包含结合PCSK9的分子的组合物和使用方法与流程
包含结合pcsk9的分子的组合物和使用方法
[0001]
相关申请
[0002]
本申请要求2018年5月16日提交的美国临时申请第62/672,187号的权益和优先权,所述申请以引用方式整体并入本文。
技术领域
[0003]
本公开涉及结合前蛋白转化酶枯草杆菌蛋白酶kexin-9(pcsk9)的基于纤连蛋白的支架结构域蛋白,以及其药物组合物和使用方法。
背景技术:
[0004]
前蛋白转化酶枯草杆菌蛋白酶/kexin 9型(pcsk9)是人类1号染色体上由pcsk9基因编码的酶。pcsk9与低密度脂蛋白颗粒(ldl)的受体结合。肝脏和其他细胞膜上的ldl受体(ldlr)结合并启动ldl颗粒从细胞外液进入细胞的内吞作用,从而降低循环ldl颗粒的浓度。如果pcsk9被阻断,则更多的ldlr被回收利用并存在于细胞表面上,由此从细胞外液中去除ldl颗粒。因此,阻断pcsk9可减低血液ldl颗粒浓度。
[0005]
当他汀类药物和其他药物疗效不佳或耐受性差时,单克隆抗体pcsk9抑制剂阿利库单抗(alirocumab)和依伏库单抗(evolocumab)被批准每两周或每月皮下注射或输注一次,用于减低ldl颗粒浓度。对于每月一次的注射,需要几毫升的药品才能达到所需剂量。尽管具有较高浓度的活性剂的配制品可提供更理想的给药排程和体积,但是这些配制品受到溶解度限制、粘度增加和生物制剂的不稳定性(包括聚集和微粒形成的倾向)的阻碍。carpenter jf等人,overlooking subvisible particles in therapeutic protein products: gaps that may compromise product quality,journal of pharmaceutical sciences第98卷,第4期(2008)。
[0006]
需要靶向pcsk9并且有潜力实现更理想的给药排程、更小的体积和/或功效方面的改善,同时维持总体上安全且良好耐受的特性的药物组合物。
技术实现要素:
[0007]
本公开提供了包含纤连蛋白支架蛋白的组合物,所述纤连蛋白支架蛋白以高亲和力结合前蛋白转化酶枯草杆菌蛋白酶kexin 9型(pcsk9),并且可以高浓度稳定地配制以实现最大的生物效应和更方便的给药排程、给药体积以及对患者友好的递送装置。结合pcsk9的融合蛋白包含结合pcsk9的基序和人血清白蛋白(hsa)的氨基酸序列。结合pcsk9的基序和hsa氨基酸序列可表达为基因融合体或者是化学缀合的。
[0008]
本文所述的结合pcsk9的基序基于adnectin,这是一种源自人第10纤连蛋白iii型结构域(10fn3)的蛋白质家族,经工程改造可实现高亲和力靶标结合。根据本公开,以高浓度稳定地配制结合pcsk9的融合蛋白,以实现最大的生物活性以及方便的给药排程和体积。组合物中结合pcsk9的融合蛋白的浓度为至少100mg/ml。在一些实施方案中,组合物中结合pcsk9的融合蛋白的浓度为至少约200mg/ml。在一些实施方案中,组合物中结合pcsk9的融
合蛋白的浓度为至少约250mg/ml,或至少约275mg/ml,或至少约300mg/ml,或至少约350mg/l。在一些实施方案中,以约275mg至约325mg(例如,约300mg)的单位剂量施用结合pcsk9的融合蛋白。
[0009]
如本文所述,结合pcsk9的基序包含(seq id no:1)的氨基酸序列或其变体或者由其组成。结合pcsk9的基序以浓度依赖性方式以亚纳摩尔亲和力与人pcsk9结合。结合pcsk9的基序与人血清白蛋白(hsa)氨基酸序列化学缀合或c端融合。在各种实施方案中,结合pcsk9的基序在c端处与hsa氨基酸序列融合,并且可在结合pcsk9的基序与hsa氨基酸序列之间包含氨基酸的连接序列。
[0010]
结合pcsk9的融合蛋白可以高浓度以溶液形式稳定地配制。在常规的长期储存条件下以及在加速和加压条件下的短期储存下,所述制剂没有表现出明显的降解或微粒形成。(除了所述的活性剂之外)示例性制剂包含以下或基本上由以下组成:l-组氨酸、l-组氨酸单盐酸盐、氯化钠和任选的聚山梨醇酯80。
[0011]
本公开的药物组合物可方便地以单位剂型存在,所述单位剂型每剂量包含预定量的本公开的活性剂。在一些实施方案中,单位剂量的体积不超过约1.5ml,或体积不超过约1ml。在一些实施方案中,单位剂量的体积不超过0.8ml,或体积不超过0.7ml。在其他实施方案中,以微剂量,例如以小于0.5ml、或小于约0.25ml、或小于约0.15ml的体积施用所述组合物。在各种实施方案中,以包含约20至约450mg的结合pcsk9的融合蛋白的单位剂量递送所述组合物。例如,在一些实施方案中,以每周微剂量(例如,以小于约0.25ml或小于约0.15ml的体积)施用20至约75mg的剂量。在其他实施方案中,约每两周、每月或每隔一个月以约0.7至1.5ml范围内的体积施用约200至约450mg的剂量。
[0012]
所述组合物或制剂适于通过皮下、肌内、皮内或静脉内施用来施用。高浓度的制剂允许较低频率的给药排程和较低的给药体积适于皮下施用。如本文所证明的,在相对低浓度的结合pcsk9的融合蛋白下实现了最大的pcsk9抑制,并且更高的浓度实现了更长的抑制持续时间。在一些实施方案中,受试者约每周一次,每2周一次,或约每3周一次,或约每4周一次(例如,约每月一次),或约每6周一次,或约每8周一次(约每2个月一次)接受单位剂量的所述组合物。
[0013]
在一些实施方案中,受试者约每周或约每两周接受微剂量,如约50至约250μl的体积的所述组合物。
[0014]
可施用所述组合物以治疗人受试者中的pcsk9相关病症。在一些实施方案中,患者需要降低ldl(例如,ldl-c)。在一些实施方案中,受试者可表现出胆固醇相关疾病,如高胆固醇血症和/或动脉粥样硬化。在一些实施方案中,受试者患有家族性高胆固醇血症。在一些实施方案中,受试者患有心血管疾病(例如,动脉粥样硬化性冠心病)或处于心血管疾病的高风险中。
[0015]
在一些实施方案中,将所述组合物与他汀类药物疗法或另一种口服降脂疗法一起施用,或者在一些实施方案中,提供所述组合物作为高胆固醇血症的唯一疗法,即,不进行口服降脂疗法(例如,他汀类药物疗法)。
附图说明
[0016]
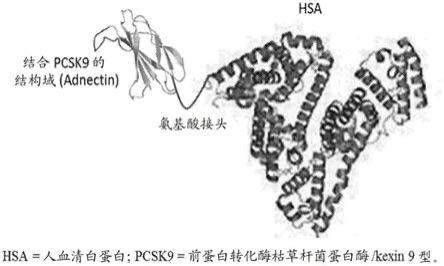
图1示出了结合pcsk9的融合蛋白的结构。
[0017]
图2a示出了在非人灵长类动物(nhp)和人中的pcsk9药代动力学和药效动力学(pk-pd)模型。lib-003是指结合pcsk9的融合蛋白。
[0018]
图2b示出了用于预测pcsk9靶向对ldlc的作用的gadkar模型。gadkar k等人,a mechanistic systems pharmacology model for prediction of ldl cholesterol lowering by pcsk9 antagonism in human dyslipidemic populations,cpt pharmacometrics syst.pharmacol.2014;3(11)。
具体实施方式
[0019]
本公开提供了包含纤连蛋白支架融合蛋白的组合物,所述纤连蛋白支架融合蛋白以高亲和力结合前蛋白转化酶枯草杆菌蛋白酶kexin-9(pcsk9),并且可以高浓度稳定地配制以实现最大的生物效应和更方便的给药排程和给药体积,并使用对患者友好的递送装置(如注射器或自动注射器)来递送。结合pcsk9的融合蛋白包含结合pcsk9的基序和在c端处的编码人血清白蛋白(hsa)的氨基酸序列。
[0020]
前蛋白转化酶枯草杆菌蛋白酶/kexin 9型是主要由肝脏分泌的循环蛋白,它在肝ldlr的回收利用中起重要作用,并已被确认为可降低ldl-c的有效药物靶标。ldlr是从循环中清除ldl-胆固醇(ldl-c)的主要途径。血浆pcsk9与ldl-c一同结合至肝ldlr,ldl-c靶向受体以便进行内吞和降解,由此降低了ldlr从循环中清除ldl-c的有效性。通过抑制pcsk9与ldlr的结合,可防止ldlr降解,增加ldlr回收利用,提高ldl-c清除率并降低循环ldl-c水平。
[0021]
本文所述的结合pcsk9的基序基于“adnectin”,这是一种源自人第10纤连蛋白iii型结构域的蛋白质家族,经工程改造可实现高亲和力靶标结合。“adnectin”是小的(≤12kda)紧凑的蛋白质,与免疫球蛋白无序列同源性,但具有β-折叠结构,具有类似于抗体可变区的多样化环。adnectin不具有二硫键并且不是糖基化的,表现出高热稳定性和单体溶液行为,并且可使用细菌、酵母或哺乳动物表达系统有效地产生。通过修改可变的环序列和长度同时保持支架残基基本上恒定,可在维持结构稳定性的同时实现亚纳摩尔级靶标结合亲和力。鉴于其大小,adnectin会被肾脏迅速过滤,因此需要进行药物动力学(pk)增强修饰以实现体内应用。示例性的结合pcsk9的基序(adnectin)公开于美国专利8,420,098;9,234,027;和9,856,309中,所述专利各自以引用方式整体并入本文。
[0022]
根据本公开,以高浓度稳定地配制结合pcsk9的融合蛋白,以实现最大的生物活性以及方便的给药排程和体积。组合物中结合pcsk9的融合蛋白的浓度为至少100mg/ml。在一些实施方案中,组合物中结合pcsk9的融合蛋白的浓度为至少约150mg/ml,或者在一些实施方案中为至少约175mg/ml,或至少约200mg/ml,或至少约225mg/ml。在一些实施方案中,组合物中结合pcsk9的融合蛋白的浓度为至少约250mg/ml,或至少约275mg/ml,或至少约300mg/ml,或至少约350mg/ml。在一些实施方案中,组合物中结合pcsk9的融合蛋白的浓度为约250mg/ml至约350mg/ml。在一些实施方案中,组合物中结合pcsk9的融合蛋白的浓度为约250mg/ml或约300mg/ml。
[0023]
结合pcsk9的基序包含(seq id no:1)的氨基酸序列或由其组成:
[0024]
vsdvprdlevvaatptslliswdapaegygyyritygetggnspvqeftvpvskgtatisglkpgvdytitvyavefdfpgagyyhrpisinyrte。
[0025]
结合pcsk9的环加下划线显示。在一些实施方案中,结合pcsk9的基序是seq id no:1的变体,相对于seq id no:1具有一至五个氨基酸取代、缺失或插入。在一些实施方案中,在结合环之外进行氨基酸改变。在一些实施方案中,在结合环内进行1个、2个或3个氨基酸改变。
[0026]
结合pcsk9的基序被设计为特异性地靶向pcsk9,同时基本上维持野生型(wt)
10
fn3序列,以将固有免疫原性减到最低。参见美国专利第8,420,098号,所述专利以引用方式整体并入本文。结合pcsk9的基序以浓度依赖性方式以亚纳摩尔亲和力与人pcsk9结合。
[0027]
在各种实施方案中,结合pcsk9的基序与人血清白蛋白(hsa)氨基酸序列在c端融合。在一些实施方案中,hsa包含与seq id no:2的氨基酸序列具有至少80%同一性、或至少85%同一性、或至少90%同一性、或至少95%同一性、或至少98%同一性或至少99%同一性的序列。例如,hsa氨基酸序列相对于seq id no:2可包含一至十个或一至五个独立地选自氨基酸取代、缺失和插入的修饰。在一些实施方案中,hsa氨基酸序列在对应于seq id no:2的位置34的位置处包含丙氨酸残基,如seq id no:2所示。在各种实施方案中,hsa氨基酸序列的长度为至少500个氨基酸。
[0028]
在一些实施方案中,使用任何已知的化学缀合方法将结合pcsk9的基序和hsa氨基酸序列化学缀合。
[0029]
hsa是由螺旋簇组成的多结构域蛋白,包含17对二硫桥;在天然hsa中只有一个半胱氨酸残基cys34作为游离巯基存在。在一些实施方案中,此cys被结合pcsk9的融合蛋白中的丙氨酸残基取代。hsa部分用于延长结合pcsk9的融合蛋白的循环半衰期。在各种实施方案中,包含结合pcsk9的基序和hsa氨基酸序列的结合pcsk9的融合蛋白具有约77kda的分子量。
[0030]
在一些实施方案中,hsa氨基酸序列是在美国专利9,493,545、美国专利9,821,039、美国专利9,944,691或美国2014/0315817中描述的变体,所述专利各自以引用方式整体并入本文。
[0031]
在一些实施方案中,结合pcsk9的基序和hsa氨基酸序列通过基因融合例如与hsa氨基酸序列在分子的c端处连接。短氨基酸接头可连接结合pcsk9的结构域和hsa氨基酸序列。例如,接头可包含2至20个氨基酸,或者在一些实施方案中包含4至10个氨基酸。在一些实施方案中,结合pcsk9的基序和hsa氨基酸序列经由6个氨基酸的接头连接。接头可主要由丝氨酸、甘氨酸、苏氨酸和丙氨酸氨基酸组成。例如,接头可以是丝氨酸/甘氨酸接头。在一些实施方案中,接头包含氨基酸序列gsgsgs或由其组成。
[0032]
结合pcsk9的融合蛋白可以高浓度以溶液形式稳定地配制。例如,在长期储存条件,如约5℃(例如2℃至8℃)的温度下或在环境条件下在短期储存条件下(例如在25
±
3℃下,1至6个月),所述配制品没有表现出明显的降解或微粒形成。结合pcsk9的融合蛋白被悬浮在适当的生理溶液(例如,盐水或其他药理学上可接受的溶剂或缓冲溶液)中,并且可任选地包含表面活性剂(例如,非离子表面活性剂)。在一些实施方案中,所述制剂包含缓冲剂、等渗剂、任选的表面活性剂和溶剂。
[0033]
药学上可接受的载体包括水、盐水、甘油。在一些实施方案中,所述制剂可包含不挥发性油、聚乙二醇、丙二醇或其他溶剂。在一些实施方案中,所述溶剂是水。
[0034]
所述制剂通常是缓冲溶液。如本文所用,“缓冲液”是指能够吸收一定量的酸或碱
而不经历ph的强烈变化的化学剂。示例性缓冲液包括柠檬酸盐缓冲液、磷酸盐缓冲液、乙酸盐缓冲液、琥珀酸盐缓冲液和碳酸氢盐缓冲液。在一些实施方案中,缓冲剂可包括l-组氨酸/l-组氨酸单盐酸盐。例如,当使用l-组氨酸/l-组氨酸单盐酸盐缓冲系统时,l-组氨酸/l-组氨酸单盐酸盐可以约1mg/ml至约10mg/ml,如约2mg/ml至约5mg/ml存在。在一些实施方案中,缓冲液将制剂的ph维持在约ph 5.5至约ph 7.2的范围内,如约ph 6.8(例如,ph 6.6至7.0)。在一些实施方案中,例如用盐酸和/或氢氧化钠调节制剂的ph。在一些实施方案中,l-组氨酸/l-组氨酸单盐酸盐的比例使得不需要进行ph调节。
[0035]
在各种实施方案中,可单独使用或组合使用的等渗剂包括右旋糖、蔗糖、甘油、海藻糖、甘露糖醇、山梨糖醇、精氨酸、氯化钠或氯化钾。在一些实施方案中,等渗剂包含氯化钠或由氯化钠组成。例如,所述制剂可包含约2至约20mg/ml的氯化钠(例如,6至12mg/ml氯化钠),或等于2至20mg/ml氯化钠(或6至12mg/ml氯化钠)的重量克分子渗透浓度的量的一种或多种等渗剂。
[0036]
在一些实施方案中,所述制剂包含表面活性剂,所述表面活性剂可充当增溶剂。在一些实施方案中,表面活性剂是非离子表面活性剂。示例性非离子表面活性剂包括聚山梨醇酯表面活性剂,如聚山梨醇酯20、40、60或80。例如,所述制剂可包含聚山梨醇酯80。也可单独或组合使用其他药学上可接受的非离子表面活性剂。在一些实施方案中,所述制剂不包含表面活性剂。
[0037]
示例性制剂包括柠檬酸盐缓冲液(例如,10-50mm,ph 5.6至6.0)、组氨酸缓冲液(例如,10mm至50mm,ph 6.0至7.0)或琥珀酸盐缓冲液(例如,10至50mm,ph 5.5至6.0)。在一些实施方案中,所述制剂包含选自精氨酸(例如,100至200mm)、nacl(例如,100至200mm)、山梨糖醇(例如,100至300mm)或蔗糖(例如,100至300mm)的赋形剂。在一些实施方案中,所述制剂包含表面活性剂,如聚山梨醇酯80(例如,0.01至0.5mg/ml)。在一些实施方案中,所述制剂不含有表面活性剂。
[0038]
在一些实施方案中,所述制剂还包含防腐剂,如苯酚、间甲酚或苯甲酸钠。
[0039]
在一些实施方案中,(除了所述的活性剂之外)结合pcsk9的融合蛋白制剂基本上由l-组氨酸、l-组氨酸单盐酸盐、氯化钠和聚山梨醇酯80组成。示例性实施方案在以下表1中显示:
[0040][0041]
本公开的药物组合物可方便地以单位剂型存在,所述单位剂型每剂量包含预定量的本公开的活性剂。
[0042]
在一些实施方案中,组合物包含在注射笔中。如“注射笔”的自动注射器是弹簧加载注射器,其被设计用于递送一定剂量的特定药物。通过设计,注射笔易于使用并且旨在由患者自行施用或由未经培训的人员施用。设计注射笔是为了克服与基于针头的药物递送装置的自行施用相关的犹豫。注射笔在注射之前使针尖保持隐蔽,并且具有被动安全机制以防止意外击发(注射)。注射深度可调节或固定,并且可结合针头护罩移除的功能。通过按下按钮,注射器针头会自动插入皮下组织中并递送药物。注射完成后,一些注射笔会出现视觉或听觉指示以便确认全剂量已被递送。
[0043]
在一些实施方案中,注射笔包含1至10个单位剂量或1至5个单位剂量。在一些实施方案中,单位剂量的体积不超过约1.5ml或约1ml(无论是否由注射笔包含或递送)。在一些实施方案中,单位剂量的体积不超过0.8ml,或体积不超过0.7ml。在一些实施方案中,注射笔递送微剂量,例如具有在约50μl至约500μl范围内的体积,或在约75μl至约250μl范围内的体积。在一些实施方案中,微剂量具有100至200μl的体积。在各种实施方案中,注射笔或用于皮下递送的其他装置提供具有30至约450mg的结合pcsk9的融合蛋白的剂量。在各种实施方案中,单位剂量为约50至约400mg、或约50至约300mg。在一些实施方案中,单位剂量为至少200mg、或至少250mg、或至少300mg。在一些实施方案中,单位剂量为约250mg至约350mg(例如,约300mg)。可基于所需的施用频率来调节每单位剂量递送的活性剂的量。例如,在一些实施方案中,以每周微剂量(例如,以小于约0.25ml或小于约0.15ml的体积)施用20至约75mg的剂量。在其他实施方案中,约每两周、每月或每隔一个月以约0.7至1.5ml范围内的体积施用约200至约450mg的剂量。在一些实施方案中,每四周通过皮下注射以约1.5ml或更小或约1ml或更小的体积施用约275mg至约350mg(例如,约300mg)的剂量。
[0044]
所述组合物或制剂适于通过皮下、肌内、皮内或静脉内施用来施用。制剂的高浓度、相对低的粘度和适当的重量克分子渗透浓度实现患者耐受、较低频率的给药排程和较
小的体积。如本文所证明的,在相对低浓度的结合pcsk9的融合蛋白下实现了最大的pcsk9抑制,并且更高的浓度实现了更长的抑制持续时间。在一些实施方案中,受试者约每1周一次(例如,通过每周施用一次微剂量),或约每2周一次,或约每3周一次,或约每4周一次(例如,约每月一次),或约每6周一次,或约每8周一次(约每2个月一次)接受单位剂量的组合物。
[0045]
可施用所述组合物以治疗人受试者中的pcsk9相关病症。pcsk9相关病症描述于美国专利8,420,098、9,238,027和9,856,306中,所述专利以引用方式整体并入本文。在一些实施方案中,患者需要降低ldl(例如,ldl-胆固醇)。在一些实施方案中,受试者可表现出胆固醇相关疾病,如高胆固醇血症和/或动脉粥样硬化。在各种实施方案中,受试者表现出选自脂质紊乱、高胆固醇血症、高脂蛋白血症、高脂血症、血脂异常、冠心病、动脉粥样硬化和糖尿病的疾患。在一些实施方案中,受试者患有家族性高胆固醇血症。在一些实施方案中,受试者患有心血管疾病(例如,动脉粥样硬化性冠心病)或处于心血管疾病的高风险中。
[0046]
高胆固醇血症是特征在于血清胆固醇升高的疾患。血清胆固醇水平升高影响相当大的一部分的人群,并且是动脉粥样硬化和心肌梗塞的重要危险因素。降低胆固醇的药物,如hmg-coa还原酶抑制剂(“他汀类药物”)通常按惯例施用给高胆固醇血症患者。“家族性高胆固醇血症”(fh)是一种遗传性病症,其特征在于血液和早期心血管疾病中的高胆固醇水平,特别是非常高的低密度脂蛋白(例如,ldl-胆固醇)水平。对于患有fh的个体,高胆固醇水平对常规胆固醇控制方法(如他汀类药物疗法)的反应较弱。
[0047]
fh是一种常染色体显性遗传性代谢病症,其特征在于ldl受体(ldl-r)基因或其他参与脂质调节的基因发生一种或多种突变,ldl-c显著升高和动脉粥样硬化早发。在一些实施方案中,高胆固醇血症是纯合子家族性高胆固醇血症或hofh,其是特征在于母系和父系ldl-r基因均突变的疾患。
[0048]
在一些实施方案中,受试者患有杂合fh。杂合fh通常用他汀类药物、胆汁酸螯合剂或其他降低胆固醇水平的降脂剂来治疗。
[0049]
在一些实施方案中,高胆固醇血症是多基因高胆固醇血症,其是特征在于胆固醇升高的疾患,是由多种遗传因素的影响所致。在某些实施方案中,饮食中摄入脂质可加剧多基因高胆固醇血症。
[0050]
在一些实施方案中,将所述组合物与他汀类药物疗法或其他口服降脂疗法一起施用。在这样的实施方案中,所述组合物将提供ldl-c的累加降低。在一些实施方案中,提供所述组合物作为高胆固醇血症的唯一疗法,即没有他汀类药物疗法或其他口服降脂疗法。例如,在这样的实施方案中,受试者可能具有他汀类药物不耐受性。当患者因在血液测试后出现副作用或指示肝脏功能或肌肉功能(肌酸激酶)的异常而无法继续使用他汀类药物时,发生“他汀类药物不耐受”。在一些实施方案中,他汀类药物不耐受可以是部分的(即,仅某些剂量的某些他汀类药物)或完全的(即,任何剂量的所有他汀类药物)。在一些实施方案中,他汀类药物不耐受导致肌肉酸痛、疼痛、虚弱或抽筋(即,肌痛);在多达15%的接受治疗的患者中发生。
[0051]
除非另有说明,否则如本文所用的术语“约”意指相关数值的
±
10%。
[0052]
现在将通过以下实施例描述本发明的实施方案。
[0053]
实施例
[0054]
实施例1:结合pcsk9的融合蛋白和pk-pd行为
[0055]
结合pcsk9的融合蛋白包含靶向前蛋白转化酶枯草杆菌蛋白酶/kexin 9型(pcsk9)的经修饰的纤连蛋白结构域(adnectin)和人血清白蛋白(图1)。融合蛋白的总分子量为大约77,000道尔顿。结合pcsk9的融合蛋白对人pcsk9的结合亲和力高,对食蟹猴pcsk9的亲和力弱>100倍。尽管在结合亲和力方面存在这种差异,但非人灵长类动物(nhp)被认为是测试结合pcsk9的融合蛋白的安全性和药理学的合适物种,因为在nhp中实现了最大程度的游离pcsk9抑制和最大程度的ldl-c降低。
[0056]
结合pcsk9的融合蛋白正在开发用于皮下(sc)施用,以便治疗例如高胆固醇血症,包括患有家族性高胆固醇血症或高胆固醇血症和动脉粥样硬化性冠心病(chd)的患者。结合pcsk9的融合蛋白的pk的两个主要决定因素是与其靶标pcsk9的相互作用,以及经由新生fc受体(fcrn)进行回收利用并最小化肾脏滤过的能力,从而降低结合pcsk9的融合蛋白的清除率并增加半衰期。在野生型小鼠中进行的研究无法解决这些决定因素对结合pcsk9的融合蛋白的pk的影响,因为结合pcsk9的融合蛋白不能有效地与小鼠pcsk9结合,并且hsa不会与小鼠fcrn相互作用。因此,食蟹猴为评估结合pcsk9的融合蛋白的pk-pd提供了最合适的体内模型,因为在该模型中与pcsk9的结合会导致游离pcsk9和ldl-c的下降;此外,hsa可经由nhp fcrn受体进行有效回收利用。
[0057]
使用hpcsk9转基因小鼠研究结合pcsk9的融合蛋白对hpcsk9的体内结合亲和力,并且还在hfcrn小鼠中评估经由hfcrn回收利用结合pcsk9的融合蛋白的能力。该人fcrn小鼠模型携带小鼠fcrn基因和在其天然人启动子的控制下表达人fcrnα链的转基因的无效突变。因此,这些小鼠充当用于评价单克隆抗体和人血清白蛋白的药代动力学的模型。
[0058]
对于抗pcsk9抗体,有大量数据集支持全身性游离pcsk9水平的抑制与血清ldl-c降低之间的关系。gadkark等人,a mechanistic systems pharmacology model for prediction of ldl cholesterol lowering by pcsk9 antagonism in human dyslipidemic populations,cptpharmacometrics syst.pharmacol.2014;3(11):1-9;squizzato a等人,pcsk9 inhibitors for treating dyslipidemia in patients at different cardiovascular risk:a systematic review and meta-analysis.intern.emerg.med.2017;七月:e1-11。参见图2b。为了估算实现期望的目标ldl-c降低水平所需的结合pcsk9的融合蛋白(lib003,seq id no:3)的人剂量,将上述研究的数据的异速比例纳入平移半机理模型中,所述模型包括pcsk9与ldl-c之间的已知关系。图2a。使用该模型构建了lib003和血清ldl-c的预期剂量效应关系。
[0059]
非人灵长类动物中的tk-pd
[0060]
在单剂量非glp剂量范围发现(drf)研究中在静脉内(iv)和皮下(sc)施用后(iv剂量10、30、100、200mg/kg;sc剂量200mg/kg)并且在glp毒性研究中在重复施用持续4周(iv剂量100mg/kg;sc剂量30和100mg/kg)、12周(sc剂量30和100mg/kg)或26周(sc剂量30和100mg/kg)后表征lib003的tk-pd行为。
[0061]
将lib003的tk测定构建为靶标捕获电化学发光测定,以hpcsk9作为捕获试剂,并以钌标记的兔抗hsa多克隆抗体作为检测试剂。在nhp中,所述测定测量总lib003(由于与nhp pcsk9相比,对捕获试剂hpcsk9的亲和力更强)。或者,tk测定使用lib003特异性mab作为捕获试剂,并使用钌标记的兔抗hsa多克隆抗体作为检测试剂。该测定形式也测量总
lib003。这些研究的结果指示,在该剂量范围内,在静脉内和皮下施用后,总结合pcsk9的融合蛋白的动力学近似线性且与剂量成比例。
[0062]
在drf研究中,在高于10mg/kg的所有剂量水平下均存在持续的抗药物抗体(ada)反应。在单剂量的lib003后,在大多数情况下,该研究中ada的存在与lib003暴露的迅速丧失有关,并且首先是靶标捕获的丧失(游离pcsk9降低)。尽管在每周施用结合pcsk9的融合蛋白(4周、12周和26周glp毒性研究)后在大多数动物中检测到ada,但对结合pcsk9的融合蛋白的暴露在整个给药间隔中都得以维持,并且ada对tk/pd的影响仅在两只动物(4周研究)、一只动物(12周研究)和一只动物(26周研究)中明显。
[0063]
在不存在ada影响的情况下,lib003清除缓慢,并且在drf研究中血清终末半衰期范围为8.3-10.4天(平均9天),在4周的glp毒性研究中在回收动物中血清终末半衰期范围为7.6-9.2天(平均8.3天);这与非人灵长类动物中白蛋白样分子的行为一致。在12周和26周的glp毒性研究中观察到了相似的结果。在drf研究中,在单次iv施用后,在可进行测定的动物中清除率(cl)范围为5.63-7.65毫升/天/千克(平均6.64毫升/天/千克)并且分布体积(vz)范围为67.6-103.8ml/kg(平均86.4ml/kg)。
[0064]
总pcsk9浓度(与lib003结合和未结合)缓慢增加,并在给药后约7天达到峰值,此时总pcsk9浓度小于循环lib003浓度的10%。如通过游离pcsk9和ldl降低所测量,在drf研究中在所有剂量下靶标捕获均最大,指示在最低测试剂量(10mg/kg)下靶标抑制最大。结果指示,在不存在ada的情况下,增加lib003的剂量会增加最大靶标捕获的持续时间。
[0065]
无论剂量或施用途径如何,血清ldl均被抑制约60%,这与所有剂量水平下的最大药效动力学效应一致。可以预知,ldl抑制的丧失和基线水平的恢复与最大pcsk9捕获的丧失一致。总的来说,这些研究表明已经达到了最大药效动力学效应,并且更高的剂量不会对游离pcsk9或ldl产生更大的抑制作用。
[0066]
与相同剂量水平下的缓慢iv推注相比,皮下给药后c
max
较低并且t
max
出现较晚,指示从注射部位吸收到体循环。在皮下施用后,皮下途径的绝对生物利用率在drf研究中估计为约76%,在4周glp毒性研究中估计为66.5%-89.7%,但这两个估计值都因ada存在以及无法表征总曲线下面积(auc)而受到影响。使用群体pk模型对这两项研究进行的综合分析估计皮下施用后的生物利用率为92%,并且可能是更可靠的估计值,因为所述模型会生成两种施用途径的总auc估计值。
[0067]
预测的人体中pk-pd行为
[0068]
结合来自多个研究的数据来预测人体中lib003的预期pk-pd行为。即,由nhp数据(drf研究和4周glp毒性研究)构建描述lib003暴露和pcsk9抑制的群体2室pk-pd结合模型。在将这些参数扩展至人时,对hpcsk9的体内结合亲和力源自hpcsk9转基因小鼠中的pk-pd研究。由于只有皮下施用后的数据可用,因此一室pk模型足以描述这些数据;所述模型在其他方面与用于nhp数据的模型相同。观察到的对nhp pcsk9和hpcsk9的lib003结合亲和力的差异在体外和体内来源的数据中都是一致的,并且将来自hpcsk9转基因小鼠的体内来源的k
d
值用于对人的预测。
[0069]
表2
[0070][0071]
nhp和hfcrn小鼠中lib003的清除率与这些动物中hsa的预期清除率一致,因此,预测lib003的清除率可模拟人体中hsa的清除率(半衰期为19天;异速指数为0.74)。使用大小为大约77kda的治疗性白蛋白融合蛋白的预期系数,对其他参数进行异速换算。
[0072]
描述对游离pcsk9的捕获和抑制的nhp和人pk-pd结合模型的总体结构显示在图2a和图2b中。先前已经建立了定量系统药理学模型来描述他汀类药物和抗pcsk9抗体在人体中的作用机理。gadkar k等人,a mechanistic systems pharmacology model for prediction of ldl cholesterol lowering by pcsk9 antagonism in human dyslipidemic populations,cpt pharmacometrics syst.pharmacol.2014;3(11)。该模型用于在首次人体研究(fih)中提供经单次皮下和静脉内给予lib003后预测的人体中pcsk9抑制与ldl-c随时间降低之间的联系。
[0073]
实施例2:制剂和稳定性评价
[0074]
评估结合pcsk9的融合蛋白的制剂中各种缓冲液和赋形剂的性能。
[0075]
为了确定在制剂中使用的适当缓冲液和ph,对2mg/ml lib003下的使用6种不同缓冲液的18种不同的缓冲液和ph条件通过dsc进行分析以评估热稳定性,并通过dls进行分析以评估聚集体形成。从这些实验中,排除了低于5和高于7的ph,这是因为对于这些ph值,蛋白质解折叠始于<50℃的温度(起始温度(tonset))。
[0076]
所分析的缓冲液/ph组合显示在下表3中。用加粗且加下划线的ph值显示经选择用于进一步筛选的组合。
[0077]
表3:缓冲液/ph筛选
[0078][0079]
确认可接受的ph范围和适当的缓冲液后,将赋形剂与限定的缓冲液和ph进行组合研究。在2mg/ml lib003下总共准备了15种不同的缓冲液/赋形剂组合,并且通过dsc进行分析以评估热稳定性,并通过dls进行分析以评估聚集体形成。由于起始温度低于55℃,因此排除了所述缓冲液/赋形剂组合中的三种。此外,dls数据表明,蔗糖、山梨糖醇和低浓度的nacl的使用会导致聚集,因此在随后的制剂中它们的使用受到限制。
[0080]
所分析的缓冲液/赋形剂组合显示在下表4中。用加粗且加下划线的赋形剂显示经选择用于进一步筛选的组合。
[0081]
表4:缓冲液/赋形剂筛选
[0082][0083]
为了确定用于维持以高浓度配制的lib003的稳定性的合适缓冲液和赋形剂组合,对上文表4中鉴定的12种不同的制剂进行溶解度筛选。将这些制剂浓缩至目标浓度为200mg/ml、250mg/ml、300mg/ml和340mg/ml。在所有制剂中浊度均等,并且当通过sec-hplc评估时未观察到聚集。基于这些参数,认为这些制剂是等效的。
[0084]
为了评价向lib003候选制剂中添加表面活性剂的潜在益处,在有或无聚山梨醇酯80(ps80)情况下对表4中的八种缓冲液/赋形剂组合进行评价,总计16种250mg/ml lib003下的制剂(表5)。由于ps80可有效防止聚集和微粒形成,因此评价了候选制剂承受反复的冻融和搅动应力的能力。基于回收率、浊度或聚集倾向(通过dls和sec评价),未观察到差异。由于ps80在长期储存和产品处理后的预期益处,所有包含ps80的制剂均取得了进展。
[0085]
表5:缓冲液/赋形剂和表面活性剂筛选
[0086][0087]
lib003具有稳定性并且在正常的储存条件和剪切应力下,即使在高lib003浓度下,制剂之间也没有差异,这表明需要一种替代方法来鉴定能够承受长期储存条件的lib003制剂。将在340mg/ml的目标浓度下从缓冲液/赋形剂筛选(表4)中鉴定出的制剂掺入0.02%ps80,在2℃-8℃或50℃下储存三周,并通过sec-hplc进行分析。通常,在50℃下储存3周后,观察到低水平的高分子量(hmw)和低分子量(lmw)物质。基于这些制剂中hmw物质的低百分比,将含柠檬酸盐的制剂缩窄至赋形剂精氨酸和山梨糖醇。在组氨酸制剂中,基于主峰物质的最高百分比,选择150mm nacl用于其他实验。最后,由于相对于2℃-8℃的对照样品,在于50℃下储存的大多数琥珀酸盐制剂中观察到的hmw和lmw物质水平增加,所以不再考虑琥珀酸盐。
[0088]
这些分析的结果显示在下表6中;优选的制剂用粗体和加下划线的字体表示。
[0089]
表6:于50℃下储存3周的高浓度lib003的sec-hplc评价结果。
[0090][0091][0092]
为了进一步探索候选制剂,将lib003在3个不同的ph下以三种缓冲液/赋形剂组合加上ps80以250mg/ml进行配制(表7)。将样品在2℃-8℃或50℃下储存三周,以诱导降解,并分析lib003的浓度/回收率(a280)、微粒(dls)、聚集(sec-hplc)、电荷分布(icief)、削波(ce-sds)、效力、浊度和热稳定性(dsc)。在50℃下储存3周后对电荷分布、聚集/微粒形成倾向以及相对效力的比较证明了所述制剂之间的某些差异(表8、表9、表10和表11)。在所述制剂中ce-sds曲线、回收率、浊度和热稳定性相当。
[0093]
表7.储存之前的候选制剂
[0094][0095][0096]
表8.在50℃下储存3周后电荷分布的icief变化
[0097][0098]
表9.在50℃下储存3周后hmw和lmw物质中的sec-hplc变化
[0099]
[0100][0101]
表10.效力与储存温度的函数关系
[0102][0103]
鉴于目标是使用带有27g或更小口径针头的自动注射器以0.25-1.5ml sc注射液的形式递送lib003,因此进行了另外的实验以便确定以250mg/ml配制的lib003是否具有适合经由自动注射器进行sc注射的特性。评价250mg/ml的带各自中心点ph的前三种制剂的粘度、重量克分子渗透浓度和微粒。目标是粘度<15cp,重量克分子渗透浓度为250-350mosm,并且微粒水平远低于法规限度。结果显示在下表11中。在所有制剂中,微粒水平均相当低。
[0104]
表11:粘度和重量克分子渗透浓度分析的结果
[0105][0106]
将结合pcsk9的融合蛋白于20mm组氨酸、150mm nacl、0.02%(w/v)聚山梨醇酯80,ph 6.8中配制为无菌注射溶液(皮下),用于进一步的稳定性研究。这些稳定性研究包括在长期储存条件、加速条件以及加压条件下的评价。结合pcsk9的融合蛋白产品的预期长期储存温度为5℃
±
3℃,并且短期储存(≤1-6个月)的温度为25℃
±
3℃。
[0107]
在预期储存温度(5℃
±
3℃)以及加速储存条件(25℃
±
2℃/60
±
5%rh)和加压储存条件(40℃
±
2℃/75
±
5%rh)下获得1个月的稳定性数据。
[0108]
对于示例性结合pcsk9的融合蛋白产品批次,稳定性方案中所有评价参数的数据(包括稳定性指示参数(icief、效力、ce-sds和sec-hplc))在检查的时间点(1个月)和每个储存条件下均处于接受标准之内。此外,在长期储存条件下未观察到聚集体或降解物物质的显著增加或效力的下降。在加速和应力条件下,通过sec-hplc观察到聚集体略微增加(约1%),但是这些结果完全在sec-hplc的主峰接受标准之内,并且未观察到效力变化。而且,在应力条件下,通过还原性ce-sds观察到碎片略微增加(约2%),但同样未观察到效力变化。在每种储存条件下通过icief观察到酸性电荷变体物质略有增加(约1%-4%),同时主峰物质随之减少(注意,对于原料药观察到相反的趋势,这指示所观察到的变化在该方法的可变性之内)。未观察到外观、物理化学参数或效力的变化。合起来,稳定性数据证明结合pcsk9的融合蛋白产品在评价的长期、加速和加压储存条件下稳定1个月。
[0109]
另外,在预期储存温度(5℃
±
3℃)下获得了18个月的长期稳定性数据,并且在加速储存条件(25℃
±
2℃/60
±
5%rh)下获得了9个月的长期稳定性数据,而且在加压储存条件(40℃
±
2℃/75
±
5%rh)下获得了三个月的长期稳定性数据。
[0110]
对于示例性结合pcsk9的融合蛋白产品批次,稳定性方案中所有评价参数的数据(包括稳定性指示参数(icief、效力、ce-sds和sec-hplc))在检查的时间点和每个储存条件下均处于接受标准之内。此外,在长期储存条件下未观察到聚集体或降解物物质的显著增加或效力的下降。在所有储存条件下,通过sec-hplc观察到聚集体略微增加(约1%),但是这些结果完全在sec-hplc的主峰接受标准之内,并且未观察到效力变化(在测定可变性之
内)。而且,在长期储存条件下,通过还原性ce-sds观察到碎片略微增加(多至约2%),但同样未观察到效力的相应变化,并且可认为该变化在该方法的可变性之内。通过还原性和非还原性ce-sds获知的碎片增加与时间和温度增加(在加压条件下有多至约5%的变化)有关。在长期储存条件下,通过icief观察到酸性物质和主峰物质的波动(多至约8%),但是该波动在该方法的可变性之内。加速和加压条件下的icief结果最初似乎指示酸性物质具有潜在的正趋势(多至约7%),而主峰物质则具有相应的负趋势,但未观察到效力随之变化。然而,其他时间点指示,观察到的微小变化也可能是与方法可变性相关的波动。亚可见颗粒似乎随时间略有波动,但仍在可接受限度之内(注意,在长期条件和加速条件下在9个月时对于≥2μm和≥5μm的颗粒均观察到非典型结果;但是,颗粒计数在12个月和18个月时间点时恢复到预期水平)。未观察到外观、物理化学参数或效力的变化。
[0111]
另外,在预期储存温度(5℃
±
3℃)下获得了12个月的长期稳定性数据,并且在加速储存条件(25℃
±
2℃/60
±
5%rh)下获得了12个月的长期稳定性数据。
[0112]
对于示例性结合pcsk9的融合蛋白产品批次,稳定性方案中所有评价参数的数据(包括稳定性指示参数(icief、效力、ce-sds和sec-hplc))在检查的时间点和每个储存条件下均处于接受标准之内。此外,在长期储存条件下未观察到聚集体或降解物物质的显著增加或效力的下降。在两种储存条件下,通过sec-hplc观察到聚集体略微增加(多至约1%),但是这些结果完全在sec-hplc的主峰接受标准之内,并且未观察到效力变化(在测定可变性之内)。此外,通过还原性ce-sds条件在长期储存条件下观察到碎片略微增加(多至约2%),在加压储存条件下观察到碎片略微增加(约3%),并且通过非还原性ce-sds在加压储存条件下观察到碎片略微增加(约3%),但同样未观察到效力的相应变化(在测定可变性之内)。在两种储存条件下通过icief均观察到酸性电荷变体物质的增加(多至约5%),同时主峰物质随之减少(这可能归因于测定可变性,因为之后的时间点具有较低的酸性物质百分比)。未观察到外观、物理化学参数或效力的变化。
[0113]
实施例3:首次人体研究
[0114]
在1期sad研究中对lib003进行研究,所述研究包括63位年龄≥18岁且≤70岁的受试者,24位女性和39位男性,45位接受lib003并且18位接受安慰剂,给药后监测至少43天。该研究是安慰剂对照的和双盲的。在9个群组中每组有7位受试者:5位接受lib-003治疗的患者和2位接受安慰剂治疗的患者,使得43位受试者接受lib003治疗,18位接受安慰剂治疗。将25mg、75mg、150mg、300mg和600mg的lib003皮下剂量施用给健康受试者,所述健康受试者采用稳定饮食,未接受降脂疗法,基线ldl-c≥100且≤190mg/dl。另外将150mg和300mg剂量sc施用给接受稳定他汀类药物疗法且基线ldl-c≥100mg/dl的患者。所有受试者的tg≤250mg/dl。另外两个未接受降脂疗法且脂质准入标准与他们的sc群组相同的健康受试者群组接受lib003300mg和600mg iv。
[0115]
安全性结果概述
[0116]
所有63位受试者都完成研究,无患者在第43天之前退出或终止研究。总的来说,在健康受试者和接受他汀类药物疗法的高胆固醇血症患者中,在单次sc和iv给药后,lib003均是安全且耐受性良好的。
[0117]
药效动力学(功效)结果概述
[0118]
游离pcsk9的平均降低在所有剂量下都是迅速的且在12小时内达到99%以上,并
且这种降低在未接受降脂疗法的接受≥150mg lib003的群组中在几乎所有受试者中都持续了至少3周(第22天)。lib003 300mg剂量在29天内维持99%的游离pcsk9抑制,但是在未接受降脂疗法的150mg剂量受试者中,游离pcsk9降至基线的12%,在接受他汀类药物的受试者中降至基线的54%。游离pcsk9的较低降低程度反映在ldl-c和载脂蛋白b的下降方面,其中未进行降脂疗法的受试者维持了较大的降低程度。但是,在接受他汀类药物的患者中,这些结果并未在4周内(第29天)持续。在经非他汀类药物和他汀类药物治疗的受试者中,单次300mg剂量可使游离pcsk9、ldl-c和载脂蛋白b的降低更为稳定且程度最大。此外,基于mab研究的先前数据,预计多次给药将使游离pcsk9抑制和ldl-c降低的持续时间更长。此外,大量的先前数据显示,接受高强度他汀类药物的患者和患有fh的患者的基线pcsk9水平较高,并且pcsk9的合成可能增加,因此将需要300mg或更高的剂量才能完全抑制游离pcsk9和ldl-c持续4周。
[0119]
药代动力学结果概述
[0120]
来自sc剂量的总lib003的c
max
、auc
0-t
和auc
inf
在75mg与300mg lib003之间以剂量成比例的方式增加,并且在25mg-75mg之间(4倍至5倍增加)和300mg-600mg之间(3倍增加)表现出微小的超剂量比例性。同样,在150mg至300mg剂量范围内,经他汀类药物治疗的受试者表现出总lib003的剂量成比例增加,但总lib003暴露(auc
0-t
和auc
inf
)总体上低于经非他汀类药物治疗的受试者中的暴露。iv施用的总lib003的c
max
、auc
0-t
和auc
inf
以剂量成比例增加。
[0121]
在所有sc剂量水平下,总lib003的中值t
max
范围为72至168小时(范围可变:36至220小时)。在所测试的sc剂量下以及当与他汀类药物一起施用lib003时,t-half、cl/f和vz/f也都相似。在两种iv剂量水平下,总lib003的中值t
max
范围为0.33至1.08小时。在所测试的两种iv剂量下,t-half、cl和vz都相似。
[0122]
在分别为300mg和600mg lib003的单次sc剂量后,总lib003的绝对生物利用率范围为67%至111%。
[0123]
2期研究剂量水平的原理阐述
[0124]
基于1期的游离pcsk9和ldl-c数据以及实现与使用自动注射器进行单次sc注射一致的体积(≤1.5ml)的至少q4w的给药的目标,计划在大约80位接受稳定他汀类药物和/或依泽替米贝的患有ascvd或有ascvd高风险或患无cvd的hefh的患者中进行2期剂量发现研究。经选择用于此2期剂量发现研究中的q4w给药的剂量包括150mg、300mg和350mg。基于在1期中多至600mg(sc和iv)的人lib003暴露以及在12周的非人灵长类动物glp毒理学研究中所达到的水平下发现的缺乏,预计所有这3种剂量都是安全的。
[0125]
实施例4:旨在评价需要进一步降低ldl-c的接受稳定降脂疗法的患者中的lib003功效和安全性的随机双盲、安慰剂对照、2期剂量发现
[0126]
在为期12周的随机、双盲、安慰剂对照、剂量发现2期研究中研究lib003,随后进行为期4周的随访评估期,以在每月(q4w)给予各种剂量的lib003下如通过friedewald公式计算的以第10周和第12周以及在第12周的平均值评估ldl-c水平相对于基线的变化百分比。该研究共纳入81位年龄≥18岁且患有动脉粥样硬化性心血管疾病(ascvd)或处于高ascvd风险(≥10%5年或≥7.5%10年风险)的男性和女性,并且对于有ascvd或cvd风险的受试者计算得出的ldl-c≥80mg/dl,或者对于患有杂合性家族性高胆固醇血症且无cvd的受试者
计算得出的ldl-c≥100mg/dl。所有受试者在接受稳定的降脂口服药物疗法(如他汀类药物加或不加依泽替米贝)下的tg≤400mg/dl。将这些受试者分为3个活性物质组和1个匹配的安慰剂治疗组。在每组中,对每一位安慰剂受试者分配三位lib003受试者,即每个治疗组20位lib003受试者和20位安慰剂受试者。每月(q4w)将150mg、300mg或350mg lib003或安慰剂皮下剂量皮下施用给接受稳定饮食和口服降ldl-c药物疗法的患有高胆固醇血症的受试者。
[0127]
该研究中招入的受试者的人口统计汇总显示在下表11中。
[0128]
表11:受试者群组的人口统计
[0129][0130][0131]
安全性结果概述
[0132]
总的来说,在该研究中,lib003作为高达350mg q4w的皮下剂量是安全的且总体上耐受性良好的。lib003的所有剂量(150mg、300mg和350mg)都具有良好的耐受性,并且没有显示出任何安全问题。在这81位受试者中,有79位受试者完成了研究,有2位受试者退出研究。退出研究不是由于不良事件。此外,在该研究中没有不良事件导致死亡。
[0133]
受试者在研究期间所经历的治疗紧急不良事件(teae)的汇总显示在下表12中。
[0134]
表12:研究期间的受试者teae
[0135][0136]
在81位受试者中共有43位(53%)有teae,其中接受安慰剂的20位受试者中有10位(50%),联合lib003治疗组中的61位受试者中有33位(54%)。大多数teae的严重程度为轻度至中度。最常报告的teae是疲劳、注射部位淤青、上呼吸道感染和呼吸困难。≤3位受试者报告了所有其他teae。
[0137]
在81位受试者中只有6位(7%)有与研究药物相关的teae,其中接受安慰剂的20位受试者中有2位(10%),联合lib003治疗组中的61位受试者中有4位(7%)。大多数与研究药物相关的teae的严重程度为轻度;没有一个被认为是严重的。最常报告的与研究药物相关的teae是注射部位红斑。≤1位受试者报告了所有其他与研究药物相关的teae。
[0138]
81位受试者中有6位(7%)有sae,安慰剂组有1位(5%),联合lib003治疗组中有5位(8%);没有一个被认为与研究药物有关。五个sae(安慰剂组中有一个(5%),联合lib003治疗组中有四个(7%))的严重程度为重度,lib003治疗组中有1个(2%)的严重程度为中度。
[0139]
没有与异常实验室值相关的teae。在任何治疗组中,肝功能测试(alt、ast或胆红素)均无临床上有意义的升高或趋势,在安慰剂组或lib003治疗组之间也没有差异。具体而言,没有受试者经历alt升高或者ast>3
×
uln,并且没有受试者的胆红素>2
×
uln。在所有治疗组中多数受试者的ck未持续增加(这与运动或活动相关),且均未超过5
×
uln。治疗组之间在肾功能、葡萄糖、其他化学或血液学参数方面无临床上显著的增加或差异。
[0140]
在生命体征、ecg和体格检查结果中无临床上有意义的发现。在研究药物施用之后的15分钟剂量后时间点,总共有五次记录的注射部位红斑事件。在第一天剂量后15分钟,安慰剂组中有一位受试者还报告了注射部位瘙痒。剂量后15分钟发生的所有注射部位反应的严重程度均为轻度,其中安慰剂组中有1个,联合lib003治疗组中有4个。
[0141]
功效结果概述
[0142]
所测试的所有lib003剂量(150mg、300mg和350mg)均导致ldl-c和游离pcsk9水平快速、持续且显著平均下降。在300mg lib003剂量群组中观察到ldl-c的最大平均下降。与以前的研究一致,较高的350mg lib003剂量不会进一步降低ldl-c。发现150mg剂量不足以在各剂量之间的整个四周内获得最大的ldl-c降低。
[0143]
该研究的功效数据的汇总显示在下表13中。
[0144]
表13:功效数据
[0145][0146][0147]
在lib003中给药后,如通过friedewald公式计算,从基线到第10周和第12周locf的平均值以及到第12周locf,ldl-c水平持续大幅平均下降。lib003群组与安慰剂组之间从基线到第10周和第12周locf平均值以及到第12周locf的ldl-c的最大ls平均变化百分比差异(95%ci)出现在300mg lib003剂量群组中,且分别为-76.1%([-86.0%,-66.2%],p<0.0001)和-77.3%([-90.5%,-64.1%],p<0.0001)。通过制备型超速离心和霍普金斯公式
对ldl-c水平的平均变化百分比进行评估得出了相似的发现。
[0148]
先前的研究表明一旦实现最大pcsk9抑制,就不会再发生ldl-c降低,与之一致的是较高剂量的350mg lib003不会导致ldl-c水平进一步降低。尽管每两周一次给药后150mg剂量实现了与300mg和350mg剂量相似的降低,但其不足以在各剂量之间在整个四周内维持最大的ldl-c降低。
[0149]
在lib003剂量后,从基线到第10周和第12周locf的平均值以及到第12周locf,游离pcsk9水平持续大幅平均下降。lib003群组与安慰剂组之间从基线到第10周和第12周locf平均值以及到第12周locf的游离pcsk9水平的ls平均变化百分比差异(95%ci)出现在300和350mg群组中,分别为-89.7%([-100.0%,-79.4%],p<0.0001)和-92.8%([-102.9%,-82.6%],p<0.0001)。lib003群组与安慰剂组之间从基线到第12周locf的游离pcsk9水平的ls平均变化百分比差异(95%ci)在300和350mg群组中分别为-84.1%([-99.5%,-68.7%],p<0.0001)和-90.2%([-105.4%,-75.0%],p<0.0001)。尽管在第12周时350mg lib0003剂量比300mg剂量抑制略多的pcsk9(分别为90.2%与84.1%),但这并未产生更大的ldl-c功效。
[0150]
另外,在lib003剂量后,从基线到第10周和第12周locf的平均值以及到第12周locf,总pcsk9水平持续且大幅平均下降。对于lib003150mg、300mg和350mg群组,从基线到第10周和第12周locf的平均值的平均变化百分比分别为95.555%、90.273%和90.080%。对于lib003150mg、300mg和350mg群组,从基线到第12周locf的平均变化百分比分别为75.165%、78.188%和86.811%。相比之下,安慰剂在同一时间点显示出最小的平均百分比变化(≤3.044%)。
[0151]
此外,从基线到第10周和第12周locf的平均值以及到第12周locf,非hdl-c水平持续大幅平均下降,并且tc水平持续中幅平均下降。但是,在lib003群组和安慰剂组中,从基线到第10周和第12周locf的平均值以及到第12周,hdl-c水平只有极小的平均变化。此外,使用anova模型比较每个lib003群组与安慰剂组之间vldl-c和tg水平的变化百分比,显示从基线到第10周和第12周locf的平均值以及到第12周locf,vldl-c和tg水平小幅平均下降。
[0152]
而且,在lib003剂量后,从基线到第12周locf,载脂蛋白b水平持续大幅平均下降。lib003群组与安慰剂组之间的载脂蛋白b最大ls平均变化百分比差异(95%ci)出现在300mg群组中并且为-58.4%([-68.9%,-48.0%],p<0.0001)。
[0153]
此外,在所有lib003剂量后,从基线到第12周locf,lp(a)水平持续中幅下降。lib003群组与安慰剂组之间的lp(a)最大ls平均变化百分比差异(95%ci)出现在300mg群组中并且为-28.7%([-42.6%,-14.8%],p<0.0001)。该降低与pcsk9单克隆抗体在等效剂量和给药频率下所实现的降低一致,后者与lib003300mg q4w相似地抑制了游离pcsk9和ldl-c。
[0154]
对于lib003人群和安慰剂组,从基线到第12周locf,载脂蛋白a1水平显示极小平均变化。
[0155]
基于上述2期研究的数据,选择lib003300mg皮下q4w剂量进行开放标签扩展和3期研究。
[0156]
序列
[0157]
结合pcsk9的基序
[0158]
vsdvprdlevvaatptslliswdapaegygyyritygetggnspvqeftvpvskgtatisglkpgvdytitvyavefdfpgagyyhrpisinyrte(seq id no:1)
[0159]
hsa氨基酸序列
[0160]
dahksevahrfkdlgeenfkalvliafaqylqqapfedhvklvnevtefaktcvadesaencdkslhtlfgdklctvatlretygemadccakqepernecflqhkddnpnlprlvrpevdvmctafhdneetflkkylyeiarrhpyfyapellffakrykaafteccqaadkaacllpkldelrdegkassakqrlkcaslqkfgerafkawavarlsqrfpkaefaevsklvtdltkvhtecchgdllecaddradlakyicenqdsissklkeccekpllekshciaevendempadlpslaadfveskdvcknyaeakdvflgmflyeyarrhpdysvvlllrlaktyettlekccaaadphecyakvfdefkplveepqnlikqncelfeqlgeykfqnallvrytkkvpqvstptlvevsrnlgkvgskcckhpeakrmpcaedylsvvlnqlcvlhektpvsdrvtkccteslvnrrpcfsalevdetyvpkefnaetftfhadictlsekerqikkqtalvelvkhkpkatkeqlkavmddfaafvekcckaddketcfaeegkklvaasqaalgl(seq id no:2)
[0161]
结合pcsk9的融合蛋白
[0162]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1