用AG10治疗TTR淀粉样变性的方法与流程
相关申请的交叉引用
本申请根据35u.s.c§119(e)要求2018年3月23日提交的美国临时申请序列号为62/647,411、2018年8月17日提交的62/765,096、2018年9月14日提交的62/731,629、2018年11月9日提交的62/758,235和2019年2月26日提交的62/810,651的优先权,各自的公开内容通过引用整体并入本文。
联邦政府资助的研究与开发所产生的发明权利的声明
不适用。
参照在光盘上提交的“序列表”,表格或计算机程序列表附录
本申请特此通过引用整体合并随本申请以计算机可读形式提交的序列表。提交的文件的标题为:“序列表–051418-505”。
发明背景
通过信号通路的蛋白质错误折叠或过度激活的异常蛋白质相互作用和聚集是许多人类退行性疾病的根本原因。因此,靶向蛋白质与蛋白质相互作用(ppi)具有治疗意义。
异常蛋白质聚集的一此类例子是可溶性蛋白质转甲状腺素蛋白(ttr或前白蛋白)。ttr是一种55kda的同源四聚体蛋白,存在于血液和脑脊液中。当从其同源四聚体形式解离时,ttr二聚体可错误折叠成淀粉样单体。已经从野生型ttr以及100多种不同的突变体中观察到了这一现象。研究表明,稳定ttr的四聚体形式抑制淀粉样蛋白单体的错误折叠以及随后ttr淀粉样蛋白的形成和沉积。
已经描述一种称为他法米迪(tafamidis)(2-(3,5-二氯苯基)-1,3-苯并噁唑-6-甲酸)的苯并噁唑衍生物抑制异常ttr聚集和原纤维形成,并且正在进行临床试验以治疗家族性和野生型ttr患者的心肌病。他法米迪仍在接受主要药物注册办事处fda的评估。
尽管在管理和改善具有异常ttr聚集和原纤维形成的受试者的治疗方面进行了持续的努力,仍几乎没有改善。例如,美国最大的淀粉样蛋白转诊中心梅奥诊所近期进行的回顾性研究表明,1965年至2013年之间,患有ttr淀粉样蛋白性心肌病(attr-cm)的受试者的总体死亡率没有明显变化(grogan等人,jamcollcardiol2016;68(10):1014–20)。
因此,本领域需要提供用于治疗异常ttr聚集和原纤维形成的方法。本公开解决了这些需求并且提供了相关的优点。
发明概述
在一些方面,本发明提供治疗有此需要的受试者的转甲状腺素蛋白(ttr)淀粉样变性的方法,所述方法包括向有此需要的受试者施用治疗有效量的具有下式的化合物1:
或其药学上可接受的盐,其中所述治疗有效量是每日总剂量约50mg至2000mg。
在一些实施方式中,化合物1的每日总剂量为约800mg。在一些实施方式中,化合物1的每日总剂量为约1600mg。在一些实施方式中,施用化合物1的盐酸盐形式。
在一些实施方式中,化合物1每天给药一次。在一些实施方式中,化合物1每天给药两次。
在一些方面,本发明提供治疗有此需要的受试者的转甲状腺素蛋白(ttr)淀粉样变性的方法,所述方法包括向有此需要的受试者施用治疗有效量的具有下式的化合物1:
或其药学上可接受的盐,其中化合物1的治疗有效量维持所需的化合物1的谷底血浆浓度(troughbloodplasmaconcentration)。
在另一方面,本文提供一种单一单位剂型,(包含)约10-1,000mg具有下式的化合物1:
或其药学上可接受的盐。
在一些实施方式中,所述单一单位剂型包含200mg化合物1。在一些实施方式中,所述单一单位剂型包含400mg化合物1。在一些实施方式中,所述单一单位剂量包含化合物1的盐酸盐。
根据以下详细描述和附图,本发明的其他目的、特征和优点对于本领域技术人员将是显而易见的。
附图的简要说明
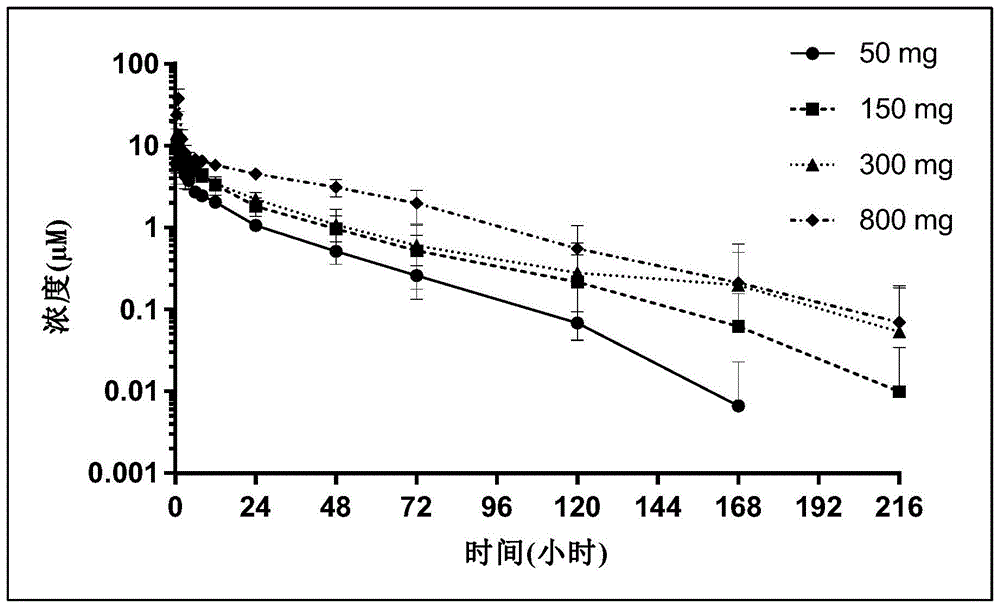
图1显示了在50mg、150mg、300mg和800mgag10hcl的口服剂量下,单次递增剂量组1-4的药代动力学(pk)曲线。
图2显示了单次递增剂量组3(300毫克ag10hcl的口服剂量)中进食与禁食受试者的pk曲线。
图3显示了12天内每12小时,在100mg、300mg、800mgag10hcl的口服剂量下多次递增剂量组1-3的pk曲线。
图4显示了单次递增剂量组3(300mg口服ag10hcl)的荧光探针排阻(fpe)分析的时间过程,展示了随时间变化的靶标结合(targetengagement)百分比。在该组中,向6名受试者施用化合物1,而向2名受试者施用安慰剂。安慰剂组是受试者1和4。
图5显示了单次递增剂量组4(800mg口服ag10hcl)的fpe分析的时间过程,展示了随时间变化的靶标结合百分比。在该组中,向6名受试者施用化合物1,而向2名受试者施用安慰剂。安慰剂组是受试者1和7。
图6显示了每个单次递增剂量组随时间变化的的平均靶标结合百分比。
图7a和图7b显示了单次递增剂量组3(300mgag10hcl)的蛋白质印迹数据。凝胶侧面的箭头指示基于分子量和ttr特异性抗体识别的ttr四聚体的位置。
图8显示了单次递增剂量组3(300mg口服ag10hcl)的蛋白质印迹数据与fpe数据之间的相关性。
图9显示了多次递增剂量组1(100mg口服ag10hclq12h)经12天的fpe分析时间过程。在该组中,向6名受试者施用化合物1,而向2名受试者施用安慰剂。安慰剂组是受试者1和8。
图10显示了多次递增剂量组2(300mg口服ag10hclq12h)经12天的fpe分析时间过程。在该组中,向6名受试者施用化合物1,而向2名受试者施用安慰剂。安慰剂组是受试者3和5。
图11显示了多次递增剂量组3(800mg口服ag10hclq12h)经12天的fpe分析时间过程。在该组中,向6名受试者施用化合物1,而向2名受试者施用安慰剂。安慰剂组是受试者1和7。
图12显示了使用fpe分析第3组(800mg口服ag10hclq12h)经12天的靶标结合百分比的峰值、平均值和谷值。
图13显示了多次递增剂量组3(800mg口服ag10hclq12h)的蛋白质印迹数据。凝胶侧面的箭头指示基于分子量和ttr特异性抗体识别的ttr四聚体的位置。受试者1和3接受ag10,而受试者2接受安慰剂。
图14显示了从单次递增剂量和多次递增剂量组汇总的药代动力学和药效学数据,存在可预测的剂量反应性pd效应。
图15显示了ag10类似物1、2、3和4的合成。a)5a,i.乙酰丙酮、dbu、苯,室温,3天;ii.水合肼、乙醇,90℃,4小时;iii.naoh、甲醇/水,50℃,14小时;b)5b,i.乙酰丙酮、dbu、苯,室温,3天;ii.水合肼、乙醇,90℃,4小时;c)i.nah、mei、dmf,室温,12小时;ii.naoh、甲醇/水,50℃,14小时;d)5b,i.3,5-庚二酮、dbu、苯,室温,3天;ii.水合肼、乙醇,90℃,4小时;e)naoh、甲醇/水,50℃,14小时。
图16a-c显示了稳定剂对缓冲液中ttr的结合亲和力和效力。(a)通过itc评估的ttr与稳定剂的相互作用。热力学数据(总结于表5);δg为蓝色条,δh为绿色条,-tδs为红色条。(b)在仅探针(对照dmso)或ttr稳定剂(2.5μm;稳定剂与ttr比率为1:1)的存在下,通过fpe探针监测缓冲液中ttr(2.5μm)的修饰引起的荧光变化。(c)相对于单独的探针,在孵育3小时后检测的在fpe探针存在下稳定剂对缓冲液中的ttr占有率的百分比的柱状图图示。误差线表示sd(n=3)。通过单向方差分析(anova),然后通过tukey多重比较检验测定差异的显著性(*p≤0.05;***p≤0.001)。
图17a-d显示了稳定剂占有和稳定人血清中ttr的功效。(a)当以指定剂量给药时,在稳定状态下在其估计的平均临床cmax下测试的在ag10(10μm)和其他稳定剂存在下,经酸介导(ph4.0)变性的人血清中ttr稳定性的代表性蛋白质印迹图像:二氟尼柳(diflunisal)(250mgbid,200μm);他法米迪(tafamidis)(80mgqd,20μm);托卡朋(tolcapone)(100mgtid,20μm)。(b)从蛋白质印迹实验获得的稳定性数据的条形图图示。误差线表示sd(n=3)。(c)在仅探针(对照dmso)、ag10(10μm)或ttr稳定剂存在下(以其估计的平均临床稳定状态cmax)检测的通过fpe探针对人血清中ttr修饰引起的荧光变化。(d)相对于单独的探针,在孵育3小时后检测的在fpe探针存在下稳定剂在人血清中ttr的占有百分比的柱状图图示。误差线表示sd(n=4)。通过单向方差分析(anova),然后通过tukey多重比较检验测定差异的显著性(n.s.,不显著;*p≤0.05;**p≤0.01;***p≤0.001)。
图18a-d显示了晶体结构,其突出显示由t119m突变和ag10与ttr结合引起的相似的相互作用。(a)与v122i-ttr结合的ag10的四级结构(pdb:4hiq)用色带表示,其中单体分别着色。两个相同的t4结合位点之一的近距视图,具有构成结合位点的四聚体的两单体的不同色带。ag10的吡唑环和s117/117'之间的关键氢键以虚线突出显示。(b)稳定的t119m-ttr变体的晶体结构(pdb:1fhn),虚线突出显示s117和s117'的羟基之间的关键相互作用。(c)ttrwt的晶体结构(pdb:3cfm)。(d)热力学稳定的r104h-ttr晶体结构(pdb:1x7t)。
图19a-e说明了ag10的吡唑环和ttr的s117/s117’之间的氢键对于有效结合ttr很重要。(a)合成的ag10类似物1、2、3和4的化学结构和计算机模拟对接研究。用于对接实验的ag10与ttr结合的共晶体结构。1是ag10的碘类似物。2是ag10的甲酯形式,不能与k15/15'形成盐桥。3是ag10的甲基吡唑形式,可能与k15或k15'仅形成一个氢键。4是ag10的二乙基吡唑类似物,其影响s117/s117’的两个氢键。(b)通过itc评估的ttr与类似物的相互作用。热力学数据;δg为蓝色条,δh为绿色条,-tδs为红色条。(c)在仅探针(对照dmso)或ttr稳定剂(2.5μm;稳定剂与ttr的比率为1:1)的存在下监测的通过fpe探针对缓冲液中ttr(2.5μm)的修饰引起的荧光变化。(d)相对于单独的探针,在孵育3小时后检测的在fpe探针存在下稳定剂对缓冲液中ttr的占有率百分比的柱状图图示。误差线表示sd(n=3)。(e)通过类似物(10μm;稳定剂与ttr的比率为2:1)稳定人血清中ttr的蛋白质印迹数据的柱状图图示。误差线表示sd(n=4)。通过单向方差分析(anova),然后通过tukey多重比较检测测定差异的显著性(n.s.,不显著;*p≤0.05;**p≤0.01;***p≤0.001)。
图20a-f说明相对于白蛋白或其他丰富的人血清蛋白,ag10对结合ttr具有高选择性。(a)凝胶过滤和透析分析,比较了与纯化的人血清白蛋白(600μm)孵育的ag10和他法米迪(各自为30μm)。将凝胶过滤后(即透析时间为0小时)与白蛋白结合的他法米迪的浓度标准化为100%。误差线表示sd(n=3)。(b)与纯化的人ttr(5μm)孵育的ag10(10μm)透析24小时时程。误差线表示sd(n=3)。(c)在单独的探针(黑圈)、探针加白蛋白(600μm)(黑色三角形)、探针加所有蛋白[纤维蛋白原(5μm)、白蛋白(600μm)、igg(70μm)、转铁蛋白(25μm)](灰色三角形)、探针和ag10(10μm)(红色方块)或探针和ag10加白蛋白(绿色菱形)、探针和ag10加所有蛋白[纤维蛋白原(5μm)、白蛋白(600μm)、igg(70μm)、转铁蛋白(25μm)](蓝色圆圈)存在的情况下,经6小时监测的通过fpe探针对纯化的人ttr(5μm)的修饰而导致的荧光变化。(d)相对于单独的探针,在孵育3小时后检测的在fpe探针和其他血清蛋白存在下,ag10对缓冲液中的ttr占有率%。(e,f)对他法米迪进行了针对ag10所述的相同实验。误差线表示sd(n=3)。通过单向方差分析(anova),然后通过tukey多重比较检验测定差异的显著性(n.s.,不显著;*p≤0.05;**p≤0.01;***p≤0.001)。
图21a-d显示了用混合的犬血清进行的fpe和蛋白质印迹分析中,ag10和他法米迪的活性。(a)在仅探针(对照dmso,黑圈)、ag10(10μm)或他法米迪(10μm)的存在下监测的用fpe探针对市售比格犬血清中犬ttr修饰引起的荧光变化。(b)相对于单独的探针,在孵育3小时后检测的在存在fpe探针的情况下,ag10和他法米迪对犬血清中的犬ttr的占有率%。误差线表示sd(n=4)。(c)在ag10(10μm)和他法米迪(10μm)存在的条件下,在混合的犬血清中ttr对酸介导变性的稳定的蛋白质印迹图像。在交联和免疫印迹之前,将血清样品与dmso或测试化合物在乙酸盐缓冲液(ph4.0)中孵育所需的时间段(0和72小时)。(d)从蛋白质印迹实验获得的稳定性数据的柱状图图示。误差线表示sd(n=3)。通过单向方差分析(anova),然后通过tukey多重比较检测测定差异的显著性(n.s.,不显著;*p≤0.05;**p≤0.01;***p≤0.001)。
图22a-d显示了口服给予的ag10可有效结合并稳定犬中的ttr。(a和b)口服逐步增加剂量的ag10(q.d.7天)后比格犬的ttr占有率。圆圈(●)表示第1天给药前,正方形(■)表示第7天给药前(在cmin的ag10浓度),三角形(▲)表示第7天给药后(在cmax的ag10浓度)。向四组动物给药:(i)0mg/kg(n=12,6雄/6雌);(ii)50mg/kg(n=4,2雄/2雌);(iii)100mg/kg(n=4,2雄/2雌);(iv)200mg/kg(n=12,6雄/6雌)。(b)柱状图,表示在3h时的ttr占有率。误差线表示sd(n=3)。(c和d)接受单次口服剂量(c)5mg/kg和(d)20mg/kg的ag10hcl的犬中ag10的药代动力学-药效学(pk-pd)分析。在不同时间点从犬获得的血清样品的浓度[ag10]相对于ttr占有率(%)的散点图(n=4,每个给药组2只雄性/2只雌性)。误差线表示sd(n=3)。通过单向方差分析(anova),然后通过tukey多重比较检测测定差异的显著性(n.s.,不显著;*p≤0.05;**p≤0.01;***p≤0.001)。
图23说明了口服给予5mg/kg的ag10hcl后,来自3只雄性食蟹猴的血清中在3小时时的ttr占有率(%)。数据是3次重复的平均值±sd。
图24说明了在猴子中口服ag10与ttr稳定之间的剂量依赖性关系。
图25显示的是使用aviva系统生物公司的前白蛋白elisa试剂盒(人)生成的标准校准曲线。
图26显示了每个mad组的ttr浓度随时间的相对变化(给药的健康志愿者的总数=24;安慰剂:活性剂=1:3;mad1组=100mgq12h,12天;mad2组=300mgq12h,12天;mad3组=800mgq12h,12天)。通过将基准值标准化来计算变化。
图27显示了所有安慰剂和ag10治疗的mad受试者中血清ttr浓度从基线到第12天的平均百分比变化。
图28绘制了所有安慰剂和ag10hcl治疗的组中基线和第12天血清ttr浓度的图表(给药的健康志愿者的总数=24;安慰剂:活性剂=1:3;mad1组=100mgq12h,12天;mad2组=300mgq12h,12天;mad3组=800mgq12h,12天)。
图29绘制了所有安慰剂组和ag10治疗组的基线和第12天血清ttr浓度(给药健康志愿者的总数=24;安慰剂:活性剂=1:3;mad1组=100mgq12h,12天;mad2组=300mgq12h,12天;mad3组=800mgq12h,12天)。
图30显示了每个治疗组中受试者的血清ttr水平的剂量反应变化。数据报告为从基线到第28天的变化百分比。
图31显示了ag10和ttr稳定剂他法米迪和二氟尼柳均会提高ttr血清浓度。报告的ag10组的ttr血清浓度是在治疗28天后得出的。报告的他法米迪的血清ttr浓度是基于报道的2周和6周值的28天内的插值。报告的二氟尼柳的ttr血清浓度是在治疗1年后。
图32表明ag10治疗将attr-cm患者的低ttr水平恢复到正常范围。在治疗前(左手边)和治疗28天后(右手边),报告了每个治疗组(安慰剂,400mgb.i.d.和800mgb.i.d.)表现出低和正常水平的血清ttr浓度的attr-cm患者的百分比。
图33绘制了参与2期研究的每个个体的基线血清ttr浓度分布。
图34说明了通过蛋白质印迹法测定的ttr的稳定性百分比,以评价四聚体ttr的稳定性。提供的数据适用于研究中的所有个体(左柱),具有野生型ttr的个体(中柱)和具有突变ttr的个体(右柱)。提供的误差线是平均值的标准误差。
图35显示了通过荧光探针测定法在第28天谷底(给药前)和第28天峰值(给药后1小时)测定的ag10的占有率%。提供的数据适用于研究中的所有个体(填充柱),具有野生型ttr的个体(白色柱)和具有突变的ttr的个体(方格柱)。
图36绘制了血浆中荧光探针结合与ag10的浓度之间的关系。当荧光探针与ttr结合时,测量探针的荧光,当荧光探针不能与ttr结合时,由于ag10的占据,不能测量到荧光。
图37绘制了荧光探针测定法中在指定的时间点测得的相对荧光单位。给药前是谷底水平,而给药1小时后是高峰水平。y轴代表针对背景校正的60分钟相对荧光单位的平均值。呈现的数据来源于接受400mgag10盐酸盐b.i.d的个体。
图38绘制了荧光探针测定法中在指定的时间点测得的相对荧光单位。y轴代表针对背景校正的60分钟相对荧光单位的平均值。给药前是谷底水平,而给药1小时后是高峰水平。呈现的数据来源于接受800mgag10盐酸盐b.i.d的个体。
图39绘制了荧光探针测定法中在指定的时间点测得的相对荧光单位。y轴代表针对背景校正的60分钟相对荧光单位的平均值。给药前是谷底水平,而给药1小时后是高峰水平。呈现的数据来源于接受安慰剂治疗的个体。
图40显示了通过荧光探针测定法在第14天谷底(给药前)和第14天峰值(给药后0.5小时)测定的ag10的占有率%。提供的数据是针对800mgb.i.d给药组中的个体。ag10hcl治疗组带有具有v30m突变的ttr蛋白。
图41显示了通过蛋白质印迹法测定的ttr的稳定性百分比,以评价四聚体ttr的稳定性。提供的数据是针对800mgb.i.d.ag10hcl治疗组的个体,其带有具有v30m突变的ttr蛋白。
图42a-d绘制了群体pk模型中的随机效应(eta)与分类协变量图。图a和b绘制了对间隙的影响,而图c和d绘制了对体积的影响。在标有“dis”的图中(图a和图c),0=健康志愿者,1=患病受试者。在标有“conmed1”的图中(图b和图d)0=接受呋塞米(furosemide)或托拉塞米(torsemide)治疗的受试者,1=未接受呋塞米或托拉塞米治疗的患者。dis:0(n=42)=来自ag10-001的所有接受ag10治疗的健康成人志愿者(sad和mad)。1(n=32)=这些都是来自ag10-201的有活力的attr-cm患者,他们均参与了群体pk分析。400mgbid组中16个,800mgbid组中16个。conmed1:0(n=49)=未同时服用利尿剂:呋塞米或托拉塞米的受试者(42健康成人志愿者,7位attr-cm患者)1(n=25)=接受呋塞米或托拉塞米的受试者。
图43绘制了来自mad3的第12天的ag10的谷底水平(研究ag10-001)与用800mgbid给药的attr-cm患者的谷底水平(ag10-201)的比较。箱线图显示了结果的第25至75百分位,每组中线的范围从最小到最大。线表示中位数,+表示平均值。两项研究均使用200mgag10片剂进行给药。
图44绘制了用400mg片剂治疗的健康受试者中ag10的谷底水平与来自ag10-201的用400mgbid给药的attr-cm患者中的谷底水平的比较。箱线图显示了结果的第25至75百分位,每组中线的范围从最小到最大。线表示中位数,+表示平均值。健康志愿者研究(ag10-003)使用400mgag10片剂进行给药,2期研究ag10-201使用200mg片剂进行给药。
图45显示了attr-cm受试者的3期临床研究的试验设计摘要。
发明详述
i.综述
本文描述了用于治疗受试者的转甲状腺素蛋白(ttr)淀粉样变性的方法。所述方法包括在治疗受试者中具有很大功效并且在受试者中良好耐受的特定给药方案。
ii.定义
尽管本文展示并描述了本发明的各种实施方式和方面,但是对于本领域技术人员而言显而易见的是,这些实施方式和方面仅以实施例的方式提供。在不脱离本发明的情况下,本领域技术人员现在将想到许多变化,改变和替换。应当理解,本文描述的本发明的实施方式的各种替代方式可以用于实施本发明。
本文使用的章节标题仅用于组织目的,而不应解释为限制所描述的主题。本申请中引用的所有文件或部分文件,包括但不限于专利、专利申请、文章、书籍、手册和专著,均明确地以任何目的通过引用全文并入本文。
除非另有定义,否则本文所使用的技术和科学术语具有与本领域普通技术人员通常理解的相同的含义。参见例如singleton等人,微生物学和分子生物学词典,第二版,约翰·威利父子公司(纽约,纽约州,1994);sambrook等人,分子克隆实验室手册,冷泉港出版社(冷泉港,纽约州,1989)。类似于或等同于本文描述的那些方法、装置和材料都可以用于实施本发明。提供以下定义以促进对本文中经常使用的某些术语的理解,但并不意味着限制本公开的范围。
“化合物1”是指化学药品3-(3-(3,5-二甲基-1h-吡唑-4-基)丙氧基)-4-氟苯甲酸(ag10),其化学式为:
或其药学上可接受的盐。当提及给予患者的化合物1的特定量时,本申请是指给予的化合物1盐酸盐的量。本领域技术人员将认识到,为了以游离碱或以不同的盐形式施用相同量的化合物1,对所施用的总量进行小的调整是必要的。
如本文所用,术语“一(a/an)”是指一或多个(种)。
术语“包括”、“包含”和“具有”及其派生词在本文中可作为综合、开放式术语互换使用。例如,“包括”、“包含”或“具有”的使用意味着包含、拥有或包括的任何元素不是包含动词的从句的主语所包含的唯一元素。
如本文所用,术语“约”是指包括指定值的值范围,本领域普通技术人员将认为合理地类似于该指定值。在一些实施方式中,术语“约”是指使用本领域通常可接受的测量的标准偏差内。在一些实施方式中,“约”是指延伸至指定值的+/-10%的范围。在一些实施方式中,“约”是指指定值。
如本文所用,“治疗”或“处理”,“缓解”或“改善”在本文可互换使用。这些术语是指获得有益或期望结果包括但不限于治疗益处的方法。治疗益处是指根除或改善所治疗的潜在疾病。而且,通过消除或改善与基础疾病相关的一种或多种生理症状获得治疗益处,从而尽管受试者仍可能患有基础疾病,但在受试者中观察到了改善。治疗包括通过给予组合物使疾病的临床症状发展缓慢;遏制疾病,即导致疾病的临床症状减轻;抑制疾病,即在症状最初出现后通过施用组合物来阻止临床症状的发展;和/或减轻疾病,即在其最初出现后通过给予组合物来引起临床症状的消退。例如,本文所述的某些方法通过减少或降低ttr原纤维形成的发生或发展来治疗转甲状腺素蛋白(ttr)淀粉样变性。或通过减少ttr淀粉样变性的症状来治疗ttr淀粉样变性。
“有效量”或“药学上有效量”是足以实现所述目的(例如,获得其给药的效果、治疗疾病、降低酶活性、减轻疾病或病状的一种或多种症状)的量。“有效量”的一例子是足以有助于治疗或减轻疾病的一或多个症状的量,也可以称为“治疗有效量”。一或多个症状的“减轻”(以及该短语的语法等价形式)是指减轻一或多个症状的严重程度或频率,或消除一或多个症状。功效也可以表示为“倍数”增加或减少。例如,相对于对照,治疗有效量可以具有至少1.2倍、1.5倍、2倍、5倍或更多的效用。
“患者”或“受试者”或“有此需要的受试者”是指患有或易患有通过使用本文提供的方法治疗的疾病或病症的活生物体。该术语不一定表示该受试者已被诊断出患有某种特定疾病,而是通常指在医学监督下的个体。非限制性实例包括人,其他哺乳动物、牛科动物、大鼠、小鼠、狗、猴、山羊、绵羊、奶牛、鹿,和其他非哺乳动物。在一些实施方式中,患者、受试者或有此需要的受试者是人。
iii.具体实施方式
方法
在一个方面,本文提供了一种治疗转甲状腺素蛋白(ttr)淀粉样变性的方法。所述方法包括向有需要的受试者施用治疗有效量的具有下式的化合物1:
或其药学上可接受的盐,其中治疗有效量是约10mg至2000mg的每日总剂量。在一些实施方式中,化合物1的每日总剂量为约10mg至50mg、50mg至300mg、50mg至150mg、150mg至800mg、800mg至1600mg或800mg至2000mg。在一些实施方式中,化合物1的每日总剂量为约10mg至50mg。在一些实施方式中,化合物1的每日总剂量为约50mg至300mg。在一些实施方式中,化合物1的每日总剂量为50mg至150mg。在一些实施方式中,化合物1的每日总剂量为150mg至800mg。在一些实施方式中,化合物1的每日总剂量为800mg至1600mg。在一些实施方式中,化合物1的每日总剂量为800mg至2000mg。应当理解,在本公开中,所列化合物1的量是所施用的化合物1的盐酸盐的量。本领域技术人员将认识到,为了施用相同量的游离碱或不同的盐形式的化合物1,对所施用的总量进行小的调整是必要的。
在一些实施方式中,化合物1的每日总剂量为约10mg。在一些实施方式中,化合物1的每日总剂量为约25mg。在一些实施方式中,化合物1的每日总剂量为约50mg。在一些实施方式中,化合物1的每日总剂量为约1000mg。在一些实施方式中,化合物1的每日总剂量为约150mg。在一些实施方式中,化合物1的每日总剂量为约200mg。在一些实施方式中,化合物1的每日总剂量为约300mg。在一些实施方式中,化合物1的每日总剂量为约600mg。在一些实施方式中,化合物1的每日总剂量为约800mg。在一些实施方式中,化合物1的每日总剂量为约16000mg。
化合物1可以每天给药一次(sid或qd)、两次(bid或q12h)、三次(tid)或四次(qid)。在一些实施方式中,化合物1每天给药一次。在一些实施方式中,化合物1每天给药两次。在一些实施方式中,化合物1每天给药三次。在一些实施方式中,化合物1每天给药四次。
在一些实施方式中,每天一次施用约50mg的化合物1。在一些实施方式中,每天一次施用约150mg的化合物1。在一些实施方式中,每天一次施用约300mg的化合物1。在一些实施方式中,每天一次施用约800mg的化合物1。
在一些实施方式中,每天两次施用约100mg的化合物1。在一些实施方式中,每天两次施用约300mg的化合物1。在一些实施方式中,每天两次施用约400mg的化合物1。在一些实施方式中,每天两次施用约800mg的化合物1。
在另一方面,本文提供了一种治疗转甲状腺素蛋白(ttr)淀粉样变性的方法。所述方法包括向有需要的受试者施用治疗有效量的具有下式的化合物1:
或其药学上可接受的盐,其中化合物1的治疗有效量维持至少5μm的化合物1的谷底血浆浓度。在一些实施方式中,化合物1的治疗有效量维持至少6μm的化合物1的谷底血浆浓度。在一些实施方式中,化合物1的治疗有效量维持至少7.5μm的化合物1的谷底血浆浓度。在一些实施方式中,化合物1的治疗有效量维持至少8μm的化合物1的谷底血浆浓度。
在一些实施方式中,化合物1的治疗有效量维持5至30μm的化合物1的谷底血浆浓度。在一些实施方式中,化合物1的治疗有效量维持5至25μm的化合物1的谷底血浆浓度。在一些实施方式中,化合物1的治疗有效量维持6至20μm的化合物1的谷底血浆浓度。在一些实施方式中,化合物1的治疗有效量维持7.5至15μm的化合物1的谷底血浆浓度。在一些实施方式中,化合物1的治疗有效量维持7.5至10μm的化合物1的谷底血浆浓度。
在一些实施方式中,接受治疗有效量的化合物1的受试者经历转甲状腺素蛋白(ttr)血清浓度相对于基线水平的增加。在一些实施方式中,在治疗28天后,接受治疗有效量的化合物1的受试者经历转甲状腺素蛋白(ttr)血清浓度相对于基线水平至少约10、15、20、25、30%或更多的增加。在一些实施方式中,在治疗28天后,接受治疗有效量的化合物1的受试者经历转甲状腺素蛋白(ttr)血清浓度相对于基线水平至少约25%的增加。在一些实施方式中,治疗前受试者的ttr血清水平低于基线血清ttr浓度(20mg/dlttr)。在一些实施方式中,接受有效量的化合物1达28天的受试者将经历血清ttr水平升高,使得血清ttr水平高于基线水平。在一些实施方式中,经历ttr水平升高的受试者是被诊断患有转甲状腺素蛋白淀粉样变性(attr)心肌病的受试者。
存在多种与转甲状腺素蛋白(ttr)淀粉样变性有关的疾病或紊乱。这些疾病或紊乱包括但不限于家族性淀粉样蛋白多发性神经病、家族性淀粉样蛋白心肌病、老年性全身性淀粉样变性、中枢性淀粉样变性、眼部淀粉样变性、软脑膜淀粉样变性、眼软脑膜淀粉样变性、玻璃体淀粉样变性、胃肠道淀粉样变性、神经性淀粉样变性、非神经性淀粉样变性、非遗传性淀粉样变性、反应性/继发性淀粉样变性、脑淀粉样变性。
在一些实施方式中,与转甲状腺素蛋白(ttr)淀粉样变性相关的疾病或紊乱是软脑膜淀粉样变性。在一些实施方式中,患有软脑膜淀粉样变性的受试者具有在第18位带有天冬氨酸至甘氨酸突变(d18g)的转甲状腺素蛋白。在一些实施方式中,患有软脑膜淀粉样变性的受试者具有在第53位带有甘氨酸至精氨酸突变(g53r)的转甲状腺素蛋白。野生型转甲状腺素蛋白(ttr)在本文中以seqidno:1提供。
在一些实施方式中,患有软脑膜淀粉样变性的受试者具有在第114位带有酪氨酸至半胱氨酸突变(y114c)的转甲状腺素蛋白。带有第114位酪氨酸至半胱氨酸突变(y114c)的受试者可表现出attrm-pn症状,软脑膜淀粉样变性症状,或两者兼有。
在一些实施方式中,患有软脑膜淀粉样变性的受试者具有在第49位带有苏氨酸到脯氨酸突变(t49p)的转甲状腺素蛋白。带有第49位苏氨酸到脯氨酸突变(t49p)的受试者可表现出attrm-pn症状,软脑膜淀粉样变性症状,或两者兼有。
在一些实施方式中,转甲状腺素蛋白(ttr)淀粉样变性病是转甲状腺素蛋白淀粉样变性(attr)心肌病或attr多发性神经病。在一些实施方式中,ttr淀粉样变性的特征在于包含在第60位苏氨酸至丙氨酸突变(t60a)的ttr蛋白。在一些实施方式中,ttr淀粉样变性的特征在于包含在第24位脯氨酸至丝氨酸突变(p24s)的ttr蛋白。在一些实施方式中,ttr淀粉样变性的特征在于包含在第38位天冬氨酸至丙氨酸突变(d38a)的ttr蛋白。在一些实施方式中,ttr淀粉样变性的特征在于包含在第58位亮氨酸至组氨酸突变(l58h)的ttr蛋白。带有这些突变的患者通常会同时出现attr心肌病和attr多发性神经病两者的症状。野生型转甲状腺素蛋白(ttr)在本文中以seqidno:1提供。
在一些实施方式中,转甲状腺素蛋白(ttr)淀粉样变性病是转甲状腺素蛋白淀粉样变性(attr)心肌病。在一些实施方式中,转甲状腺素蛋白(ttr)淀粉样变性病是转甲状腺素蛋白淀粉样变性(attr)多发性神经病。
attr心肌病包括野生型attr心肌病(attrwt-cm)和遗传性(家族性)attr心肌病(attrm-cm)。attrm-cm是由ttr蛋白中的突变引起,而attrwt-cm不是由突变引起。相反,attrwt-cm通常是与年龄相关的过程。在一些实施方式中,attr心肌病是attrwt-cm。在一些实施方式中,attr心肌病是attrm-cm。在一些实施方式中,患有attrm-cm的受试者在ttr蛋白的第122位具有缬氨酸至异亮氨酸突变(v122i)。在一些实施方式中,患有attrm-cm的受试者具有在第49位带有苏氨酸到脯氨酸突变(t49p)的转甲状腺素蛋白。带有第49位苏氨酸到脯氨酸突变(t49p)的受试者可表现出attrm-cm症状,软脑膜淀粉样变性症状,或两者兼有。野生型转甲状腺素蛋白(ttr)在本文中以seqidno:1提供。
attr多发性神经病包括野生型和遗传性(家族性)attr多发性神经病。如针对心肌病所讨论,attrm-pn是由ttr蛋白中的突变引起,而attrwt-pn不包含遗传成分。在一些实施方式中,attr-pn是attrwt-pn。在一些实施方式中,attr-pn是attrm-pn。在一些实施方式中,attrm-pn的特征在于包含在第30位缬氨酸至甲硫氨酸突变(v30m)的ttr蛋白。在一些实施方式中,attrm-pn的特征在于包含在第64位苯丙氨酸至亮氨酸突变(f64l)的ttr蛋白。在一些实施方式中,attrm-pn的特征在于包含在第114位酪氨酸至半胱氨酸突变(y114c)的ttr蛋白。带有第114位酪氨酸至半胱氨酸突变(y114c)的受试者可表现出attrm-pn症状,软脑膜淀粉样变性症状,或两者兼有。野生型转甲状腺素蛋白(ttr)在本文中以seqidno:1提供。
attr心肌病(野生型和家族性两者)是一种缓慢进行性疾病,会在患病受试者中引起心力衰竭和死亡。本公开的方法通过停止或减慢ttr原纤维在心肌中的积累,为患有attr心肌病的受试者提供临床改善。通过该过程,当前描述的方法为attr心肌病患者提供了临床改善。临床改善包括但不限于纽约心脏协会(nyha)功能分级的改善,堪萨斯城心肌病问卷反应的改善,euroqol-5维度(eq-5d-5l)的改善,6分钟步行测试性能的改善,与心脏健康相关的标志物的改善,例如肌钙蛋白t、肌钙蛋白i、b型利钠肽(bnp)和n末端bnp前体,降低心血管相关住院的频率,和/或降低死亡率。
在一些实施方式中,本文提供的方法改善、稳定或延迟受试者的纽约心脏协会(nyha)功能分级中的恶化。nyha功能分级将心力衰竭症状的严重程度分级为四个功能级别之一。nyha功能分级在临床实践和研究中被广泛使用,因为它提供了严重程度的标准描述,可用于评估对治疗的反应并指导管理。nyha功能分级基于症状的严重程度和身体活动的限制:
第i级:没有身体活动的限制。日常身体活动不会引起过度的呼吸困难、疲劳或心悸。
第ii级:身体活动的轻微限制。休息时舒适,但日常身体活动会导致过度的呼吸困难、疲劳或心悸。
第iii级:身体活动明显受限。休息时舒适,但少于日常的身体活动会导致过度的呼吸困难、疲劳或心悸。
第iv级:无法在没有不适感的情况下进行任何身体活动。休息时可能会出现症状。如果进行任何身体活动,会增加不适感。
在一些实施方式中,给予治疗有效量的化合物1降低了受试者的纽约心脏协会(nyha)功能分级。在一些实施方式中,nyha功能分级从iv级降低至iii级,从iv级降低至ii级,或从iv级降低至i级。在一些实施方式中,nyha功能分级从iv级降低至iii级。在一些实施方式中,nyha功能分级从iv级降低至ii级。在一些实施方式中,nyha功能分级从iii级降低至ii级。在一些实施方式中,nyha功能分级从iii级降低至i级。在一些实施方式中,nyha功能分级从ii级降低至i级。
在一些实施方式中,本文提供的方法改善、稳定或延迟受试者的堪萨斯城心肌病问卷(kccq)分级中的恶化。在一些实施方式中,本文所述的方法提供提高的堪萨斯城心肌病问卷(kccq)的评分(greencp等人,(2000)美国心脏病学会杂志35:1245-55),其内容出于所有目的通过引用并入本文。kccq包含与心脏健康有关的特定问题,并提供针对疾病特异性健康相关生活质量的有效、可靠和敏感的测量。
kccq的问题要求受试者评估自己在正常生活中的受限制程度(例如,严重受限、相当受限、中等受限、轻微受限或完全不受限)。在一些实施方式中,在用化合物1治疗后,受试者在问卷的所有问题上具有至少一个水平的平均改善(例如,严重受限到相当受限,相当受限到中等受限,中等受限到轻微受限)。
在一些实施方式中,本文提供的方法改善、稳定或延迟受试者的euroqol-5维度(eq-5d-5l)评分的恶化。eq-5d-5l是一种简短的自我管理的通用健康状况工具,大约需要5分钟去完成。该仪器包括两个部分。在第一部分中,要求受访者从5个维度(移动性、自我护理、日常活动、疼痛或不适、焦虑或抑郁)中评估其当前的健康状况,每个维度都具有五个功能级别(1-没有问题,2-轻微问题,3-中等问题,4-严重问题和5-极端问题)。第二部分是被调查者在视觉模拟量表(eqvas)上对当前健康状况的自我评估,其端点标记为“最佳可想象的健康状态”(得分为100)和“最差可想象的健康状态”(得分为0)。来自5个维度的分数可用于计算单个索引值,也称为效用分数。eq-5d-5l问卷属于公共领域,可以从euroqol获得。
在一些实施方式中,接受本文所述治疗方法的患者在euroqol-5维度(eq-5d-5l)效用得分中具有至少1、2、3、4、5、6、7、9、10个点的平均改善。在一些实施方式中,接受本文所述的治疗方法的患者在euroqol-5维度(eq-5d-5l)效用得分中具有至少五个点的平均改善。
在一些实施方式中,本文描述的方法改善了受试者的六分钟步行测试性能。六分钟步行测试是六分钟的自定步速定时步行,以评估受试者的功能能力水平。如果运动水平超过其舒适水平,则允许受试者在测试期间停止并休息。在治疗之前、之后和期间的评估相对容易进行,包括评估受试者在六分钟时间内的行走距离。因此,在一些实施方式中,受试者在用化合物1治疗后增加了六分钟步行测试中涵盖的总距离。在一些实施方式中,受试者比用化合物1治疗之前测得的基线距离多行走至少25m。在一些实施方式中,受试者比用化合物1治疗之前测得的基线距离多行走至少30m。在一些实施方式中,受试者比用化合物1治疗之前测得的基线距离多行走至少50m。在一些实施方式中,受试者比用化合物1治疗之前测得的基线距离多行走至少75m。在一些实施方式中,受试者比用化合物1治疗之前测得的基线距离多行走至少100m。在一些实施方式中,接受本文所述的治疗方法的受试者在六分钟的步行距离上具有缓慢的减小。例如,在一些实施方式中,受试者保持与治疗之前相同的六分钟步行距离。在一些实施方式中,受试者在六分钟的步行测试中少覆盖10m。在一些实施方式中,使用六分钟步行测试将治疗组与非治疗组进行比较。在一些实施方式中,相对于基线的组间平均变化为至少10米。在一些实施方式中,相对于基线的组间平均变化为至少20米。在一些实施方式中,相对于基线的组间平均变化为至少30米。在一些实施方式中,与不接受治疗的个体相比,本文提供的治疗方法减少了六分钟步行测试距离的下降。
肌钙蛋白t、肌钙蛋白i、脑利钠肽(bnp)和n末端bnp前体是在心肌健康较差的受试者的血清血液中升高的多肽。在一些实施方式中,在用化合物1治疗后,肌钙蛋白t、肌钙蛋白i、bnp和/或n末端bnp前体的水平降低。在一些实施方式中,与用化合物1治疗之前的所述受试者中肌钙蛋白t、肌钙蛋白i、bnp和/或n末端bnp前体的基线水平相比,肌钙蛋白t、肌钙蛋白i、bnp和/或n末端bnp前体的水平降低了约10%。在一些实施方式中,与在用化合物1治疗之前的所述受试者中肌钙蛋白t、肌钙蛋白i、bnp和/或n末端bnp前体的基线水平相比,肌钙蛋白t、肌钙蛋白i、bnp和/或n末端bnp前体的水平降低了约15%。
如上所述,与未接受治疗的受试者相比,在所公开方法的一些实施方式中提供的临床改善正在降低接受治疗的受试者中与心血管有关的住院治疗的比率。在一些实施方式中,与未接受治疗的患者相比,患者每年平均至少减少0.5、1、1.5、2、3、4、5次心血管相关住院。
在本文公开的方法的一些实施方式中提供的另一临床益处是与未接受治疗的个体相比死亡率降低。在一些实施方式中,与未接受治疗的受试者相比,死亡率降低了约2、4、6、8、10、12、14、16、18、20、22、24、26、28、30或更多百分比。
attr多发性神经病是ttr淀粉样蛋白(attr)沉积损害或损害正常神经功能的疾病。attr多发性神经病是一种进行性疾病,其在患病受试者中引起恶病质和死亡。本公开的方法通过停止或减慢ttr原纤维的积累,为患有attr多发性神经病的受试者提供了临床改善。通过此过程,当前所述的方法为attr多发性神经病患者提供了临床改善。临床改善包括但不限于神经病损害评分(nis)或改良的神经病损害评分+7(mnis+7)的改善,诺福克生活质量糖尿病性神经病问卷的改善,综合自主神经症状评分(compass-31)的改善,通过改良的体重指数(mbmi)测得的改善的营养状况和/或受试者10米步行测试的改善。
在一些实施方案中,本文描述的方法提供了改善的神经病损害评分(nis)。nis是指一种评分系统,用于测量无力、感觉和反射。nis评分评估一标准组的肌肉无力(1为25%弱,2为50%弱,3为75%弱,3.25为克服重力运动,3.5为消除重力运动,3.75为无运动的肌肉颤动,以及4为麻痹),一标准组的肌肉拉伸反射(0为正常,1为减少,2为缺失),触摸压力,振动,关节位置和运动,以及针刺(所有均在食指和大脚趾上分级:0表示正常,1表示减小,2表示缺失)。评估会针对年龄,性别和身体素质进行校正。
在一些实施方式中,与不服用化合物1的受试者相比,本文所述的方法减慢了疾病的进展,从而降低了nis评分的增加速率。在一些实施方式中,本文所述的方法停止疾病的进展,使得在用化合物1治疗后nis评分没有变化。
在一些实施方式中,在用化合物1治疗后本文所述的方法降低了nis评分。在一些实施方式中,与用化合物1治疗之前测量的基线水平相比,本文所述的方法使nis评分降低了至少5、10、15、20、25、30、35、40、45、50、55或60%。在一些实施方式中,与用化合物1治疗之前测量的基线水平相比,本文所述的方法使nis评分降低了至少5%。在一些实施方式中,与用化合物1治疗之前测量的基线水平相比,本文所述的方法使nis评分降低了至少10%。在一些实施方式中,与用化合物1治疗之前测量的基线水平相比,本文所述的方法使nis评分降低了至少15%。
在一些实施方式中,本文描述的方法提供了改善的改良神经病损害评分(mnis+7)。mnis+7是指基于临床检查的神经功能损害(nis)结合小型和大型神经纤维功能(ncs和qst)的电生理测量以及自主神经功能(姿势性血压)测量的评估。mnis+7评分是nis+7评分的修改(代表nis加上七个测试)。mnis+7分析了肌肉无力和肌肉伸展反射。七项测试中有五项包含神经传导属性。这些属性是指腓神经复合肌肉动作电位幅度,运动神经传导速度和运动神经末梢潜伏期(mndl),胫骨mndl和腓肠感觉神经动作电位幅度。这些值已针对年龄、性别、身高和体重的变量进行了校正。七个测试中的其余两个测试包括振动检测阈值和伴随深呼吸的心率降低。mnis+7评分对nis+7进行了修改,以考虑使用智能体位定量感觉测试,新的自主神经评估,以及使用尺、腓、胫神经的振幅的复合肌肉动作电位,和尺神经和腓神经感觉神经动作电位(suanprasert,n.等人,用于治疗试验的ttrfap组修改nis+7的回顾性研究(retrospectivestudyofattrfapcohorttomodifynis+7fortherapeutictrials),j.neurol.sci.,2014.344(1-2):121-128)。mnis+7检测的更多详细信息可以在us2017/0307608中找到,出于所有目的,其内容通过引用并入本文。
在一些实施方式中,本文描述的方法减慢了疾病的进展,使得与不服用化合物1的受试者相比,降低了mnis+7评分的增加速率。在一些实施方式中,本文所述的方法停止疾病的进展,使得在用化合物1治疗后mnis+7评分没有变化。
在一些实施方式中,在用化合物1治疗后本文所述的方法降低了mnis+7分数。在一些实施方式中,与用化合物1处理之前测定的基线水平相比,本文描述的方法将mnis+7评分降低了至少5%、10%、15%、15%、20%、25%、30%、35%、40%、45%、50%、55%或60%。在一些实施方式中,与在用化合物1治疗之前测量的基线水平相比,本文描述的方法使mnis+7评分降低了至少5%。在一些实施方式中,与在用化合物1治疗之前测量的基线水平相比,本文描述的方法使mnis+7评分降低了至少10%。在一些实施方式中,与在用化合物1治疗之前测量的基线水平相比,本文描述的方法使mnis+7评分降低了至少15%。
在一些实施方式中,本文所述的方法提供了诺福克生活质量糖尿病性神经病(qol-dn)调查表中改善的评分。所述调查表是本领域技术人员众所周知的,并且是经过验证的调查表,其捕获与大纤维、小纤维和自主神经病有关的疼痛。所述问卷包括与受试者经历的症状有关的事项以及与神经病对受试者的日常活动的影响有关的问题。
在一些实施方式中,本文所述的方法减慢了疾病的进展,使得与不服用化合物1的受试者相比,诺福克qol-dn评分的下降速率减缓。在一些实施方式中,本文所述的方法停止疾病的进展,使得在用化合物1治疗后诺福克qol-dn评分没有变化。
在一些实施方式中,本文描述的方法减慢疾病的进展,使得在用化合物1治疗后诺福克qol-dn评分没有变化。
在一些实施方式中,在用化合物1治疗后,本文所述的方法改善了诺福克qol-dn评分。在一些实施方式中,与用化合物1治疗之前测量的基线水平相比,本文所述的方法将诺福克qol-dn提高了至少5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%或60%。在一些实施方式中,与用化合物1治疗之前测量的基线水平相比,本文所述的方法将诺福克qol-dn评分提高了至少5%。在一些实施方式中,与用化合物1治疗之前测量的基线水平相比,本文所述的方法将诺福克qol-dn得分提高了至少10%。在一些实施方式中,与用化合物1治疗之前测得的基线水平相比,本文所述的方法将诺福克qol-dn评分提高了至少15%。在一些实施方式中,与受试者的基线评分相比,本文所述的方法提供受试者的诺福克qol-dn中约-1.5、-2.0、-2.5、-3.0、-3.5、-4.0、-4.5、-5.0、-5.5、-6.0、-6.7、-7.0、-7.5、-8.0、-8.5、-9.0、-9.5或-10.0的变化。
在一些实施方式中,本文公开的方法提供了改善的综合自主神经症状评分(compass-31)。综合自主神经症状评分(compass-31)是一种评估自主神经功能障碍症状的患者问卷。在一个实施方式中,本发明的方法向受试者提供相对于基线的compass-31评分的改善。这样的改善可以表现为增加受试者的compass-31得分的至少0.1个点的形式,例如至少0.2、至少0.3、至少0.4或至少0.5个点,例如0.1、0.2、0.3、0.4或0.5个点。在一些实施方式中,所述方法减慢疾病的进展,使得compass-31评分没有变化。在其他实施方式中,本发明的方法减慢compass-31评分降低的速率,例如,与未使用ag10治疗的受试者的compass评分的降低速率相比,用ag10治疗的受试者中compass-31得分的降低速率。
在一些实施方式中,本文公开的方法提供如通过修正体重指数(mbmi)测量的改善的营养状况,所述修正体重指数通过个体的bmi乘以他们的血清白蛋白水平来确定。mbmi的计算考虑了水肿对总重量的贡献。在一个实施方式中,本公开的方法向受试者提供了相对于基线的mbmi的改善。所述改善可以表现为mbmi评分下降约2、5、7、10、12、15、20或约25的形式。在其他实施方式中,所述方法阻止mbmi指数评分增加,例如,所述方法导致mbmi得分增加0%。在其他实施方式中,本发明的方法减慢了magmi分数增加的速率,例如,与未使用ag10治疗的受试者mbmi分数的增加速率相比,使用ag10治疗的受试者中的mbmi分数的增加速率。
在一些实施方式中,本文公开的方法提供了对10米步行测试的改善。该测试可测量个体历经10米的步行速度。在一个实施方式中,本公开的方法向受试者提供了在10米步行测试中相对于基线的增加。在一些实施方式中,在10米步行测试中相对于基线的增加是大约0.025、0.03、0.04、0.05、0.06、0.07、0.08、0.09、1.0、1.5、2.0、2.5、3.0、3.5、4.0、4.5或约5.0米/秒。
在一些实施方式中,本发明公开的方法提供改善的dyck/rankin评分。dyck/rankin评分是本领域已知的,由医师在评估患者的症状、神经性损伤、测试结果以及评估患者的日常生活行为能力后赋值。仅对与周围神经病相关的残疾进行分级。在确定患者是否有困难或无法执行某些日常生活的任务或行动时,应不仅使用患者报告进行判断;医生应使用客观标准。阶段(0-8)概述如下:
0.无神经病
·无症状(nss<1),体征(nis<2分);或神经病变异常的测试(例如7个测试<97.5th)。
1.最小的神经病变(只有a、b、c中之一异常)
a)测试是唯一的异常(例如7次测试>97.5th);或者
b)神经病征是唯一的异常(例如nis>2分);或者
c)神经病症状是唯一的异常(例如nss>1)
2.最小的神经病变:1a+1b。
3.有症状的神经病变:1a,1a+1c;1a+1c或1b+1c。患者能够继续进行日常生活、工作或休闲活动的通常行为,并能够履行日常家庭和社会责任。
*神经病的症状:nss≥1肌肉无力,萎缩或痉挛症状;阴性或阳性神经病感官症状(n-nns,p-nss);或神经性自主神经症状。
-日常生活、工作、休闲以及社会和家庭活动的通常行为:尽管有神经病症状,患者仍能够按其日常活动工作,维持其日常家庭义务并参加休闲活动。通常,尽管有一些运动、感觉或自主神经症状,患者仍可以继续行动。患者可能无法进行特殊活动,例如竞技体育,耐力运动等。
4.症状性神经病变(3中定义),其干扰和限制工作、平常的日常活动、休闲活动或家庭和社会义务,但在没有他人帮助的情况下可能具有独立的功能。在这个评分上,由于神经病,对日常工作*、生活或休闲行为、家庭或社会义务*有明确的限制。
*运动,感觉或自主神经症状或损伤的程度足以限制工作能力去进行通常的日常活动、休闲活动或履行家庭和通常的社会责任。使用罐头或矫形器可能会将患者归为此类(或更高级别),除非患者能够执行“通常”的日常生活、休闲活动并履行社会和家庭责任(则他们会落入更低级别)。
5.症状性神经病变(3中定义),其限制了日常生活、工作和休闲活动。需要其他护理人员*(<21/2小时/天)的帮助。如果在日常的生活、休闲活动或社会和家庭责任中必须使用轮椅,则患者通常会落入这个或更高的评分(>5)。
*需要家庭成员或来访护士提供日常生活行为(沐浴、剃须、刷牙、摄食等),每日使用镇痛药或鸦片制剂,或帮助管理患者无法充分地或安全地进行的自主神经功能障碍。
6.症状性神经病变(在3中定义),需要护理人员≥21/2小时至<8小时/天的帮助,如5中所述。
7.症状性神经病变(在3中定义),需要护理人员>8小时/天的帮助,但不像第8阶段那样连续。
8.症状性神经病变(在3中定义),需要在重症监护病房中持续护理。
给药时间取决于许多因素,包括所治疗的特定疾病。例如,在特定转甲状腺素蛋白(ttr)淀粉样变性病或病状中,存在遗传成分,因此可能需要长期(即持续,长期)给药。然而,在一些实施方式中,当受试者表现出或经历与ttr淀粉样变性病或病状有关的症状时,或在遇到特定结点(例如症状减轻或完全消除)之后的一段设定的时间段内,将继续向患有遗传性ttr淀粉样变性病的受试者给予化合物1。如果ttr淀粉样变性病的症状恢复或开始重新出现,则重新开始化合物1的给药。
对于患有非遗传性相关的ttr淀粉样变性病的受试者,有多种给药选择,这取决于疾病的严重程度和所呈现的临床症状。在一些实施方式中,需要化合物1的长期给药。在一些实施方式中,需要化合物1的短期或急性给药。在一些实施方式中,在受试者表现出或经历与ttr淀粉样变性病或病状有关的症状时,或在遇到特定结点(例如症状减轻或完全消除)之后的一段特定的时间段内,继续向患有非遗传性ttr淀粉样变性病的受试者给予化合物1。如果ttr淀粉样变性病的症状恢复或开始重新出现,则重新开始化合物1的给药。
在一些实施方式中,化合物1被施用至少7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99、100天或更长时间。在一些实施方式中,化合物1被施用7、14、21、28、35、42、49或56天。在一些实施方式中,化合物1被施用28天。在一些实施方式中,化合物1被施用56天。在一些实施方式中,化合物1被施用84天。
在一些实施方式中,化合物1被施用至少4、5、6、7、8、9、10、11、12、13、14、15、16、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、或50个月。在一些实施方式中,化合物1被施用10、15、20、25、30、35、40、45或50个月。在一些实施方式中,化合物1被施用6个月。在一些实施方式中,化合物1被施用12个月。在一些实施方式中,化合物1被施用18个月。在一些实施方式中,化合物1被施用24个月。在一些实施方式中,化合物1被施用30个月。在一些实施方式中,化合物1被施用36个月。在一些实施方式中,化合物1被施用42个月。
有利地,在利尿剂治疗中使用的药剂在治疗期间不会改变ag10的暴露。因此,接受利尿剂治疗的患者可以在不更改或特殊给药方案的情况下使用ag10。因此,在一些实施方式中,接受ag10的受试者也正在接受另外的利尿治疗剂。利尿剂治疗包括但不限于依他尼酸、布美他尼(bumetanide)、呋塞米(furosemide)和托拉塞米(torsemide)。在一些实施方案中,利尿治疗剂选自呋塞米(furosemide)或托拉塞米(torsemide)。
药物组合物
化合物1可以制备成适于递送至受试者的各种组合物。适用施用于受试者的组合物通常包含化合物1或其药学上可接受的盐和药学上可接受的赋形剂。
用于施用化合物1的药物组合物可以方便地以单位剂型存在,并且可以通过药学和药物递送领域中已知的任何方法来制备。所有方法都包括使活性成分与含有一种或多种辅助成分的运载体结合的步骤。通常,通过将活性成分与液体运载体或细分的固体运载体或两者均匀和紧密地结合在一起,然后如果需要,将产品塑形为所需制剂,来制备药物组合物。
在《雷明顿:药学的科学与实践》,第21版(由gennaro编辑,利平科特威廉姆斯.威尔金斯(lippincottwilliams&wilkins)出版社(2003)出版)中发现了用于本发明的合适制剂,在此通过引用并入本文。本文所述的药物组合物可以以本领域技术人员已知的方式制造,即通过常规的混合、溶解、制粒、糖衣制造、浸出,乳化、包囊、包埋或冻干方法。以下方法和赋形剂仅是示例性的,没有任何限制性。
可以将化合物1掺入用于治疗施用的多种制剂中。更特别地,化合物1可以与适当的药学上可接受的运载体或稀释剂一起或分别配制成药物组合物,并且可以配制成固体、半固体、液体或气体形式的制剂,例如片剂、胶囊剂、丸剂、粉剂、颗粒剂、糖衣丸剂、凝胶剂、浆剂、软膏剂、溶液剂、栓剂、注射剂、吸入剂和气雾剂。这样,可以以多种方式来实现本发明化合物的给药,包括口服、颊、肠胃外、静脉内、皮内(例如皮下,肌内)、透皮等给药。此外,化合物1可以以局部而非全身的方式给药,例如以长效制剂或缓释制剂的形式给药。
口服制剂也可以以硬明胶胶囊剂的形式存在,其中活性成分与惰性固体稀释剂(例如碳酸钙、磷酸钙或高岭土)混合,或者以软明胶胶囊剂的形式存在,其中活性成分与水或油介质,例如花生油,液体石蜡或橄榄油混合。另外,可用非水混溶性成分如油制备乳液,并用表面活性剂如单甘油二酸酯、peg酯等稳定性乳液。
水性悬浮液含有与适于制备水性悬浮液的赋形剂混合的活性物质。这些赋形剂是悬浮剂,例如羧甲基纤维素钠、甲基纤维素、羟丙基甲基纤维素、藻酸钠、聚乙烯吡咯烷酮、黄芪胶和阿拉伯胶;分散剂或湿润剂可以是天然存在的磷脂,例如卵磷脂,或环氧烷与脂肪酸的缩合产物,例如聚氧乙烯硬脂酸酯,或环氧乙烷与长链脂族醇的缩合产物,例如十七烷-乙烯氧基鲸蜡醇,或环氧乙烷与衍生自脂肪酸和己糖醇的偏酯的缩合产物,例如聚氧乙烯山梨醇单油酸酯,或环氧乙烷与衍生自脂肪酸和己糖醇酐的偏酯的缩合物,例如聚乙烯脱水山梨糖醇单油酸酯。水性悬浮液还可包含一种或多种防腐剂(例如对羟基苯甲酸乙酯或对羟基苯甲酸正丙酯)、一种或多种着色剂、一种或多种调味剂以及一种或多种甜味剂(例如蔗糖或糖精)。
适用于通过加水制备水性悬浮液的可分散粉末和颗粒,提供与分散剂或湿润剂、助悬剂和一种或多种防腐剂混合的活性成分。适合的分散剂或湿润剂和助悬剂由上述已经提到的例子说明。也可以存在其他赋形剂,例如甜味剂、调味剂和着色剂。
药物剂型
本公开包括化合物1或其药学上可接受的形式的药物剂型。本文所述的剂型适合于口服给药于受试者。所述剂型可以是适合口服给药的任何形式,包括但不限于胶囊剂或片剂。
在一些实施方式中,本公开提供了包含10-1000mg具有下式的化合物1或其药学上可接受的盐的单一单位剂量的胶囊或片剂形式:
在一些实施方式中,化合物1的量为约100至800mg。在一些实施方式中,化合物1的量为约150至600mg。在一些实施方式中,化合物1的量为约200至400mg。在一些实施方式中,化合物1的量为约200mg。在一些实施方式中,化合物1的量为约400mg。在一些实施方式中,单一剂量胶囊或片剂包含化合物1的盐酸盐。
在一些实施方式中,化合物1的单一单位剂型是片剂。
在一些实施方式中,化合物1的单一单位剂型是胶囊。
在一些实施方式中,单一单位剂型在尺寸为#0、#1、#2、#3、#4或#5的胶囊中。在一些实施方式中,单一单位剂型在大小为#0的胶囊中。在一些实施方式中,单一单位剂型在大小为#1的胶囊中。在一些实施方式中,单一单位剂型在大小为#2的胶囊中。在一些实施方式中,单一单位剂型在大小为#3的胶囊中。在一些实施方式中,单一单位剂型在大小为#4的胶囊中。在一些实施方式中,单一单位剂型在大小为#5的胶囊中。
试剂盒
本公开内容还包括包含本发明的药物组合物和剂型的试剂盒。
在一些方面,本发明提供了包含化合物1或其药学上可接受的盐的试剂盒。本文所述的某些试剂盒包括描述化合物1施用方法的标签。本文所述的某些试剂盒包括描述治疗转甲状腺素蛋白(ttr)淀粉样变性的方法的标签。在一些实施方式中,本文所述的试剂盒包括描述治疗野生型转甲状腺素蛋白淀粉样心肌病(attr-cm,也称为老年性系统性淀粉样变性)的方法的标签。在一些实施方式中,本文所述的试剂盒包括描述治疗家族性淀粉样心肌病(attr-mcm)的方法的标签。在一些实施方式中,本文所述的试剂盒包括描述治疗家族性淀粉样多发性神经病(attr-pn,也称为fap)的方法的标签。
本发明的组合物,包括但不限于在瓶子、罐、小瓶、安瓿瓶、试管、泡罩包装,或食品和药物管理局(fda)或其他监管机构批准的其他容器密闭系统中包含化合物1的组合物,其可以提供包含化合物1或其药学上可接受的盐的一或多个单位剂量。包装或分配器还可以伴随有与容器相关的注意事项,容器的形式由管理药品制造、使用或销售的政府机构规定,注意事项表明被机构批准。在某些方面,试剂盒可包括如本文所述的制剂或组合物,包括该制剂的容器封闭系统或包括该制剂的一或多个剂量单位形式,以及描述如本文所述的使用方法的注意事项或说明。
诸如泡罩包装的包装系统包括适用于药物包装的可热成型的硬质薄膜或pvc,以及推入式盖。所述盖子可以包括由底漆/铝/热封涂层组成的箔,或者可以是纸基的。本领域技术人员将容易地制备包含化合物1的泡罩包装。本文所述的瓶系统可以制成各种尺寸(例如75cc、100cc、200cc等),并且通常包括可以由聚丙烯制成的防止儿童接触的封闭物。在一些实施方式中,将化合物1的药物剂型包装在75cc瓶中,带有防止儿童接触的封闭物。本领域技术人员可以容易地制备本文所述的瓶系统。
在一些实施方式中,本公开提供了用于每日两次给药的试剂盒。这些试剂盒为每次给药提供一或多个包含化合物1的单位剂量。
在一些实施方式中,化合物1的每日总剂量为800mg,意指在第一次给药时400mg,而在第二次给药时400mg。在一些实施方式中,在第一次给药时给予包含200mg化合物1的两个单位剂量,在第二次给药时给予包含200mg化合物1的两个单位剂量。在一些实施方式中,在第一次给药时给予包含400mg化合物1的一个单位剂量,在第二次给药时给予包含400mg化合物1的一个单位剂量。在一些实施方式中,化合物1的盐酸盐形式被施用。
在一些实施方式中,化合物1的每日总剂量为1600mg,意指在第一次给药时800mg,而在第二次给药时800mg。在一些实施方式中,在第一次给药时给予包含200mg化合物1的四个单位剂量,在第二次给药时给予包含200mg化合物1的四个单位剂量。在一些实施方式中,在第一次给药时给予包含400mg化合物1的两个单位剂量,在第二次给药时给予包含400mg化合物1的两个单位剂量。在一些实施方式中,化合物1的盐酸盐形式被施用。
iv.实施例
提供以下实施例以说明但不限制要求保护的发明。
材料和方法
以下通用材料和方法在指示的位置使用,或者可以在以下实施例中使用:
ag10血浆浓度的测定
使用蛋白质沉淀提取包含ag10和内标ag10-d6的人血浆,并通过配备hplc柱的sciexapi4000lc-ms-ms进行分析。针对ag10-d6内标产物离子的峰面积检测ag10产物离子的峰面积。使用从提取当天制备的校准标准品产生的加权1/x2线性最小平方回归分析进行定量。
荧光探针排阻法(fpe)
通过荧光探针(探针)在6小时反应时间内与血清中游离四聚ttr结合位点共价结合的能力检测四聚ttr的甲状腺素结合口袋中ag10的占有率。
将每个血清样品的等分试样铺在96孔板中。在室温下,使用可荧光的微孔板,每隔15分钟监测添加探针后荧光的变化(λex=328nm,λem=384nm),共监测6小时。
用于评估四聚ttr稳定性的蛋白印迹
如通过蛋白印迹凝胶的光密度测量法测定的,通过将酸变性72小时后剩余的四聚ttr蛋白的量与四聚ttr蛋白的初始量进行比较,来确定ag10对ttr四聚体的稳定作用。
在0和72小时的时间点,用酸化缓冲液(乙酸钠、kcl、edta、dtt,ph约4.0)稀释来自受试者的血浆样品。0小时的样品与戊二醛直接交联,然后淬灭。72小时的样品在室温下孵育72小时,然后通过相同的方案交联并淬灭。然后通过添加sds凝胶上样缓冲液使所有样品变性,并在凝胶上样前煮沸。每个样品均在sds-page凝胶中分离,并使用抗ttr抗血清(多克隆兔抗人类前白蛋白,dako目录号a0002)通过免疫印迹进行分析。使用红外licor成像系统或荧光成像系统对所有ttr条带的密度进行定量并报告。使用licor925-32232或英杰(invitrogen)84546通过igg带进行标准化。
在下面进一步详细讨论的实验、表格和图中,抽血的标称时间用于所有药代动力学和药效学数据。
实施例1:ag10·hcl的制备。
将式iiia的化合物(100g,495mmol1.0当量)溶解在丙酮(1l)中。将式ii的化合物(49.59g,495mmol,1.0当量)添加到上述溶液中,然后在室温搅拌下添加k2co3(82.14g,594.38mmol,1.2当量)和ki(41.11g,247mmol,0.5当量)。将反应混合物加热至60±5℃,并在该温度下搅拌40h。过滤反应混合物,然后在减压下浓缩以提供呈粘性橙色液体的式iv的化合物(102g)。
将式iv的化合物(100g,632mmol,1.0当量)溶解在乙醇(1l)中。在室温下将水合肼(87g,1738mmol,2.75当量)和浓hcl(4.6ml,0.2当量)加入上述溶液中。将反应混合物加热至75±5℃,并在该温度下搅拌3h。通过tlc(70%乙酸乙酯:正己烷,在碘中可见)完成反应并在质谱中观察到产物峰后,将反应混合物在减压下浓缩,以得到呈无色液体糖浆的式v的化合物(70g),直接用于下一步骤。
将式v的化合物(35g,227mmol,1.0当量)溶于1,2-二氯乙烷(525ml)。在室温下经30分钟分批加入pbr3(64.67ml,681mmol,3当量)。将反应混合物加热至75±5℃,并在该温度下搅拌3小时。通过tlc(50%乙酸乙酯:正己烷,在碘中可见)完成反应并在质谱图中观察到产物峰后,将反应混合物用二氯甲烷(350ml)稀释,并用饱和nahco3溶液淬灭至ph=7到8。分离并收集有机层和水层。有机层用mgso4干燥并过滤。减压浓缩滤液,得到呈粘稠橙色液体的式via的化合物(38g)。
将4-(3-溴丙基)-3,5-二甲基-1氢-吡唑氢溴酸盐(via)和dmso装入容器中,并在20±10℃下搅拌10分钟。然后在搅拌下将混合物加热至55±5℃。向该混合物中转移包含4-氟-3-羟基-苯甲酸甲酯(viia)、碳酸钾和无水dmso的搅拌溶液。缓慢转移烷基溴的dmso溶液,以保持内部温度为55.0±5℃。6小时后添加完成,并将混合物在55.0±5℃下再搅拌1小时。在30分钟的过程中将混合物冷却至25±5℃,并在保持温度低于25℃的同时添加水。混合物用乙酸乙酯萃取,水层用乙酸乙酯反萃取。合并的乙酸乙酯溶液用盐水洗涤。将合并的乙酸乙酯洗涤液在真空下浓缩至最小体积,并加入庚烷,其沉淀出viiia。将混合物加热至75±5℃,并在搅拌下老化1小时。在两个小时的过程中将混合物冷却至25±5℃,并通过过滤收集所得固体。用乙酸乙酯的庚烷溶液(30%)洗涤滤饼。用氮气流干燥分离的固体。将固体装入容器中,并与乙酸乙酯和庚烷混合。将所得混合物加热至75±5℃以溶解固体。在两个小时的过程中将溶液冷却至25±5℃,并通过过滤收集所得固体。用30%乙酸乙酯/庚烷溶剂混合物洗涤固体,并在55℃的真空烘箱中干燥,得到纯度>99.5%的viiia。
有夹套的玻璃容器中装有式viiia(1.0当量)的化合物和甲醇。将混合物在搅拌下冷却至10±5℃,并在20分钟的过程中加入氢氧化钠水溶液(3当量)。将该混合物在20±5℃下搅拌不少于2小时老化,此时反应完成。停止搅拌并加水。然后在内部温度不大于35℃下通过真空蒸馏除去甲醇。将所得的浓缩、澄清的水溶液冷却至10℃,并添加浓hcl,直到ph降低至1.4-1.6(ph计)以沉淀盐酸盐为止。通过过滤收集固体,用0.2nhcl洗涤,并在50℃下真空干燥,得到纯度为不少于99.5%的式ia化合物。
实施例2:1期临床研究
ag10-001研究是一项由两部分组成的随机、双盲、安慰剂对照、单次和多次递增剂量的研究,首次人体试验在健康成人志愿者中进行,以评估ag10在单次和多次给药后的安全性、耐受性、pk和pd。这项研究还旨在评估食物对ag10的pk的影响。
a部分是单次递增剂量(sad)设计,其中将4个组的8名健康男性和/或女性按3:1的总比例随机分为ag10或匹配的安慰剂。
b部分为多次递增剂量(mad)设计,其中将3个组的8名健康男性和/或女性以3:1的比例随机分为ag10或其安慰剂,给药12天。总计,每组包括8名健康受试者的4个组(总共32个受试者,其中24名受试者服用了ag10hcl,8名受试者服用了安慰剂以匹配),各自服用50mg、150mg、300mg和800mg的盲研究药物的递增剂量完成了研究的sad部分。其中一个sad组接受了两次300mg剂量,其中一次在禁食条件下,另一次在适当的清除期后服用了高脂测试餐。每组包括8名健康受试者的3个组(共24名受试者,其中18名服用ag10hcl,6名服用安慰剂以匹配)每12小时分别接受100mg,300mg或800mg的盲研究药物,共12天,完成了研究的mad部分。
单次递增剂量
对于服用了50mgag10hcl(sad组1),150mgag10hcl(sad组2),300mgag10hcl(sad组3)和800mgag10hcl(sad组4)的健康受试者,观察到的pk曲线揭示ag10具有快速的口服吸收能力,平均tmax低于1小时,消除半衰期约为22-27小时。表1列出了sad组1-4的pk参数的几何平均值。此外,如图1所示,在sad组1-4中,pk中存在中等程度的受试者间差异,cmax的%cv范围为10.0%-41.9%,而aucinf的%cv范围为12.5%–40.4%。
表1:sad组1-4的pk参数的几何平均值
数值=几何平均值±sd
此外,如图2(示出组3)所示,sad研究的食物影响部分显示对ag10的药代动力学的最小食物影响,进食与禁食状态相比,cmax略有降低,tmax略有延长,但总体而言,auc0-24定义的暴露没有明显变化。组3的pk数据如下表2中所示。
表2:sad组3的进食与禁食pk参数
所有数据均以几何平均值表示。
多次递增剂量
mad组1(健康受试者每12小时服用100mgag10hcl,连续12天),组2(健康受试者每12小时服用300mgag10hcl,连续12天)和组3(健康受试者每12小时服用800mgag10hcl,连续12天)的药物动力学曲线在图3中显示,pk参数在表3中列出。即使在每个剂量组的cmax值中观察到一些受试者间的差异,auc0-12值显著相似,%cv值在8.8%–22.2%之间。在给药的12天内没有观察到明显的积累。
表3:mad组1-3pk参数
ag10药效学
已经通过使用荧光探针排阻(fpe)和蛋白质印迹测定法(如上所述)评估了ag10的药效学(pd)特性,这两种方法都是ttr靶标结合和ttr稳定性的既定测定法。
如图4和图5中所示,来自fpe分析的数据证实了300mg和800mg单剂量的靶标结合,在峰值浓度下ttr完全稳定,并持续稳定长达12小时,在300mg时范围从29%到62%,在800mg时为56%至82%。图6显示了每个单次递增剂量组的平均靶标结合百分比随时间的变化;该数据表明剂量递增增加了稳定性。类似地,蛋白质印迹分析(图7a和7b)确认ttr在峰值浓度下完全稳定,并在300mgag10hcl单剂量下持续稳定长达12小时。sad组3的fpe和蛋白质印迹分析结果与>0.9的r2系数密切相关(图8)。药理活性的两种检测彼此紧密相关。
另外,如图9、图10和图11所示,连续12天服药ag10hcl100mgq12h、300mgq12h和800mgq12h后,fpe分析数据证实了稳定状态下持续的靶标结合,在给药的最后一天,在给药后12小时时间点ttr的平均稳定范围从100mgq12h剂量的33%到800mgq12h剂量的89%。图12显示了在800mgq12h的组中使用ag10hcl的12天内,ttr靶标结合百分比的峰值、平均值和谷值。
图13显示了用800mgag10hclq12h治疗的组3中三名受试者的ttr蛋白质印迹。受试者1和3服用ag10,受试者2服用安慰剂。在第12天给药前标记为0小时的泳道包含在给药11天后从受试者收集的ag10谷水平的样品(总共22剂)。第12天给药后标记的泳道显示了在第23剂ag10给药后在峰值水平采集的样品。经过72小时酸化后进行交联,然后如上所述的sds-page和免疫印迹用于检测四聚ttr蛋白的稳定性。如该实验方案所检测,受试者2显示没有剩余的ttr四聚体。相比之下,受试者1和受试者3在ag10的谷值和峰值水平均显示四聚ttr的完全稳定。
来自sad和mad组的pk-pd汇总数据如图14所示,表明了在用ag10给药的人类受试者中通过fpe测定法测量到的ag10的可预测和剂量响应性pd效应。
总结
本文为ag10提供的数据证实了靶标结合,单次和多次剂量后在峰值浓度下ttr完全稳定并持续稳定长达12小时。
实施例3:计划的2期临床研究
2期研究计划作为一项随机、多中心、双盲、平行组、安慰剂对照,剂量范围研究,以评估具有稳定心力衰竭治疗背景的attr-cm患者中ag10的安全性、耐受性、pk和pd。筛选和随机分组后将进行为期28天的盲目、安慰剂对照治疗。计划大约有45名受试者参加2期研究。2期研究有关的概要列于表4中。
表4:2期研究概要
实施例4:由ag10的热焓驱动的转甲状腺素蛋白稳定性模仿避免转甲状腺素蛋白淀粉样变性的自然发生的遗传变异
以下实施例描述了ag10的焓结合与稳定多蛋白复合物的增强效力之间的相关性。
材料与方法
等温滴定量热法(itc)使用microcalpeaq-itc在25℃下进行结合实验。制备配体溶液(25μm,在pbsph7.4,100mmkcl,1mmedta,2.5%dmso中),滴定到在相同的缓冲液中含有2μmttr的itc池中。19份配体的注射液(每次2.0μl)被注入itc池中(25℃),至ttr完全被配体饱和。绘制量热数据,并使用标准单点结合模型进行拟合。作为对照,我们测试了将缓冲液中的空白dmso滴定到ttr中引起的焓变化,得到的结合焓<0.4kcal/mol。我们还使用itc来对人血清白蛋白(hsa)滴定他法米迪和ag10。他法米迪的kd值(2.3μm)与先前报道的相似(kd=2.5μm;ema评估报告ema/729083/2011)。计算出的ag10的结合亲和力约为8μm,这也符合我们在图20中的数据(其中ag10与他法米迪相比对白蛋白的结合较低)。
结合缓冲液和人或犬血清中ttr的fpe检测法。ag10和其他稳定剂与缓冲液和血清中ttr的结合亲和力和选择性是通过它们与荧光探针排阻物(fpe探针)的结合竞争结合在缓冲液和人血清中的ttr的能力来确定的。fpe探针是本身不发荧光的硫酯ttr配体,但是与ttr的t4结合位点结合后,它会共价修饰赖氨酸15(k15),形成荧光偶联物。结合到ttrt4位点的配体会降低fpe探针的结合,如通过较低的荧光所观察到的。fpe测定法也适用于犬血清。fpe与缓冲液中的ttr:将98μlpbs(ph7.4,终浓度:2.5μm)中ttr的等分试样与1μl测试化合物(2.5μm)和1μlfpe探针(0.18mm的dmso储备溶液:最终浓度:1.8μm)混合。使用微板分光光度计读数器(spectramaxm5)在室温下监测荧光(λex=328nm和λem=384nm)变化6小时。fpe与人和犬血清中的ttr:将98μl合并的人血清(由人类男性ab血浆,西格玛;目录号h4522;ttr浓度5μm制备)或犬血清(创新研究,目录号:ibg-ser;ttr浓度4.6μm)的等分试样与1μl测试化合物[所有化合物均制成dmso中的10mm储备液,并相应地用dmso稀释(血清中的最终浓度为:ag1010μm;二氟尼柳200μm;他法米迪20μm;托卡朋20μm)]和1μl的fpe探针(dmso中0.36mm的储备溶液:最终浓度:3.6μm)混合。对于犬血清(用ag10口服治疗后),将1μl的fpe探针和1μl的dmso加入每个孔中,并与98μl适当的犬血清样品混合。使用微板分光光度计读数器(spectramaxm5)在室温下监测荧光变化(λex=328nm和λem=384nm)6小时。
通过免疫印迹检测血清中ttr的稳定性。如先前报道进行蛋白质印迹。所有化合物均制成dmso中的10mm储备液,并相应地用dmso稀释(血清中的最终浓度为:ag1010μm;二氟尼柳200μm;他法米迪20μm;托卡朋20μm)。将2μl的每种化合物添加到98μl的人血清中(ttr浓度为5μm)。样品在37℃孵育2小时,然后用酸化缓冲液(ph4.0、100mm乙酸钠,100mmkcl,1mmedta,1mmdtt)将10μl样品以1:10稀释。如先前报道的,蛋白质印迹法也在尿素缓冲液(ph7.4)中进行。将样品在室温下孵育72小时,与戊二醛(最终浓度为2.5%)交联5分钟,然后用10μl的7%硼氢化钠溶液(在0.1mnaoh中)淬灭。加入100μlsds凝胶上样缓冲液使所有样品变性,并煮沸5分钟。将10μl的每个样品在12%sds-page凝胶中分离,并使用抗ttr抗血清(dakoa0002,人血清以1:10,000稀释,犬血清以1:2,000稀释)进行免疫印迹分析。通过使用odyssey红外成像系统(li-cor生物科学)量化ttr条带(ttr四聚体和结合到rbp的四聚体)的组合强度,并报告为0小时(认为100%稳定)和72小时(介于10%和35%ttr剩余之间的范围)时ttr四聚体相对于dmso对照的ttr四聚体密度的百分比。四聚物稳定性百分比计算为100×[(四聚物和四聚物+rbp强度,72小时)/(dmso的四聚物和四聚物+rbp强度,0小时)]。
计算机模拟的结构和建模研究。对从rcsbpdb位点获得的四个ttr晶体结构进行了ttr的晶体结构分析。使用pdb文件中提供的x射线晶体学单元信息,构建ttr四聚体的生物组装体。当建议多个模型时,将使用首选模型。使用molden38构建了ag10及其四个衍生物(1,2,3和4)的初始几何结构,并使用gaussian’09程序包(美国康涅狄格州沃灵福德:高斯公司,2009年)用6-311+g(d)基组在杂化密度函数b3lyp级别上进行了几何优化。在优化的几何结构上进行了频率计算,以确保它们没有虚频率。dock6程序用于对接实验。具有ag10的v122i突变ttr复合物的晶体结构(pdbid:4hiq)用作受体。利用晶体学数据建立四聚ttr,除去溶剂和其他杂原子,并选择一个包括t4结合位点的大对接网格。对于所有对接实验,使用相同的受体和网格。进行柔性配体对接以允许围绕扭转角旋转。ucsfchimera软件包用于3d结构的可视化和分析。
ag10和他法米迪与人血清白蛋白的结合。将测试化合物(ag10或他法米迪;均为30μm)与人血清白蛋白(hsa;600μm;人血清白蛋白;西格玛奥德里奇,目录号:a3782)在分析缓冲液(10mm磷酸钠,100mmkcl和1mmedta,ph7.6)中在37℃孵育1小时。通过重力在pdminitrapg25色谱柱(ge生命科学,目录号45-001-529)上对500μl的has和ag10或他法米迪混合物在测定缓冲液中的溶液进行凝胶过滤,并通过nanodroptm鉴定含有hsa的组分。还使用nanodroptm(基于已知has浓度的校准曲线)确定hsa的浓度(即在0时的浓度)。他法米迪样品的hsa浓度为351μm,ag10样品的hsa浓度为345μm。使用hplc(基于已知浓度的测试化合物的校准曲线)评估这些组分中的测试化合物的浓度(即在0时的浓度)。然后将500μl的各hsa/测试化合物样品添加到slide-a-lyzer透析盒g2(3.5kmwco,赛默飞科学,目录号pi87722)中。将透析盒置于100ml测定缓冲液中,并在室温下搅拌。24小时后,从透析盒中取出样品并测量体积。如上所述,使用nanodroptm和hplc测定has和测试化合物的浓度。
ag10:ttr复合物的透析。将ag10(10μm)与人野生型ttr(5μm;从人血浆中纯化;西格玛奥德里奇,目录号p1742)在测定缓冲液(10mm磷酸钠,100mmkcl和1mmedta,ph7.6)中于37℃孵育1小时。然后将500μl各ag10/ttr溶液添加到slide-a-lyzer透析盒g2中。将透析盒置于100ml测定缓冲液中,并在室温下搅拌。在不同的时间点(0、0.5、1、2、6和24小时)从透析缓冲液中取样。24小时后,从透析盒中取出样品,测量体积并将结果标准化。使用nanodroptm和lcms分别确定从测定缓冲液中获得的ttr和ag10的浓度。
与其他血清蛋白相比,ag10和他法米迪对ttr的选择性。修改了fpe分析,并用纯化的人ttrwt(5μm)进行。将其他血清蛋白单独或组合[纤维蛋白原(5μm)、白蛋白(600μm)、igg(70μm)、转铁蛋白(25μm)]添加到tgf和fpe混合物中,并如上所述在6小时内监测荧光。在孵育3小时后测量的在血清蛋白存在下fpe探针与ttr结合的百分比用于计算%ttr占有率。
对犬重复7天ag10口服给药。将16只雄性(m)和16只雌性(f)比格犬分成四个治疗组,并用媒介(6m/6f,0mg/kg)或0.5%甲基纤维素制剂中的ag10(2m/2f,50mg/kg;2m/2f,100mg/kg;6m/6f,200mg/kg)口服灌胃给药,总共32只犬。在研究第1天(d1给药前),研究第7天给药前(d7给药前)和研究第7天给药后1小时(d7给药后),从颈静脉收集血液(约1.5ml)到血清分离管中。使用上述fpe分析法分析这些血清样品的ttr占有率。
给犬单次口服ag10,以确定与ttr结合和稳定有关的暴露效应(pk-pd)关系。将4只雄性和4只雌性比格犬分为两个治疗组(n=2/性别/组),总共8只犬被评估以获取ag10结合和ttr稳定的同时药代动力学(pk)和药效动力学(pd)数据。每只动物以5或20mg/kg的单剂量接受一次在0.5%甲基纤维素中的ag10的口服灌胃(po)。在给药前以及给药后2、4、6、8、12和24小时收集血液并进行分析。通过lcms分析这些血清样品中ag10的浓度,并通过fpe分析分析ttr的占有率。
统计分析所有结果均表示为平均值±sd。所有统计分析均使用graphpadprism软件进行。通过单向方差分析(anova),然后通过tukey多重比较检测测定差异的显著性(n.s.,不显著;*p≤0.05;**p≤0.01;***p≤0.001)。
化学反应.综述.除非另有说明,否则所有反应均在氩气中使用干燥溶剂在无水条件下进行。所使用的溶剂是费希尔(fisher)的acs等级。试剂购自奥德里奇和费希尔公司,无需进一步纯化即可使用。通过薄层色谱法(tlc)监测反应,在0.20mm
关键化合物的纯度hplc分析在c18和c4反相柱上进行。所有关键化合物的纯度均>95%。纯度分析的描述已包含在实验部分中。修订后的手稿的补充信息中包含了关键化合物的详细hplc信息(痕量、保留时间和纯度%)。
合成程序如先前报道的那样,合成了ag10和他法米迪。托卡朋和二氟尼柳购自费希尔。所有ag10类似物如下制备。
3-(3-(3,5-二甲基-1h-吡唑-4-基)丙氧基)-4-碘苯甲酸(1a);将3-(3-溴丙氧基)-4-碘代苯甲酸甲酯(5a)(834mg,2.1mmol,1当量)的苯溶液(3ml)滴加到乙酰丙酮(0.43ml,4.2mmol,2当量)和dbu(0.627ml,4.2mmol,2当量)的苯(7ml)溶液中。将反应混合物在室温搅拌3天。将混合物过滤并浓缩。向该中间体的乙醇(5ml)溶液中加入水合肼(0.28ml,5.25mmol,2.5当量),并将该反应加热回流4小时。浓缩反应物,并通过快速柱色谱法(硅胶,1-20%meoh/ch2cl2)纯化,得到化合物1a的甲酯;将氢氧化钠(79mg,1.98mmol,2当量)的水(2.5ml)溶液加入到酯中间体(412mg,0.99mmol)的甲醇(10ml)溶液中,并将反应混合物加热回流4小时(50℃)。将反应物浓缩并通过快速柱色谱法(硅胶,1-5%meoh/etoac)纯化,得到化合物1a(183mg,三步产率为22%);(通过hplc获得98.3%纯度):tr(柱)(c18)=25.72分钟;tr(c4)=16.06分钟。1hnmr(cd3od,600mhz)δ7.86(d,1h,j=8.4hz),7.41(d,1h,j=1.2hz),7.34(dd,1h,j=1.2hz和8.4hz),4.0(t,2h,j=6.0hz),2.67(t,2h,j=7.2hz),2.13(s,6h),1.97-1.93(m,2h)。13cnmr(cd3od,600mhz)δ168.5,157.6,142,139.2,133.2,123.1,114,117.8,91.7,67.5,29.6,18.7,9.3;(hrms(dart)m/z:c15h17in2o3+h+401.0362计算得出;实测值401.0347(m+h+)。
3-(3-(3,5-二甲基-1h-吡唑-4-基)丙氧基)-4-氟苯甲酸甲酯(2);将3-(3-溴丙氧基)-4-氟苯甲酸甲酯(5b)(780mg,2.69mmol,1当量)的苯(3ml)溶液滴加到乙酰丙酮(0.552ml,5.38mmol,2当量)和dbu(0.804ml,5.38mmol,2当量)的苯(7ml)溶液中。将反应混合物在室温搅拌3天。将混合物过滤并浓缩。残余物通过快速柱色谱法(硅胶,1-10%etoac/己烷)纯化,得到烷基化的中间体,其直接用于下一步。向该中间体的乙醇(5ml)溶液中加入水合肼(0.36ml,6.73mmol,2.5当量),并将该反应加热回流4小时。将反应物浓缩并通过快速柱色谱法(硅胶,1-20%meoh/ch2cl2)纯化,得到化合物2(288mg,35%产率);(通过hplc获得96.3%纯度):tr(柱)(c18)=25.11分钟;tr(c4)=14.03分钟。1hnmr(cd3od,600mhz)δ7.63-7.58(m,2h),7.19-7.15(m,1h),4.00(t,2h,j=6.0hz),3.86(s,3h),2.58(t,2h,j=7.2hz),2.12(s,6h),1.97-1.92(m,2h)。13cnmr(cd3od,600mhz)δ168.1,158.4,156.7,148.9,128.5,124.6,117.6,117.0,115.6,69.4,53.3,31.1,20.2,10.9;hrms(dart)m/z:c16h19fn2o3+h+307.1458计算得出;实测值307.1463(m+h+)。
4-氟-3-(3-(1,3,5-三甲基-1h-吡唑-4-基)丙氧基)苯甲酸(3):向2(21mg,0.07mmol,1当量)的dmf(3ml)溶液中添加氢化钠(5mg,0.21mmol,3当量)和甲基碘(17μl,0.28mmol,4当量)。将反应混合物在室温搅拌2小时。将混合物用盐水萃取,过滤并浓缩。残余物通过快速柱色谱法(硅胶,0.5-2%meoh/etoac)纯化,得到烷基化的中间体,其直接用于下一步。将氢氧化钠(5.6mg,0.14mmol,2当量)的水(0.5ml)溶液加入到烷基化中间体的甲醇(2ml)溶液中,并将该反应加热回流4小时(50℃)。浓缩反应物并通过快速柱色谱法纯化(硅胶,1-5%meoh/etoac),得到化合物3(11mg,两步产率52%);(通过hplc获得纯度为97.8%):tr(柱)(c18)=25.25分钟;tr(c4)=15.71分钟。1hnmr(cd3od,600mhz)δ7.58-7.51(m,2h),7.10-7.06(m,1h),3.92(t,2h,j=6.0hz),3.56(s,3h),2.49(t,2h,j=7.2hz),2.05(s,3h),2.01(s,3h),1.83-1.88(m,2h)。13cnmr(cd3od,600mhz)δ168.1,154.6,146.8,145.3,137.2,128.1,122.8,115.5,115.4,114.8,67.4,34.3,29.4,18.8,10.1,7.9;hrms(dart)m/z:c16h19fn2o3+h+307.1458计算得出;实测值307.1449(m+h+)。
3-(3-(3,5-二乙基-1h-吡唑-4-基)丙氧基)-4-氟苯甲酸(4):将氢氧化钠(3.2mg,0.08mmol,2当量)的水(0.5ml)溶液加入6(13mg,0.04mmol,1当量)的甲醇(2ml)溶液中,并将反应加热回流4小时(50℃)。将反应物浓缩并通过快速柱色谱法纯化(硅胶,1-5%meoh/etoac),得到化合物4(10mg,80%产率);(通过hplc获得纯度为96.0%):tr(柱)(c18)=25.16分钟;tr(c4)=15.56分钟。1hnmr(cd3od,600mhz)δ7.57-7.49(m,2h),7.08-7.04(m,1h),3.94(t,2h,j=6.0hz),2.51-2.43(m,6h),1.87-1.82(m,2h),1.06(t,6h,j=7.8hz)。13cnmr(cd3od,600mhz)δ169.8,157.9,156.3,149.3,148.5,124.6,117.2,117.1,114.1,69.4,31.8,20.1,19.9,14.7;hrms(dart)m/z:c17h21fn2o3+h+321.1614计算得出;实测值321.1601(m+h+)。
3-(3-溴丙氧基)-4-氟苯甲酸甲酯(5):化合物5如先前报道合成。向4-氟-3羟基苯甲酸甲酯(1.0g,5.87mmol,1当量)和1,3-二溴丙烷(3.0ml,29.4mmol,5当量)的dmf(15ml)溶液中添加k2co3(0.98g,7.1)mmol,1.2当量)。将反应混合物在室温搅拌16小时。将混合物用etoac(500ml)稀释,用盐水(3×200ml)洗涤,并用na2so4干燥。将溶液过滤并浓缩。残余物通过快速柱色谱法(硅胶,1-10%etoac/己烷)纯化,得到化合物5(1.3g,76%产率);1hnmr(cd3od,600mhz)δ7.67-7.61(m,2h),7.14-7.07(m,1h),4.21(t,2h,j=5.89hz),3.89(s,3h),3.62(t,2h,j=6.38hz),2.38-2.31(m,2h);(esi+)m/z:c11h12brfo3+h+290.00计算得出;实测值290.01(m+h+)。
3-(3-(3,5-二乙基-1h-吡唑-4-基)丙氧基)-4-氟苯甲酸甲酯(6):将5b(100mg,0.35mmol,1当量)的苯(2ml)溶液滴加到3,5-庚二酮(0.095ml,0.7mmol,2当量)和dbu(0.104ml,0.7mmol,2当量)的苯(5ml)溶液中。将反应混合物在室温搅拌3天。将混合物过滤并浓缩。残余物通过快速柱色谱法(硅胶,1-10%etoac/己烷)纯化,得到烷基化的中间体,其直接用于下一步。将水合肼(0.047ml,0.875mmol,2.5当量)加入到烷基化中间体的乙醇(4ml)溶液中,并将该反应加热回流4小时。将反应浓缩并通过快速柱色谱法(硅胶,1-5%meoh/etoac)纯化,得到化合物6(75mg,两步产率65%);1hnmr(cd3od,600mhz)δ7.59-7.54(m,2h),7.15-7.11(m,1h),3.98(t,2h,j=6.0hz),3.81(s,3h),2.56-2.47(m,6h),1.91-1.86(m,2h),1.13(t,6h,j=7.8hz)。cnmr(cd3od,600mhz)δ167.9,156.6,156.2,148.8,148.7,124.4,117.5,117.3,116.9,113.9,69.5,53.1,31.8,20.1,14.7;hrms(dart)m/z:c18h23fn2o3+h+335.1771计算得出;实测值335.1773(m+h+)。
结果
稳定剂和ttr之间相互作用的结合亲和力和热力学的确定。我们使用等温滴定量热法(itc)来确定以及临床开发中所有ttr稳定剂(即ag10、他法米迪、二氟尼柳和托卡朋)和ag10类似物1、2、3和4的结合亲和力(kd)以及分子相互作用的潜在机制。大多数报道的ttr配体以很强的负协同性结合到ttr的两个相同的t4结合位点,因此第一个配体的结合将主导总结合能以及稳定作用。尽管在itc温度记录图中可以观察到一些协作性差异,但是这些差异将对结合能以及稳定作用产生较小的影响。因此,表5中报告的kd值是基于适合独立单点结合模型的数据得出的。ag10和他法米迪对缓冲液中ttr的结合亲和力(分别为kd=4.8±1.9和4.4±1.3nm)比托卡朋(kd=20.6±3.7nm)高4倍,比二氟尼柳(kd=407±35nm)高约100倍。化合物1-4的kd值范围为90至1250nm,结果总结于表5。稳定剂与ttr结合的kd由吉布斯结合自由能的变化(δg)表示,其中δg=δh–tδs。通过分析每个分子的热力学特征,我们可以评估焓(δh;代表化学键的形成或破坏)和熵力(δs;与系统中的混乱程度相关并经常受到由于疏水相互作用而释放结合的水分子的主导和支持)的相对贡献。尽管在缓冲液中ag10和他法米迪与ttr的结合亲和力相似(即类似的δg值),但它们与ttr的结合能却明显不同。ag10的结合(δh=-13.60kcal/mol和tδs=-2.26kcal/mol)是由焓驱动的,而他法米迪的结合约为50%的熵和50%的焓(δh=-5.00kcal/mol,tδs=6.39kcal/mol)驱动(图16a和表5)。托卡朋(δh=-10.1kcal/mol和tδs=0.4kcal/mol)与二氟尼柳(δh=-8.38kcal/mol和tδs=0.34kcal/mol)的结合在熵上是有利的,但主要受焓相互作用的驱动。ag10对ttr不利的熵结合能(tδs=-2.26kcal/mol)可能是由于其极性和/或构象柔韧性高于其他ttr稳定剂。化合物1-4和ttr之间的结合相互作用的热力学讨论如下。
表5:ttr稳定剂的比较
焓力预测ttr稳定剂在缓冲液中的效能及在人血清中的功效。最近一项对二氟尼柳和其他非甾体类抗炎药(nsaid)的研究发现,与具有较低δh影响的配体相比,具有有利(即较大的阴性)δh的配体具有较高的ttr选择性。尽管该研究描述了焓力与ttr稳定剂选择性之间的相关性,但尚未报道配体的结合焓与稳定ttr的能力之间的相关性,或者据我们所知,还没有与其他多聚体蛋白的相关性。为了评估稳定剂占据和稳定在缓冲液中ttr的效能,我们使用了荧光探针排阻(fpe)分析。fpe测定使用本身不发荧光的荧光探针(fpe探针),但是与ttr的t4结合位点结合后,它会共价修饰赖氨酸15(k15),从而形成荧光偶联物。结合到ttrt4位点的配体将降低fpe探针的结合,通过较低的荧光观察到。已经报道了在fpe测定中的荧光程度和ttr的稳定之间存在线性相关性。因此,我们首先使用fpe测定法来测量稳定剂结合和稳定缓冲液中ttr的效能(以稳定剂与ttr四聚体的1:1比例进行测试;图16b,c和表5)。稳定剂对缓冲液中ttr的效能顺序为ag10>托卡朋>他法米迪>二氟尼柳。
然后,我们使用fpe和蛋白质印迹分析来评估稳定剂(10μm)在占据和稳定人血清中(ttr浓度为5μm)ttr的功效(代表效能和选择性)(表5)。蛋白质印迹法测量在存在和不存在稳定剂的情况下,酸处理72小时后完整的ttr四聚体的量。稳定剂在人血清中的功效顺序与我们观察到的在缓冲液中ttr的功效相似(ag10>托卡酮>他法米迪>二氟尼柳;表5)。由于与所有其他稳定剂相比,其对ttr的结合亲和力明显较低(kd=407±35nm)(亲和力比其他稳定剂低20至80倍),因此预测二氟尼柳的效能和功效最低。令人惊讶的是,其他三种稳定剂的kd值与它们在占据和稳定缓冲液和人血清中ttr的效能和功效之间没有相关性。例如,尽管事实上托卡朋对ttr的结合亲和力(kd=20.6±3.7nm)比他法米迪对ttr的结合亲和力(kd=4.4±1.3nm)稍低,但托卡朋的效能和功效却高于他法米迪。有趣的是,这些稳定剂对在缓冲液和血清中ttr的效能和功效与它们的结合焓(对于ag10、托卡朋和他法米迪,分别为δh=-13.6、-10.1和-5.0kcal/mol)密切相关(r2=0.98)。该数据表明,与其他稳定剂相比,由焓驱动的ag10和托卡朋与ttr的结合(下文详细讨论)是它们有效稳定ttr的主要驱动力。
然后,我们使用蛋白质印迹分析法比较了ag10(10μm)与其他稳定剂在报道的人体内平均最大血浆浓度(对于80mg他法米迪qd,cmax为20μm;对于250mgbid的二氟尼柳,为200μm;对于100mg剂量tid的托卡朋,则为20μm)时的功效。10μm的ag10完全稳定人血清中的ttr(%ttr稳定度:95.4±4.8%);其他化合物在其报告的临床cmax下稳定了约50-75%的四聚ttr(图17a,b)。ag10的pka值(pka=4.13)和他法米迪的pka值(pka=3.73)高于二氟尼柳的pka值(pka=2.94)。因此,稳定剂的羧酸基团的电离百分数在ph4时可能会有所不同。这可能会影响羧酸基团与t4结合位点顶部的赖氨酸15(k15)和k15'的ε-氨基之间的静电相互作用强度,这可能会影响稳定剂的效能。为了解决这个问题,我们使用尿素缓冲液(ph7.4)进行了蛋白质印迹分析。尿素缓冲液中ttr的稳定性数据类似于在酸性ph下从蛋白质印迹和在生理ph下从fpe测定获得的数据。与蛋白质印迹ttr稳定性实验数据一致,在fpe分析中10μmag10的t4结合位点率基本上是完全的(%ttr占有率为96.6±2.1%),并高于其报告的临床cmax下的所有稳定剂。托卡朋在20μm的靶占有率(%ttr占有率86±3.2%)高于他法米迪和二氟尼柳的(分别在20μm和200μm时%ttr占有率~65%)(图17c,d)。
疾病防护性t119m突变内的ag10和ttr模拟分子的s117/s117'相互作用之间的结合相互作用。我们通过比较稳定剂与ttr的所报道共晶体结构与稳定的ttr变体(t119m和r104h)的晶体结构,研究了结合焓和ttr稳定之间的相关性。我们假设这可以使我们识别ttr的t4结合位点内的氨基酸官能团,这些官能团对于ttr的结合和稳定性很重要。ag10、他法米迪、二氟尼柳的羧酸部分和托卡朋上的羟基均与位于t-结合位点顶部的赖氨酸15(k15)和k15’的α-氨基团发生静电相互作用。ag10和托卡朋与ttr的焓驱动结合是由两个分子在t4结合位点形成的额外氢键驱动的。托卡朋的羰基与ttrwt的t119的羟基侧链形成一个氢键(距离
t119m变体ttr中的单体a和b的两个s117侧链羟基形成直接氢键,其距离为
ag10关键功能基团的表征对ttr稳定性至关重要。为了研究ag10各个官能团对ttr结合和稳定的焓贡献,我们合成并测试了四种ag10类似物(化合物1、2、3和4;图15),并评估了它们结合和稳定ttr的能力(图19)。ag10以不利的熵结合ttr(tδs=-2.26kcal/mol)。ag10的氟原子被置于ttr的hbp1中,因此我们假设可以通过用碘(化合物1a)代替ag10的氟原子来优化ag10与ttr的熵结合。建模研究表明,1的碘置入ttr的hbp1(t4的碘结合处),其可以通过从hbp1置换更多的水分子来改善熵结合(图19a)。与ag10(kd=4.8±1.9nm)相比,化合物1a对缓冲液中的ttr显示出显著较低的结合亲和力(kd=90±14nm)。itc分析表明,尽管1与ttr的熵相互作用比ag10更为有利(tδs分别为-0.21kcal/mol和-2.26kcal/mol),但焓值对结合的贡献显著下降(δh=-9.82kcal/mol和-13.6kcal/mol)(图19b)。正如模型所示,结合焓的降低可以用1的羧酸部分和k15/k15'之间的盐桥强度降低来解释(距离约为
ag10的羧酸部分在t4结合位点的外围直接与k15和k15’的ε-氨基形成两个盐桥,这些盐桥用来封闭ag10周围的t4口袋,并部分地将其与溶剂隔离。我们合成了ag10的甲酯类似物(化合物2,图19a),以测试修饰ag10在t4结合位点外围形成的两个盐桥的效果。与ag10(kd=4.8±1.9nm)相比,化合物2对在缓冲液中ttr的亲和力显著更低(kd=258±17nm),这可以解释为与ag10中的盐桥相比,2的酯基与k15/k15'的之间的潜在氢键的强度更低(δh=-6.49kcal/mol)(图19b)。与ag10和化合物1a相比,化合物2也显示出对缓冲液和人血清中的ttr的效能降低(图19c-e和表5)。
ag10的3,5-二甲基-1h-吡唑环位于t4结合位点的内腔深处,并与相邻亚基的s117和s117'形成两个氢键。通过阻断这些相互作用,我们可以分别使用itc和fpe分析有效地观察它们的焓贡献。因此,我们合成了具有n-甲基吡唑的化合物3。n-甲基将限制3的吡唑环与相邻的一个ttr亚基仅形成一个氢键(图19a)。我们还合成了化合物4,其中ag10的二甲基吡唑被二乙基吡唑代替。模型研究表明,大部分二乙基会阻止分子深入t4结合位点,从而降低其潜在地与s117/s117’形成任何氢键的能力(图19a)。如通过建模预测的,3(kd=251±12nm)和4(kd=1253±79nm)均显示出大大降低对缓冲液中的ttr的结合亲和力。降低的亲和力导致ttr在缓冲液和人血清中的效能显著降低,尤其是对于化合物4。稳定ttr的效能顺序在缓冲液和血清中相似(1>2>3>4;图19c-e和表5)。正如我们在临床attr稳定剂中观察到的那样,ag10和化合物1、2、3和4在占据和稳定ttr中的效能与这些分子的结合焓(δh分别为-13.6、-9.82、-6.49、-4.73和-2.1kcal/mol)密切相关(r2=0.98)。有趣的是,尽管2和3具有相似的结合亲和力,但它们的效能却大不相同(表5)。与3相比,2具有更高的效能,可以通过其良好的焓结合来解释(δh分别为-6.49kcal/mol和-4.73kcal/mol)(图19b)。该观察结果与从ag10和他法米迪获得的数据类似(即相似的kd值,但效能显著不同)(表5)。这些结果突出了吡唑环发挥的关键作用,以及它与两个ttr二聚体形成的氢键的重要性,模仿了保护性t119m-ttr突变中的相互作用,并增强了ttr四聚体的动力学稳定性。
研究焓对ag10对ttr选择性的影响。为了检验焓对ag10相比于其他丰富的血清蛋白对ttr的选择性的作用,我们在整个人血清中的fpe分析中测试了ag10和他法米迪的浓度-效应关系。我们测试了ag10和他法米迪,因为它们对缓冲液中ttr的结合亲和力非常相似(kd分别为4.8±1.9nm和4.4±1.3nm),但是它们与ttr结合的热力学(尤其是焓组分)显著不同。因此,从血清中获得的数据将在很大程度上反映出选择性。ag10表现出渐进的浓度依赖性占有率,当ag10浓度≥10μm时获得完全占有。即使在低于化学计量的浓度下,ag10仍能占据并稳定大多数ttr(在5μm时,fpe获得的ttr占有率为69.2%,蛋白质印迹获得的稳定率为74.5%)。相反,当浓度超过20μm时,他法米迪的占有或稳定活性的增加幅度更适中。当评估ag10的活性时,观察到ttr占有率(通过fpe)与ttr稳定度(通过蛋白质印迹)之间具有良好的相关性(r2=1.0)。对于他法米迪,浓度达到10μm有很好的相关性(r2=0.87),但是,在更高的浓度下,fpe分析中存在一平稳段。
通过在存在或不存在纯化的血清蛋白的情况下在缓冲液中重复这些测定,进一步研究了ag10和他法米迪对ttr的选择性。将ag10或他法米迪(30μm)与纯化的人血清白蛋白(在其600μm的生理浓度)预孵育后进行凝胶过滤,然后进行透析。在时间为0时(紧接在凝胶过滤之后),与他法米迪相比,更少的ag10与白蛋白结合(18.3±0.98μm相对于24.1±1.1μm;图20a)。对缓冲液透析24小时后,与hsa结合的ag10的浓度低于他法米迪的浓度(7.8±0.1μm相对于18.8±2.1μm)。这些数据表明,与他法米迪相比,ag10对白蛋白的结合亲和力较低。同时,还在该凝胶过滤/透析测定中研究了ag10与ttr的结合。将ag10(10μm)与等摩尔比的ttr(5μm四聚ttr,代表10μm的ttrt4结合位点)预孵育。在最初的6小时内,ag10从ttr的解离很慢(ag10-ttr摩尔比约为1.2:1),并且在24小时的孵育过程中保持1:1摩尔比(图20b)。
最后,使用改良的fpe测定法评估了ag10和他法米迪与人血清中ttr结合的选择性,其中用缓冲液(pbs缓冲液,ph7.4)中纯化人ttr代替人血清。除纯化的ttr(5μm)外,还向缓冲液中的fpe分析中添加了四个独立的代表性和丰富的血浆蛋白。白蛋白、转铁蛋白、纤维蛋白原或免疫球蛋白(igg)的添加不影响ag10的ttr占有率(在不存在或存在任何这些蛋白质的情况下>97%的ttr占有率,图20c,d)。白蛋白,而不是其他测试的血清蛋白,干扰了他法米迪对ttr的占有(41.5±0.9%相对没有白蛋白的情况下68.2±0.1%;图20e,f)。同时添加所有测试的血浆蛋白对ag10产生相同的结果。ag10对ttr的较高选择性可以归因于许多特性,包括焓结合和与更亲脂性的他法米迪(clogp=4.2)相比,ag10的更大亲水性(clogp=2.78)。
健康的比格犬是评估ttr稳定剂功效的合适实验模型。然后,我们研究了能否在体内维持ag10对ttr的高效能和选择性。忠实复制人类attr-cm病理的转基因动物模型尚不可用。因此,我们采用了一种类似于临床上目前使用的方法来检查ag10与其他ttr动力学稳定剂的功效。ttr稳定剂在占据和稳定ttr中的活性通常在从稳定剂给药前后的患者获得的血液样品中离体评估。为了探索ag10的体内活性,在健康的比格犬中使用了相同的方法。选择健康的比格犬作为实验模型有几个原因。ag10和其他稳定剂结合的ttrt4结合位点中的所有氨基酸在犬和人之间是保守的。我们还测试了犬血清中ttr的浓度(约4.6μm),发现它与健康人的相似。为了确认与基于人的试剂一起使用的测定用于犬研究的适用性,使用与上述实验相同的fpe和蛋白质印迹测定法,在混合的犬血清中评估ag10和他法米迪的活性。在使用犬血清重复进行的两种测定中,ag10和他法米迪的体外ttr结合和稳定浓度-效应关系与在人血清中观察到的相似(图21)。这些特征使健康犬成为后续研究的合适系统。
在口服给药后,ag10有效且选择性地与犬ttr结合。为了探索体内的药代动力学-药效学(pk-pd)关系,每天通过口服灌胃对健康的比格犬施用ag10共7天。共有16只雄性(m)和16只雌性(f)比格犬组成四个治疗组:(i)6m/6f,0毫克/千克/天(媒介对照);(ii)2m/2f,50毫克/千克/天;(iii)2m/2f,剂量为100毫克/千克/天;以及(iv)6m/6f,200毫克/千克/天。在研究第1天(基线)给药前,研究第7天给药前(代表稳态时的谷底浓度或cmin)以及研究第7天给药后1小时(代表稳态下的峰值浓度或cmax)采集定时血清样品。通过fpe测定评估ag10对ttr的结合占有率(图22a,b)。来自仅用媒介处理的犬的所有样品,以及在暴露于ag10之前从主动治疗组收集的样品,其ttr占有率为零。经ag10治疗的犬的血清在稳态谷值时显示出结合占有率呈剂量比例反应(第7天给药前;ttr占有率~81-94%),并且所有ag10治疗组在稳态cmax(给药后第7天)时均显示完全(>97%)ttr占有。随后测试了较低剂量的ag10,以进一步探索pk-pd(暴露-效果)关系,以确定可能仍有效结合并稳定ttr的最低有效剂量的ag10。分成两个积极治疗组的八只犬接受5或20mg/kg的ag10hcl的单次口服剂量。这些结果表明,与5mg/kg剂量组相比,20mg/kg的ttr占有率增加(图22c,d)。对于两种剂量,在cmax时的ttr占有率(%)均显著高于(p≤0.001)在cmin时的。在cmin时20mg/kg剂量的ttr占有率显著高于(p≤0.001)5mg/kg剂量。数据还显示,ag10的循环血浆浓度与ttr占有率(%)密切相关。
总之,比格犬证明ag10在特定剂量水平上口服有效,并达到了剂量依赖性血浆浓度,该浓度有效且选择性地结合并稳定四聚ttr。
实施例5:猴子通过静脉内给药或口服灌胃ag10hcl的单剂量研究
向3只雄性食蟹猴以1mg/kg静脉内或每次5mg/kg口服的剂量给予ag10hcl。在两个阶段之间有两个星期的清除期。在给药前和给药后约0.083(仅iv)、0.25、0.5、1、2、4、8、12、24、48、72和96小时收集血液样品。测定血浆样品中的ag10和ag10酰基葡糖苷酸,并在fpe测定中测试血清。fpe测定结果如图23所示,表明口服ag10可有效结合猴血清中的ttr。
fpe分析还表明,口服ag10以剂量依赖性方式稳定了ttr(图24)。
实施例6:服用ag10的健康个体的ttr血清浓度升高。
为了测量血清ttr浓度,使用了来自奥维亚系统生物学(avivasystemsbiology)(目录号okia00081-96w,批次号kc0699)的前白蛋白elisa试剂盒(人)。
根据elisa试剂盒制造商提供的方案进行研究。通过在制造商建议的校准曲线中添加三个标准浓度来改良该方法。将提供的ttr校准物溶解在1ml蒸馏水中,获得8.85μg/ml的浓度。第一个附加标准液为1000ng/ml,通过将178.4μl校准物添加到1400μl的1x稀释剂中制备。第二个附加标准液为200ng/ml,通过将32.4μl校准物添加到1400μl的1x稀释剂中制备。第三个附加标准液为0.78125ng/ml,通过将600μl的1.5625ng/ml标准液添加到600μl的1x稀释剂中制备。elisa试剂盒利用山羊多克隆抗ttr抗体进行捕获和检测。产生了针对天然人ttr蛋白的抗体。混合人血清购自创新研究公司(目录号ipla-ser,批次号24453)。
将混合人血清和mad血清样品在37℃的水浴中融化10分钟。分两步将所有样品以1:10000的比例稀释。首先将5μl血清与995μl的elisa试剂盒中提供的1x稀释剂混合。然后,将5μl的混合物加入到非结合微孔板中的245μl的1x稀释剂中。将100μl每个最终稀释样品加到elisa板的每个孔中。所有标准品和样品均一式两份进行测试。ttr研究从此时起遵循制造商的方案,没有任何进一步的修改。简而言之,将标准品和血清样品在elisa板中于室温孵育1小时。将elisa板用1x洗涤缓冲液洗涤四次,然后在室温下与1x辣根过氧化物酶偶联物一起避光孵育30分钟。然后将elisa板洗涤四次。添加tmb底物并使其显色10分钟,然后添加终止溶液。最后,测量每个孔在450nm的吸光度。
通过获取重复的0ng/ml孔在450nm处的平均吸光度并从每个孔的总450nm吸光度中减去该吸光度,对吸光度测量值进行参考校正。
使用graphpadprism软件为每个elisa板生成标准曲线。标准物的log(ng/ml)绘制在x轴上,参考校正的450nm吸光度值绘制在y轴上。使用s形4参数曲线拟合数据。从标准曲线内插混合人血清和mad血清样本的log(ng/ml)。将log(ng/ml)值转换为以mg/l为单位的血清ttr浓度,并校正样品稀释度。使用提供的校准物生成的标准曲线是可重现的(图25),该试剂盒成功地以从1:5000到1:20000的增加的稀释比将混合人血清样品进行了区分。因此,将奥维亚elisa试剂盒用于测试。
使用奥维亚elisa试剂盒测试了来自mad研究的三个组的人类正常志愿者样本(100、300、800mgag10hcl,连续十二天每天施用两次),以及安慰剂组。每个组的ttr浓度随时间的相对变化是通过相对基线值标准化来计算的(图26)。图27显示了所有安慰剂和ag10治疗组从基线到第12天血清ttr浓度的平均百分比变化。图28和29绘制了所有安慰剂和ag10hcl治疗组的基线和第12天血清ttr浓度(给药的健康志愿者的总数=24;安慰剂:活性剂=1:3;mad1组=100mgq12h,12天;mad2组=300mgq12h,12天;mad3组=800mgq12h,12天)。
arup前白蛋白测定(免疫比浊法测定)
还使用了arup前白蛋白测定法来分析被测mad组中的ttr血清浓度。arup使用罗氏诊断提供的前白蛋白试剂盒分析样品中的前白蛋白,并在罗氏诊断c702(rochediagnosticsc702)模块上运行。最低可报告限值为3mg/dl。
表6总结了每个组测得的基线和给药后(24小时后)血清ttr浓度。在每个测试组中,在24小时的测量期内ttr浓度都有可测量的增加。
表6:mad组1-3pk参数——血清ttr浓度
实施例7:2期临床研究结果–患有attr-cm的个体
基本上如实施例3所述进行了2期临床研究。总共包括49位患者而非45位:16位接受400mgbid;16位接受800mgbid;以及17位接受安慰剂。
参与研究的个体的基线特征显示在下表7中。
表7:基线特征
结果
使用在实施例部分开始时描述的fpe和蛋白质印迹测定离体测量ttr稳定度。通过监测试验参与者的ttr血清浓度在体内测量ttr的稳定性。
图30显示了每个治疗组中受试者的血清ttr水平的剂量反应变化。数据报告为从基线到第28天的变化百分比。每天两次接受400mgag10hcl的个体的血清ttr水平平均增加36%,每天两次接受800mgag10hcl的个体的血清ttr水平平均增加50%。相比之下,安慰剂治疗组的血清ttr水平平均降低了7%。
在ag10hcl给药组(400mg和800mgb.i.d.)中血清ttr水平的增加与先前测试的ttr稳定剂的先前报道观察结果一致。图31绘制了本研究中每个治疗组从基线到给药第28天的平均百分比变化。还绘制了他法米迪2期(fdacder咨询委员会会议背景资料)和二氟尼柳观察研究(hanson,j.l.s.等人,circheartfail201811:e004000)报道的血清ttr含量百分比变化。从图中可以看出,ag10、他法米迪和二氟尼柳均提高了血清ttr水平。
治疗前,400mgb.i.d.治疗组的40%受试者和800mgb.i.d.治疗组的56%受试者的血清ttr水平低于正常ttr水平(正常ttr水平为20-40mg/dl(3.6-7.3μm))。治疗28天后,每个活跃组100%的血清ttr浓度均在正常范围内(即在28天治疗方案结束时,所有接受治疗的患者具有正常的血清ttr水平)。比较而言,有18%的安慰剂个体的血清ttr水平低于治疗前的正常ttr水平。经过28天的治疗,该数字上升至31%的没有正常水平血清ttr浓度的安慰剂个体。参见图32。图33显示了该研究中个体的血清ttr浓度的基线分布。
体外蛋白质印迹分析证实ag10hcl的每种剂量水平(400mgb.i.d.和800mgb.i.d.)有效稳定ttr。参见图34,显示了与在给药前时间点(d1h0)的低水平的稳定相比,在第14和28天谷底(h0)和高峰(h1)时间点的ttr稳定的高水平。提供的误差线是平均值的标准误差。
荧光探针测定进一步证实了该蛋白质印迹结果,该结果显示具有野生型和突变型ttr个体在第28天谷底(给药前)和第28天高峰(给药后1小时)具有高水平的ttr稳定性。参见图35。
当绘制四聚ttr的甲状腺素结合口袋中ag10的占有率与ag10循环血浆浓度之间的关系时,确定在约5μm的血浆浓度发生了显著的靶标结合(ag10占有),并且在约7.5μm的血浆浓度时发生了完全的靶标结合。参见图36。
图37和图38绘制了在第1天给药前、第14天给药前(谷)、第14天给药后1小时(峰)、第28天给药前(谷)和第28天给药后1小时(峰)在荧光探针测定中测得的400mgb.i.d.组(图37)和800mgb.i.d.组(图38)的相对荧光单位。从两个图中都可以看出,荧光探针测定表明,在谷和峰时间点时,在400mgb.i.d和800mgb.i.d.两组中到第14天都接近完全的靶标结合。比较而言,图39绘制了安慰剂对照组在上述每个参考时间点测得的相对荧光单位。该图表明安慰剂对照组缺乏靶标结合。
总体而言,本文提供的数据表明,有症状的attr-cm患者在28天中对ag10的耐受性良好,ag10以剂量依赖的方式增加血清ttr浓度,ag10将低ttr水平恢复至正常水平,ag10在测试的两种剂量水平中完全稳定ttr。
当查看受试组中的野生型和特定突变型ttr种群时,与起始水平相比,每个活性给药组(无论ttr基因型如何)在给药结束时血清ttr浓度均升高。使用如实施例6所述的arup前白蛋白测定法分析ttr血清数据,如下表8所示。
表8:ii期组中的血清ttr浓度
特别查看具有v30mttr突变(与家族性attr多发性神经病(attrm-pn)相关的高度普遍的突变)的个体的数据,分别使用fpe和蛋白质印迹分析法观察到此类患者群体在治疗的第14天接近完全靶标结合并在治疗的第28天接近完全ttr稳定。参见图40和图41。
实施例8:常用利尿剂不干扰ag10的暴露
具有心力衰竭临床证据的attr-cm患者通常表现出容量超负荷或心内压升高的体征和症状。这需要用利尿剂治疗(ruberg和berk,circulation2012126:1286-300)。利尿剂(如呋塞米或托拉塞米)是用于attr-cm患者的ag10-2012期研究中最常用的药物。参见表9。
表9:接受利尿治疗的受试者的概况
对于群体pk分析,采用正式的3步协变量选择过程来研究其对ag10药代动力学的影响。尽管在向前添加过程中多个协变量被认为是显著的,在向后消除期间仅发现中央分布体积上的疾病状态为不显著,显著性水平α为=0.01。因此,最终模型保留在中央分布体积上的疾病状态。
使用群体pk分析来确定利尿剂(如呋塞米)的共同施用是否对ag10的药代动力学有任何影响。在2期中接受了ag10给药的32位受试者,其中25位正在接受呋塞米或托拉塞米治疗。在服用安慰剂的17名受试者中,有14名服用呋塞米或托拉塞米。
基于患者状态或利尿剂(呋塞米/托拉塞米),在1期的健康成人志愿者与2期的受试者之间,未观察到ag10清除率的差异(图42a,b)。
如前所述,中央分布体积受attr-cm疾病状态的影响。在以下中观察到较低的体积(图42c,d):
a)2期的attr-cm患者相对于1期的健康成人志愿者
b)接受呋塞米或托拉塞米的attr-cm患者相对于未接受呋塞米或托拉塞米的attr-cm患者。
因此,与健康成人志愿者相比,接受呋塞米/托拉塞米治疗的受试者中分布体积更低。在药物-疾病相互作用(即慢性心力衰竭对分布体积的影响)与药物-药物相互作用(利尿剂对分布体积的影响)之间没有区别。分布体积的变化主要影响药物的峰值血浆浓度。1期(ag10-001)健康成人志愿者研究(mad3,800mgq12h)和attr-cm患者(ag10-201)的2期均已证明约8μmag10的循环谷底浓度是稳定ttr的合适目标。如图43所示,在接受呋塞米或托拉塞米治疗的患者以及未同时使用利尿剂的受试者中,ag10的800mgbid剂量均可达到合适的浓度。
在重复给药的健康志愿者中,血浆ag10cmax的平均累积率在1.3到1.6之间。累积率用于预测稳定状态下施用ag10400mg片剂的药代动力学特征。图44比较了单一400mg的ag10片剂给药后12小时的ag10的血浆水平,假设ag10的累积比率为1.45(400mg片剂重复给药后),和在2期服用ag10400mgbid的attr-cm患者中ag10的实际循环浓度。与每日800mgbid剂量(4x200mgag10片剂,每日两次)获得的结果相似,使用较高剂量强度片剂(400mg)的预测谷底水平也与2期400mgbid(2x200mgag10片剂,每日两次)的更低每日剂量组的谷曲线相匹配。
因此,常用的利尿剂不会干扰ag10的暴露。
实施例9:3期临床研究–attr-cm
这项前瞻性、随机、多中心、平行组研究将评估在稳定心力衰竭治疗背景下与安慰剂相比,施用ag10在有症状受试者中的功效和安全性。筛选和随机分组后将进行总共30个月的盲法、安慰剂对照治疗。在治疗的12个月(a部分)结束时,将通过对功能性(6mwt)和健康相关的qol(通过hf专用仪器kccq进行测量)终点的分析来评估ag10的功效。在治疗30个月(b部分)结束时,将通过分析全因死亡率和与cv相关的住院治疗来进一步评估ag10的功效。
目前尚无批准用于治疗attr-cm的疗法。禁止在适应症外使用或作为非处方补充剂用于attr-cm治疗的其他研究性治疗或疗法。但是,如果在试验过程中,其他疗法获得了监管部门的批准专门指定用于治疗一个或多个地区的attr-cm,那么对于研究受试者来说,有几种可能的途径:
·无论是否有任何批准的指定产品,都应鼓励受试者在试验中保留至少12个月的盲研究治疗。如果受试者在任何时候选择退出试验,将鼓励他们完成提前终止随访和相关程序。
·任何已经完成至少24个月的盲法研究治疗并随后获得批准的指定产品的研究受试者,都将被鼓励留在试验中,甚至在开始使用该产品进行治疗后也继续进行盲法研究治疗。使用批准的指定产品开始治疗且仍在试验中的受试者,在开始伴随治疗之前应进行不定期的随访和研究评估。
所有完成30个月盲研究治疗以及双盲治疗期最终评估的受试者都可能有资格参加长期ag10治疗的开放标签扩展研究。
合格受试者将以2:1的比例随机分配成口服bid给药的ag10800mg或匹配的安慰剂组。根据受试者是否患有野生型attr-cm(attrwt-cm)或突变型attr-cm(attrm-cm)将受试者随机分组,目标最低为20%的患有attrm-cm的受试者。将通过基因分型尽一切努力来确认ttr的状态(野生型或变体)。在特殊情况下(当受试者拒绝进行基因检测时),可以要求医学监督局或指定人员批准在没有记录基因分型的情况下招募受试者。如果批准,并且该受试者已纳入研究,则此类ttr状态未知的受试者被划分为“野生型ttr”阶层。在筛选时,受试者还将根据nt-bnp前体水平(≤3000相对>3000pg/ml)和egfr定义的肾功能(≥45相对<45ml/min/1.73m2)进行分级。
血浆pk和血清/血浆pd的样品将在poppk-pd子研究中收集。
整个研究期间将收集有关ae和伴随用药的信息。研究的安全性和实施办法将由独立的数据监控委员会(dmc)进行监控。
图45显示了试验设计的概要。
本研究的a部分将通过评估ag10和安慰剂组在治疗12个月后6分钟步行试验(6mwt)中与基线相比的变化差异,确定ag10在治疗有症状的转甲状腺素淀粉样心肌病(atr-cm)的受试者中的功效。
本研究的b部分将通过评估ag10和安慰剂组经30个月全因死亡率和心血管(cv)相关住院的累积频率的联合终点上的差异,确定ag10在治疗有症状的atr-cm的受试者中的功效。
受试者群体和分级
在本研究中,约510名年龄≥18岁且≤90岁且患有慢性、稳定、有症状的attr-cm(nyhai-iii级)的男性和女性,将按2:1的比例进行随机分组(340名受试者接受活性治疗,170名接受相匹配的安慰剂)。根据受试者是否患有attrm-cm或attrwt-cm将受试者随机分组,目标最低为20%患有attrm-cm的受试者。将通过基因分型尽一切努力来确认ttr的状态(野生型或变体)。在特殊情况下(当受试者拒绝进行基因检测时),可以要求医学监督局或指定人员批准在没有记录基因分型的情况下招募受试者。如果批准,并且该受试者已纳入研究,则此类ttr状态未知的受试者被划分为“野生型ttr”阶层。在筛选时,受试者还将根据nt-bnp前体水平(≤3000相对>3000pg/ml)和egfr定义的肾功能(≥45相对<45ml/min/1.73m2)进行分级。
治疗持续时间
除非耐受性不佳,否则将使用研究药物(ag10或安慰剂)治疗受试者30个月。完成30个月治疗的合格受试者可根据研究者的判断继续ole以接受ag10。
a部分,b部分和ole将分别报告。在将完成治疗后最终访视的所有受试者的数据包括在最终数据库中并且完成了该试验的最终报告之后,该试验完成。
施用治疗
符合资格标准的受试者将以2:1方式(ag10:安慰剂)随机分组,以双盲方式接受以下治疗方案:
·口服800mgag10bid(两片400mgag10片剂,bid)
·口服匹配的安慰剂bid(两片匹配的安慰剂片剂,bid)
如果研究者根据受试者的ae报告(可能表明研究药物不具有良好耐受性)确定需要调整剂量,则可将盲法剂量减至施用400mgag10或匹配的安慰剂一天两次(bid)。这将通过让研究人员指示受试者一天两次服用一片研究药物而非两片来实现。任何剂量调整将记录在数据库中。
违禁药物
1.在研究过程中,禁止将帕替司南(patisiran)、伊诺特森(inotersen)、他法米迪[参见以下说明]或任何其他研究药物用于治疗attr-cm。
2.禁止使用二氟尼柳、多西环素、用于attr-cm未经证实的疗法的天然产物或衍生物(例如绿茶提取物、牛磺熊去氧胆酸[tudca]/熊去氧胆酸)。
3.禁止使用钙通道阻滞剂(例如维拉帕米、地尔硫卓)或洋地黄。
注意:如果在参与本研究期间,他法米迪变为可商购,并且受试者可以使用它,那么如果受试者已经完成了至少24个月的盲法研究治疗,则允许他们开始使用他法米迪作为伴随药物进行治疗。
研究程序
评估时间表
下面提供了整个研究过程中要执行的步骤的说明。
筛选(第-35天到第-1天)
在首次服用imp之前的35天内进行筛选。筛选时将执行以下过程:
·知情同意书管理
·查看纳入/排除标准以确认受试者符合条件
·诊断确认委员会所需的原始文件的提交应在筛选期间尽早完成,并且必须包括:
1.心内膜活检报告;或
2.阳性99mtc-焦磷酸酯或-双膦酸酯扫描的平面图像,
和不包括al淀粉样变性诊断的临床实验室证据(基于血清和/或尿液两者的免疫固定电泳(ife),和无血清轻链(sflc)分析);
注意:患有并发意义不明的单克隆丙种球蛋白病(mgus)的受试者可能需要通过心内膜心肌活检和质谱分析来确认attr-cm的诊断。
·医疗和手术史评估
·nyha等级评估
·身体检查,包括体重和身高测量
·生命体征评估
·12导联静息心电图(ecg)
·静息经胸超声心动图(echo),如果没有根据超声心动图或cmr将lv壁(室间隔或lv后壁)厚度记录在病史中
·六分钟步行测试(6mwt),两次评估间隔>24小时至≤2周
·血液样本采集,用于血液学、血清化学(包括循环生物标志物)、尿液分析
·血液样本采集,用于血清和血浆探索性试验
·尿液妊娠试验,仅针对有生育能力的女性受试者
·事先药物使用评估
治疗天数
下面按研究天数列出研究程序,每天最好按以下列出的顺序执行。
第1天及每3个月(±7天)
这些评估将在第1天和第3、6、9、15、18、21、24和27个月进行:
·查看纳入/排除标准以确认受试者是否合格(第1天)
·将受试者随机分配至治疗组并分配随机编号(第1天)
·nyha等级评估
·身体检查,包括体重测量
·生命体征评估
·12导联静息ecg
·堪萨斯城心肌病问卷(kccq)
·euroqol-5维度(eq-5d-5l)
·第6、9、18、24个月的6分钟步行测试(6mwt)
·血液样本采集,用于血液学、血清化学(包括循环生物标志物)、尿液分析
·尿液妊娠试验,仅针对有生育能力的女性受试者
·pd血样采集,用于poppk-pd子研究中分析ttr稳定性(给药前)
·poppk-pd子研究中的pk血液样本采集(给药前)
·前白蛋白血样采集(给药前)
·在指定见证人(即现场人员)下分配/收集和施用imp
·伴随用药评估
·ae/生命状态评估/住院判定
·imp顺应性评估(除第1天外的所有随访)
第28天(±3天)
·nyha等级评估
·身体检查,包括体重测量
·生命体征评估
·服药前和服药后1小时的12导联静息ecg
·血液样本采集,用于血液学、血清化学(包括循环生物标志物)、尿液分析
·尿液妊娠试验,仅针对有生育能力的女性受试者
·pd血样采集,用于在poppk-pd子研究中分析ttr稳定性(给药前和给药后1小时)
·poppk-pd子研究中的pk血液样本采集(给药前和给药后1小时)
·前白蛋白血样采集(给药前)
·在指定见证人(即现场人员)下分配/收集和施用imp
·伴随用药评估
·ae/生命状态评估/住院判定
·imp顺应性评估
第12个月(±7天)
·nyha等级评估
·身体检查,包括体重测量
·生命体征评估
·12导联静息ecg
·堪萨斯城心肌病问卷(kccq)
·euroqol-5维度(eq-5d-5l)
·六分钟步行测试(6mwt)
·血液样本采集,用于血液学、血清化学(包括循环生物标志物)、尿液分析
·尿液妊娠试验,仅针对有生育能力的女性受试者
·pd血样采集,用于在poppk-pd子研究中分析ttr稳定性(给药前)
·poppk-pd子研究中的pk血液样本采集(给药前)
·前白蛋白血样采集(给药前)
·在指定见证人(即现场人员)下分配/收集和施用imp
·伴随用药评估
·ae/生命状态评估/住院判定
·imp顺应性评估
每月电话联系(±7天)
这些电话联系发生在第2、4、5、7、8、10、11、13、14、16、17、19、20、22、23、25、26、28和29个月:
·伴随用药评估
·ae/生命状态评估/住院确定
·imp顺应性评估
如果受试者停止研究药物和研究评估,则必须尽一切努力通过完成每月的生命状态联系以继续跟踪受试者的研究过程,或直到撤回同意书。
第30个月(±7天)和开放标签扩展(ole)开始
完成30个月的双盲治疗期的受试者将继续在ole中接受ag10。
·nyha等级评估
·身体检查,包括体重测量
·生命体征评估
·12导联静息ecg
·堪萨斯城心肌病问卷(kccq)
·euroqol-5维度(eq-5d-5l)
·六分钟步行测试(6mwt)
·血液样本采集,用于血液学、血清化学(包括循环生物标志物)、尿液分析
·尿液妊娠试验,仅针对有生育能力的女性受试者
·pd血样采集,用于在poppk-pd子研究中分析ttr稳定性(给药前)
·poppk-pd子研究中的pk血液样本采集(给药前)
·前白蛋白血样采集(给药前)
·在指定的见证人(即现场人员)下分配/收集和施用imp
·伴随用药评估
·ae/生命状态评估/住院判定
·imp顺应性评估
开放标签扩展开始1个月后(±3天)
·nyha等级评估
·身体检查,包括体重测量
·生命体征评估
·12导联静息ecg
·血液样本采集,用于血液学、血清化学(包括循环生物标志物)、尿液分析
·尿液妊娠试验,仅针对有生育能力的女性受试者
·pd血样采集,用于在poppk-pd子研究中分析ttr稳定性(给药前)
·poppk-pd子研究中的pk血液样本采集(给药前)
·前白蛋白血样采集(给药前)
·在指定的见证人(即现场人员)下分配/收集和施用imp
·伴随用药评估
·ae/生命状态评估/住院判定
·imp顺应性评估
开放标签扩展开始后每3个月(±7天)
·nyha等级评估
·身体检查,包括体重测量
·生命体征评估
·12导联静息ecg
·堪萨斯城心肌病问卷(kccq)
·euroqol-5维度(eq-5d-5l)
·每6个月进行六分钟步行测试(6mwt)
·血液样本采集,用于血液学、血清化学(包括循环生物标志物)、尿液分析
·尿液妊娠试验,仅针对有生育能力的女性受试者
·pd血样采集,用于在poppk-pd子研究中分析ttr稳定性(给药前)
·poppk-pd子研究中的pk血液样本采集(给药前)
·前白蛋白血样采集(给药前)
·在指定的见证人(即现场人员)下分配/收集和施用imp
·伴随用药评估
·ae/生命状态评估/住院判定
·imp顺应性评估
药物浓度测量
pk抽血时间表
在参与地点处在受试者亚组中,将在以下时间收集pk样品以确定ag10血浆浓度:
·第1天,每3个月进行研究问诊:给药前
·第28天:给药前和给药后1小时
·ole开始后1个月以及ole的每3个月:给药前
·et
pd抽血时间表
在参与地点处在受试者亚组中,ag10的pd特性将通过已建立的ttr稳定性分析(包括荧光探针排阻(fpe)分析和蛋白质印迹)进行评估。将在以下时间进行采样以执行这些pd检测:
·第1天,和每3个月进行研究问诊:给药前
·第28天:给药前和给药后1小时
·ole开始后1个月以及ole的每3个月:给药前
·et
前白蛋白血液取样程序
将在以下时间取样以测量前白蛋白的浓度:
·第1天,第28天以及每3个月进行研究问诊:给药前
·ole开始后1个月以及ole的每3个月:给药前
·et
六分钟步行测试(6mwt)
在随机分组之前,两次6mwt将间隔>24小时至≤2周进行。步行距离必须≥150米,并且在不同的日期进行两次连续测试时,步行差距必须在15%以内。如果2个测试结果不在15%之内,则应在其中一个6mwt的24小时至2周内重复进行一次额外的6mwt。如果最后一次尝试仍不在一个6mwt的15%之内,则该受试者将不具备参加资格。
在必要时应在访问中在完成kccq和eq-5d-5l之后进行6mwt。
使用柏格量表(borgscale)的6mwt将根据美国胸科学会的指南进行。学习程序手册中提供了有关6mwt程序的完整详细信息。
堪萨斯城心肌病问卷(kccq)
kccq是一项23项问卷,旨在测量心力衰竭患者的健康状况和与健康相关的生活质量。项目包括心力衰竭症状、对身体和社会功能的影响以及他们的心力衰竭如何影响生活质量(qol)。应由受试者在服药前完成。研究程序手册中提供了完整的详细信息。
euroqol-5维度(eq-5d-5l)
eq-5d-5l是一种简短的自我管理的通用健康状况工具,大约需要5分钟完成,应在kccq完成后进行。该工具包括两个部分。在第一部分中,要求受访者从5个维度(移动性、自我护理、日常活动、疼痛或不适、焦虑或抑郁)中评估其当前的健康状况,每个维度都具有五个功能级别(1-没有问题,2-轻微问题,3-中等问题,4-严重问题和5-极端问题)。第二部分是被调查者在视觉模拟量表(eqvas)上对当前健康状况的自我评估,其端点标记为“最佳可想象的健康状态”(得分为100)和“最差可想象的健康状态”(得分为0)。来自5个维度的分数可用于计算单个索引值,也称为效用分数。研究程序手册中提供了完整的细节给药和评分。
临床实验室测定
将收集用于临床实验室测试的血液和尿液样本。在筛选时,研究人员将评估实验室提供的参考范围以外的任何值的临床意义,并将被判定为具有临床意义的异常的受试者将被排除在研究范围之外。
将进行以下临床实验室测试:
生命体征
研究中心的工作人员将在休息5分钟后评估服药前后的生命体征。任何被认为具有临床意义的异常生命体征(即与症状相关和/或需要医学干预)都将记录为ae。
心电图
将评估标准的12导联ecg。在服药前休息5分钟后,在仰卧位进行心电图检查。在第28天给药1小时后进行ecg,直到收集和分析了有关pk-pd与qtc关系的其他1期数据为止。根据pk-pd数据的结果,可能不再需要在第28天进行给药后1小时的心电图,以减轻受试者的负担。将通过常规交流将所有更改告知所有研究人员。
研究人员或合格的辅助研究人员将复查所有ecg解释和间隔持续时间测量的临床意义。任何被认为具有临床意义(即与症状相关和/或需要医学干预)的ecg解释都将报告为ae。
体格检查
受试者将接受包括体重和身高测量在内的完整体格检查(pe),由医生或经过适当培训的保健专业人员完成。任何被认为具有临床意义的异常体格检查发现(即与症状相关和/或需要医学干预)都将记录为ae。
cv相关住院的定义
心血管相关住院是指非选择性地入院接受急性护理环境进行医疗治疗,这导致至少需要住院24小时(如果入院/出院时间不可用,则需要更改日期),或如果出院诊断和干预措施表明住院目的是静脉利尿剂疗法以治疗失代偿性心力衰竭,则住院时间少于24小时。研究人员有责任确保收集并记录潜在的研究终点,包括入院和出院日期;提供研究人员评估住院是否是cv相关;并提交导致死亡或住院的所有ae的不良事件通知。
实施例10:3期临床研究–attr-pn
这项前瞻性、随机、多中心、平行组研究将评估与安慰剂相比在有症状的有attr-pn的受试者中ag10的安全性和有效性。筛选和随机分组后将进行18个月的双盲、安慰剂对照的治疗期。
符合条件的受试者将以1:1的比例随机分配为口服bid给药的ag10800mg或匹配的安慰剂组。将根据筛查神经病变损害评分(nis)临界值(<30分且≥30分),并根据他们是否正在服用他法米迪(
整个研究期间将收集有关ae和伴随用药的信息。研究的安全性和实施办法将由独立的数据监控委员会(dmc)进行监控。
图46显示了试验设计的概要。
受试者群体和分级
需要有阳性基因型的文档确认已建立的attr-pn诊断。选择关键的纳入标准来确定一个受试者群体,该人群的疾病进展程度足够晚期以在安慰剂组中显示病情进展,但尚未晚到排除疾病状态变化的检测(例如,pnd评分≤iiia和karnofsky表现评分≥60%)。由于许多attr-pn患者也有心脏受累,考虑到与该程度的心肌病相关的高死亡率,将具有nyhaiv级症状的受试者排除在外。为了更好地阐明可归因于ag10的功效和安全性,排除了调节转甲状腺素蛋白生产或稳定性的其他疗法的同时使用(他法米迪20mg/d可能除外,如可用)。
神经病变损害评分(nis)是一种相对容易实施的神经系统评估,它使用肌肉、反射和感觉模式以及特定部位的标准组来代表临床损伤(虚弱、反射减弱和感觉丧失)的总和。它以0到244的等级计算,得分越高,表明疾病越严重。由于已表明nis与疾病严重程度和预后的其他衡量指标相关,并且由于有大量attr-pn患者组报道中位数约为30,因此筛查时将nis评分分级(<30和≥30)包括在内,以减轻各治疗组之间神经病变严重程度的潜在失衡(adams2015)。受试者还将根据他们是否服用他法米迪20mg/天,如在它可用的某些国家或地区用于attr-pn治疗所指示的,进行分级。
治疗持续时间
受试者将使用研究药物(ag10或安慰剂)治疗18个月。
根据在多个公开组中观察到的纵向评估,在mnis+7上,第18个月该受试者群体的进展速度预计为12.5至17分(adams2017,berk2013)。由于以mnis+7测量的安慰剂受试者的疾病进展是渐进的,因此在18个月的试验期内,预计至少12分的恶化(adams2017,berk2013)。由于预计ag10通过防止持续的淀粉样蛋白生成来阻止疾病进展,因此18个月可能长至足以检测mnis+7评分中具有临床意义的经安慰剂调整的变化。
施用治疗
受试者将以1:1的方式随机分组(ag10:安慰剂),以双盲方式接受以下治疗组:
·口服800mgag10bid(两片400mgag10片剂,bid)
·口服匹配的安慰剂bid(两片匹配的安慰剂片剂,bid)
如果研究者根据受试者的ae报告(可能表明研究药物不具有良好耐受性)确定需要调整剂量,则可将盲法剂量减至400mgag10或匹配的安慰剂一天两次(bid)。这将通过让研究人员指示受试者一天两次服用一片研究药物而非两片来实现。任何剂量调整将记录在数据库中。
违禁药物
1.在研究过程中,禁止将帕替司南、伊诺特森或任何其他批准的或研究性的药物用于治疗attr-pn(除他法米迪20mg剂量)。
2.在研究过程中,禁止使用未指定用于治疗attr的批准产品(例如,二氟尼柳、多西环素),或未经验证的用于attr治疗的天然产品或衍生物(例如,绿茶提取物、牛磺熊去氧胆酸[tudca]/熊去氧胆酸)。
研究程序
评估时间表
下面提供了整个研究过程中要执行的步骤的说明。
筛选(第-28天到第-1天)
在首次服用imp之前的28天内进行筛选。筛选时将执行以下过程:
·知情同意书管理
·查看纳入/排除标准以确认受试者符合条件
·医疗和手术史评估
·nyha等级评估
·卡氏评分(karnofskyperformancestatus)
·体格检查,包括mbmi
·生命体征评估
·12导联静息ecg
·pnd评分
·nis
·mnis+7
·10米步行测试(10mwt),两次评估间隔>24小时至<1周
·血液样本采集,用于血液学、血清化学(包括循环生物标志物)、尿液分析
·采集血液样本以进行血清和血浆探索性试验
·尿液妊娠试验,仅针对有生育能力的女性受试者
·事先药物使用评估
治疗天数
下面按研究天数列出研究程序,每天最好按以下列出的顺序执行。
第1天及每3个月(±7天)
这些评估将在第1天和第3、6、9、15个月进行:
·查看纳入/排除标准以确认受试者是否合格(第1天)
·将受试者随机分配至治疗组并分配随机编号(第1天)
·nyha等级评估
·体格检查,包括mbmi
·生命体征评估
·12导联静息ecg
·pnd评分
·戴克/兰金得分
·mnis+7
·诺福克qol-dn
·compass-31
·10米步行测试(10mwt)
·血液样本采集,用于血液学、血清化学(包括循环生物标志物)、尿液分析
·尿液妊娠试验,仅针对有生育能力的女性受试者
·pd血样采集以分析ttr稳定性(给药前)
·pk血液样本采集(给药前)
·前白蛋白血样采集(给药前)
·在指定的见证人(即现场人员)下分配/收集和施用imp
·伴随用药评估
·ae/生命状态评估
·imp顺应性评估(除第1天外的所有随访)
第18个月(±7天)
·nyha等级评估
·体格检查,包括mbmi
·生命体征评估
·12导联静息ecg
·pnd评分
·戴克/兰金得分
·mnis+7
·诺福克qol-dn
·compass-31
·10米步行测试(10mwt)
·血液样本采集,用于血液学、血清化学(包括循环生物标志物)、尿液分析
·尿液妊娠试验,仅针对有生育能力的女性受试者
·pd血样采集以分析ttr稳定性(给药前)
·pk血液样本采集(给药前)
·前白蛋白血样采集(给药前)
·在指定的见证人(即现场人员)下分配/收集和施用imp
·伴随用药评估
·ae/生命状态评估
·imp顺应性评估
每月电话联系(±7天)
这些电话联系发生在第2、4、5、7、8、10、11、13、14、16、17个月:
·伴随用药评估
·ae/生命状态评估
·imp顺应性评估
如果受试者停止研究药物和研究评估,则必须尽一切努力通过完成每月的生命状态联系以继续跟踪受试者的研究过程,或直到撤回同意书。
药物浓度测量
pk抽血时间表
在参与地点在受试者亚组中,将在以下时间收集pk样品以确定ag10血浆浓度:
·第1天,和每3个月进行研究问诊:给药前
pd抽血时间表
在参与地点在受试者亚组中,ag10的pd特性将通过已建立的ttr稳定性分析(包括荧光探针排阻(fpe)分析和蛋白质印迹)进行评估。将在以下时间进行采样以执行这些pd检测:
·第1天,和每3个月进行研究问诊:给药前
前白蛋白血采样程序
将在以下时间取样以测量前白蛋白的浓度:
·第1天、第28天以及每3个月进行研究问诊:给药前
评估
将完成以下评估:
·10米步行测试(10mwt)
·神经病变损害评分(nis)
·改良的神经系统损害评分(mnis+7)
·compass-31
·戴克/兰金得分
·营养状况(根据mbmi计算)
10米步行测试(10mwt)
10mwt是一项性能指标,用于评估短距离内以米/秒为单位的步行速度。它用于确定功能性活动性,步态和前庭功能。在随机分组之前,两次10mwt将间隔>24小时至<1周进行。
神经病变损害评分(nis)
nis是一种神经学评估,代表了使用肌肉、反射和感觉方式以及特定部位的标准组的临床损伤(虚弱、反射降低和感觉丧失)的总和。
改良的神经系统损害评分(mnis+7)
mnis+7神经测试是一种综合评分,可部分评估肌肉无力、感觉丧失和肌肉伸展反射降低。
综合自主神经症状评分-31(compass-31)
使用compass-31量化ttr淀粉样变性对每个受试者的自主神经症状的影响。
戴克/兰金得分
生活质量将通过戴克/兰金得分进行评估。
营养状况
营养状况将根据mbmi的变化进行评估。
临床实验室测定
将收集用于临床实验室测试的血液和尿液样本。在筛选时,研究人员将评估实验室提供的参考范围以外的任何值的临床意义,并将被判定为临床显著异常的受试者排除在研究范围之外。
将进行以下临床实验室测试:
生命体征
研究中心的工作人员将在休息5分钟后评估服药前后的生命体征。任何被认为具有临床意义的异常生命体征(即与症状相关和/或需要医学干预)都将记录为ae。
心电图
将评估标准的12导联心电图。在服药前休息5分钟后,在仰卧位进行心电图检查。研究人员或合格的辅助研究人员将复查所有ecg解释和间隔持续时间测量的临床意义。任何被认为具有临床意义(即与症状相关和/或需要医学干预)的ecg解释都将报告为ae。
体格检查
受试者将接受包括mbmi在内的完整体格检查(pe),该检查将由医生或经过适当培训的保健专业人员完成。任何被认为具有临床意义的异常体格检查发现(即与症状相关和/或需要医学干预)都将记录为ae。
卡氏评分
多发性神经病残疾评分(pnd评分)
尽管出于清楚理解的目的已经通过图示和实施例的方式详细地描述了前述发明,但是本领域技术人员将理解,可以在所附权利要求的范围内进行某些改变和修改。另外,本文提供的每篇参考文献通过引用整体并入本文,其程度与每个参考文献通过引用单独并入的程度相同。在本申请与本文提供的参考文献之间存在冲突的情况下,以本申请为准。
序列表
<110>文涵治疗有限公司
u·辛哈
s·拉奥
<120>用ag10治疗ttr淀粉样变性的方法
<130>051418-505001wo
<140>未分配
<141>2019-03-23
<150>62/647,411
<151>2018-03-23
<150>62/765,096
<151>2018-08-17
<150>62/731,629
<151>2018-09-14
<150>62/758,235
<151>2018-11-09
<150>62/810,651
<151>2019-02-26
<160>1
<170>siposequencelisting1.0
<210>1
<211>127
<212>prt
<213>智人(homosapiens)
<400>1
glyprothrglythrglygluserlyscysproleumetvallysval
151015
leuaspalavalargglyserproalaileasnvalalavalhisval
202530
phearglysalaalaaspaspthrtrpgluprophealaserglylys
354045
thrsergluserglygluleuhisglyleuthrthrgluglugluphe
505560
valgluglyiletyrlysvalgluileaspthrlyssertyrtrplys
65707580
alaleuglyileserprophehisgluhisalagluvalvalphethr
859095
alaasnaspserglyproargargtyrthrilealaalaleuleuser
100105110
protyrsertyrserthrthralavalvalthrasnprolysglu
115120125
- 还没有人留言评论。精彩留言会获得点赞!