘤衍生细胞(图14,c图,右边直方图)。随着大的hos-frα肿瘤停止对第3周期的治疗作出响应(图14,b图),在第47天,瘤内cart细胞也减少(图14,c图)。值得注意地,在疾病进程后通过3h-fa放射性配体测定分析的hos-frα肿瘤在存在及不存在ec17治疗下展示类似frα水平。因此,似乎nsg小鼠中的hos-frα肿瘤快速地增长,超出cart细胞的肿瘤浸润能力且可如体外共培养研究表明,因t细胞耗竭所致而具有减小的细胞溶解活性。
含肿瘤及无肿瘤小鼠体内的ec17剂量发现研究
在携带mda-mb-231肿瘤的小鼠中进行初始ec17剂量发现研究,如实验性布局的示意图所示(图12,a图)。在第0天,不具有或具有mda-mb-231肿瘤(约211±65mm3)的nsg小鼠经移植约10百万“临床传真”批次的e2-cart细胞(约39%egfrt+,51:49cd4/cd8)。此批次cart细胞由大部分tscm及tcm表型组成(参见图9)。在car-t细胞注射后两天,一个群组的肿瘤携带小鼠保持未治疗,同时三个群组经不同ec17方案治疗,包括以500nmol/kg每周单次注射(siw),或作为5/50/500(递增-1)或5/100/1000nmol/kg(递增-2)的递增ec17剂量水平,按星期一/星期四/星期一方案给予,其中具有1周的无药物间隔。为了比较,两个无肿瘤群组未经治疗或以500nmol/kgec17siw治疗。
使用动物外周组织,在第11天及第12天(在先前ec17剂量之后约20及42小时)来测量在所有群组中小鼠血液中的人类t细胞衍生的ifnγ水平。与仅接受cart细胞的肿瘤携带小鼠相比,所有经ec17治疗的肿瘤群组具有约30×(第11天)及约10×(第12天)更高的小鼠血浆中的ifnγ产生,且此细胞因子的水平稍后自20小时至42小时自然地降低(图12,b图)。根据ifnγ释放,ec17亦触发经鉴别为小鼠血液中的人类cd3ε+egfrt+事件的car-t细胞扩增,且细胞在肿瘤携带动物中存留多达54天(最末测量)(图12,c图)。在无肿瘤群组中,通过流式细胞术未检测到car-t细胞扩增,但在第11天及第12天于接受相同数目的cart细胞加ec17的动物中检测到低水平的ifnγ(图12,b至c图)。此外,在以500nmol/kg接受两个每周剂量的ec17的无肿瘤小鼠中未观测到crs症状(0至5个分级中的0级)或体重损失。
不依赖于方案,在持续ec17给药的肿瘤携带群组中观测到中度至重度crs症状(2至3级)及显著体重损失(图12,d图)。在500nmol/kg下的ec17siw导致crs及体重损失的最早开始,而激进的ec17递增-2方案(多达1000nmol/kg)导致持久性体重损失,其中动物恢复出现在ec17剂量停止之后(图12,d图,顶部列)。值得注意地,移植物抗宿主病(gvhd)的症状包括红色皮肤瘙痒及无毛发且在car-t细胞移植之后约1个月时变得明显。尽管仅接受cart细胞的动物展示出非特异性car-t细胞/肿瘤同种异体反应性的体征,但仅经ec17治疗的群组产生100%治愈(图12,d图,底部列)。因此,在fr+肿瘤存在下的ec17施用为驱动i)car-t细胞活化,ii)细胞因子产生,及iii)体内car-t细胞扩增及存留的关键。然而,在特定条件下,通过高car-t细胞剂量与等于或大于500nmol/kg的ec17剂量组合而触发严重crs(≥3级)。
抗白血病活性的ec17cam给药控制
经静脉内植入的表达gfp的thp1-frβ肿瘤细胞在nsg小鼠中产生多发性疾病,具有循环中的肿瘤细胞和全身的肝/非肝转移。thp1-frβ肿瘤细胞亦可定位于小鼠卵巢,该卵巢在肿瘤进展的初期阶段出现发炎。因此,研究群组中的各动物中的总肿瘤负荷是通过对血液中的循环gfp+肿瘤细胞、肝重量及裸眼可见的全包含性非肝巨型转移进行定量来评估。尽管thp1-frβ在体外表达低水平的fr,但发现thp1-frβ肿瘤转移表达高于预期的在约8.9pmol/mg±2.8pmol/mg膜蛋白的功能性fr水平。因此,以约6百万/动物向thp1-frβ肿瘤携带小鼠移植研究批次的egfrt-未分选的e2-cart细胞(约23%egfrt+,1:1cd4:cd8)且随后用3种不同ec17给药方案来治疗(图13)。在car-t细胞注射之后3天起,开始ec17给药方案,其作为在500nmol/kg持续的siw,以5/50/500nmol/kg在星期一/星期三/星期五进行一周三次(tiw),随后中断9天,或通过加速剂量递增方案在第1周期中以5/10/100nmol/kg、在第2周期中以5/30/300nmol及在第3周期中以5/50/500nmolkg在星期一/星期四/星期一(m/th/m)进行,随后中断6天(图13,a图)。在接受ec17siw治疗的动物体内观测到周期2及周期3中的一些体重损失及1至2级crs,而接受ec17tiw的动物体重损失最少,仅为0至1级crs(图13,b图)。在接受3个周期的ec17m/th/m剂量递增的动物当中,在300nmol/kg的ec17的最末剂量之后的第2周期中观测到0至1级crs及极轻微体重损失。在第31天使用动物外周组织,对血液中的cart细胞进行计数,且数据展现出在所有经治疗群组中的ec17依赖型car-t细胞扩增及存留(图13,c图)。与仅接受肿瘤细胞或肿瘤细胞加cart细胞而无ec17的对照动物相比,经3个“患者内”递增格式中的任一者给药的ec17有效地降低血液中的循环thp1-frβ肿瘤细胞,且展示出针对肝肿瘤转移的类似活性(图13,d图,左侧及中间条形图)。在仅接受cart细胞的小鼠中,仅发现微量的针对thp1-frβ肝转移的同种异体反应性。以10倍剂量递增(开/关)的ec17siw及tiw成功地控制非肝巨型转移,以每周期缓慢步调的ec17m/th/m剂量递增(图13,a图)未能控制非肝巨型转移(图13,d图,最右侧图)。在研究结束(即car-t细胞注射后39天)时,自肝thp1-frβ肿瘤转移分离的cart细胞显示具有最少的双重及三重阳性t细胞抑制受体pd1、lag3及tim3的表达(图13,e图)。总体而言,在任何经ec17治疗的群组中未观测到严重crs(即等级≥3)。然而,更激进的ec17tiw剂量递增(开/关)排程倾向于为用于降低这些小鼠中的整体thp1-frβ肿瘤负荷的最佳方案。
实施例24
功能性fr评估
这些功能性fr评估适用于本文中所描述的实施例。除经frα(hos-frα)及frβ(thp1-frβ)转染的儿科癌细胞系外,亦包括具有不同组织学及fr表达水平的癌细胞系(图11)。如通过放射性配体结合测定(在37℃下,100nm3h-fa,1h)所估计,在这些细胞系上总体可获得的fr的分级次序为:9×104(ov90,低-fr表达卵巢癌细胞系)、1.9×105(thp1-frβ)、2.4×105(hos-frαfluc)、7×105(hos-frα)、2.1×106(mda-mb-231)及4.8×106(kb)fa分子/细胞。还包括作为fr-阴性对照的hos-143b(fluc)及thp1-fg12亲本细胞系。因此,共培养fr+癌细胞系上的功能性fr表达的一般分级为:kb>mda-mb-231>hos-frα>hos-frαfluc>thp1-frβ(aml)>ov90。
实施例25
统计
使用计算机程序graphpadprism(graphpadsoftware公司.,sandiego,ca)来执行统计分析。使用学生t测试或单向anova对数据进行分析。如果适用,使用适当的多重比较事后测试跨治疗组对数据作进一步分析。在所有测试中将*=p<0.05视为统计学上显著的。
- 还没有人留言评论。精彩留言会获得点赞!