包含细菌源微细胞的组合物及其使用方法与流程
包含细菌源微细胞的组合物及其使用方法
1.相关申请的交叉引用
2.本申请按照35 usc
§
119的规定要求2018年7月23日提交的美国临时申请62/702,172和2019年1月4日提交的美国临时申请62/788,265的优先权,其全部内容通过引用整体合并于此。
背景技术:
3.当前,大多数用于治疗癌症的药物是全身性给予的。尽管细胞毒性抗癌药物的全身递送在癌症治疗中起着至关重要的作用,但其也带来了严重的问题。例如,正常组织/器官对所给予的药物的全身暴露可引起严重的毒性。这因全身递送的癌症化疗药物通常必须以非常高的剂量递送以克服药物的不良生物利用度和患者体内的大体积分布的事实而加剧。而且,全身性药物给予可能是侵入性的,因为其通常需要在主要血管中使用固定的导管。由于全身性药物给予通常需要使用周围或中央静脉,其可引起局部并发症,如静脉炎。药物的外渗还可在局部给予位点引起起疱/组织损伤,如在长春花生物碱和蒽环类的给予中常见。
4.癌症治疗中的另一挑战是内在性或获得性临床肿瘤化疗抗性。内在性抗性存在于对一线化疗无法响应的肿瘤的诊出时。获得性抗性发生于可对初始治疗响应良好但在复发时呈现抗性表型的肿瘤。这样的肿瘤获得了对在前使用的药物和对新药物(包括具有不同结构和作用机制的药物)两者的抗性。术语mdr(多药抗性)描述了这种现象,其中肿瘤细胞在暴露于一种药物后变得对数种结构不相关的药物具有交叉抗性。多药抗性的机制是复杂的和多因素的,很大程度上归因于癌细胞中高水平的基因组不稳定性和突变。示例性机制是药物灭活、药物通过细胞膜泵挤出、药物流入减少、药物靶标突变和凋亡无法启动。bredel,2001;chenet al.,2001;sun et al.,2001;和white&mccubrey,2001。
5.免疫系统和恶性细胞之间的相互作用在肿瘤发生中也起重要作用。免疫系统无法检测和排斥转化的细胞可导致癌症发展。肿瘤利用多种机制逃避免疫介导的排斥。现在细胞和分子水平上已知这些机制中的多个。尽管存在这种已知,癌症免疫疗法仍不是临床上的已确立的治疗方法。
6.因此,仍非常需要能够提供可以降低药物抗性,促进凋亡并诱导免疫响应,同时避免全身递送这些药物相关的问题的药物靶向递送的递送系统。本发明满足了这些需求。
技术实现要素:
7.本发明的一个实施方式涉及组合物,其包含(a)治疗有效剂量的包含抗肿瘤剂的纯化的完整的细菌源微细胞(bacterially derived minicels),和(b)i型干扰素激动剂、ii型干扰素激动剂或i型干扰素激动剂和ii型干扰素激动剂的组合。i型干扰素激动剂和/或ii型干扰素激动剂可以任选地存在于完整的细菌源微细胞中。
8.在一个实施方式中,所述组合物包含(a)治疗有效剂量的包含抗肿瘤剂的纯化的完整的细菌源微细胞,和(b)治疗有效剂量的包含i型干扰素激动剂的纯化的完整的细菌源
微细胞。在另一实施方式中,所述组合物包含(a)治疗有效剂量的包含抗肿瘤剂的纯化的完整的细菌源微细胞,和(b)治疗有效剂量的包含ii型干扰素激动剂的纯化的完整的细菌源微细胞。在再另一实施方式中,所述组合物包含(a)治疗有效剂量的包含抗肿瘤剂的纯化的完整的细菌源微细胞;(b)治疗有效剂量的包含i型干扰素激动剂的纯化的完整的细菌源微细胞;和(c)治疗有效剂量的包含ii型干扰素激动剂的纯化的完整的细菌源微细胞。
9.在一个实施方式中,抗肿瘤剂和i型干扰素激动剂、ii型干扰素激动剂或i型干扰素激动剂和ii型干扰素激动剂的组合被包装(packaged)在两个或更多个纯化的完整的细菌源微细胞中。在一个实施方式中,抗肿瘤剂和i型干扰素激动剂、ii型干扰素激动剂或i型干扰素激动剂和ii型干扰素激动剂的组合被包装在三个单独的纯化的完整的细菌源微细胞群中。
10.在一个实施方式中,组合物包含抗肿瘤剂、i型干扰素激动剂和ii型干扰素激动剂,其中:(a)抗肿瘤剂、i型干扰素激动剂和ii型干扰素激动剂被包含在相同微细胞中;(b)抗肿瘤剂和i型干扰素激动剂被包含在第一微细胞中,并且ii型干扰素激动剂被包含在第二微细胞中;(c)抗肿瘤剂和ii型干扰素激动剂被包含在第一微细胞中,并且i型干扰素激动剂包含在第二微细胞中;(d)抗肿瘤剂被包含在第一微细胞中,并且i型干扰素激动剂和ii型干扰素激动剂被包含在第二微细胞中;或(e)抗肿瘤剂被包含在第一微细胞中,i型干扰素激动剂被包含在第二微细胞中,并且ii型干扰素激动剂被包含在第三微细胞中。
11.在一个实施方式中,组合物不包含i型干扰素激动剂。
12.在一个实施方式中,抗肿瘤剂选自放射性核素、化疗药物、功能性核酸和转录得到功能性核酸的多核苷酸。在一个实施方式中,抗肿瘤剂是超毒性化疗药物。在一个实施方式中,超毒性化疗药物选自吗啉基蒽环类、美登素类(maytansinoid)、杜卡霉素(ducarmycin)、奥瑞他汀(auristatins)、加利车霉素(calicheamicins)(dna损伤剂)、α
‑
鹅膏菌素(rna聚合酶ii抑制剂)、centanamycin、吡咯并苯并二氮杂链霉黑素(streptonigtin)、氮芥、亚硝基脲、烷烃磺酸酯、嘧啶类似物、嘌呤类似物、抗代谢物、叶酸类似物、蒽环类、紫杉烷、长春花生物碱、拓扑异构酶抑制剂、激素剂及其组合。在一个实施方式中,吗啉基蒽环类选自奈莫柔比星(nemorubicin)、pnu
‑
159682、伊达比星(idarubicin)、柔红霉素(daunorubicin);卡米霉素(caminomycin)和多柔比星(oxorubicin)。在一个实施方式中,超毒性化疗药物是pnu
‑
159682。
13.在一个实施方式中,功能性核酸选自sirna、mirna、shrna、lincrna、反义rna和核酶。在一个实施方式中,功能性核酸抑制促进肿瘤细胞增殖、血管生成或化疗抗性的和/或抑制凋亡或细胞周期停滞的基因。在一些实施方式中,sirna抑制核糖核苷酸还原酶m1(rrm1)表达。在一些实施方式中,sirna抑制polo样激酶1(plk1)表达。在一些实施方式中,mirna是mirna 16a。
14.在一个实施方式中,i型干扰素激动剂、ii型干扰素激动剂或i型干扰素激动剂和ii型干扰素激动剂的组合是寡核苷酸。在一个实施方式中,寡核苷酸包含至少约40个核苷酸、至少约50个核苷酸或至少约60个核苷酸的序列。在一些实施方式中,寡核苷酸是pnpasel、聚(i:c)、聚
‑
iclc、咪喹莫特(imiquimod)、咪唑并喹啉瑞喹莫德(imidazoquiolineresquimod)、cgamp或cpg
‑
寡脱氧核苷酸的多核苷酸产物。
15.在一个实施方式中,i型干扰素激动剂选自双链rna(dsrna)、聚(da:dt)dna、双链
z
‑
dna和b
‑
dna、长于36bp的dna(dsdna)和dna
‑
rna杂交体、细菌第二信使环
‑
二gmp、tlr3、tlr4、tlr7、tlr8和tlr9激动剂、sting激动剂及其组合。
16.在一个实施方式中,ii型干扰素激动剂选自α
‑
半乳糖神经酰胺(α
‑
c
‑
galcer)的c
‑
糖苷形式、α
‑
半乳糖神经酰胺(α
‑
galcer)、半乳糖神经酰胺的12碳酰基形式(β
‑
galcer)、β
‑
d
‑
吡喃葡萄糖神经酰胺(β
‑
glccer)、1,2
‑
二酰基
‑3‑0‑
半乳糖基
‑
sn
‑
甘油(bbgl
‑
ii)、含有二酰基甘油的糖脂(glc
‑
dag
‑
s2)、神经节苷脂(gd3)、神经节三酰基神经酰胺(gangliotriaosylceramide)(gg3cer)、糖基磷脂酰肌醇(gpi)、α
‑
葡萄糖醛酰神经酰胺(gsl
‑
1或gsl
‑
4)、异球蛋白三己糖基神经酰胺(isoglobotrihexosylceramide)(igb3)、磷脂聚糖(lipophosphoglycan)(lpg)、溶血磷脂酰胆碱(lyosphosphatidylcholine)(lpc)、α
‑
半乳糖神经酰胺类似物(och)、苏糖醇神经酰胺和其组合。在一个实施方式中,ii型干扰素激动剂是α
‑
半乳糖神经酰胺(α
‑
galcer)。
17.在一个实施方式中,组合物还包含与包含抗肿瘤剂的微细胞结合的双特异性配体。在一个实施方式中,组合物还包含与包含i型干扰素激动剂的微细胞结合的双特异性配体。在一个实施方式中,组合物还包含与包含ii型干扰素激动剂的微细胞结合的双特异性配体。
18.在一个实施方式中,双特异性配体包含对微细胞表面结构具有特异性的第一臂和对非吞噬性哺乳动物细胞表面受体具有特异性的第二臂。在一个实施方式中,微细胞表面结构是微细胞表面上脂多糖的o
‑
多糖组分。在一个实施方式中,非吞噬性哺乳动物细胞表面受体能够激活微细胞的受体介导型内吞作用。
19.在一个实施方式中,双特异性配体包含双特异性抗体或抗体片段。在一个实施方式中,该抗体或抗体片段包含对细菌源微细胞表面结构具有特异性的第一多价臂和对癌细胞表面受体具有特异性的第二多价臂,其中癌细胞表面受体能够激活微细胞的受体介导型内吞作用。
20.在一个实施方式中,组合物包含每107个微细胞少于约1个污染性(contaminating)亲本细菌细胞、每108个微细胞少于约1个污染性亲本细菌细胞、每109个微细胞少于约1个污染性亲本细菌细胞、每10
10
个微细胞少于约1个污染性亲本细菌细胞、或每10
11
个微细胞少于约1个污染性亲本细菌细胞。
21.在一个实施方式中,组合物还包含药学上可接受的载体。在一实施例中,微细胞的直径为约400nm。在一个实施方式中,组合物不含通过200nm过滤可除去的亲本细菌细胞污染。
22.在一个实施方式中,组合物包含以下量的微细胞或被杀死的细菌细胞:(a)至少约109个;(b)至少约1
×
109个;(c)至少约2
×
109个;(d)至少约5
×
109个;(e)至少8
×
109个;(f)不超过约10
11
个;(g)不超过约1
×
10
11
个;(h)不超过约9
×
10
10
个;或(i)不超过约8
×
10
10
个。
23.本发明的一个实施方式涉及治疗有需要的对象的方法,该方法包括向该对象给予有效量的本文公开的组合物。在一个实施方式中,所述对象是人、非人灵长类、犬、猫、牛、绵羊、马、兔、小鼠或大鼠。在一个实施方式中,所述对象是人。
24.在一个实施方式中,该对象患有癌症。在一个实施方式中,所述癌症选自肺癌、乳腺癌、脑癌、肝癌、结肠癌、胰腺癌和膀胱癌。在一个实施方式中,所述癌症选自急性成淋巴细胞性白血病;急性髓性白血病;肾上腺皮质癌;aids相关癌症;aids相关淋巴瘤;肛门癌;
阑尾癌;星形细胞瘤;非典型畸胎样/横纹肌样肿瘤;基底细胞癌;膀胱癌;脑干神经胶质瘤;脑肿瘤;乳腺癌;支气管肿瘤;burkitt淋巴瘤;原发位点未知的癌症;类癌;原发位点未知的癌;中枢神经系统非典型畸胎样/横纹肌样肿瘤;中枢神经系统胚胎肿瘤;宫颈癌;儿童期癌症;脊索瘤;慢性淋巴细胞性白血病;慢性髓性白血病;慢性骨髓增生性障碍;结肠癌;结肠直肠癌;颅咽管瘤;皮肤t细胞淋巴瘤;内分泌胰腺胰岛细胞瘤;子宫内膜癌;成室管膜细胞瘤;室管膜瘤;食道癌;成感觉神经细胞瘤;ewing肉瘤;颅外生殖细胞肿瘤;性腺外生殖细胞肿瘤;肝外胆管癌;胆囊癌;胃癌;胃肠类癌肿瘤;胃肠道间质细胞肿瘤;胃肠道间质肿瘤(gist);妊娠滋养层肿瘤;神经胶质瘤;毛细胞白血病;头颈癌;心脏癌症;hodgkin淋巴瘤;下咽癌;眼内黑色素瘤;胰岛细胞肿瘤;kaposi肉瘤;肾癌;langerhans细胞组织细胞增生症;喉癌;唇癌;肝癌;恶性纤维组织细胞瘤骨癌;髓母细胞瘤;髓上皮瘤;黑色素瘤;merkel细胞癌;merkel细胞皮肤癌;间皮瘤;原发隐匿的转移性鳞状颈癌;口癌;多发性内分泌肿瘤综合征;多发性骨髓瘤;多发性骨髓瘤/浆细胞瘤;蕈样真菌病;骨髓增生异常综合征;骨髓增生性肿瘤;鼻腔癌;鼻咽癌;成神经细胞瘤;非hodgkin淋巴瘤;非黑素瘤皮肤癌;非小细胞肺癌;口唇癌;口腔癌;口咽癌;骨肉瘤;其他脑和脊髓肿瘤;卵巢癌;卵巢上皮癌;卵巢生殖细胞肿瘤;卵巢低恶性潜在肿瘤;胰腺癌;乳头状瘤病;鼻旁窦癌;甲状旁腺癌;骨盆癌;阴茎癌;咽癌;中间分化的松果体实质性肿瘤;成松果体细胞瘤;垂体瘤;浆细胞肿瘤/多发性骨髓瘤;胸膜肺母细胞瘤;原发性中枢神经系统(cns)淋巴瘤;原发性肝细胞肝癌;前列腺癌;直肠癌;肾癌;肾细胞(肾)癌;肾细胞癌;呼吸道癌;视网膜母细胞瘤;横纹肌肉瘤;唾液腺癌;sezary综合征;小细胞肺癌;小肠癌;软组织肉瘤;鳞状细胞癌;鳞状颈癌;胃癌;幕上原发性神经外胚层肿瘤;t细胞淋巴瘤;睾丸癌;咽喉癌;胸腺癌;胸腺瘤;甲状腺癌;过渡细胞癌;肾盂和输尿管过渡细胞癌;滋养层肿瘤;输尿管癌;尿道癌;子宫癌;子宫肉瘤;阴道癌;外阴癌;waldenstrom巨球蛋白血症;和wilm肿瘤。
25.在一个实施方式中,脑癌或肿瘤选自脑干神经胶质瘤、中枢神经系统非典型畸胎样/横纹肌样肿瘤、中枢神经系统胚胎肿瘤、星形细胞瘤、颅咽管瘤、成室管膜细胞瘤、室管膜瘤、髓母细胞瘤、髓上皮细胞瘤、中间分化的松果体实质性肿瘤、幕上原发性神经外胚层肿瘤和成松果体细胞瘤。
26.在一个实施方式中,组合物经数周的过程被给予至少一周一次。在一个实施方式中,组合物经数周至数月被给予至少一周一次。在一个实施方式中,组合物至少一周一次被给予约2周、约3周、约4周、约5周、约6周、约7周、约8周、约9周、约10周、约11周、约12、约13周、约14周、约15周、约16周、约17周、约18周、约19或约20周或更长。在一个实施方式中,组合物被给予每周约两次。在一个实施方式中,组合物一周两次被给予约2周、约3周、约4周、约5周、约6周、约7周、约8周、约9周、约10周、约11周、约12周、约13周、约14周、约15周、约16周、约17周、约18周、约19周或约20周或更长。
27.前文概述和以下附图描述以及详细描述都是示例性和说明性的。其旨在提供本发明的更多细节,但不应解释为是限制性的。根据本发明的以下详细描述,其他目的、优点和新颖特征对于本领域技术人员将是显而易见的。
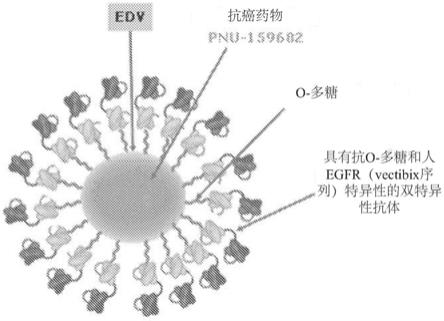
附图说明
28.图1是engeneic dream vehicle(edv)(例如细菌微细胞)的图示,其包含针对o
‑
多
糖和人表皮生长因子受体抗原的双特异性抗体并负载有抗癌药pnu
‑
159682(蒽环类类似物)。
29.图2是engeneic dream vehicle(edv)的图示,其包含表面上的o
‑
多糖并负载有免疫调节性60mer双链dna。
30.图3是engeneic dream vehicle(edv)的图示,其包含表面上的o
‑
多糖并负载有免疫调节性α半乳糖基神经酰胺(αgc)。
31.图4是评价用于治疗间皮瘤患者的靶向egfr的且负载有mirna16a的edv的临床试验的图形总结。
32.图5显示了所示化疗药物对a549肺癌细胞系的细胞毒性作用。图4a比较了所示化疗药物与超毒性药物pnu
‑
159682的效果。图4b比较了多柔比星和pnu
‑
159682的效果。
33.图6显示了所示化疗药物对肾上腺皮质癌细胞系acc01(图6a)和acc07(图6b)的效果。
34.图7显示了所示化疗药物对mda
‑
mb
‑
468乳腺癌细胞系的效果。
35.图8显示了所示化疗药物对人结肠直肠癌细胞系caco
‑
2(图8a)和hct116(图8b)的效果。
36.图9显示了所示化疗药物对成胶质细胞瘤细胞系u87
‑
mg的效果。
37.图10显示了所示化疗药物对人胰腺细胞系miapaca
‑
2(图10a)和吉西他滨抗性miapaca
‑
2 gemr细胞(图10b)的效果。
38.图11显示了靶向egfr并负载有pnu
‑
159682(
egfr
edv
s682tm
)或多柔比星(
egfr
edv sdoxtm
)的edv对小鼠的a549异种移植肿瘤生长的效果。阴性对照是仅盐水或负载有pnu
‑
159682的非靶向edv(edv
s682tm
)。箭头指示何时用所示盐水或edv组合物治疗小鼠。星号表示最初用盐水治疗的小鼠何时被给予
egfr
edv
s682tm
组合物。
39.图12显示所示nsclc细胞系中相对于gapdh表达的gapdh(3
‑
磷酸甘油醛脱氢酶)(g)、ksp(驱动蛋白纺锤体蛋白)、plk1(polo样激酶1)(p)和rrm1(核糖核苷酸还原酶1)(r)的表达。
40.图13显示了将靶向egfr的、包装sirrm1的edv递送至间皮瘤细胞系(msto,图13a)或肾上腺皮质癌细胞系(h295r,图。13b)的效果。
41.图14显示将靶向egfr的mirna16a(
egfr
edv
mirna16atm
)或靶向egfr的包装sirrm1的edv(
egfr
edv
sirrm1tm
)对balb/c nu/nu小鼠的间皮瘤异种移植肿瘤生长的效果。阴性对照是盐水或负载有加扰(scrambled)sirna的靶向egfr的edv。
42.图15显示了与盐水处理或负载有加扰sirna的靶向egfr的edv(处理)相比,用靶向egfr的mirna16a(
egfr
edv
mirna16atm
)或靶向egfr的包装sirrm1的edv(
egfr
edv
sirrm1tm
)处理的从间皮瘤异种移植物balb/c nu/nu小鼠分离的肿瘤。
43.图16a和图16b示出了通过负载有靶向polo样激酶1(
egfr
edv
tmsiplktm
)和核糖核苷酸还原酶1(
egfr
edv
sirrm1tm
)的sirna的靶向egfr的edv在肾上腺皮质癌细胞(acc01)中诱导的凋亡——基于测量细胞碎片数量(图16a)和annexin5与碘化丙锭(pi)阳性细胞的比例(图16b)。作为阴性对照,包括未经处理的acc01细胞、用负载有无关sirna的edv(
egfr
edv
tmsiluciferasetm
)处理的acc01细胞和无负载edv(
egfr
edv
tm
)的凋亡。
44.图17a
‑
d显示了通过负载有靶向polo样激酶1(
egfr
edv
tmsiplktm
)(图17d)和核糖核苷
酸还原酶1(
egfr
edv
sirrm1tm
)的sirna的靶向egfr的edv在肾上腺皮质癌细胞(acc01)中诱导的亚g1阻滞(图17d)——基于测量细胞碎片数量和annexin5与碘化丙锭(pi)阳性细胞的比例。未经处理的acc01(图17a)、用负载有不相关sirna的edv(
egfr
edv
tmsiluciferasetm
)处理的acc01细胞(图17c)和无负载edv(
egfr
edv
tm
)(图17b)的凋亡被包括作为阴性对照。
45.图18显示了用以下治疗的balb/c nu/nu小鼠中a549(肺癌)异种移植肿瘤生长的效果:(i)实心三角形=
egfr
edvs
pnu
‑
159682tm
+edvs
40mertm
,(ii)实心圆形=
egfr
edvs
pnu
‑
159682tm
(iii)空心正方形=
egfr
edvs
pnu
‑
159682tm
+edv,(iv)空心三角形=
egfr
edvs
pnu
‑
159682tm
+edvs
50mertm
,以及(v)实心正方形=盐水。在异种移植物植入后第24、27、29、31、34、36和38天用这些edv组合治疗小鼠,如上箭头所示。在第36和38天,用
egfr
edvs
pnu
‑
159682tm
+edvs
50mertm
治疗肿瘤体积~650mm3的盐水组小鼠,如下箭头所示。
46.图19显示了用包含40mers的edv(
egfr
edvs
40merstm
)与包含pnu
‑
159682的edv(
egfr
edvs
pnutm
)的组合治疗的balb/c nu/nu小鼠中对a549(肺癌)异种移植肿瘤生长的效果。三角形表示治疗天数。
47.图20显示了用盐水(阴性对照)、ifn
‑
γ(每剂量0.5x104iu)、负载多柔比星的靶向egfr的edv(
egfr
edvs
doxtm
)和
egfr
edvs
doxtm
+ifn
‑
γ治疗的balb/c nu/nu小鼠中对a549(肺癌)异种移植肿瘤生长的效果。三角形表示治疗天数。
48.图21显示了用盐水(阴性对照)、ifn
‑
γ(每剂量0.5x104iu)、负载有多柔比星的靶向egfr的edv(
egfr
edvs
doxtm
)和
egfr
edvs
doxtm
+ifn
‑
γ治疗的balb/c nu/nu小鼠中对mda
‑
mb 468(乳腺癌)异种移植肿瘤生长的效果。三角形表示治疗天数。
49.图22显示了另一实验,示例用盐水(阴性对照),ifn
‑
γ(每剂量0.5x104iu)、负载多柔比星的靶向egfr的edv(
egfr
edvs
doxtm
)和
egfr
edvs
doxtm
+ifn
‑
γ治疗的balb/c nu/nu小鼠中对mda
‑
mb 468(乳腺癌)异种移植肿瘤生长的效果。三角形表示治疗天数。
50.图23显示了用盐水(阴性对照组,第1组)、负载有多柔比星的靶向egfr的edv(
egfr
edvs
doxtm
,第2组)、
egfr
edvs
doxtm
+ifn
‑
γ(每剂量0.75x104iu)(第3组)和
egfr
edvs
doxtm
+ifn
‑
γ(每剂量0.5x104iu)(第4组)治疗的balb/c nu/nu小鼠中对多柔比星抗性a549异种移植肿瘤生长的效果。第1
‑
3组的小鼠每周两次接受治疗,如实心三角形所示。第4组的小鼠每周3次被治疗,如空心三角形所示。
51.图24a
‑
k显示了来自首次人类临床研究的患者的细胞因子概况,其中给予了不同剂量的负载有紫杉醇的
egfr
edvs
tm
。(图24a)=对于5个剂量(5剂,5doses)中的每一个所测量的il
‑
6的pg/ml;(图24b)=对于5个剂量中的每一个所测量的il
‑
8的pg/ml;(图24c)=对于5个剂量中的每一个测量的il
‑
10的pg/ml;(图24d)=对于5个剂量中的每一个所测量的tnf
‑
α的pg/ml;(图24e)=对于5个剂量中的每一个测量的ifn
‑
α的pg/ml;(图24f)=对于5个剂量中的每一个测量的ifn
‑
γ的pg/ml;(图24g)=对于5个剂量中的每一个所测量的il
‑
1β的pg/ml;(图24h)=对于5个剂量中的每一个所测量的il
‑
2的pg/ml;(图24i)=对于5个剂量中的每一个所测量的il
‑
4的pg/ml;和(图24j)=对于5个剂量中的每一个所测量的il
‑
12的pg/ml。最后,(图24k)显示了测试的五个剂量。
52.图25a
‑
k显示了来自首次人类临床研究的患者的细胞因子概况,其中给予了不同剂量的负载有多柔比星的
egfr
edvs
tm
。(图25a)=对于8个剂量中的每一个所测量的il
‑
6的pg/ml;(图25b)=对于8个剂量中的每一个所测量的il
‑
8的pg/ml;(图25c)=对于8个剂量
中的每一个所测量的il
‑
10的pg/ml;(图25d)=对于8个剂量中的每一个所测量的tnf
‑
α的pg/ml;(图25e)=对于8个剂量中的每一个测量的ifn
‑
α的pg/ml;(图25f)=对于8个剂量中的每一个测量的ifn
‑
γ的pg/ml;(图25g)=对于8个剂量中的每一个所测量的il
‑
1β的pg/ml;(图25h)=对于8个剂量中的每一个所测量的il
‑
2的pg/ml;(图25i)=对于8个剂量中的每一个所测量的il
‑
4的pg/ml;和(图25j)=对于8个剂量中的每一个所测量的il
‑
12的pg/ml。最后,(图25k)显示了测试的另外三个剂量,其中前五个剂量与(图24k)中所示的剂量相同。
53.图26显示了具有dna挑战(challenge)的胞质dna传感器的信号传导途径。迄今为止,多种胞质dna传感器已被限定以检测胞内双链dna。rna聚合酶iii将富at的dna转录成被rna传感器rig
‑
i识别的rna,然后进行sting和irf3激活。dna传感器dai、ifi16、ddx41和lsm14a直接感测dsdna,以激活用于i型ifn生成的sting。在存在dsdna的情况下,cgas催化csamp(sting的强激活剂)的合成。通过dsdna,lrrfip1以sting依赖性方式启动β
‑
连环蛋白和irf3激活。其他dna传感器独立于sting引发免疫响应。识别dsdna后,sox2触发嗜中性粒细胞中tab2/tak1复合物的激活。当通过dsdna检测时,dhx9/36通过myd88激活nfκb和irf7。dna传感器ku70触发irf1和irf7的激活。aim2通过具有dna结合的asc启动炎性体的激活。
54.图27显示了响应edv处理的raw264.7细胞和骨髓源树突状细胞(bmdc)激活。(图27a)用lμg/ml lps、ep
‑
edv、ep
‑
edv682或682直接培育的raw细胞中的cd86表达。(图27b)与用ep
‑
edv、ep
‑
edv682或682处理的4t1或ct26ep12.1细胞共培养的raw细胞中的cd86表达。与ep
‑
edv682处理的肿瘤细胞共培养的raw细胞导致cd86表达显著增加。(图27c)在raw细胞/肿瘤细胞共培养物中的tnfα产生,显示与edv处理的肿瘤细胞一起培育的raw细胞的tnfα产生显著增加。(图27d)raw细胞/肿瘤细胞共培养物中的il
‑
6产生,显示与edv处理的肿瘤细胞一起培育的raw细胞的il
‑
6产生显著增加。(图27e)bmdc/4t1共培养物中的ifnα和ifnβ表达的pcr定量。(图27f)bmdc/ct26ep12.1共培养物中的ifnα和ifnβ表达的pcr定量。(图27g)cd86
hi
和mhc ii类
hi
表达以及(图27h)cd86
hi
表达在bmdc/肿瘤细胞共培养物中的定量。(图27i)在bmdc与edv和药物处理的ct26ep12.1细胞的共培养物中mhc ii类vs cd86表达的流式细胞术密度作图。(图27j)tnfα(图27k)il
‑
12p40和(图27l)il
‑
6的elisa分析——来自bmdc/肿瘤细胞共培养物的上清液。数据表示平均值
±
s.e.m.并且通过单向anova和tukey多重比较测试来分析。
55.图28显示了响应edv治疗的肿瘤响应和巨噬细胞激活。在携载(图28a)4t1或(图28b)ct26ep12.1肿瘤的balb/c小鼠中响应于ep
‑
edv和ep
‑
edv682治疗的肿瘤生长。(图28c)在携载t84异种移植物的balb/c裸鼠中响应edv
‑
682和edv
‑
egfr682的肿瘤生长和(图28d)在携载a549/mdr异种移植的balb/c裸鼠中edv
‑
682、edv
‑
egfrdox和edv
‑
egfr682的肿瘤生长。绿色箭头指示正式盐水治疗的小鼠的edv
‑
egfr682何处开始。数据(图28a
‑
d)表示平均值
±
s.e.m.,并且通过双向anova和tukey多重比较测试来分析。(图28e)从4t1肿瘤分离并与4t1细胞以5:1(e:t)的比例共培养的cd11b
+
的xcelligence rtca。作图表示标准化的、与细胞粘附和生长/死亡vs时间相关的细胞指数。来自4t1肿瘤的cd11b
+
细胞经历初始粘附和沉降阶段,如细胞指数增加所示,然后生长或死亡,由细胞指数增加或减少表示。(图28f)被治疗小鼠的4t1肿瘤中m1(cd86
+
):m2(cd206
+
)巨噬细胞的比例。(图28g)从ct26ep12.1肿瘤分离并与ct26ep12.1细胞以5∶1(e∶t)的比例共培养的cd11b
+
的xcelligence rtca。(图
28h)被治疗的小鼠的ct26ep12.1肿瘤中的m1(cd86
+
):m2(cd206
+
)巨噬细胞的比例。数据(图28f和28h)表示平均值
±
s.e.m.,并且通过单向anova和tukey多重比较测试来分析。
56.图29显示nk细胞对edv处理的响应。(图29a)从携载4t1肿瘤的小鼠的脾脏分离的与4t1细胞以20∶1(e∶t)的比例共培养的nk细胞的xcelligence rtca。作图表示随时间的细胞活力,由标准化细胞指数计算。(图29b)在添加nk后70小时,与来自盐水、ep
‑
edv或ep
‑
edv
‑
682治疗的小鼠的nk细胞共培养的4t1细胞的%活力。(图29c)从携载ct26ep12.1肿瘤的小鼠的脾脏分离的与ct26ep12.1细胞以20:1(e:t)比例共培养的nk细胞的xcelligence rtca。(图29d)在添加nk细胞后50小时,与来自盐水、ep
‑
edv或ep
‑
edv
‑
682治疗的小鼠的nk细胞共培养的ct26ep12.1细胞的%活力。(图29e)4t1肿瘤内的nk细胞cd45
+
、cd11b
+
、dx5
+
)中的nkg2d表达,显示ep
‑
edv
‑
682治疗的小鼠中的nkg2d表达增加。从携载4t1肿瘤的edv治疗小鼠的脾脏分离的nk细胞与4t1细胞的共培养物中的(图29f)rantes和(图29g)tnfα的产生。(图29h)4种不同小鼠肿瘤细胞系表面上的nkg2d配体rae
‑
1、h60a和mult
‑
1的定量。(图29i)在存在rae
‑
1和/或h60a抑制抗体的情况下与4t1细胞以20:1(e:t)比例共培养的从携载4t1肿瘤的epedv
‑
682治疗小鼠的脾脏分离的nk细胞的xcelligence rtca,证明两者均在nk肿瘤细胞的细胞溶解中非常重要。(图29j)nk细胞添加后80小时的nk细胞溶解的定量,显示单独的h60a抗体与rae
‑
1抑制抗体结合对nk细胞溶解的显著抑制。数据(图29b、29d、29e
‑
g和29j)表示平均值
±
s.e.m.,并且通过单向anova和tukey多重比较测试来分析。
57.图30显示了响应edv治疗脾细胞/肿瘤细胞共培养物的间质肿瘤细胞因子/趋化因子的产生。在携载(图30a)4t1或(图30b)ct26ep12.1肿瘤的balb/c小鼠中响应ep
‑
edv和ep
‑
edv682治疗产生的间质细胞因子和趋化因子的elisa分析。ep
‑
edv
‑
682治疗主要导致th1细胞因子增加。数据表示平均值
±
s.e.m.。各个细胞因子数据通过单向anova和tukey多重比较测试来分析。分离自盐水、ep
‑
edv和ep
‑
edv
‑
682治疗的携载4t1和ct26ep12.1肿瘤的小鼠的脾细胞与其相应肿瘤细胞的共培养物的上清液的(图30c)tnfα、(图30d)il
‑
2、(图30e)il
‑
1β、(图30f)ifnγ和(图30g)il
‑
10的elisa分析。数据表示平均值
±
s.e.m.。单向anova分析和tukey多重比较用于比较+或
‑
肿瘤细胞的组。t检验用于比较有无肿瘤细胞的个体治疗。
58.图31显示了响应edv治疗的t细胞功能和表型。(图31a)从携载4t1肿瘤的小鼠脾脏中分离的与4t1细胞以30:1(e:t)的比例共培养的cd8
+
t细胞的xcelligence rtca。作图表示标准化的与细胞粘附和生长/死亡vs时间相关的细胞指数。(图31b)在添加cd8
+
t细胞后30小时,与来自盐水、ep
‑
edv或ep
‑
edv
‑
682治疗的小鼠的cd8
+
t细胞共培养的4t1细胞的%活力。(图31c)从携载ct26ep12.1肿瘤的小鼠的脾分离的与ct26ep12.1细胞以30:1(e:t)的比例共培养的cd8
+
t细胞的xcelligence rtca。作图表示标准化的与细胞粘附和生长/死亡vs时间相关的细胞指数。(图31d)在添加cd8
+
t细胞后20小时,与来自盐水、ep
‑
edv或ep
‑
edv
‑
682治疗的小鼠的cd8
+
t细胞共培养的ct26ep12.1细胞的%活力。(图31e)在4t1肿瘤中检测到的cd8
+
t细胞(限定为cd45
+
、cd3
+
、cd8
+
)的百分比。(图31f)在4t1肿瘤中检测到的t
‑
regs(限定为cd45
+
、cd3
+
、cd4
+
、cd25
+
)的百分比。(图31g)携载4t1肿瘤的小鼠的肿瘤引流淋巴结中的t细胞数目显示为总细胞的百分比。(图31h)在携载4t1肿瘤的小鼠的肿瘤引流淋巴结中的树突状细胞中的%cd80/mhc ii类表达。(图31i)来自ep
‑
edv
‑
682治疗的小鼠的分离的cd8
+
t细胞与4t1细胞的相互作用的共焦图像。红色
‑
肌动蛋白,绿色
‑
穿孔素,蓝色
(深)
‑
dapi;比例尺10μm。数据(图31b、31d和31e
‑
h)表示平均值
±
s.e.m.,并通过单向anova和tukey多重比较测试(图31b、31d和31e
‑
g)或t检验(图31h)来分析。
59.图32显示了患者外周血单核细胞(pbmc)的预后指标和免疫分型,揭示了剂量12下树突状细胞和单核细胞抗原呈递增加以及细胞毒性cd8
+
t细胞含量升高的证据。预后指标(图32a)ca19
‑
9和(图32b)c反应蛋白血清水平。关于(图32c)单核细胞和(图32d)中间(cd14+cd16++)抗原呈递单核细胞亚型,利用duraclone免疫分型图进行的pbmc分析。以%白细胞表示。树突状细胞亚型包括(图32e)驱动cd8
+
效应t细胞响应的髓样树突状细胞(clec9a
+
)(mdc)和(图32f)浆细胞样树突状和髓样树突状(细胞)(专职抗原呈递dc)。如示,以%dc或%mdc表示。(图32g)cd8
+
t细胞亚型。细胞毒性cd8
+
t细胞包括效应亚型和衰竭(pd1
+
)亚型。
60.图33显示了edv如何首先通过将细胞毒性剂直接递送至肿瘤来建立免疫原性肿瘤微环境,然后直接或间接刺激先天免疫系统趋向抗肿瘤表型,并且最后产生适应性响应(其中肿瘤特异性细胞毒性t细胞产生)的示意图。(图33a)ep
‑
edv
‑
682通过渗漏的脉管系统进入肿瘤微环境,导致肿瘤凋亡和免疫激活damp释放。(图33b)肿瘤微环境内巨噬细胞直接通过吞食凋亡细胞或甚至edv而进行的相互作用,导致m1巨噬细胞极化和炎性细胞因子tnfα和il
‑
6释放。(图33c)m1巨噬细胞能够进一步溶解肿瘤细胞并释放mip
‑
1α,mip
‑
1α可以募集更多的免疫细胞。(图33d)未成熟的树突状细胞吞食凋亡细胞体和释放的肿瘤抗原——其响应ep
‑
edv
‑
682治疗而存在,并成熟,释放1型干扰素tnfα、il
‑
12p40和il
‑
6。(图33e)成熟的dc然后迁移到淋巴结以将抗原呈递至t细胞。(图33f)nk细胞激活也发生在肿瘤微环境中,导致ifnγ和tnfα以及rantes释放以吸引另外的免疫细胞。此外,激活的nk细胞有效地溶解肿瘤细胞。(图33g)rantes和mip
‑
1α的释放将另外的t细胞、nk细胞和巨噬细胞募集到肿瘤中,其中(图33h)肿瘤特异性cd8
+
t细胞然后通过肿瘤细胞溶解而促进响应。(图33i)这些步骤全部组合以产生有效的抗肿瘤免疫响应。
61.图34显示了响应edv处理的raw264.7和jaws ii细胞激活。(图34a)直接与edv制剂一起培育的raw264.7细胞的tnfα生产。(图34b)直接与edv制剂一起培育的raw264.7细胞的il
‑
6生产。(图34c)jaws ii细胞与未处理的ct26ep12.1和4t1细胞或用单独ep
‑
edv、ep
‑
edv
‑
682或682处理的ct26ep12.1和4t1细胞的jaws ii细胞的共培养物中的cd86和mhc ii类表达的流式细胞术直方图叠加。(图34d)通过流式细胞术确定的、与经处理的肿瘤细胞共培养的jaws ii细胞上的cd86表达的定量。(图34e)通过流式细胞术确定的、与经处理的肿瘤细胞共培养的jaws ii细胞上的mhc ii类表达的定量。数据表示平均值
±
s.e.m.并通过单向方差分析和tukey多重比较测试来分析。
62.图35显示了balb/c和balb/c裸异种移植物中响应edv治疗的体重变化和巨噬细胞激活。携载(图35a)ct26ep12.1、(图35b)4t1、(图35c)t84和(图35d)a549/mdr肿瘤的小鼠响应治疗的%体重变化。在初始剂量下观察到不超过5%的体重减轻,然后体重恢复并随着后续用药而稳定化。数据表示平均值
±
s.e.m.。edv治疗的小鼠的(图35e)a549/mdr和(图35f)t84肿瘤中的巨噬细胞的m1/m2(cd86:cd206)比例。(图35g)从a549/mdr肿瘤分离并与a549/mdr细胞以5∶1(e∶t)的比例共培养的cd11b
+
的xcelligence rtca。作图表示标准化的与细胞粘附和生长/死亡vs时间相关的细胞指数。(图35h)与来自盐水或egfr
‑
edv
‑
682治疗的小鼠的肿瘤的cd11b
+
细胞共培养的a549/mdr细胞在cd11b
+
细胞加入后6.5小时的%细胞溶解。
(图35i)从经治疗的4t1肿瘤分离的cd11b
+
细胞与4t1细胞的共培养物中的mip
‑
1α生产。数据(图35e、35f、35h和35i)表示平均值
±
s.e.m.并通过单向anova和tukey多重比较测试(图35e、35h、35i)或t检验(图35f)来分析。
63.图36显示nk细胞对edv处理的响应。(图36a)从携载t84肿瘤的balb/c裸鼠脾脏分离的与t84细胞以10∶1(e∶t)的比例共培养的nk细胞的xcelligence rtca。作图表示由标准化细胞指数计算的随时间的细胞活力。(图36b)从经盐水和egfr
‑
edv
‑
682治疗的小鼠的脾脏分离的nk细胞和t84细胞的共培养物中的粒酶b(granzyme b)生产。数据表示平均值
±
s.e.m.并且通过t检验来分析。(图36c)从携载a549/mdr肿瘤的balb/c裸鼠脾脏分离的与a549/mdr细胞以10∶1(e∶t)的比例共培养的nk细胞的xcelligence rtca。作图表示由标准化细胞指数计算的随时间的细胞活力(盐水n=5;egfr
‑
edv
‑
682n=4)。
64.图37显示了患者源胰管腺癌细胞的受体表达和药物敏感性筛选。(图37a)来自肿瘤头部的细胞的egfr表面受体定量。(图37b)来自肿瘤尾部的细胞的egfr表面受体定量。(图37c)与682敏感性相比,一线和二线化疗药物/药物组合的药物敏感性和ic 50。
65.图38显示来自总pbmc池(fsc v ssc)的单个细胞基于前向散射宽度(fsc
‑
w)vs前向散射面积(fsc
‑
a)进行门控,然后分析cd45染色(fsc v cs45
+
上的cd45
+
门控)。细胞活力为96%(计数和活力试剂盒#c00162,beckman coulter,数据未显示),基于fsc阈值判别器80排除死细胞。总树突状细胞(dc)在白细胞(cd45
+
)上门控,并被限定为hla
‑
dr
+
和谱系
‑
(#b53351,beckman coulter)。谱系阴性标记物由与相同的荧光团(pe)缀合的抗体群组成,该群抗体分别针对用于t细胞、单核细胞、b细胞和nk细胞阴性选择的cd3、cd14、cd19、cd20和cd56产生。其余的hla
‑
dr
+
细胞被门控为树突状细胞,并细分为浆细胞样dc(cd11c
‑
cd132+)或抗原呈递髓样dc(cd11c
+
cd123
‑
)。髓样dc(mdc)分为三个主要子集,cdlc+mdcl、cd141+mdc2(此处示为clec9a+)和cd16+mdc。cd14+的表达限定了在白细胞上门控的单核细胞(#b93604,beckman coulter),然后细分为经典(cd14+cd16
‑
)、中间(cd14+cd16+)和非经典(cd14+cd16++)。t细胞(#b53328,beckman coulter)是cd3+并在淋巴细胞(cd45+ssc低)上门控。然后限定了cd3
+
t细胞亚群、t辅助细胞(cd4
+
)和细胞毒性t细胞(cd8
+
)。针对ssc绘制pd1+cd8+细胞毒性t细胞门,并在cd8+t细胞上门控。所有fsc和ssc轴都是线性的,而荧光通道轴(所有cd标志物)是对数或双指数的(“logicle”,kaluza软件,beckman coulter)。
66.图39显示了流式细胞术分析,显示用于xcelligence rtca实验的细胞分离的纯度。(图39a)从小鼠肿瘤分离cd11b
+
细胞。密度图显示同种型对照vs.fsc和cd11b vs.fsc。样品的cd11b
+
细胞纯度为~80%。(图39b)从小鼠脾脏分离nk细胞。密度图显示同种型对照vs.fsc、nkp46 vs.fsc和cd11b vs.fsc。样品的nk细胞纯度为~90%。(图39c)从小鼠脾脏分离cd8
+
t细胞。密度图显示同种型对照vs.fsc、cd3e vs.fsc和cd8 vs.fsc。样品的t细胞(cd3e
+
)纯度为~90%,其中超过98%的那些t细胞为cd8
+
。
67.图40显示了在同系小鼠模型(balb/c小鼠中的
ep
ct26结肠肿瘤)中使用
ep
微细胞
dox
和微细胞
α
‑
gc
的组合治疗。
68.图41显示
ep
微细胞
dox
和微细胞
α
‑
gc
的组合治疗有效减少携载ct26同种异系移植物的balb/c小鼠中的大肿瘤。
69.图42显示
ep
微细胞
dox
和微细胞
α
‑
gc
对具有ct26同系移植物的balb/c小鼠的肿瘤消退的效果。
70.图43显示用(图43a和43b)
ep
微细胞
dox
和微细胞
α
‑
gc
、(图43d和43e)仅微细胞
α
‑
gc
、(图43f)仅
ep
微细胞
dox
和(图43c)盐水处理的不同尺寸的ct26同系移植物。
71.图44显示了用
ep
微细胞
dox
和微细胞
α
‑
gc
处理的不同尺寸的ct26同系移植物。
72.图45显示了微细胞
αgc
处理后不同时间点的jawsii细胞的αgc
‑
cd1d呈递(图45a
‑
e)
具体实施方式
73.i.概述
74.本发明基于以下发现:其包含下列组合的组合物可以协同性地改善癌症治疗策略:(i)抗肿瘤剂和i型干扰素激动剂;(ii)抗肿瘤剂和ii型干扰素激动剂;或(iii)抗肿瘤剂、i型干扰素激动剂和ii型干扰素激动剂,其中至少所述抗肿瘤剂被包装在完整细菌源微细胞中。
75.活性剂和免疫调节剂(一种或多种)的组合——其中至少抗肿瘤剂以及任选地i型和/或ii型干扰素激动剂被包装在完整的细菌源微细胞中——导致针对癌细胞的显著功效,以及惊地不存在对象体内抗药性的发展。所述组合物避免了抗肿瘤药物与免疫调节药物如i型和/或ii型干扰素激动剂组合的全身递送相关的毒性,以提供协同性改善的癌症治疗策略。
76.癌症免疫疗法的最新进展已导致特定癌症的空前的持久的临床响应(emens等,2017;farkona等,2016;oiseth和aziz,2017;sharma等,2017;ventola,2017)。然而,目前的免疫疗法策略跨越多种肿瘤类型造成的成功率有限,并且大部分最初显示令人鼓舞的肿瘤消退的患者随着时间的推移复发(emens等,2017;mellman等,2011;oiseth和aziz,2017;sharma等,2017;ventola,2017)。
77.在下面的实施例16中描述的数据阐明了肿瘤靶向的纳米细胞治疗剂的细胞免疫治疗功能机制,其中其通过递送与免疫系统的多臂接合组合的超细胞毒素对肿瘤发起双重攻击。该方法通过创建免疫原性肿瘤微环境和作用于多个免疫细胞亚群,从而避免患者可能出现的原发性和/或适应性抗性,而规避免疫疗法的当前一些陷阱。
78.此外,一部分患者由于缺乏肿瘤细胞抗原或缺乏免疫细胞浸润而缺乏肿瘤免疫原性,因此对目前可用的策略被呈现初始反应(emens等,2017;oiseth和aziz,2017;sharma等,2017)。因此,新的有力的免疫治疗方法的鉴定可导致临床结果显著改善,并且仍然是高度优先的领域。
79.为了增加有效的抗肿瘤免疫响应,必须自发地或在治疗上实现某些步骤。首先,可通过肿瘤细胞死亡原位得到的或外源递送的肿瘤细胞抗原必须被树突状细胞(dc)摄取(anguille等,2015;emens等,2017;jung等,2018;mellman等,2011)。结合抗原摄取,dc需要接收适当的成熟化信号,促进分化和抗原的加工和呈递增强,从而促进抗肿瘤功能而不是耐受性(anguille等,2015;emens等,2017;jung等,2018;mellman等,2011;simmons等,2012)。然后,这些成熟的、肿瘤抗原负载的dc必须有效产生抗肿瘤t细胞响应,该响应可通过肿瘤特异性细胞毒性t细胞的产生、nk和/或nkt细胞响应的触发以及1型t辅助细胞响应的增强等而发生(emens等,2017;fang等,2017;mellman等,2011;sharma等,2017;zitvogel等,2015)。抗肿瘤t细胞必须最终进入可能存在免疫抑制信号的肿瘤微环境,并有效执行其抗肿瘤功能(emens等,2017;mellman等,2011)。这些步骤中任何一个出现的问题都会阻碍
免疫治疗的功效,并且甚至可导致治疗完全失败(emens等,2017;mellman等,2011;sharma等,2017)。
80.目前,临床上最受关注的免疫治疗策略包括免疫检查点抑制剂和嵌合抗原受体t细胞疗法(car
‑
t)(emens等,2017;mellman等,2011;oiseth和aziz,2017;sharma等,2017;ventola,2017)。检查点抑制剂(如细胞毒性t淋巴细胞抗原4(ctla
‑
4)和程序性细胞死亡i/程序性细胞死亡1配体(pd
‑
1/pdl
‑
1))通过阻断免疫抑制信号的传递和直接刺激激活肿瘤微环境中的细胞毒性t淋巴细胞而发挥作用(dine等,2017;jenkins等,2018;sharpe,2017)。这些途径的抑制剂在特定癌症中已显示出显著临床结果,但跨不同癌症的总体响应率仍然很低(~15
‑
25%),并且这些疗法相关的免疫相关毒性可能很高(dine等,2017;emens等,2017;jenkins等,2018;sharpe,2017;ventola,2017)。随着不断地发现新的检查点作为潜在免疫靶标,很明显肿瘤能够利用一套复杂而多样的免疫抑制途径(dine等,2017;emens等,2017;farkona等,2016;jenkins等,2018;jenkins,2017)。因此,对检查点抑制剂的抗性的发展仍然是一个障碍,并且正在尝试利用多于一种检查点抑制剂的组合来克服这些问题,尽管这通常会加剧相关的毒性(dine等,2017;jenkins等,2018;sharma等,2017;ventola,2017)。
81.受到广泛关注的第二种疗法是car
‑
t细胞疗法,其需要对患者的t细胞进行基因改造,以表达具有限定的肿瘤抗原特异性并能够引发强大的t细胞激活以启动对目标肿瘤细胞的杀伤的膜融合受体(d'aloia等,2018';farkona等,2016;mellman等,2011;sharma等,2017)。这种治疗方法在“液体”血液学癌症的治疗中产生了空前的临床结果,但是迄今在靶向实体恶性病方面尚未产生具有可比性的响应——由于缺乏良好的特异性抗原靶标、肿瘤归巢性不良、肿瘤中的外渗不良和缺乏在不利肿瘤微环境中的持久性相关的限制(d'aloia等,2018';sharma等,2017)。还存在来自重度预治疗患者的淋巴细胞的可用性和制造时间长对于疾病快速进展的患者而言不是可行的治疗选择相关的实际限制(oiseth和aziz,2017;rezvani等,2017)。
82.engeneic dream vector(edv)是由直径400
±
20nm的无活力纳米细胞组成的一种细菌源传递系统,其通过重新激活细菌中细胞分裂的极体位点而产生(macdiarmid等,2007b)。已经证明这些纳米细胞可以与细胞毒性药物、sirna或mirna一起包装,并通过将双特异性抗体附着到纳米细胞的表面多糖而特异性靶向肿瘤细胞表面受体(macdiarmid等,2009;macdiarmid等,2007b;reid等,2013)。小鼠和犬的后静脉内给予研究表明,由于其尺寸,其被保留在血管系统中,但随后通过肿瘤相关的渗漏脉管系统迅速外渗到肿瘤中(macdiarmid等,2007b;sagnella等,2018)。通过相关双特异性抗体进行的肿瘤后细胞表面受体接合导致内体中的巨胞饮作用和有效载荷通过溶酶体中的细胞内降解而释放(macdiarmid等,2009;macdiarmid等,2007b;sagnella等,2018)。这些纳米细胞疗法的安全性已在三项i期临床试验中得到证实,其中已在各种晚期癌症患者中给予了上千剂量,并且负载682的edv目前正在i期试验中被递送给患者并且迄今显示有前景的安全性概况(2017;kao等,2015;solomon等,2015;van zandwijk等,2017;whittle等,2015)。
83.a.细菌微细胞递送方法概述
84.此前已经描述了细菌微细胞将化疗剂递送至癌细胞的应用。治疗癌症的这种递送方法将毒性化疗剂或药物或功能性核酸包装到一般约400nm直径的细菌源微细胞中。一般,
微细胞携带靶向特定癌细胞的抗体。该抗体附着至癌细胞表面,并且微细胞被癌细胞内化。以此方式,毒性化疗剂不会广泛遍布全身,因此减少了在毒性药物或化合物被递送到癌细胞内时的副作用和不耐受的机会。使用抗体靶向微细胞作为毒性化疗剂的递送载体导致杀死癌细胞所需的药物显著更少,从而提高治疗指数。
85.实际上,本发明人已经证明,微细胞(或engeneic dream vehicle(edv)可以将化疗药物如紫杉醇或多柔比星递送至小鼠(实施例1)、犬(实施例2)和猴子(实施例3)的异种移植肿瘤中。靶向的递送确保癌细胞接收到大部分化疗剂,导致低水平的毒性。参见实施例1
‑
3;还参见macdiarmid等,2007b;macdiarmid等,2007a;macdiarmid等,2009;和macdiarmid等,2016。此外,微细胞在异种移植模型中不诱导显著的免疫响应,并且微细胞被良好耐受(实施例4)。因此,完整的细菌源微细胞是被良好耐受的用于向患者递送抗癌药物的媒介物,实例包括靶向晚期实体肿瘤的多柔比星(实施例5)、靶向成胶质细胞瘤的多柔比星(实施例6)和靶向间皮瘤的microrna
‑
16a(实施例7)。
86.但是,这些治疗策略并未导致所有患者的所有癌症完全缓解或治愈。因此,需要改进的癌症治疗疗法。本发明人发现,使用具有三种不同类型的有效载荷的微细胞的组合产生惊人的显著和有效的临床功效。
87.具体地,本发明人发现,包含化疗剂(在下面的实例中,例如,该剂是pnu
‑
159682,一种超毒性化疗药物)的微细胞与包含i型干扰素激动剂和/或ii型干扰素激动剂的微细胞组合产生协同性的抗肿瘤效果,并且被患有晚期胰腺癌的患者良好耐受。参见实施例12。实际上,晚期胰腺癌患者在该治疗后呈现显著改善的生活质量,这对于该阶段的患者而言是显著的。这种三重或二重组合策略可协同性地改善癌症的治疗,尤其是晚期末期癌症。发明人还发现,包含化疗剂的微细胞与包含ii型干扰素激动剂的微细胞组合产生协同性的抗肿瘤效果。
88.还惊人地发现,微细胞包装的抗肿瘤剂与ii型干扰素激动剂组合的双重组合,并且在不存在i型干扰素激动剂的情况下,产生了针对大尺寸肿瘤的显著功效。这样的结果以前没有被描述过。从理论上讲,在某些患者中,i型干扰素激动剂和ii型干扰素激动剂组合可能是反效果的,因为这两种类型的干扰素激动剂可能是竞争而不是协同地作用。下文将更详细地描述此数据。
89.以下描述概述了与这些发现相关的本发明,而不将本发明限于所述特定实施方式、方法、方案或试剂。同样,这里使用的术语仅描述具体实施例,而不限制本发明的范围。
90.b.实验结果总结
91.(i)微细胞包装的抗肿瘤剂与微细胞包装的i型干扰素激动剂组合
92.在第一实施方式中,描述了涉及细菌微细胞包装的抗肿瘤剂与包装在细菌微细胞中的i型干扰素激动剂组合的组合的组合物和方法。
93.实施例11和图18描述了数据,显示在用各种微细胞(edv)组合物治疗的小鼠的肺癌异种移植模型中的结果,如下表总结。第1组和第5组的动物被给予了被包装在完整细菌源微细胞中的化疗剂(pmj 159682)和也被包装在完整细菌源微细胞中的i型干扰素激动剂(40mer双链dna或50mer双链dna)的组合。所有微细胞组合物均导致肿瘤生长稳定化。然而,最显著的结果是在对因实验第1部分中的盐水治疗而造成的大肿瘤尺寸进行治疗后获得。在后续在实验第2部分中用包含微细胞包装的抗肿瘤剂+微细胞包装的i型干扰素激动剂的
组合的组合物治疗盐水治疗对照组时,肿瘤尺寸经5天时间减少62%。
[0094][0095]
在实施例11的跟进中(结果示于图19),包装在微细胞中的i型干扰素激动剂的添加导致肿瘤尺寸显著减小,这是在没有i型干扰素激动剂佐剂的情况下使用包装在微细胞中的抗肿瘤剂时未观察到的。结果总结在下表中。这些结果清楚地证明了添加微细胞包装的i型干扰素激动剂对微细胞包装的抗肿瘤剂的佐剂作用。
[0096][0097][0098]
(ii)微细胞包装的抗肿瘤剂与微细胞包装的i型干扰素激动剂和任选地ii型干扰素激动剂(非微细胞包装)组合
[0099]
第二实施方式涉及利用微细胞包装的抗肿瘤剂与微细胞包装的i型干扰素激动剂组合或微细胞包装的抗肿瘤剂与微细胞包装的i型干扰素激动剂以及ii型干扰素激动剂(无细菌微细胞)的方法和组合物。
[0100]
实施例12中显示的临床结果反映了本发明组合物的显著和惊人的有效性的进一步证据。具体地,实施例12涉及当用包含以下的组合物治疗患有晚期实体肿瘤的患者时发生的情况:(1)微细胞包装的抗肿瘤剂和微细胞包装的i型干扰素激动剂的组合;(2)微细胞包装的抗肿瘤剂、微细胞包装的i型干扰素激动剂、和ii型干扰素激动剂的组合。
[0101]
具体地,实施例12中详述的人类临床数据证明了在人类患者中用作微细胞包装的抗肿瘤剂的佐剂的i型和ii型ifn激动剂的安全性概况。参见下表3中的数据,相关的包装在完整细菌衍生微细胞的i型干扰素激动剂是40mer双链dna(ed v
s40mer
)或60mer双链dna(edv
s60mer
)。
[0102]
另外,关于用尽所有其他治疗选择的4期胰腺癌患者获得的结果是显著的。患者的
肿瘤标志物(ca 19
‑
9)水平在初始三个剂量(相当于仅10天的治疗)后下降超过90%。十个剂量后,其还进一步下降,肿瘤标志物水平降低几乎95%。该患者还显示与大多数iv期胰腺癌患者经历的恶病质状态相比显著的增重,并报告了生活质量的显著改善。这些结果是显著的,特别是考虑到晚期胰腺癌相关的预后不良。
[0103]
总之,五名患者共接受了69个剂量的
egfr(v)
edvs
pnu/dox
或
egfr(v)
edvs
pnu
+edvs
40mer/60mer
(i型ifn激动剂)
±
imukin(ii型ifn激动剂)。该治疗耐受良好,并且添加免疫调节佐剂未呈现改变单药负载和靶向edv的安全性概况。
[0104][0105][0106]
(iii)微细胞包装的抗肿瘤剂与ii型干扰素激动剂组合
[0107]
实施例13描述了为评价包装在微细胞中的抗肿瘤剂与ii型干扰素激动剂(例如ifn
‑
γ)组合的有效性而进行各种研究的结果。结果表明,添加ii型干扰素激动剂在包括肺癌和乳腺癌在内的各种癌症的异种移植模型中增加或增强包装在微细胞中的抗肿瘤剂的抗癌效果。此外,在实施例13中提出并在下表4中列出的数据表明,在通常对单独抗肿瘤剂具有抗性的肿瘤的治疗中ii型干扰素激动剂添加至包含包装在微细胞中的抗肿瘤剂的组合物是实现肿瘤稳定化所必不可少的。因此,组合微细胞包装的抗肿瘤剂与ii型干扰素激动剂可以克服药物抗性。
[0108][0109]
(iv)微细胞包装的抗肿瘤剂、微细胞包装的i型干扰素激动剂、和ii型干扰素激动剂(单独或微细胞包装式)的三重组合
[0110]
本发明人还发现,微细胞包装的抗肿瘤剂、微细胞包装的i型干扰素激动剂、和ii型干扰素激动剂(单独或微细胞包装式)的三重组合可产生显著的抗癌效果。具体地,实施例14详述了用微细胞包装的抗肿瘤剂、微细胞包装的i型干扰素激动剂和ii型干扰素激动剂的组合治疗患有晚期内源性肿瘤(脑癌、肉瘤或黑素瘤)的犬。结果表明,该组合式组合物耐受良好。此外,在7只可评价动物中的6只(85.7%)中该疾病被稳定化,虽然一只犬实现了近部分响应(肿瘤尺寸减少29.8%)。
[0111]
(v)在不存在i型干扰素激动剂的情况下,微细胞包装的抗肿瘤剂与微细胞包装的ii型干扰素激动剂组合的二重组合
[0112]
在另一实施方式中,本发明涉及惊人的发现:包含微细胞包装的抗肿瘤剂和微细胞包装的ii型干扰素激动剂如例如α
‑
半乳糖基神经酰胺(α
‑
gc)的组合的组合物,在没有i型干扰素激动剂的情况下,显示出惊人的抗癌功效。
[0113]
具体地,实施例23描述了示例微细胞包含的治疗剂和微细胞包含的ii型干扰素激动剂的二重组合对抗肿瘤的功效的数据。该结果表明缺乏i型干扰素激动剂的组合物可用于有效治疗肿瘤。还参见图40和42。实验结果表明,接受
ep
微细胞
dox+
微细胞
α
‑
gc
(ii型干扰素激动剂)的组合治疗组,与盐水和
ep
微细胞
dox
治疗相比,肿瘤进展明显停止。该结果支持向
ep
微细胞
dox
添加微细胞
‑
gc处理导致免疫佐剂效果的理论。
[0114]
进一步的数据表明,盐水治疗的对照肿瘤在治疗变为药物和α
‑
gc edv介导的双重组合疗法之后显示显著的肿瘤消退(图41);例如,微细胞包装的抗肿瘤剂和微细胞包装的ii型干扰素激动剂的组合。具体地,已达到800mm3的肿瘤在实验终止前3天内下降到600mm3以下——短时间内肿瘤尺寸显著急剧减少(~25%)。双重组合的组合物在短时间内显著减小大肿瘤的能力在本发明之前是未知的。
[0115]
在本发明的一个实施方式中,双重组合的组合物(例如,微细胞包装的抗肿瘤剂与微细胞包装的ii型干扰素激动剂组合)可使肿瘤尺寸(包括大肿瘤的尺寸)减少约5%、
10%、约15%、约20%、约25%、约30%、约35%、约40%、约45%、约50%、约55%、约60%、约65%、约70%、约75%、约80%、约85%、约90%、约95%或约100%。肿瘤尺寸的减少可以经任何合适的时间段测量,如约3天、约5天、约1周、约2周、约3周、约1个月、约2个月、约3个月、约4个月、约5个月、约6个月、约7个月、约8个月、约9个月、约10个月、约11个月、约12个月、约1.5年、约2年或更长。
[0116]
c.免疫疗法数据
[0117]
实施例16详述了证明靶向肿瘤细胞表面受体的微细胞包装的抗肿瘤剂作为癌症免疫疗法,例如作为细胞免疫疗法的数据。具体地,该实施例示例了细菌微细胞激活先天免疫系统细胞的能力,包括巨噬细胞,nk细胞和树突状细胞。其后是树突状细胞成熟和抗原呈递,导致适应性t细胞响应,其中产生肿瘤特异性细胞毒性t细胞,并导致其他免疫细胞进一步募集到肿瘤微环境。这种方法通过创建免疫原性肿瘤微环境以及作用于多个免疫细胞亚群,从而避免患者体内可能产生的原发性和/或适应性抗性,而规避了免疫疗法的当前一些陷阱。
[0118]
因此,该实施例显示了细菌微细胞在肿瘤细胞内递送细胞毒性药物和同时引发特异性针对肿瘤的先天性和适应性免疫响应的能力。
[0119]
进一步的免疫疗法数据显示在实施例18中,该实施例描述了数据证明在用包含抗肿瘤剂的靶向微细胞治疗后nk细胞在体内采用抗肿瘤表型。这是非常重要的,因为nk细胞是先天免疫系统的主要效应细胞,并且受到激活和抑制信号的平衡的严格调控(morvan和lanier,2016;wallace和smyth,2005)。nk细胞功能受损与肿瘤发生、生长和转移增加相关,因此其在促进抗肿瘤免疫响应中的重要性被充分证明(fang等,2017;morvan和lanier,2016;rezvani等,2017;wallace和smyth,2005)。
[0120]
值得关注地,实施例19详述了数据,显示在用微细胞包封的(encapsulated)抗肿瘤剂(例如,pnu
‑
159682)治疗后呈现在肿瘤微环境内主要th1细胞因子响应。肿瘤微环境中细胞因子和趋化因子的产生使免疫细胞有效地相互交流以生成配合响应,该配合响应可以是肿瘤促进性或抑制性的(belardelli和ferrantini,2002;lee和margolin,2011)。个体细胞因子对免疫响应的效果取决于多种因素,包括局部浓度、细胞因子受体表达模式和周围细胞的激活状态(lee和margolin,2011)。因此,已证明多种细胞因子能够引起对肿瘤生长的相反作用(dredge等,2002;landskron等,2014;lee和margolin,2011)。
[0121]
此外,实施例20详述了数据,表明用微细胞包封的抗肿瘤剂(例如,pnu
‑
159682)治疗导致肿瘤特异性cd8+t细胞产生。初始体外实验表明,edv治疗可导致树突状细胞成熟——通过直接相互作用或缘于响应于负载有有效化疗剂的靶向edv的细胞死亡。因此,此实验旨在考察这个结果是否可以转化为体内dc成熟和抗原呈递,导致肿瘤特异性cd8
+
细胞毒性t细胞产生。所得数据表明,微细胞治疗成功引起肿瘤特异性cd8
+
t细胞产生。此外,在用微细胞包封的抗肿瘤剂(例如,pnu
‑
159682)治疗的小鼠的淋巴结中观察到总t细胞数(cd3+)显著增加以及cd4+和cd8+t细胞数均显著增加。)(图31g)。还检测到在被治疗小鼠的淋巴结中成熟树突状细胞显著增加(图31h),并且分离自被治疗小鼠的cd8
+
t细胞与4t1细胞之间的相互作用的可视化显示,这些t细胞能够将穿孔素(绿色)附着至并排入肿瘤细胞(图31i)。
[0122]
实施例21证明了负载有抗肿瘤剂(例如,超细胞毒素pnu
‑
159682)的靶向细菌微细
胞不仅有效地将此药物递送至肿瘤位点,而且还可以通过刺激多个免疫细胞亚群而充当免疫治疗剂的能力。该实施例证实了微细胞包裹(capsulated)的抗肿瘤剂治疗将包括巨噬细胞、nk细胞和cd8
+
t细胞在内的免疫细胞亚群推向能够有效消除肿瘤细胞的抗肿瘤表型的能力。当结合抗肿瘤剂的有效性时,这导致对肿瘤的双重攻击。
[0123]
虽然癌症免疫疗法的想法已经存在了数十年,但只有最近其潜能才开始通过多种免疫疗法的批准被实现(farkona等,2016;ventola,2017)。细菌微细胞代表一种独特的组合细胞免疫疗法,其首先通过将细胞毒性剂直接递送至肿瘤来创建免疫原性肿瘤微环境,其中其直接或间接刺激先天免疫系统趋向抗肿瘤表型。这种先天免疫激活然后触发适应性响应,其中产生肿瘤特异性细胞毒性t细胞(图33)。
[0124]
在静脉内给予后,微细胞通过肿瘤的渗漏脉管系统外渗到肿瘤中,其中携带其毒性有效载荷的靶向微细胞的给予剂量的≥30%在2小时时间内直接沉积到肿瘤微环境中(macdiarmid等,2007b)。靶向的细菌微细胞结合肿瘤细胞上的受体(在实施例21的情况下为4t1和ct26ep12.1),然后被内化,有效地将其有效载荷(抗肿瘤剂)直接在肿瘤细胞内递送。pnu
‑
159682是高度有效的超级细胞毒素,导致在递送至肿瘤细胞后24小时内快速凋亡(图33a)。然后,通过细菌微细胞(例如ep
‑
edv
‑
682)处理产生的凋亡细胞和damp信号可以与先天性免疫细胞(如肿瘤相关巨噬细胞(tam))相互作用,并刺激cd86的上调和th1促炎性细胞因子如tnfα和il
‑
6的产生(图33b)。这些改变是能够溶解肿瘤细胞和释放细胞因子以传导其他免疫细胞亚群激活的信号的巨噬细胞的m1极化的一般(特征),因此已被证明具有抗肿瘤特性(sawa
‑
wejksza和kandefer
‑
szerszen,2018;yuan等,2015)。
[0125]
此外,细菌微细胞本身也可以与tam直接相互作用产生相似的m1极化,尽管预期这将会在当前系统中以非常低的水平发生。tam总体上是肿瘤微环境中最丰富的免疫细胞,并且已证明tam数量增加与预后不良和肿瘤生长增加相关(sawa
‑
wejksza和kandefer
‑
szerszen,2018)。这很大一部分是由于tam主要由抗炎性m2巨噬细胞组成,该抗炎性m2巨噬细胞已被证明具有肿瘤促进特性,而炎性m1巨噬细胞呈现抗肿瘤特性(sawa
‑
wejksza和kandefer
‑
szerszen,2018;yuan等,2015)。实施例21在4种不同的肿瘤模型中证明了细菌微细胞治疗转变肿瘤微环境内的m1:m2平衡的能力。尽管在不同的肿瘤模型中这种转变的程度存在差异,但显示m1极化的增加转化为从已用细菌微细胞治疗的小鼠的肿瘤分离的tam造成的肿瘤细胞溶解的增加。除了表型向m1的转变外,来自细菌微细胞治疗的小鼠的肿瘤的tam还分泌增加量的mip
‑
1α(图33c)——一种已被证实在促进免疫细胞募集和具体地nk细胞、cd4
+
t细胞和cd8
+
t细胞的肿瘤浸润中发挥作用(allen等,2018)。
[0126]
除tam激活外,未成熟树突状细胞(dc)与细菌微细胞直接相互作用,或更可能与凋亡细胞和被细菌微细胞所治疗的肿瘤产生的damp信号相互作用,导致树突状细胞成熟化和迁移至淋巴结进行抗原呈递。dc已被作为癌症免疫治疗的潜在靶标探索,因为dc被认为是最有效的抗原呈递细胞并构成先天性和适应性免疫系统之间的桥梁(allen等,2018)。
[0127]
基于dc的免疫疗法的大多数当前策略涉及离体操作和dc或dc前体的启动,但是该策略的成功因多种因素而受到限制,所述因素包括:免疫耐受性的产生、数量不足的cd8
+
细胞毒性t细胞(ctl)或抗肿瘤效果不良的cd8
+
细胞毒性t细胞(ctl)的诱导以及肿瘤微环境的抑制属性(anguille等,2015;jung等,2018;landskron等,2014;oiseth和aziz,2017)。细菌微细胞治疗实现响应于垂死肿瘤细胞的肿瘤微环境内体内的dc引发和成熟(图33d)。不
成熟的dc能够吞食damp和/或响应靶向的、药物负载的细菌微细胞而产生的凋亡肿瘤细胞体。这些damp和垂死肿瘤细胞然后被加工通过mhc i类和ii类分子在dc表面进行抗原呈递,并伴随dc成熟。共刺激分子cd86、cd80和mhc ii类(其已被鉴定为dc成熟过程的标志物)的上调显示在与细菌微细胞处理的肿瘤细胞共培养的dc中发生,并且细菌微细胞治疗的小鼠的肿瘤引流淋巴结中检测到的成熟dc的百分比增加(anguille等,2015;cauwels等,2018;simmons等,2012)。在成熟过程中,dc迁移至肿瘤引流淋巴结,以将抗原呈递至t细胞,从而增加cd4
+
t辅助细胞和肿瘤特异性cd8
+
ctl的产生,启动对肿瘤的适应性免疫响应(图33e)。随后检测到与细菌微细胞处理过的肿瘤细胞共培养的dc的ifnα/β、tnfα、il
‑
12p40和il
‑
6的产生增加,此外还在4t1和ct26ep12.1肿瘤模型中均观察到肿瘤微环境中的ifnα浓度显著增加(实施例21)。已显示肿瘤微环境中1型ifn(ifnα/β)和ifn刺激基因的表达水平与有利的疾病结果相关,并且实际上甚至可能对包括免疫疗法在内的癌症疗法的成功是必要的(cauwels等,2018);fitzgerald
‑
bocarsly和feng,2007;zitvogel等,2015)。1型ifn的抗肿瘤活性通过dc、t和b淋巴细胞、nk细胞和巨噬细胞的免疫细胞激活而间接产生(cauwels等,2018;fitzgerald
‑
bocarsly和feng,2007;showalter等,2017;zitvogel等,2015)。
[0128]
结合增强巨噬细胞和dc抗肿瘤功能,用包含抗肿瘤剂的细菌微细胞进行处理能够引起nk细胞激活,导致细胞毒性增加(图33f)。nk细胞具有以抗原独立性方式溶解恶性细胞的固有能力,因此必须严格控制其激活和功能状态,以避免对宿主的潜在不利影响。在肿瘤微环境中吸引nk细胞并激活nk细胞的能力对于其发挥其抗肿瘤功能的能力至关重要。已知在ep
‑
edv
‑
682治疗的肿瘤的微环境中显著增加的包括il
‑
2、ifnγ和ifnα在内的细胞因子会激活nk细胞以增加细胞因子产生并增强细胞溶解功能(fang等,2017;ferlazzo和munz,2004;lee和margolin,2011;morvan和lanier,2016;rezvani等,2017)。实际上,证据表明激活nk细胞的细胞毒性需要1型ifn(ferlazzo和munz,2004;muller等,2017)。此外,1型ifn能够诱导细胞衰老,然后上调肿瘤细胞中nkg2d配体的表达,从而促进其被nk细胞清除(muller等,2017)。在用ep
‑
edv
‑
682治疗的小鼠的肿瘤内的nk细胞上观察到nkg2d受体的上调,并且该受体被证明对分离自ep
‑
edv
‑
682治疗的小鼠的nk细胞的细胞溶解能力有重大贡献。此外,未成熟的、中间的和成熟的小鼠nk细胞表达可与趋化因子miplα和rantes结合的ccr1和ccr5趋化因子受体,二者均在ep
‑
edv
‑
682治疗的肿瘤中以及被edv
‑
682治疗的小鼠的巨噬细胞和nk细胞被上调(bernardini等,2016)。
[0129]
趋化因子,如miplα和rantes,负责将辅助和效应免疫细胞(包括nk细胞、巨噬细胞和t细胞)进一步募集到肿瘤微环境(图33g)(allen等,2018;bernardini等,2016;zibert等,2004)。在因包括巨噬细胞、nk细胞和dc在内的edv处理而导致的初始先天免疫响应之后,进行适应性免疫响应,其中肿瘤特异性ctl和t辅助细胞产生,然后被募集到肿瘤位点(图33h)。然后,肿瘤特异性ctl靶向并溶解肿瘤细胞,进一步促进已被其他免疫细胞亚群与靶向的、药物负载的edv结合产生的主要抗肿瘤环境。靶向的、药物负载的edv治疗引起主要th1响应,如肿瘤微环境中th1细胞因子(tnfα、ifnα、ifnγ、il
‑
2和il
‑
6)的增加所证明。如前所述,先天免疫细胞亚群在被激活时成为这些具体细胞因子中一种或多种的主要来源。t细胞同样能够产生所有上述细胞因子(belardelli和ferrantini,2002;lee和margolin,2011)。先天免疫细胞或t细胞释放这些细胞因子负责其他免疫细胞的共同刺激、激活、生长和抗原呈递增加,形成反馈环路,其进一步增强免疫系统的抗肿瘤活性(图33i)(lee和
margolin,2011)。
[0130]
细菌微细胞治疗代表了一种独特的癌症治疗策略,能够将常规的和新型的药物疗法直接递送至肿瘤位点,并随后引发抗肿瘤免疫响应。对肿瘤的双重攻击首先通过响应所递送的治疗剂的细胞死亡,然后先天免疫细胞激活导致适应性免疫响应而发生。这种类型的治疗(疗法,therapy)相对于当前免疫治疗策略具有某些优势,因为免疫细胞激活发生在体内,并且主要发生在肿瘤位点,肿瘤位点是快速变化的动态环境。此外,其创造了免疫原性肿瘤环境,并引起对多个免疫细胞亚群的效果,避免与对其肿瘤显示极少甚至无免疫响应的患者或对仅靶向单一免疫细胞亚群的疗法的适应相关的问题。实施例21中描述的研究突出了细菌微细胞作为一种新型的癌症免疫治疗剂的潜力,并且鉴于这种技术在有效载荷和靶向能力方面的多功能性,未来的细菌微细胞制剂可以进一步利用其固有的免疫原性(macdiarmid等,2007a)。
[0131]
d.超毒性抗肿瘤剂
[0132]
实施例17详述了证明有效递送超毒性抗肿瘤剂例如pnu
‑
159682的数据,由于与该化合物相关的严重毒性而其不能利用常规手段递送。具体而言,实施例17详述了pnu
‑
159682是怎样的对于甚至抗药性癌细胞的ic50也在pm范围内的超级细胞毒素(quintieri等,2005),这意味着该化合物由于严重的全身性毒性而不能在临床上使用(staudacher和brown,2017)。然而,当包封在细菌微细胞中时,诸如pnu
‑
159682的超细胞毒素可以有效地被递送至肿瘤而几乎没有副作用。
[0133]
ii.组合物组分
[0134]
如上所述,本发明的组合物包含至少两种不同的活性剂,抗肿瘤剂和i型干扰素激动剂、ii型干扰素激动剂、或i型干扰素激动剂和ii型干扰素激动剂以及抗肿瘤剂。该三种不同的活性剂可以包装在一个、两个或三个不同的微细胞中。ii型干扰素激动剂也可以在不被包装在微细胞中的情况下被包括在本发明的方法和组合物中。
[0135]
a.可用于治疗癌症的抗肿瘤或细胞毒性活性剂
[0136]
短语“抗肿瘤剂”表示预防或抑制肿瘤细胞的生长、发育、成熟或扩散的化学或生物学药物。术语“抗肿瘤剂”与“抗癌剂”和“化疗剂”被可互换使用。
[0137]
在本公开的上下文中,与常规医学实践一致,选择用于治疗给定脑肿瘤患者的抗肿瘤剂取决于几个因素。这些因素包括但不限于患者的年龄、karnofsky评分以及患者可能接受过的无论任何在前治疗。一般参见principles and practice of neuro
‑
oncology,m.mehta(demos medical publishing 2011)和principles of neuro
‑
oncology,d.schiff and p.o’neill,eds.(mcgraw
‑
hill 2005)。
[0138]
该组合物可以包含至多约1mg的抗肿瘤剂或化疗药物。可选地,化疗药物量可以是至多约750μg、约500μg、约250μg、约100μg、约50μg、约10μg、约5μg、约1μg、约0.5μg或约0.1μg。在另一方面,组合物包含化疗药物,该化疗药物的量小于不被包装到微细胞中使用时的治疗有效药物量的约1/1,000,或可选地小于约1/2,000、1/5,000、1/10,000、1/20,000、1/50,000、1/100,000、1/200,000或1/500,000。根据本公开的再另一方面,组合物可包含至少约1nmol的化疗药物。因此,本公开还包括其中化疗药物量分别为至少约2nmol、约3nmol、约4nmol、约5nmol、约10nmol、约20nmol、约50nmol、约100nmol或约800nmol的实施方式。
[0139]
在本公开的上下文中,选择用于治疗给定肿瘤的抗肿瘤剂取决于几个因素。这些
因素包括但不限于患者的年龄、肿瘤的阶段以及患者可能已接受的任何在前疗法。
[0140]
根据本公开,可以从以下详述的类别中选择一种药物,以包装到完整的细菌源微细胞中。这些药物也可以是通过药物设计和发现工作而设计的合成类似物。任何已知的化疗剂均可用于本发明的组合物中。已知化疗剂的例子包括但不限于:
[0141]
(1)烷化剂,如芥子气衍生物(二氯甲基二乙胺(mechlorethamine)、环磷酰胺(cytoxan)、苯丁酸氮芥(chlorambucil)(leukeran)、美法仑(melphalan)和异环磷酰胺(ifosfamide))、亚乙基亚胺(噻替哌(thiotepa)(thioplex)和六甲基三聚氰胺)、烷基磺酸酯(盐)(白消安(busulfan)(myleran))、肼和三嗪(六甲密胺(altretamine)(hexalen)、丙卡巴嗪(procarbazine)(matulane)、达卡巴嗪(dacarbazine)(dtic)和替莫唑胺)、亚硝基脲(卡莫司汀(carmustine)、洛莫司汀(lomustine)和链脲霉素(streptozocin))、和金属盐(卡铂(carboplatin)、顺铂(platinol)和奥沙利铂(oxaliplatin))、二氯甲基二乙胺和马法兰(alkeran);
[0142]
(2)植物生物碱、萜类和拓扑异构酶抑制剂,如长春花生物碱(长春新碱(oncovin)、长春碱(velban)、长春地辛和长春瑞滨)、紫杉烷(紫杉醇(taxol)和多西他赛(taxotere))、鬼臼毒素(podophyllotoxins)(etoposide和tenisopide)和喜树碱(camptothecan)类似物(伊立替康(irinotecan)和拓扑替康(topotecan));
[0143]
(3)抗肿瘤抗生素,如蒽环类(多柔比星(doxorubicin)(adriamycin、鲁比克斯(rubex)、多西尔(doxil))、柔红霉素(daunorubicin)、表柔比星(epirubicin)、米托蒽醌(mitoxantrone)、依达比星(idarubicin)、多卡霉素(duocarmycin))、更生霉素(dactinomycin)(cosmegen))、色霉素(dactinomycin和普卡霉素(plicamycin)光神霉素(mithramycin))和各种(丝裂霉素(mitomycin)和博来霉素(bleomycin)(blenoxane));
[0144]
(4)抗代谢物,如叶酸拮抗剂(甲氨蝶呤)、嘧啶拮抗剂(5
‑
氟尿嘧啶、foxuridine、阿糖胞苷(cytarabine)、氟尿嘧啶(5
‑
fu)、卡培他滨(capecitabine)和吉西他滨(gemcitabine))、嘌呤拮抗剂(6
‑
巯基嘌呤(purinethol)和6
‑
硫鸟嘌呤)、6
‑
硫嘌呤和腺苷脱氨酶抑制剂(克拉屈滨(cladribine)(leustatin)、氟达拉滨(fludarabine)、奈拉拉滨(nelarabine)和戊他汀(pentostatin))、阿扎胞苷(azacitidine)、硫鸟嘌呤(thioguanine)和阿糖胞苷(cytarabine)(ara
‑
c);
[0145]
(5)拓扑异构酶抑制剂,如拓扑异构酶i抑制剂(伊洛替康(ironotecan)、拓扑替康(topotecan))和拓扑异构酶ii抑制剂(氨苄菌碱(amsacrine)、依托泊苷(etoposide)、依托泊苷磷酸酯、替尼泊苷(teniposide));
[0146]
(6)激素剂,例如雌激素和雄激素抑制剂(他莫昔芬(tamoxifen)和氟他胺(flutamide))、促性腺激素释放激素激动剂(亮丙瑞林(leuprolide)和戈瑟瑞林(goserelin)(zoladex))、芳香酶抑制剂(氨基谷氨酰胺(aminoglutethimide)和阿那曲唑(anastrozole)(arimidex));
[0147]
(7)dna低甲基化剂,例如阿扎胞苷、地西他滨;
[0148]
(8)聚(二磷酸腺苷[adp]
‑
核糖)聚合酶(parp)途径抑制剂,如伊尼帕利布(iniparib)、奥拉帕尼(olaparib)、维利帕尼(veliparib);
[0149]
(9)pi3k/akt/mtor途径抑制剂,例如依维莫司(everolimus);
[0150]
(10)组蛋白脱乙酰基酶(hdac)抑制剂,例如伏立诺他(vorinostat)、恩替司他
(entinostat)(sndx
‑
275)、莫西司他(mocetinostat)(mgcd0103)、帕诺比司他(panobinostat)(lbh589)、罗米地辛(romidepsin)、丙戊酸(valproic acid)、细胞周期蛋白依赖性激酶(cdk)抑制剂,例如夫拉平度(flavopiridol)、奥洛莫卡因(olomoucine)、罗斯科维汀(roscovitine)、肯帕龙(kenpaullone)、ag
‑
024322(pfizer)、fascaplysin、ryuvidine、purvalanol a、nu2058、bml
‑
259、su 9516、pd
‑
0332991、p276
‑
00。热休克蛋白(hsp90)抑制剂,例如格尔德霉素(geldanamycin)、坦西霉素(tanespimycin)、阿维斯霉素(alvespimycin)、拉迪霉素(radicicol)、德盖林(deguelin)和biib021;
[0151]
(11)鼠双微体2(mdm2)抑制剂,例如顺式咪唑啉、苯二氮卓二酮(benzodiazepinedione)、螺
‑
羟吲哚、异喹啉酮、噻吩、5
‑
脱氮黄素、色胺;
[0152]
(12)间变性淋巴瘤激酶(alk)抑制剂,例如氨基吡啶、二氨基嘧啶、吡啶基异喹啉、吡咯并吡唑、吲哚并咔唑、吡咯并嘧啶、二苯胺并嘧啶;
[0153]
(13)聚[adp核糖]聚合酶(parp)抑制剂,例如苯甲酰胺、酞菁酮(phthalazinone)、三环吲哚、苯并咪唑、吲唑、吡咯并咔唑、酞嗪酮、异吲哚啉酮;和
[0154]
(14)各种抗癌药物,例如氨曲林(amsacrine)、天冬酰胺酶(elspar)、羟基脲、米托蒽醌(novantrone)、米托坦(mitotane)(lysodren)、美登素类、视黄酸衍生物、骨髓生长因子(沙格司亭和非格司亭)、氨磷汀(amifostine)、叶酸代谢破坏剂例如培美曲塞(pemetrexed)、核糖核苷酸还原酶抑制剂(羟脲)、肾上腺皮质类固醇抑制剂(米托坦)、酶(天冬酰胺酶(asparaginase)和培门冬酶(pegaspargase))、抗微管剂(雌莫司汀(estramustine))和类维生素a(贝沙罗汀(bexarotene),异维a酸(isotretinoin)、维甲酸(tretinoin)(atra))。
[0155]
示例小分子药物亚类的化疗药物为放线菌素d、马法兰(alkeran)、ara
‑
c、阿那曲唑(anastrozole)、bicnu、比卡鲁胺(bicalutamide)、博来霉素、白消安、卡培他滨、卡铂(carboplatin)、卡铂(carboplatinum)、卡莫斯汀(carmustine)、ccnu、苯丁酸氮芥(chlorambucil)、顺铂、克拉屈滨(cladribine)、cpt
‑
11、环磷酰胺、阿糖胞苷、胞嘧啶阿拉伯糖苷(cytosine arabinoside)、环磷酰胺(cytoxan)、达卡巴嗪(dacarbazine)、更生霉素(dactinomycin)、柔红霉素、右雷佐生(dexrazoxane)、多西他赛(docetaxel)、多柔比星、dtic、表柔比星(epirubicin)、乙撑亚胺(ethyleneimine)、依托泊苷(etoposide)、氟尿苷(floxuridine)、氟达拉滨(fludarabine)、氟尿嘧啶、氟他胺(flutamide)、福莫司汀(fotemustine)、吉西他滨、六甲胺、羟基脲、依达比星(idarubicin)、异环磷酰胺(ifosfamide)、伊立替康(irinotecan)、洛莫斯汀(lomustine)、二氯甲基二乙胺、美法仑、巯基嘌呤、甲氨蝶呤、丝裂霉素、米托坦、米托蒽醌(mitoxantrone)、奥沙利铂、紫杉醇、帕米膦酸(pamidronate)、戊他汀、普卡霉素(plicamycin)、丙卡巴肼(procarbazine)、类固醇、链脲霉素、sti
‑
571、链脲霉素、他莫昔芬、替莫唑胺、替尼泊苷(teniposide)、四嗪、硫鸟嘌呤、噻替哌、拓优得(tomudex)、拓扑替康(topotecan)、曲奥舒凡(treosulphan)、曲美沙特(trimetrexate)、长春碱、长春新碱、长春地辛、长春瑞滨、vp
‑
16、和希罗达(xeloda)。
[0156]
美登素类(分子量:~738道尔顿)是美登素的一组化学衍生物,具有强细胞毒性。尽管由于毒性考虑而被认为对于人类患者使用是不安全的,但根据本发明,美登素类适于通过微细胞递送至脑肿瘤患者。
[0157]
杜卡霉素(分子量:~588道尔顿)是一系列相关天然产物,首次从链霉菌(是
streptomyces)细菌分离出来。其也具有强细胞毒性,但被认为对人类使用是不安全的。像美登素类一样,杜卡霉素是适合用于本发明的化疗药物。
[0158]
生物学化疗药物的亚类包括但不限于天冬酰胺酶、ain
‑
457、巴比单抗(bapineuzumab)、贝利木单抗(belimumab)、布伦妥昔单抗(brentuximab)、布雷奴单抗(briakinumab)、卡那单抗(canakinumab)、西妥昔单抗(cetuximab)、达洛妥珠单抗(dalotuzumab)、地诺单抗(denosumab)、依帕妥珠单抗(epratuzumab)、estafenatox、法妥组单抗(farletuzumab)、figitumumab、加利昔单抗(galiximab)、吉妥单抗(gemtuzumab)、吉伦妥昔单抗(girentuximab)(wx
‑
g250)、赫塞汀(herceptin)、替伊莫单抗(ibritumomab)、诺妥珠单抗(inotuzumab)、伊匹单抗(ipilimumab)、美泊珠单抗(mepolizumab)、莫罗单抗cd3(muromonab
‑
cd3)、纳他单抗(naptumomab)、尼珠单抗(necitumumab)、尼妥珠单抗(nimotuzumab)、奥珠单抗(ocrelizumab)、奥法单抗(ofatumumab)、奥替利珠单抗(otelixizumab)、奥佐米星(ozogamicin)、帕格巴昔单抗(pagibaximab)、帕尼单抗(panitumumab)、帕妥珠单抗(pertuzumab)、雷莫昔单抗(ramucirumab)、瑞珠单抗(reslizumab)、利妥昔单抗(rituximab)、regn88、索拉珠单抗(solanezumab)、他尼珠单抗(tanezumab)、替利组单抗(teplizumab)、tiuxetan、托西单抗(tositumomab)、曲妥珠单抗(trastuzumab)、曲美单抗(tremelimumab)、维多珠单抗(vedolizumab)、扎鲁单抗(zalutumumab)和扎诺莫单抗(zanolimumab)。
[0159]
在一些实施方式中,抗肿瘤剂包括选自下列的化合物:放线菌素d、马法兰(alkeran)、ara
‑
c、阿那曲唑、bicnu、比卡鲁胺、博来霉素、白消安、卡培他滨、卡铂(carboplatin)、卡铂(carboplatinum)、卡莫斯汀(carmustine)、ccnu、苯丁酸氮芥、顺铂、克拉屈滨、cpt
‑
11、环磷酰胺、阿糖胞苷、胞嘧啶阿拉伯糖苷、环磷酰胺(cytoxan)、达卡巴嗪、更生霉素(dactinomycin)、柔红霉素、右雷佐生、多西他赛、多柔比星、dtic、表柔比星、乙撑亚胺、依托泊苷、氟尿苷(floxuridine)、氟达拉滨、氟尿嘧啶、氟他胺、福莫司汀、吉西他滨、六甲胺、羟基脲、依达比星、异环磷酰胺、伊立替康、洛莫斯汀、二氯甲基二乙胺、美法仑、巯基嘌呤、甲氨蝶呤、丝裂霉素、米托坦、米托蒽醌、奥沙利铂、紫杉醇、帕米膦酸、戊他汀、普卡霉素、丙卡巴肼、类固醇、链脲霉素、sti
‑
571、他莫昔芬、替莫唑胺、替尼泊苷、四嗪、硫鸟嘌呤、噻替哌、拓优得、拓扑替康、曲奥舒凡(treosulphan)、曲美沙特、长春碱、长春新碱、长春地辛、长春瑞滨、vp
‑
16、希罗达、天冬酰胺酶、ain
‑
457、巴比单抗(bapineuzumab)、贝利木单抗(belimumab)、布伦妥昔单抗(brentuximab)、布雷奴单抗(briakinumab)、卡那单抗(canakinumab)、西妥昔单抗(cetuximab)、达洛妥珠单抗(dalotuzumab)、地诺单抗(denosumab)、依帕妥珠单抗(epratuzumab)、estafenatox、法妥组单抗(farletuzumab)、figitumumab、加利昔单抗(galiximab)、吉妥单抗(gemtuzumab)、吉伦妥昔单抗(girentuximab)(wx
‑
g250)、赫塞汀(herceptin)、替伊莫单抗(ibritumomab)、诺妥珠单抗(inotuzumab)、伊匹单抗(ipilimumab)、美泊珠单抗(mepolizumab)、莫罗单抗cd3(muromonab
‑
cd3)、纳他单抗(naptumomab)、尼珠单抗(necitumumab)、尼妥珠单抗(nimotuzumab)、奥珠单抗(ocrelizumab)、奥法单抗(ofatumumab)、奥替利珠单抗(otelixizumab)、奥佐米星(ozogamicin)、帕格巴昔单抗(pagibaximab)、帕尼单抗(panitumumab)、帕妥珠单抗(pertuzumab)、雷莫昔单抗(ramucirumab)、瑞珠单抗(reslizumab)、利妥昔单抗(rituximab)、regn88、索拉珠单抗(solanezumab)、他尼珠单抗
(tanezumab)、替利组单抗(teplizumab)、tiuxetan、托西单抗(tositumomab)、曲妥珠单抗(trastuzumab)、曲美单抗(tremelimumab)、维多珠单抗(vedolizumab)、扎鲁单抗(zalutumumab)、扎诺莫单抗(zanolimumab)、5fc、霍夫曼
‑
罗氏公司的异维甲酸(accutane hoffmann
‑
la roche)、aee788 novartis、amg
‑
102、抗瘤酮(anti neoplaston)、aq4n(巴诺蒽醌(banoxantrone))、avandia(马来酸罗格列酮(rosiglitazone maleate))、基因泰克公司的阿瓦斯汀(avastin genetech)(贝伐单抗)、bcnu、bicnu卡莫司汀、cci
‑
779、ccnu、ccnu洛莫司汀、塞来昔布(celecoxib)(全身)、氯喹(chloroquine)、西仑吉肽(cilengitide)(emd 121974)、cpt
‑
11(camptosar、伊立替康)、达沙替尼(dasatinib)(bms
‑
354825、sprycel)、树突状细胞疗法、依托泊苷(eposin、etopophos、vepesid)、gdc
‑
0449、格列卫(甲磺酸伊马替尼)、格立得植入剂(gliadelwafer)、羟氯喹、il
‑
13、imc
‑
3g3、免疫疗法、易瑞沙(iressa)(zd
‑
1839)、拉帕替尼(lapatinib)(gw572016)、用于癌症的甲氨蝶呤(systemic)、novocure、osi
‑
774、pcv、诺华公司(novartis)的rad001(mtor抑制剂)、雷帕霉素(拉帕姆(rapamune)、西罗莫司(sirolimus))、rmp
‑
7、rta 744、辛伐他汀(simvastatin)、西罗莫司(sirolimus)、索拉非尼(sorafenib)、su
‑
101、set5416 sugen、柳氮磺胺吡啶(sulfasalazine)(azulfidine)、索坦(sutent)(pfizer)、tarceva(盐酸厄洛替尼(erlotinib hcl))、紫杉醇(taxol)、temodar schering
‑
pough、tgf
‑
b反义、thalomid(沙利度胺)、拓扑替康(topotecan)(systemic)、vegf trap、vegf
‑
trap、伏立诺他(vorinostat(saha)、xl 765、xl 184、xl765、zarnestra(替匹法尼(tipifarnib))、zocor(辛伐他汀(simvastatin)、环磷酰胺(cytoxan)、马法兰(alkeran)、苯丁酸氮芥(leukeran)、(噻替派(thiopeta)(thioplex))、马勒兰(busulan)(myleran)、丙卡巴肼(matulane)、达卡巴嗪(dtic)、奥曲胺(altretamine)(hexalen)、苯丁酸氮芥、顺铂(platinol)、异环磷酰胺、甲氨蝶呤(mtx)、6
‑
硫嘌呤(巯基嘌呤[6
‑
mp]、硫鸟嘌呤[6
‑
tg])、巯基嘌呤(purinethol)、磷酸氟达拉滨、(leustatin)、氟尿嘧啶(5
‑
fu)、阿糖胞苷(ara
‑
c)、阿扎胞苷、长春碱(velban)、长春新碱(oncovin)、鬼臼毒素(依托泊苷{vp
‑
16}和替尼泊苷(teniposide){vm
‑
26})、喜树碱(camptothecins)(拓扑替康和伊立替康)、紫杉烷如紫杉醇(taxol)和多西他赛(taxotere)、(多柔比星、rubex、doxil)、更生霉素(dactinomycin)(cosmegen)、普卡霉素(光神霉素)、丝裂霉素:(mutamycin)、博来霉素(blenoxane)、雌激素和雄激素抑制剂(tamoxifen)、促性腺激素释放激素激动剂(leuprolide和goserelin(zoladex))、芳香酶抑制剂(氨鲁米特(aminoglutethimide)和阿那曲唑(arimidex))、氨曲林(amsacrine)、天冬酰胺酶(el
‑
spar)、米托蒽醌(novantrone)、米托坦(lysodren)、视黄酸衍生物、骨髓生长因子(沙格司亭(sargramostim)和非格司亭(filgrastim))、氨磷汀(amifostine)、培美曲塞(pemetrexed)、地西他滨、伊尼帕利布(iniparib)、奥拉帕尼(olaparib)、维利帕尼(veliparib)、依维莫司(everolimus)、伏立诺他(vorinostat)、恩替司他(entinostat)(sndx
‑
275)、莫西司他(mocetinostat)(mgcd0103)、帕诺比司他(panobinostat)(lbh589)、罗米地辛(romidepsin)、丙戊酸、夫拉平度(flavopiridol)、奥洛莫卡因(olomoucine)、罗斯科维汀(roscovitine)、肯帕龙(kenpaullone)、ag
‑
024322(pfizer)、fascaplysin、ryuvidine、purvalanol a、nu2058、bml
‑
259、su 9516、pd
‑
0332991、p276
‑
00、格尔德霉素、坦西霉素(tanespimycin)、阿维斯霉素(alvespimycin)、拉迪霉素(radicicol)、德盖林(deguelin)、biib021、顺式咪唑啉、苯并二氮杂二酮、螺
‑
羟吲哚、异环喹啉酮、噻吩、5
‑
脱氮黄素、色胺、氨基吡啶、二氨基嘧啶、吡啶基异喹啉、吡咯并吡唑、二苯胺并嘧啶、苯甲酰胺、酞嗪酮、三环吲哚、苯并咪唑、吲唑、吡咯并咔唑、异吲哚酮、吗啉基蒽环类、美登素类、杜卡霉素(ducarmycin)、澳瑞他汀(auristatins)、加利车霉素(calicheamicins)(dna破坏剂)、α
‑
鹅膏蕈碱(α
‑
amanitin)(rna聚合酶ii抑制剂)、centanamycin、吡咯并苯并二氮杂链黑菌素(streptonigtin)、氮芥、亚硝基脲、烷烃磺酸盐、嘧啶类似物、抗代谢物、叶酸类似物、蒽环类、紫杉烷类、长春花生物碱、拓扑异构酶抑制剂、激素剂及其任意组合。
[0160]
根据本公开可用的活性剂不限于以上列举的那些药物类别或具体试剂。不同的发现平台持续产生针对癌细胞独特分子特征的新药物。实际上,已经发现了数千种这样的化学和生物学药物,此处仅列出其中一些。然而,完整细菌源微细胞和被杀死的细菌细胞适应包装各种亲水性或疏水性活性剂的惊人能力意味着,根据本公开的发现,基本上任何此类药物在被包装在微细胞中时都有治疗癌症的潜力。
[0161]
抗肿瘤剂类别的示例是放射性核素、化疗药物和功能性核酸,包括但不限于调控性rna。该类的成员将在下面被进一步讨论。
[0162]
i.放射性核素
[0163]“放射性核素”是具有不稳定核的原子,即,其特征在于过量能量可用于赋予核内新产生的辐射粒子或原子电子。本文中的放射性核素也可以称为“放射性同位素”、“放射成像剂”或“放射标记”。放射性核素可用于成像和/或治疗目的。其可被包含在微细胞中,也可以附接至微细胞外表面上的配体、肽或糖脂。附接可以是直接的或通过连接体,该连接体包含螯合部分,该螯合部分包含螯合剂,如巯基乙酰基三甘氨酸(mag3)、dota、edta、hynic、dtpa,或可使用冠状醚。螯合剂可以直接附接至微细胞表面组分,或通过连接体附接至微细胞。多种放射性核素在本领域中是已知的,并且已知其中多种适于医学用途、如钇
‑
90、锝
‑
99m、碘
‑
123、碘
‑
124、碘
‑
125、碘
‑
131、铷
‑
82、铊201、镓67、氟18、氙133和铟111。
[0164]
因此,在一些实施方式中,放射性同位素包括选自以下的放射性同位素:钇90、钇86、锝
‑
152、锝
‑
155、锝
‑
149、锝
‑
161、锝
‑
99m、碘
‑
123、碘
‑
131、铷
‑
82、铊
‑
201、镓
‑
67、氟
‑
18、铜
‑
64、镓
‑
68、氙
‑
133、铟
‑
iii、镥
‑
177、及其任意组合。
[0165]
可用于附接于微细胞以用于成像和治疗目的的放射性同位素包括,例如,碘131和镥
‑
177,其是γ和β发射体。因此,这些试剂可用于成像和治疗。
[0166]
相同元素的不同同位素,例如碘123(γ发射体)和碘1331(γ和β发射体)也可以用于成像和治疗目的(gerard和cavalieri,2002;alzahrani等,2012)。
[0167]
较新的实例是钇
‑
86/钇
‑
90或:铽同位素(tb):
152
tb(β+发射体)、
155
tb(γ发射体)、
149
tb(α发射体)和
161
tb(β
‑
粒子)(miiller等,2012;walrand等,2015)。
[0168]
核成像利用γ和正电子发射体(β
+
)。γ发射体,如锝
‑
99m(
99m
tc)或碘
‑
123(
123
i),可以使用γ相机(平面成像)或spect(单光子发射计算机断层扫描)进行定位(holman和tumeh,1990)。
[0169]
这些粒子的组织渗透与放射性同位素的能量成正比(kramer
‑
marek和capala,2012)。β粒子具有潜在的杀细胞作用,但其也因具有仅几毫米的组织穿透而留出周围的健康组织。常规核肿瘤学实践中常用的β发射体包括镥
‑
177(
177
lu,组织穿透:0.5
‑
0.6mm,最大:2mm,497kev,半衰期:6.7天)和钇
‑
90(
90
y,组织穿透:平均2.5mm:最大:11mm,935kev,半衰期:64小时)(teunissen等,2005;kwekkeboom等,2008;ahmadzadehfar等,2010;pillai
等,2013;ahmadzadehfar等,2016)。
[0170]
放射性核素已在核医学中得到广泛使用,特别是作为破坏肿瘤细胞的β射线发射体。在一些实施方式中,放射性核素适合用作抗肿瘤剂。
[0171]
放射性核素可以通过任何已知技术与完整的细菌源微细胞结合。因此,可以利用商业可得的标记手段,如使用pierce biotechnology inc.的产品pierce碘化试剂,用放射性核素标记蛋白质或其他微细胞表面部分(见下文),详述于rice et al.,semin.nucl.med.,41,265
‑
282(2011)。可选地,可以将放射性核素掺入微细胞内部的蛋白质中。
[0172]
在后一种情况下,用编码外源蛋白的质粒dna转化产生微细胞的细菌菌株。当在不对称细胞分裂过程中形成微细胞时,若干拷贝的质粒dna分离到微细胞质中。将所得的重组微细胞在存在放射性标记的氨基酸的情况下在这样的条件下培育:使得在微细胞中表达的来自质粒dna的外源蛋白质掺入携载放射性核素的氨基酸。根据例如clark
‑
curtiss and curtiss,methods enzymol.,101:347
‑
362(1983)的方案,重组微细胞在含有
35s
甲硫氨酸的最低限度生长培养基中被培育,因此新表达的质粒编码的蛋白包含
35s
甲硫氨酸。可以使用类似的方法,使得重组微细胞按照需要与其他放射性标记一起被包装。
[0173]
微细胞表面的低聚糖也可以被放射性标记,利用例如fukuda,curr.protocols molec.biol.(suppl.26),17.5.1
‑
17.5.8(1994)描述的完善建立的方案进行。微细胞特有的这种低聚糖的示例是在源自革兰氏阴性细菌的微细胞的表面上发现的脂多糖(lps)的o
‑
多糖组分(见下文)。
[0174]
在这方面,优选的方法是放射性标记用作肿瘤靶向配体的双特异性抗体,该双特异性抗体用于将微细胞靶向至特定肿瘤。参见美国专利公开2007/0237744,其内容通过引用并入本文。也就是说,“包覆”在微细胞上的双特异性抗体暴露大量另外的表面蛋白用于放射性标记。因此,可以实现与抗体包覆的微细胞结合的放射性标记的更高的比活性。相比之下,无包覆的微细胞的放射性标记,即当放射性核素仅标记特有部分时,可导致标记较弱(比活性较低)。在一个实施方式中,认为这种较弱的标记发生是因为源自革兰氏阴性细菌的微细胞的外膜结合蛋白被lps掩盖,lps如下所述包含覆盖微细胞表面的o
‑
多糖的长链。
[0175]
为了治疗肿瘤,本公开的组合物将以提供足以至少缩小肿瘤质量(如不完全消除肿瘤)的肿瘤内照射水平的单剂量或多剂量递送。可以逐案依此监测治疗进展。然而,总体而言,组合物中包装的放射性量通常将为约30至约50gy,尽管本发明还考虑较高的放射性,如例如约50至约200gy,其总范围在约30gy和约200gy之间。
[0176]
在一些情况下,考虑到微细胞负荷的放射性核素向肿瘤的高效和特异性递送,包装在组合物中的放射性量可以甚至低于上述。因此,一方面,组合物包含约20至约40gy,或约10至约30gy,或约1至约20gy,或小于约10gy。
[0177]
一些靶向肿瘤的配体可以包括放射性同位素,该放射性同位素在配体结合肿瘤细胞时起到向肿瘤递送辐射的效果。在一些实施方式中,配体包含arg
‑
gly
‑
asp(rgd)肽、铃蟾肽(bombesin)(bbn)/胃泌素释放肽(grp)、胆囊收缩素(cck)/胃泌素肽、α
‑
黑素细胞刺激激素(α
‑
msh)、神经肽y(npy)、神经降压素(nt)、[
68
ga]ga
‑
psma
‑
hbed
‑
cc([
68
ga]ga
‑
psma
‑
11[pet])、[
177
lu]lu/[
90
y]y
‑
j591、[
123
i]i
‑
mip
‑
1072、[
131
i]i
‑
mip
‑
1095、
68
ga或
177
lu标记的psma
‑
i&t、
68
ga或
177
lu标记的dkfz
‑
psma
‑
617(psma
‑
617)、生长抑素(sst)肽、物质p、t140、肿
瘤分子靶向肽1(tmtp1)、血管活性肠肽(vip)或其任何组合。
[0178]
在一些实施方式中,放射性同位素缀合至肿瘤靶向配体。在一些实施方式中,缀合是通过接头。在一些实施方式中,肿瘤靶向配体包含含有用于放射性同位素或螯合放射性同位素的螯合剂部分的缀合的官能团(一个或多个)的肽。缀合可用的肽的官能团包括但不限于赖氨酸侧链上的ε
‑
氨基、精氨酸侧链上的胍基、天冬氨酸或谷氨酸上的羧基、半胱氨酸巯基和酪氨酸上的苯酚。最常见的缀合反应是碳二亚胺/n
‑
羟基琥珀酰亚胺(edc/nhs)介导的羧基和胺偶联、马来酰亚胺与硫醇基的偶联以及酪氨酸上苯酚的重氮鎓修饰。将肽与成像部分偶联的代表性化学方法可在多个综述中找到(erathodiyil和ying,2011;takahashi等,2008)。
[0179]
在一些实施方式中,放射性同位素用作放射成像剂。几种放射性同位素已被用于肽标记、包括
99m
tc、
123
i和
111
in用于spect成像,以及
18
f、
64
cu和
68
ga用于pet成像(chatalic等,2015)。总体上,这些放射性同位素通过螯合剂附接至肽。一些广泛使用的螯合剂被描述于(sun等,2017)。大多数治疗性放射性药物用β发射同位素标记(β
‑
)。
[0180]
靶向肿瘤细胞的本发明的微细胞还将从放射性同位素向与微细胞结合的肿瘤细胞递送靶向辐射。在一些实施方式中,放射性同位素用作治疗性辐射发射剂,并且其中放射性同位素提供的辐射量足以对肿瘤提供治疗作用。在一些实施方式中,治疗效果是肿瘤尺寸减小。肿瘤尺寸可减小约100%、约90%、约80%、约70%、约60%、约50%、约40%、约30%、约20%、约10%或约5%。
[0181]
放射性标记的膦酸酯具有高骨亲和力,并且可用于对疼痛的骨转移进行成像和缓解。取决于骨代谢程度,示踪剂通过粘附至骨并且优选粘附至成骨细胞的骨转移而积累。治疗计划需要使用锝
‑
99m
‑
羟乙叉基二膦酸((hedp)进行骨闪烁显像,以估测新陈代谢和转移波及程度。双膦酸hedp可以用铼
‑
186(β
‑
发射体,半衰期:89小时,最大能量1.1mev,最大范围:4.6mm)或铼
‑
188(β
‑
发射体[至85%,2.1mev]和γ发射体[至15%,155kev],半衰期:16.8小时,软组织中的最大范围:10mm(palmedo,2007)标记用于治疗。用于骨缓解治疗的新的有前景的放射性药物包括唑来膦酸(zoledronic acid)的放射性标记复合物。唑来膦酸属于具有环状侧链的新一代最有效的双膦酸酯(盐)。用钪
‑
46或镥
‑
177标记的唑来膦酸的骨亲和力显示出极好的吸收性(唑来膦酸lu[177lu]为98%,唑来膦酸sc[46sc]为82%),远高于用钐
‑
153标记的双膦酸盐(最大:67%)(majkowska等,2009)。这些双膦酸盐可以与完整的微细胞缀合,用作骨转移的诊断或治疗。
[0182]
ii.化疗药物
[0183]
本公开中使用的抗肿瘤剂也可以是化疗药物。在本说明书中,“化疗药物”、“化疗剂”和“化学疗法”被可互换地使用,以表示具有杀死或破坏肿瘤细胞的能力的药物。化疗剂可以是小分子药物或生物学药物,如下文进一步详述。
[0184]“小分子药物”亚类包括特征在于(i)对生物过程具有效果和(ii)与蛋白质或聚合物大分子相比具有低分子量的化合物。小分子药物通常为约800道尔顿以下,下限为约150道尔顿,例如(替莫唑胺),约194道尔顿,其用于治疗成胶质细胞瘤和其他脑癌类型。在这种情况下,“约”表示符合条件的分子量值经受测量精度变异和几道尔顿或几十道尔顿量级的实验误差。因此,小分子药物的分子量可以为例如约900道尔顿以下、约800以下、约700以下、约600以下、约500以下或约400道尔顿以下,例如在约150至约400道尔顿范
围内。更具体地,小分子药物的分子量可以为约400道尔顿以上、约450道尔顿以上、约500道尔顿以上、约550道尔顿以上、约600道尔顿以上、约650道尔顿以上、约700道尔顿以上、或约750道尔顿以上。在另一个实施方式中,包装到微细胞中的小分子药物的分子量在约400至约900道尔顿之间、约450至约900道尔顿之间、约450至约850道尔顿之间、约450至约800道尔顿之间、约500至约800道尔顿之间、或约550至约750道尔顿之间。
[0185]
具体而言,合适的小分子药物包括但不限于以上所列的那些,如氮芥、亚硝基脲、乙撑亚胺、烷烃磺酸酯(盐)、四嗪、铂化合物、嘧啶类似物、嘌呤类似物、抗代谢物、叶酸类似物、蒽环类、紫杉烷类、长春花生物碱和拓扑异构酶抑制剂等。因此,用于本发明的小分子药物可以选自以下任意种等:烯二炔,如达尼霉素a(dynemicin a)、尤卡霉素(unicalamycin)、加利车霉素γ1和加利车霉素θ
‑
1;米亚霉素(meayamicin)、fr901464的合成类似物;苯并环庚烯(benzosuberene)衍生物——如例如tanpure et al.,bioorg.med.chem.,21:8019
‑
32(2013)所述;澳瑞他汀,如澳瑞他汀e、单甲基澳瑞他汀e(mmae)和澳瑞他汀f,其是多拉司他汀(dolastatin)的合成类似物;多卡霉素类,如多卡霉素sa和cc
‑
1065;美登素及其衍生物(美登素类),如dm1和dm4;伊立替康和其他拓扑异构酶抑制剂,如拓扑替康(topotecan)、依托泊苷、米托蒽醌和替尼泊苷(teniposide);和谷田霉素(yatakemycin),其合成由okano等,2006详述。
[0186]
更具体而言,本文详述的任何一种或多种或全部具体小分子药物是适用于本发明的那些药物的示例:放线菌素d、马法兰(alkeran)、ara
‑
c、阿那曲唑、bicnu、比卡鲁胺、比生群(bisantrene)、博来霉素、白消安、卡培他滨卡铂(carboplatin)、卡铂(carboplatinum)、卡莫斯汀(carmustine)、ccnu、苯丁酸氮芥、顺铂、克拉屈滨、cpt
‑
11、环磷酰胺、阿糖胞苷、胞嘧啶阿拉伯糖苷、环磷酰胺(cytoxan)、达卡巴嗪、更生霉素(dactinomycin)、柔红霉素、右雷佐生、多西他赛、多柔比星、dtic、表柔比星、乙撑亚胺、依托泊苷、氟尿苷(floxuridine)、氟达拉滨、氟尿嘧啶、氟他胺、福莫司汀、吉西他滨、六甲胺、羟基脲、依达比星、异环磷酰胺、伊立替康、洛莫斯汀、二氯甲基二乙胺、美法仑、巯基嘌呤、甲氨蝶呤、丝裂霉素、米托坦、米托蒽醌、奥沙利铂、紫杉醇、帕米膦酸、戊他汀、普卡霉素、丙卡巴肼、链脲霉素、sti
‑
571、他莫昔芬、替莫唑胺、替尼泊苷、四嗪、硫鸟嘌呤、噻替哌、拓优得、拓扑替康、曲奥舒凡(treosulphan)、曲美沙特、长春碱、长春新碱、长春地辛、长春瑞滨和vp
‑
16。
[0187]
出于本描述的目的,“生物学药物”相比之下被定义表示可以通过生物过程产生的任何生物学活性大分子,不包括下文讨论的“功能性核酸”和尺寸符合如上定义的小分子药物的多肽。因此,“生物学药物”亚类不包括小分子药物和功能性核酸亚类,并且不与之重叠。生物学药物的示例是治疗性蛋白质和抗体,无论是天然的还是重组的还是合成制备的,例如利用药物化学和药物设计工具制备。
[0188]
iii.超毒性化疗药物
[0189]
被设计用于化疗目的的某些分子在临床前或临床试验中由于不可接受的毒性而失败。本发明人已经表明,将高毒性或“超毒性”化疗药物包装在微细胞中,然后全身递送至肿瘤患者,导致药物递送至肿瘤细胞。此外,即使在肿瘤细胞破裂并且含药物细胞质被释放到附近的正常组织之后,结果也不是对正常组织造成毒性。这是因为该药物已经与肿瘤细胞结构(如dna)结合,并且不能再攻击正常细胞。因此,本发明对于将高毒性(“超毒性”)化
疗药物递送至癌症患者特别有用。
[0190]
当癌症对象已经用尽所有治疗选择时,肿瘤可能已经达到相当异质的阶段,并且对常规细胞毒性药物具有高度抗性。在本说明书中“高毒性化疗药物”或“超毒性化疗药物”指的是可以克服对常规药物的抗性的化疗药物,因其对正常细胞的致死剂量相对低于其对癌细胞的有效剂量。
[0191]
因此,一方面,高毒性化疗药物具有的中位致死剂量(ld
50
)低于其针对靶向癌症的中位有效剂量(ed
50
)。例如,高毒性或超毒性化疗药物的ld
50
可低于该药物对靶向癌症的ed
50
的约500%、400%、300%、250%、200%、150%、120%或100%。在另一方面,高毒性或超毒性化疗药物的最大亚致死剂量(即,不会引起严重或不可逆毒性的最高剂量)低于其最低有效剂量,例如最低有效剂量的约500%、约400%、约300%、约250%、约200%、约150%、约120%、约100%、约90%、约80%、约70%、约60%或约50%。在一个实施方式中,靶向的癌症可以是,例如,(1)设计该药物所针对的癌症类型,(2)对该药物进行临床前或临床试验的第一种癌症类型,或(3)在所有测试癌症中,该药物显示出最高功效的癌症类型。
[0192]
超毒性化疗药物的示例性非限制性实例包括但不限于美登素类、多卡霉素类、吗啉基蒽环及其衍生物。美登素类(分子量:约738道尔顿)是美登素的一组化学衍生物,具有强细胞毒性。尽管由于毒性考虑而被认为对于人类患者使用是不安全的,但根据本发明,美登素类适于通过微细胞递送给肿瘤患者。多卡霉素类(分子量:约588道尔顿)是一系列相关的天然产物,首先从链霉菌属(streptomyces)细菌分离出来。其也具有强细胞毒性,但被认为对人类使用是不安全的。像美登素类一样,多卡霉素是适合用于本发明的化疗药物。
[0193]
示例性的还有国际专利申请wo 1998/002446中描述的吗啉基蒽环类衍生物的化合物。这类衍生物中有奈莫柔比星(nemorubicin)(3'
‑
脱氨基
‑
3'
‑
[2(s)
‑
甲氧基
‑4‑
吗啉基]多柔比星)(mmdx)及其主要代谢产物pnu
‑
159682(3'
‑
脱氨基
‑3”‑
4'
‑
脱水
‑
[2”(s)
‑
甲氧基
‑
3”(r)
‑
羟基
‑4”‑
吗啉基
‑
]多柔比星,以及美国专利号8,470,984中所述的四种其他这种衍生物,该专利的内容通过引用并入本文:3
′‑
脱氨基
‑3″‑4′‑
脱水
‑
[2”(s)
‑
甲氧基
‑
3”(r)
‑
羟基
‑4”‑
吗啉基]
‑
伊达比星;3
′‑
脱氨基
‑3″‑4′‑
脱水
‑
[2”(s)
‑
甲氧基
‑
3”(r)
‑
羟基
‑4”‑
吗啉基]
‑
柔红霉素;3
′‑
脱氨基
‑3″‑4′‑
脱水
‑
[2”(s)
‑
甲氧基
‑
3”(r)
‑
羟基
‑4”‑
吗啉基]
‑
卡米霉素;3
′‑
脱氨基
‑3”‑4’‑
脱水
‑
[2”(s)
‑
乙氧基
‑
3”(r)
‑
羟基
‑4”‑
吗啉基]
‑
多柔比星。
[0194]
在本公开的示例性实施方式中,微细胞包含超毒性化疗药物3
′‑
脱氨基
‑
3”,4
’‑
脱水
‑
[2”(s)
‑
甲氧基
‑
3”(r)
‑
氧
‑4”‑
吗啉基]
‑
多柔比星(pnu
‑
159682)。本发明人发现,pnu
‑
159682是呈现在许多不同的肿瘤细胞系中克服抗药性并且在针对多种不同肿瘤细胞系的细胞毒性试验中比一系列常规化学治疗剂远更有效的有效药物。参见实施例8和9。此外,在体内小鼠异种移植实验中显示,对多柔比星具有抗性的人肿瘤异种移植物可以通过iv施用靶向egfr并且负载pnu
‑
159682的edv而被有效治疗。参见实施例11。显著地,发现pnu
‑
159682负载的edv与i型干扰素激动剂组合被良好地耐受,并在晚期胰腺癌患者中提供协同和提高的抗癌效果。参见实施例12。因此,在本发明的一个实施方式中,组合物包含靶向egfr的小细胞,所述小细胞包含pnu
‑
159682作为活性抗癌药物。
[0195]
其他合适的可呈现超毒性化疗性质的癌症化疗药物包括澳瑞他汀、加利车霉素(dna破坏剂)、α
‑
鹅膏菌素(rna聚合酶ii抑制剂)、降霉素、格尔德霉素、吡咯并苯并二氮杂链黑菌素、氮芥、亚硝基脲、乙撑亚胺、烷烃磺酸酯(盐)、四嗪、铂化合物、嘧啶类似物、
嘌呤类似物、抗代谢物、叶酸类似物、蒽环类、紫杉烷类、长春花生物碱、拓扑异构酶抑制剂和激素剂等。
[0196]
iv.生物化疗药物
[0197]
在另一方面,微细胞可以包含生物学化疗药物。这种药物的实例包括但不限于天冬酰胺酶、ain
‑
457、巴比单抗(bapineuzumab)、贝利木单抗(belimumab)、布伦妥昔单抗(brentuximab)、布雷奴单抗(briakinumab)、卡那单抗(canakinumab)、西妥昔单抗(cetuximab)、达洛妥珠单抗(dalotuzumab)、地诺单抗(denosumab)、依帕妥珠单抗(epratuzumab)、estafenatox、法妥组单抗(farletuzumab)、figitumumab、加利昔单抗(galiximab)、吉妥单抗(gemtuzumab)、吉伦妥昔单抗(girentuximab)(wx
‑
g250)、替伊莫单抗(ibritumomab)、诺妥珠单抗(inotuzumab)、伊匹单抗(ipilimumab)、美泊珠单抗(mepolizumab)、莫罗单抗cd3(muromonab
‑
cd3)、纳他单抗(naptumomab)、尼珠单抗(necitumumab)、尼妥珠单抗(nimotuzumab)、奥珠单抗(ocrelizumab)、奥法单抗(ofatumumab)、奥替利珠单抗(otelixizumab)、奥佐米星(ozogamicin)、帕格巴昔单抗(pagibaximab)、帕尼单抗(panitumumab)、帕妥珠单抗(pertuzumab)、雷莫昔单抗(ramucirumab)、瑞珠单抗(reslizumab)、利妥昔单抗(rituximab)、regn88、索拉珠单抗(solanezumab)、他尼珠单抗(tanezumab)、替利组单抗(teplizumab)、tiuxetan、托西单抗(tositumomab)、曲妥珠单抗(trastuzumab)曲美单抗(tremelimumab)、维多珠单抗(vedolizumab)、扎鲁单抗(zalutumumab)和扎诺莫单抗(zanolimumab)。
[0198]
v.功能性核酸
[0199]“功能性核酸”是指在引入宿主细胞后特异性地干扰蛋白质表达的核酸分子。关于治疗癌症,根据本发明,优选地,经由完整的细菌源微细胞递送至癌细胞的功能性核酸有效载荷抑制促进肿瘤细胞增殖、血管生成或化疗抗性和/或抑制凋亡或细胞周期停滞的基因;即“促进癌症的基因”。
[0200]
总体上,本公开中使用的功能性核酸分子具有通过与蛋白质的转录物相互作用来减少蛋白质表达的能力。用于本公开的微细胞有效载荷的这种类别包括调控性rna,如sirna、shrna、短rna(通常长度小于400个碱基)、微小rna(mirna)、核酶和诱饵rna、反义核酸和lincrna等。就这点而言,“核酶”是指具有酶活性的可以以核苷酸碱基序列特异性的方式重复切割其他rna分子的rna分子。“反义寡核苷酸”表示与具体基因转录物的一部分互补的核酸分子,使得该分子可以与该转录物杂交并阻断其翻译。反义寡核苷酸可包括rna或dna。“lincrna”或“长基因间非编码rna”专栏涵盖超过200个核苷酸的非蛋白质编码转录本。lincrna可以调控基因的转录、剪接和/或翻译,如khalil等(2009)所述。
[0201]
各类型的调控性rna可以是如上所述抑制肿瘤促进基因的功能性核酸分子并且因此适合根据本发明使用的来源。因此,在本公开的一个实施方式中,完整的微细胞携载介导转录后基因沉默rna干扰(rnai)机制的sirna分子,该sirna分子可被用于靶向促肿瘤基因。例如,参见macdiarmid等,2009(抗体呈递微细胞递送化疗药物和对抗药物抗性产生的sirna),以及oh和park,advanced advanced drug delivery rev.,61:850
‑
62(2009)(治疗性sirna的递送,分别治疗乳腺癌、卵巢癌、宫颈癌、肝癌、肺癌和前列腺癌。
[0202]
如述,“sirna”总体上是指长约10至约30个核苷酸的双链rna分子,因其特异性地干扰蛋白质表达的能力而被命名。优选地,sirna分子约12至约28个核苷酸长,更优选约15
至约25个核苷酸长,还更优选约19至约23个核苷酸长,最优选约21至约23个核苷酸长。因此,sirna分子可以是,例如,约12、约13、约14、约15、约16、约17、约18、约19、约20、约21、约22、约23、约24、约25、约26、约27、约28或约29个核苷酸长。
[0203]
一条链的长度表示sirna分子的长度。例如,被描述为21个核糖核苷酸长(2
‑
mer)的sirna可以包含两条相对的rna链,该链可以退火19个连续的碱基配对。每条链上的两个剩余核糖核苷酸将形成“突出部”。当sirna包含两条不同长度的链时,链中的较长者表示sirna的长度。例如,包含21个核苷酸长的一条链和20个核苷酸长的第二条链的dsrna构成2
‑
mer。
[0204]
辅助设计sirna和调控性rna的工具总体上是容易获得的。例如,基于计算机的sirna设计工具是可从互联网上的www.dharmacon.com获得的。
[0205]
在另一个优选的实施方式中,本公开的完整微细胞携载mirna,其像sirna一样能够介导转录后基因沉默rna干扰(rnai)机制。而且像sirna一样,mirna介导的基因沉默效应可用于靶向肿瘤促进基因。例如,参见kota等,2009(通过转染递送mirna导致癌细胞增殖抑制、肿瘤特异性凋亡以及在鼠肝癌模型中无毒性的显著保护防止疾病进展),以及takeshita等,2010(通过瞬时转染递送合成的mirna抑制转移性前列腺肿瘤细胞在骨组织上的生长)。
[0206]
尽管两者均介导rna干扰,但mirna和sirna已被注意到差异。在这方面,“mirna”总体上是指一类约17至约27个核苷酸的单链rna分子(代替sirna情况下的双链)。因此,mirna分子的长度可以是例如约17、约18、约19、约20、约21、约22、约23、约24、约25、约26或约27个核苷酸。优选地,mirna分子为约21至约25个核苷酸长。
[0207]
mirna和sirna之间的另一差异是,前者总体上不完全互补mrna靶标。相比之下,sirna必须与mrna靶标完全互补。因此,sirna总体上导致单个特定靶标沉默,而mirna是随意性的(promiscuous)。
[0208]
此外,尽管两者都组装成risc(rna诱导的沉默复合体),但sirna和mirna在risc组装之前其对应的初始处理不同。这些差异被详细描述于chu等,2006;和gregory等,2006。多个数据库充当mirna存放处。例如,参见mirbase(www.mirbase.org)和tarbase(http://diana.cslab.ece.ntua.gr/dianatoolsnew/index.php?r=tarbase/index)。在常规用法中,mirna通常以前缀
“‑
mir”命名,并带有序号。例如,在小鼠mir
‑
352之后发现的新mirna将被命名为“mir
‑
353”。同样,辅助设计包括mirna在内的调控性rna工具是容易获得的。在这方面,基于计算机的mirna设计工具是在互联网上wmd2.weigelworld.org/cgi
‑
bin/mirnatools.pl可获得的。
[0209]
本发明人发现,mirna16a可以通过靶向微细胞介导的递送至间皮瘤和肾上腺皮质癌细胞来给予。参见实施例7。在被癌细胞内化后,发现mirna16a有效抑制癌细胞增殖。因此,在一些实施方式中,本公开的微细胞包含mirna16a。可用于抑制肿瘤细胞增殖的其他微小rna包括mir
‑
34家族和let
‑
7家族。
[0210]
如上所述,本发明的组合物中使用的功能性核酸可以抑制促进肿瘤细胞增殖、血管生成或化疗抗性的基因。被抑制的基因本身也可以抑制凋亡或细胞周期停滞。下面提供了可以被功能性核酸靶向的基因的实例。
[0211]
本公开的功能性核酸优选地靶向促进药物抗性、抑制凋亡或促进肿瘤表型的蛋白
质的基因或转录物。功能性核酸策略在这些情况下的成功应用已经在本领域中实现,但是没有微细胞载体的益处。参见例如sioud,trends pharmacol.sci.,2004;caplen,expert opin.biol.ther.,2003;nieth等,2003;caplen和mousses,2003;duxbury等,2004;yague等,2004;和duan等,2004。
[0212]
促进抗药性的蛋白质构成功能性核酸的优选靶标。该蛋白质可促进获得性抗药性或固有性抗药性。当患病的细胞(如肿瘤细胞)起初对药物有响应,但在随后的治疗周期中变得难治时,获得了抗性表型。涉及获得性抗药性的有用靶标包括atp结合盒转运蛋白,如p糖蛋白(p
‑
gp、p
‑
170、pgy1、mdr1、abcb1、mdr相关蛋白、多药抗性蛋白1)、mdr
‑
2和mdr
‑
3。mrp2(多药抗性相关蛋白)、bcr
‑
abl(断点簇区域
‑‑
abelson原癌基因)、sti
‑
571抗性相关蛋白、肺抗性相关蛋白、环氧合酶
‑
2、核因子κ、xrcc1(x
‑
射线交叉互补组1)、ercc1(切除交叉互补基因)、gstp1(谷胱甘肽s
‑
转移酶)、突变体β
‑
微管蛋白和生长因子(如il
‑
6)是获得性抗药性中涉及的其他靶标。
[0213]
促进抗药性的特别有用的靶标包括atp结合盒转运蛋白,如p
‑
糖蛋白、mdr
‑
2、mdr
‑
3、bcrp、apt11a和lrp。有用的靶标还包括促进凋亡抗性的蛋白质。这些包括bcl
‑
2(b细胞白血病/淋巴瘤)、bc1
‑
x
l
、a1/bfl 1、粘着斑激酶、二氢二醇脱氢酶和p53突变蛋白。
[0214]
有用的靶标还包括致癌和突变肿瘤抑制蛋白。这些的示例是β
‑
连环蛋白pkc
‑
α(蛋白激酶c)、c
‑
raf、k
‑
ras(v12)、dp97 dead盒rna解旋酶、dnmt1(dna甲基转移酶1)、flip(flice样抑制蛋白)、c
‑
sfc、53bpi、多梳组蛋白ezh2(zeste同源物增强剂)、erbb1、hpv
‑
16e5和e7(人乳头瘤病毒早期5到早期7)、fortilin&mci1p(骨髓细胞白血病1蛋白)、dip13α(ddc相互作用蛋白13a)、mbd2(甲基cpg结合结构域)、p21、klf4(kruppel样因子4)、tpt/tctp(翻译控制肿瘤蛋白)、spk1和spk2(鞘氨醇激酶)、p300、plk1(polo样激酶
‑
1)、trp53、ras、erbb1、vegf(血管内皮生长因子)、bag
‑
1(bcl2相关的永生基因1))、mrp2、bcr
‑
abl、sti
‑
571抗性相关蛋白、肺抗性相关蛋白、环氧合酶
‑
2、核因子κ、xrcc1、ercc1、gstp1、突变体
‑
β
‑
微管蛋白和生长因子。
[0215]
还可用作靶标的是全局调控元件,其以细胞质聚腺苷酸化元件结合蛋白(cepb)为例。例如,cepb4在成胶质细胞瘤和胰腺癌中过表达,其中该蛋白激活数百种与肿瘤生长相关的基因,并且其在健康细胞中检测不到(oritz
‑
zapater等,2011)。因此,根据本说明书,成胶质细胞瘤的治疗可以通过给予包含完整的细菌源微细胞的组合物来实现,所述微细胞包含对抗cepb4过表达的试剂,如sirna或破坏肿瘤细胞的cepb4表达的其他功能性核酸分子。
[0216]
功能性核酸有用靶标的另一实例包括复制蛋白a(rpa),由70
‑
kda(rpa1)、32
‑
kda(rpa2)和14
‑
kda(rpa3)亚基构成的三聚体复合物——其对于所有生物体中的dna复制都是至关重要。iftode等,1999。
[0217]
其他有用的靶标是对有丝分裂和维持基因组稳定性重要的那些。实例包括polo样激酶(plk1),其被发现在宽范围的癌细胞中过表达。参见实施例3,图12。本公开的发明人还发现,抑制plk1的sirna(siplk1)表达抑制间皮瘤和肾上腺皮质癌细胞的增殖。参见实施例10。因此,在一些实施方式中,本公开的微细胞包括plk1。
[0218]
其他有用的靶标是那些涉及dna复制和修复的那些。实例包括核糖核苷酸还原酶(rr),其是癌症的潜在治疗靶标,因为其催化核糖核苷5'
‑
二磷酸转化为dna复制和修复所
必需的其相应2'
‑
脱氧核糖核苷5'
‑
三磷酸。参见d'angiolella等,2012。人rr包含两个亚基rrm1和rrm2,并且靶向这两个亚基的功能性核酸可用于本发明。本公开的发明人证明,靶向rrm1的sirna(sirrm1)在与微细胞一起递送时有效地抑制间皮瘤和肾上腺皮质癌细胞增殖。参见实施例10。因此,在一些实施方式中,微细胞包含抑制核糖核苷酸还原酶m1(rrm1)表达的sirna。
[0219]
b.i型干扰素激动剂
[0220]
本发明的组合物可以包含1型干扰素激动剂,即增加1型干扰素水平(例如活性或表达水平)的试剂。人类i型干扰素(ifn)是有助于调控免疫系统活性的干扰素蛋白的一大亚组。干扰素与干扰素受体结合。所有i型ifn与特定细胞表面受体复合物结合,该细胞表面受体复合物称为ifn
‑
α受体(ifnαr),由ifnαr1和ifnαr2链组成。哺乳动物i型ifn被称为ifn
‑
α(alpha)、ifn
‑
β(beta)、ifn
‑
κ(kappa)、ifn
‑
δ(delta)、ifn
‑
ε(epsilon)、ifn
‑
τ(tau)、ifn
‑
ω(omega)、和ifn
‑
ζ(zeta,也称为limitin)。
[0221]
i.寡核苷酸
[0222]
图2显示了包含免疫调节的60mer双链dna的微细胞的示例性实施方式的图示。本发明人发现,用靶向egfr的微细胞进行的i型干扰素激动剂如双链dna的递送充当负载有细胞毒性药物的微细胞的抗肿瘤功效的佐助(即,增强)。参见实施例11。因此,组合包装有超毒性药物pnu
‑
159682的微细胞导致抗肿瘤效果增强,并且这种治疗被晚期胰腺癌患者很好地耐受。参见实施例12。
[0223]
i型干扰素(ifn)的表达可以通过将双链dna传递至靶标细胞来诱导。具体而言,来自微生物病原体的胞质dna的先天性免疫激活是由胞质dna传感器(如cgamp、环状gmp
‑
amp合成酶(cgas)和ifnγ诱导因子16(ifi16))介导的i型ifn和促炎性细胞因子的有效触发剂。参见,例如,hansen等,2014;和unterholzner等,2013。与双链dna结合后,cgas具有产生第二信使环状gmp
‑
amp的酶促能力,该第二个信使环gmp
‑
amp对接在ifn基因的内质网结合的蛋白刺激物(sting)上。barber等,2011。这会诱导构象变化,该变化使sting同源二聚化,从er迁移(dobbs等,2015),并募集磷酸化sting的tank结合激酶1,产生启动ifn表达的转录因子ifn调控因子3。参见dobbs等,2015;wang等,2014;和liu等,2015。因此,可以通过将双链dna递送至可以被胞质dna传感器识别的靶标细胞来诱导i型ifn的表达,如上文和引用的参考文献中所述。
[0224]
在一些实施方式中,本文公开的组合物包括完整的微细胞,其包含i型ifn激动剂。在一些实施方式中,如本文所述,i型ifn激动剂是适于dna传感器介导的i型ifn诱导的寡核苷酸。在一些实施方式中,寡核苷酸包含至少约10、至少约20、至少约30、至少约40、至少约50、至少约60、至少约70、至少约80、至少约90、至少约100、至少约110、至少约120、至少约130、至少约140、至少约150、至少约160、至少约170、至少约180、至少约190或至少约200个核苷酸的序列。在另一个实施方式中,寡核苷酸包含约10上至约200个核苷酸或这两个值之间的任何量的序列。在一些实施方式中,寡核苷酸包含至少约40个核苷酸、至少约50个核苷酸或至少约60个核苷酸的序列。
[0225]
在其他实施方式中,酶多核苷酸磷酸化酶(pnpase 1)的多核苷酸产物可以用作ifn活性的合成诱导剂。field等,1967。类似地,dsrna模拟物聚肌苷酸:聚胞苷酸(poly(i:c))显示充当tlr3和mda5的激动剂。alexopoulou等,2001;和gitlin等,2006。因此,在一些
实施方式中,该寡核苷酸是pnpase 1、聚(i:c)、聚
‑
iclc、咪喹莫特(imiquimod)、咪唑并喹啉瑞喹莫德(imidazoquioline resquimod)或cpg
‑
寡脱氧核苷酸的多核苷酸产物。
[0226]
也可以设计合成的寡核苷酸并将其用作核酸传感器的激动剂。例如,基于细菌dna的免疫刺激特性,设计tlr9刺激性合成cpg寡脱氧核苷酸(cpg
‑
odn),其与人dna相比,富有未甲基化的cpg基序。krieg等,1995。序列特征和骨架修饰的优化导致优先激活b细胞或pdc的cpg
‑
odn亚型。因此,本文考虑cpg
‑
odn可以是甲基化的或未甲基化的或两者的组合。
[0227]
有多种分子已知是i型ifn分泌的刺激物,并且这些分子及其激动剂适合通过微细胞递送以引发i型ifn分泌。这些分子包括但不限于双链rna(dsrna)、聚(da:dt)dna、双链z
‑
dna和b
‑
dna、长于36bp的dna(dsdna)和dna
‑
rna杂交体、细菌第二信使环状二gmp、tlr3、tlr4、tlr7、tlr8和tlr9激动剂和sting激动剂,其将在下面被更全面地描述。
[0228]
ii.双链rna(dsrna)
[0229]
双链rna是i型ifn的诱导物。rna解旋酶视黄酸诱导基因i(rig
‑
1)和黑素瘤分化相关基因5(mda5)是触发i型ifn分泌的胞质受体。这些受体(rig
‑
i样受体)通过线粒体定位的接头分子ips
‑
1或mavs以及激酶tbk1和ikki传递信号来激活irf3并诱导i型ifn基因的转录(kawai和akira,2010)。rig
‑
1和mda5响应于5'端三磷酸化的病毒rna(leung和amarasinghe,2016;lu等,2010;marq等,2011;wang等,2010)。
[0230]
iii.聚(da:dt)dna
[0231]
rna聚合酶iii是聚(da:dt)dna的胞质dna传感器(ablasser等,2009)。在细胞质中,rna聚合酶iii将聚(da:dt)转化为5'三磷酸化的rna。然后,转化后的5'
‑
ppp rna启动rig
‑
i
‑
mavs途径和nfκb激活以引发i型ifn分泌。
[0232]
iv.双链z
‑
dna和b
‑
dna
[0233]
胞质dna传感器、irf的dna依赖性激活剂(dai)或z
‑
dna结合蛋白1已知在tbk1和irf3介导的机制中会响应右手dsdna构象(b
‑
dna)诱导i型ifn(kawai和akira,2010)。rna聚合酶iii还将b
‑
dna转录成5'
‑
ppp rna,其然后通过rig
‑
i激活i型ifn转录(chiu等,2009)。在被磷酸化后,这些转录因子有助于驱动i型ifn家族的所有基因的表达,从而扩大i型ifn的产生。多种胞质dna传感器已被报道识别细胞内的病原性dna。参见例如图26,摘自xia等,“dna sensor cgas
‑
mediated immune recognition,”protein cell,7(11):777
‑
791(2016))。
[0234]
例如,ddx41(zhang等,2011b)、ifi16(orzalli等,2012;unterholzner等,2010)和dai(takaoka等,2007)检测双链dna(dsdna)并激活sting
‑
tbk1
‑
irf3途径。lrrfip1结合dsdna,并通过β
‑
连环蛋白触发irf3激活(yang等,2010)。dhx9和dhx36与dsdna缔合,并通过myd88导致nfκb激活(kim等,2010)。ku70与dsdna结合,以通过激活irf1和irf7诱导i型干扰素(ifn)(zhang et al.,2011a)。aim2与dsdna相互作用,并通过募集asc和半胱天冬酶原
‑
1(procaspase
‑
1)激活炎性体(burckstummer等,2009;fernandes
‑
alnemri等,2009;homung等,2009)。值得注意的是,sox2在嗜中性粒细胞的细胞质中表达,并在以序列依赖性方式与dsdna结合后激活tab2/tak1复合物(xia等,2015)。
[0235]
v.长于36bp的dna(dsdna)和dna
‑
rna杂交体
[0236]
cgas是识别细胞质dna的dna传感器(ablasser等,2013a;ablasser等,2013b;gao等,2013a;li等,2013b;schoggins等,2014;sun等,2013;wu等,2013)。长于36bp的双链dna
(dsdna)对于cgas激活是最优的(gao等,2013b)。dna结合后,cgas发生构象变化,其使atp和gtp进入催化口袋,导致cgamp的合成成为sting
‑
tbk1轴的强激活剂(civril等,2013;gao等,2013b;kranzusch等,2013;wu等,2013;zhang等,2014)。cgas可以被dsdna和dna
‑
rna杂交体激活(mankan等,2014)。
[0237]
vi.细菌第二信使环状二gmp
[0238]
细菌第二信使环状二gmp通过独立于dai或其他已知胞质受体但需要tbk1和irf3的机制有效诱导i型ifn(mcwhirter等,2009)。
[0239]
vii.tlr3、tlr4、tlr7、tlr8和tlr9激动剂
[0240]
在一些细胞类型例如巨噬细胞和dc中,分别响应dsrna和脂多糖触发跨膜受体toll样受体3(tlr3)和tlr4而产生i型ifn。tlr3和tlr4通过接头分子trif传导信号,该接头分子与tbk1缔合并激活irf3(kawai和akira,2010)。
[0241]
天然ifn产生细胞,浆细胞样dc(pdc)(colonna等,2004)优先表达细胞内内体受体tlr7和tlr9,使其分别通过接头蛋白myd88触发信号转导而响应单链rna和dna病毒(colonna等,2004)。这些受体有效诱导仅pdc中的i型ifn,因为这些细胞组成性表达irf7和irf8,并且myd88
‑
irf7复合物在tlr连接后经历时空调控,使得其保留在内体区室中,在此其诱导i型ifn产生(colonna等,2004)。
[0242]
tlr4激动剂吡喃葡萄糖基脂质佐剂(gla)被单独测试或与抗pd
‑
1mab组合测试[immune design 2016](j.meulen and s.brady,“immune design,”hum.vaccin.immunother.,13(1):15
‑
16(2017))。tlr3激动剂聚iclc(hiltonol
tm
)和tlr7/8激动剂medi9197也在患有晚期可及实体肿瘤的患者中被测试(medlmmune 2016;oncovir 201)。(“activating the natural host defense;hiltonol(poly
‑
iclc)and malignant brain tumors,a.salzar,oncovir,inc.,www.oncovir.com/id2(于2018年7月11日访问);以及gupta等,“abstract ct091:safety and pharmacodynamic activity of medi9197,a tlr 7/8agonist,administered intratumorally in subjects with solid tumors,”cancer research,aacr annual meeting 2017;2017年4月1
‑
5日(2017年7月发布))。肿瘤内注射tlr激动剂如富cpg寡脱氧核苷酸(cpg odn,pf
‑
3512676)以及低剂量放疗在i/ii期临床研究中显示了晚期非hodgkin淋巴瘤患者的临床响应[dynavax 2016](adamus等,2018)。
[0243]
viii.sting激动剂
[0244]
环状二核苷酸(cdn)[环状二gmp(鸟苷5'
‑
单磷酸)、环状二amp(腺苷5'
‑
单磷酸)和环状gmp
‑
amp(cgamp)]是通过干扰素基因的细胞质模式识别受体刺激物(sting)激活tbk1/irf3/1型干扰素信号传导轴的一类与病原体相关的分子模式分子(pamp)。
[0245]
正在开发新的sting激动剂以引发i型干扰素响应。一种主要方法涉及对cdn进行合理修饰以提高效率,其导致合成二硫混合连接cdn的产生(corrales等,2015)。一种化合物(ml rr
‑
s2 cd a或adlt
‑
s100)结合人和小鼠sting,并在多种动物模型中显示出有效的抗肿瘤效果(corrales等,2015)。adu
‑
s100在患有皮肤可及的实体肿瘤和淋巴瘤的患者中的1期临床试验正在进展中(aduro biotech 2016)。
[0246]
对1000genome project数据库(http://www.1000genomes.org/)的分析确定了五个人类sting变体,包括wt等位基因、参考(ref)等位基因(r232h)、haq等位基因(r71h、
g230a、r293q)、aq等位基因(g230a、r293q)和q等位基因(r293q)(yi等,2013)。
[0247]
合理设计的合成cdn激动剂ml rr
‑
s2 cda已被开发,并呈现与细菌或宿主细胞cgas产生的天然sting配体相比增强的稳定性、人类sting激活、细胞摄取和抗肿瘤功效以及低反应原性(corrales等,2015;fu等,2015)。
[0248]
与不含二硫修饰的cdn相比,rp、rp(r,r)二硫取代的非对映异构体cdns抵抗磷酸二酯酶的消化,刺激培养的人细胞中ifn
‑
b的更高表达,并诱导更有效的抗肿瘤免疫力(corrales等,2015;fu等,2015)。为了增加对人sting的亲和力,ml rr
‑
s2 cda包含由具有一个2'
‑
5'和一个3'
‑
5'混合磷酸二酯键(2',3'cdn)的磷酸酯桥限定的非典型结构。2',3'混合键结构赋予增加的sting结合亲和力(gao等,2013b),并且还被发现于真核cgas产生的内源性cgamp中。ml rr
‑
s2 cda被证明在hek293t细胞sting信号传导测定中广泛激活所有已知的人类sting等位基因,并在从多个具有不同sting基因型的供体分离的人外周血单核细胞(pbmc)中诱导ifn
‑
β的剂量依赖性表达,该供体包括ref等位基因纯合的供体,其已知对细菌3',3'cdn诱导的信号传导是难治的(corrales等,2015;fu等,2015)。
[0249]
c.ii型干扰素激动剂
[0250]
本发明的组合物和方法可以包括ii型ifn激动剂,即增加ii型干扰素水平(例如活性或表达水平)的试剂。该类ii型干扰素(ifn)目前包括被称为ifn
‑
γ(gamma)的成员。成熟的ifn
‑
γ是反平行同源二聚体,其与ifn
‑
γ受体(ifngr)复合物结合以在其靶标细胞内引发信号。ifngr由两个亚基构成,各亚基被命名为ifngr1和ifngr2。ifn
‑
γ参与免疫和炎症响应的调控;在人类体内,只有一种类型的干扰素
‑
γ。其在激活的t细胞和自然杀伤细胞中产生。ifn
‑
γ增强i型ifn的效果。th1细胞释放的ifn
‑
γ将白细胞募集到感染位点,导致炎症增加。其还刺激巨噬细胞以杀死被吞食的细菌。th1细胞释放的ifn
‑
γ在调控th2响应方面也很重要。由于ifn
‑
γ与免疫响应的调控密切相关,其产生可导致自身免疫障碍。
[0251]
因此,本发明的一个实施方式包括组合物,其包含微细胞,该微细胞包含ii型ifn激动剂。尽管微细胞源自细菌,但微细胞本身不激活人类患者的ii型干扰素响应。参见实施例15。本发明人发现,ifnγ的添加增强了负载有多柔比星的靶向egfr的edv的抗肿瘤功效,并且在异种移植模型中引起肿瘤消退。参见实施例13。此外,包含(i)负载有超毒性化疗药物pnu
‑
159682的efgr靶向微细胞;(ii)负载有包含60个核苷酸的双链dna的非靶向微细胞;和(iii)包含ifnγ产品imukin的微细胞的组合物被耐受,并且在患有晚期内源性肿瘤的犬中引起抗癌效果。参见实施例14。
[0252]
ii型干扰素通过激活细胞毒性t细胞在抗肿瘤免疫中发挥重要作用。参见例如chikuma等,2017。先天性自然杀伤细胞在天然抗原结合后释放ifnγ细胞因子,但是糖鞘脂化合物可充当先天性和获得性免疫响应的有效激活剂。本发明人发现,暴露于糖鞘脂会通过先天性自然杀伤t(inkt)细胞(包括ii型干扰素、ifn
‑
γ和多种白介素(th1、th2和/或th17型细胞因子)诱导强细胞因子响应。参见,例如,carreno等,2016。inkt细胞然后诱导dc成熟并显示出t细胞辅助因子样功能,其导致细胞毒性t细胞响应的产生。
[0253]
可用于诱导ii型ifn响应的糖鞘脂的实例在本文中被描述,并且包括α
‑
半乳糖苷神经酰胺的c
‑
糖苷形式(ac
‑
galcer)、α
‑
半乳糖苷神经酰胺(α
‑
galcer)、半乳糖苷神经酰胺的12碳酰基形式(β
‑
galcer)、β
‑
d
‑
吡喃葡萄糖神经酰胺(β
‑
glccer)、1,2
‑
二酰基
‑3‑0‑
半乳糖基
‑
sn
‑
甘油(bbgl
‑
ii)、含有二酰基甘油的糖脂(glc
‑
dag
‑
s2)、神经节苷脂(gd3)、神经节
三酰基神经酰胺(gangliotriaosylceramide)(gg3cer)、糖基磷脂酰肌醇(gpi)、α
‑
葡萄糖醛酸神经酰胺(α
‑
glucuronosylceramide)(gsl
‑
1或gsl
‑
4)、异珠三己糖基神经酰胺(isoglobotrihexosylceramide)(igb3)、脂质磷酸聚糖(lipophosphoglycan)(lpg)、溶血磷脂酰胆碱(lyosphosphatidylcholine)(lpc)、α
‑
半乳糖基神经酰胺类似物(och)和苏糖醇神经酰胺。在具体实施方式中,本文公开的微细胞包含α
‑
半乳糖苷神经酰胺(α
‑
galcer)作为ii型ifn激动剂。
[0254]
α
‑
gc,一种inf ii型激动剂,已知通过激活一种称为自然杀伤t细胞(nkt细胞)的白细胞类型来刺激免疫系统(birkholz等,2015)。已知微细胞能够促进α
‑
gc在靶标细胞上的呈递,如实施例17中进一步讨论,申请人进而研究利用微细胞
α
‑
gc
的微细胞促进的免疫激活可以补充由微细胞促进的化疗药物递送组成的治疗。
[0255]
如实施例18中所示,申请人发现,与仅给予
ep
微细胞
dox
的小鼠相比,被给予含有化疗性多柔比星(
ep
微细胞
dox
)的微细胞和含有α
‑
gc的微细胞(微细胞
α
‑
gc
)的含肿瘤小鼠显示出肿瘤进展的明显中止。这些观察结果表明,不包括i型inf激动剂并且取而代之包含ii型inf激动剂的微细胞组合物有效治疗小鼠的肿瘤。
[0256]
微细胞可以将ii型ifn激动剂直接递送至免疫系统的细胞,以增强体内的inkt细胞激活和ii型干扰素ifn
‑
γ产生。可选地,非靶向的edv被免疫系统的吞噬细胞摄取,并在内体中被分解,并且αgc被呈递给inkt细胞进行免疫激活。因此,在一些实施方式中,微细胞提供ii型干扰素激动剂的靶向递送。在其他实施方式中,本文公开的组合物包含非靶向微细胞,该非靶向微细胞包含ii型干扰素激动剂。
[0257]
ifn
‑
γ的产生受控于抗原呈递细胞(apc)分泌的细胞因子,最显著地白介素(il)
‑
12和il
‑
18。这些细胞因子在先天免疫响应中充当联系感染与ifn
‑
γ产生的桥梁。多种病原体的巨噬细胞识别诱导il
‑
12和趋化因子的分泌。这些趋化因子将nk细胞吸引到炎症位点,并且il
‑
12促进这些细胞中的ifn
‑
γ合成。在巨噬细胞、自然杀伤细胞和t细胞中,il
‑
12和il
‑
18刺激的组合进一步增加ifn
‑
γ的产生。因此,这些蛋白质中的任一种或其组合是适于本公开目的的试剂。
[0258]
ifn
‑
γ产生的负调控因子包括il
‑
4、il
‑
10、转化生长因子β和糖皮质激素。抑制这些因素的蛋白质或核酸将能够刺激ifn
‑
γ的产生。
[0259]
还适用于此情况的是编码ifn
‑
γ的多核苷酸或激活ifn
‑
γ的产生和/或分泌的基因。
[0260]
增加ifn
‑
γ水平的试剂也可以是病毒疫苗。多种可以诱导ifn
‑
γ的产生而不引起感染或其他类型的不利影响的病毒疫苗是可获得的。此类病毒疫苗剂的示例是flu(流感)疫苗。
[0261]
数据显示,当患者还接受负载药物的、靶向双特异性抗体的微细胞或被杀死的细菌细胞的给予时,有效激活宿主对肿瘤细胞的免疫响应所需的ifn
‑
γ血清浓度低。因此,一方面,本发明的方法导致不高于约30,000pg/ml的血清ifn
‑
γ浓度增加。在另一方面,血清ifn
‑
γ浓度增加至不高于约5000pg/ml、1000pg/ml、900pg/ml、800pg/ml、700pg/ml、600pg/ml、500pg/ml、400pg/ml、300pg/ml、200pg/ml、或100pg/ml。在另一方面,所得血清ifn
‑
γ浓度为至少约10pg/ml,或至少约20pg/ml、30pg/ml、40pg/ml、50pg/ml、60pg/ml、70pg/ml、80pg/ml、90pg/ml、100pg/ml、150pg/ml、200pg/ml、300pg/ml、400pg/ml或500pg/ml。
[0262]
根据一些方面,所述试剂是ifn
‑
γ蛋白或改造蛋白或类似物。在一些方面,所述给予达到每ml宿主血液约0.02ng至1微克ifn
‑
γ。一方面,宿主血液中达到的ifn
‑
γ浓度为每毫升约0.1ng至约500ng、每毫升约0.2ng至约200ng、每毫升约0.5ng至约100ng、每毫升1ng至约50ng、或每毫升约2ng至约20ng。
[0263]
iii.完整的细菌源微细胞
[0264]
术语“微细胞”在此用于表示这样的细菌细胞衍生物:缺乏染色体(“无染色体”),并且是通过在二元裂变期间细胞分裂与dna分离的协调中的扰动而产生的。微细胞区别于其他小囊泡,如所谓的“膜泡”(尺寸约0.2μm或更小),其在某些情况下自发产生和释放但并不是由于特定基因重排或游离基因表达所致。出于同样的原因,完整的微细胞区别于细菌幽灵,细菌幽灵不是由于特定基因重排或游离基因表达而产生的。在本公开中采用的细菌源微细胞是完全完整的,因此区别于特征在于外膜或限定膜被破坏或降解甚至被去除的细菌细胞衍生物的其他无染色体形式。参见美国专利号7,183,105第111栏第54行及其以下。表征本发明的微细胞的完整膜允许治疗有效载荷保留在微细胞中,直到有效载荷在摄取后在肿瘤细胞内被释放。
[0265]
微细胞或edv是无核的非活体纳米粒子,其是通过使控制正常细菌细胞分裂的基因失活,从而去抑制细胞的极体位点而产生的。ma等,2004。去抑制意味着细菌在中心和极点处分裂;本公开的发明人所证实的产生微细胞的极体分裂可以充当允许有效包装各种不同的化疗药物的抗泄漏微储库载体。此外,与例如当前隐形脂质体药物载体如doxil(脂质体多柔比星)(每个粒子只能包装~14,000个分子)(park et al.,breast cancer res.,4(3):95
‑
99(2002))或“武装抗体”(可以携载少于5个药物分子)相比,edv可以轻松容纳上至100万个药物分子的有效载荷。此外,edv可以利用双特异性抗体靶向癌细胞表面上的过表达受体,请参见下文d章节,这允许体内和体外均高度显著的肿瘤生长抑制和/或消退。
[0266]
本发明中使用的微细胞可以从细菌细胞如大肠杆菌(e.coli)和鼠伤寒沙门氏菌(s.typhymurium)制备。原核染色体复制与正常的二元裂变相关,后者涉及中细胞隔膜形成。例如,在大肠杆菌中,min基因(如mincd)的突变可以去除细胞分裂过程中细胞极点处隔膜形成的抑制,导致正常的子细胞和缺少染色体的微细胞产生。参见de boer et al.,j.bacteriol.,174:63
‑
70(1992);raskin&de boer,j.bacteriol.,181:6419
‑
s24(1999);hu&lutkenhaus,mol.microbio.,34:82
‑
90(1999);harry,mol.microbiol.,40:795
‑
803(2001)。
[0267]
除最小操纵子突变外,无染色体微细胞还在一系列影响隔膜形成的其他遗传重排或突变后生成,例如在枯草芽孢杆菌(b.subtilis)中的divivb1中。参见reeve and cornett,j.virol.,15:1308
‑
16(1975)。在参与细胞分裂/染色体分离的蛋白质的基因表达水平扰动后,也可以形成微细胞。例如,mine的过度表达导致极体分裂和微细胞产生。类似地,无染色体的微细胞可由染色体分离的缺陷引起,例如枯草芽孢杆菌中的smc突变(britton et al.,genes dev.,12:1254
‑
9(1998))、枯草芽孢杆菌中的spooj缺失(ireton et al.,j.bacteriol.,176:5320
‑
29(1994))、大肠杆菌中的mukb突变(hiraga et al.,j.bacteriol.,171:1496
‑
1505(1989))、以及大肠杆菌中的parc突变(stewart and d’ari,j.bacteriol.,174:4513
‑
6(1992))。此外,cafa可在复制后提高细胞分裂速度和/或抑制染色体分配(okada et al.,j.bacteriol.,176:917
‑
22(1994)),导致链状细胞和无染色体微
细胞形成。
[0268]
因此,由于这些细菌中细菌细胞分裂的保守性质,可以从任何细菌细胞制备本发明的微细胞,无论其是革兰氏阳性还是革兰氏阴性来源。此外,如上所述,本公开中使用的微细胞应具有完整的细胞壁(即“完整的微细胞”),并且应与其他小囊泡(例如膜泡)区别开并与之分开,该其他小囊泡不属于特定遗传重排或游离基因表达。
[0269]
在给定的实施方式中,微细胞的亲本(源)细菌可以是革兰氏阳性的,或者其可以是革兰氏阴性的。一方面,亲本细菌是选自以下的一种或多种:terra
‑
/glidobacteria(bv1)、变形菌门(proteobacteria)(bv2)、bv4——包括螺旋体门(spirochaetes)、鞘脂杆菌门(sphingobacteria)和浮游细菌门(planctobacteria)。根据另一方面,所述细菌是选自以下的一种或多种:厚壁菌门(firmicutes)(bv3)如杆菌(bacilli)、梭菌(clostridia)或软壁菌(tenericutes)/柔膜菌(mollicutes)或放线菌门(actinobacteria)(bv5),例如放线菌目(actinomycetales)或双歧杆菌目(bifidobacteriales)。
[0270]
根据本发明,被杀死的细菌细胞是细菌、蓝细菌、真细菌和古细菌的非活体原核细胞,如bergey的manual of systematic biology第二版中定义。这种细胞如果其拥有完整的细胞壁和/或细胞膜并且包含细菌物种内源的遗传物质(核酸),则被认为是“完整的”。制备被杀死的细菌细胞的方法被描述于例如美国专利申请公开号2008/0038296,其内容通过引用并入本文。
[0271]
在又一方面,细菌是选自以下的一种或多种:原细菌(eobacteria)(绿弯菌门(chloroflexi)、异常球菌
‑
栖热菌门(deinococcus
‑
thermus))、蓝细菌(cyanobacteria)、热脱硫杆菌门(thermodesulfobacteria)、嗜热菌(thermophiles)(产水菌门(aquificae)、热袍菌门(thermotogae))、α、β、γ(肠杆菌科(enterobacteriaceae))、δ或ε变形菌门、螺旋体门、纤维杆菌门(fibrobacteres)、绿菌门(chlorobi)/拟杆菌门(bacteroidetes)、衣原体门(chlamydiae)/疣微菌门(verrucomicrobia)、浮霉菌门(planctomycetes)、酸杆菌门(acidobacteria)、产金菌门(chrysiogenetes)、脱铁杆菌门(deferribacteres)、梭杆菌门(fusobacteria)、芽单胞菌门(gemmatimonadetes)、硝化螺旋菌门(nitrospirae)、互养菌门(synergistetes)、网团菌门(dictyoglomi)、lentisphaerae bacillales、芽孢杆菌科(bacillaceae)、李斯特氏菌科(listeriaceae)、葡萄球菌科(staphylococcaceae)、乳杆菌目(lactobacillales)、肠球菌科(enterococcaceae)、乳杆菌科(lactobacillaceae)、明串珠菌科(leuconostocaceae)、链球菌科(streptococcaceae)、梭菌目(clostridiales)、盐厌氧菌目(halanaerobiales)、嗜热厌氧菌目(thermoanaerobacterales)、支原体目(mycoplasmatales)、虫原体目(entomoplasmatales)、厌氧原体目(anaeroplasmatales)、无胆甾原体目(acholeplasmatales)、haloplasmatales、放线菌亚目(actinomycineae)、放线菌科(actinomycetaceae)、棒杆菌亚目(corynebacterineae)、诺卡氏菌科(nocardiaceae)、棒状杆菌科(corynebacteriaceae)、弗兰克氏菌亚目(frankineae)、弗兰克氏菌科(frankiaceae)、微球菌亚目(micrococcineae)、短杆菌科(brevibacteriaceae)和双歧杆菌科(bifidobacteriaceae)。
[0272]
对于药物用途,本公开的组合物应包含与免疫原性组分和其他毒性污染物尽可能彻底分离的微细胞或被杀死的细菌细胞。纯化细菌源微细胞以去除游离内毒素和亲本细菌细胞的方法被描述于例如wo 2004/113507,其通过引用整体并入本文。简而言之,纯化过程
实现了(a)较小的囊泡(如膜泡,总体上尺寸小于0.2μm)、(b)从细胞膜释放的游离内毒素、以及(c)不论是活还是死的亲本细菌及其碎片(其也是游离内毒素的来源)的去除。这种去除可以通过以下来实现:去除较小囊泡和细胞碎片的0.2μm滤器、在诱导亲本细胞形成细丝后去除亲本细胞的0.45μm滤器、杀死活细菌细胞的抗生素和抗游离内毒素抗体。
[0273]
纯化程序的基础是本发明人的如下发现:尽管其细菌来源不同,但所有完整的微细胞的尺寸均为约400nm,即大于膜泡和其他较小的囊泡,但小于亲本细菌。可以通过使用固态(例如电子显微镜)或通过基于液体的技术(例如动态光散射)来完成微细胞的尺寸确定。通过每种这样的技术得出的尺寸值可具有误差范围,并且这些值在不同技术之间可有所不同。因此,干燥状态下的微细胞的尺寸可以通过电子显微镜测量为约400nm
±
50nm。动态光散射可以测量相同的微细胞尺寸为约500nm
±
50nm。而且,再次使用动态光散射,可以测量包装药物的、靶向配体的微细胞为约400nm至600nm
±
50nm。
[0274]
这种尺寸分散(scatter)值在实践中容易适应于例如将微细胞从免疫原性组分和其他毒性污染物分离的目的,如上所述。即,完整的细菌源微细胞的特征在于细胞质被刚性膜包围,这给予微细胞刚性的球形结构。这种结构在透射电子显微照片中很明显,其中在刚性膜的外部界限之间测量跨越微细胞的微细胞直径。该测量提供上述400nm
±
50nm的尺寸值。
[0275]
源自革兰氏阴性细菌的被杀死的细菌细胞或微细胞的另一个结构元素是脂多糖(lps)的o多糖组分,其通过脂质a锚定体嵌入外膜中。该组分是重复的糖残基单元链,每个链重复单元具有4至5个糖的多达70至100个重复单元。由于这些链在液体环境中如在体内一样不是刚性的,因此其可以采用波浪状的柔性结构,该结构给出珊瑚海环境中的海藻的总体外观。即,链与随液体移动,同时保持锚定在微细胞膜上。
[0276]
如上所述,受o
‑
多糖组分的影响,动态光散射可提供约500nm至约600nm的微细胞尺寸值。然而,来自革兰氏阴性和革兰氏阳性细菌的微细胞都容易通过0.45μm的滤器,这证明有效的微细胞尺寸为400nm
±
50nm。上述尺寸分散被包括在本发明中,并且特别是通过短语“约400nm的尺寸”等中的限定词“约”来表示。
[0277]
关于毒性污染物,本公开的组合物优选包含少于约350eu的游离内毒素。在这方面的示例是如下游离内毒素水平:约250eu或更少、约200eu或更少、约150eu或更少、约100eu或更少、约90eu或更少、约80eu或更少、约70eu或更少、约60eu或更少、约50eu或更少、约40eu或更少、约30eu或更少、约20eu或更少、约15eu或更少、约10eu或更少、约9eu或更少、约8eg或以下、约7eg或以下、约6eg或以下、约5eg或以下、约4eg或以下、约3eg或以下、约2eg或以下、约1eg或以下、约0.9eu或更少、约0.8eu或更少、约0.7eu或更少、约0.6eu或更少、约0.5eu或更少、约0.4eu或更少、约0.3eu或更少、约0.2eu或更少、约0.1eu或更少、约0.05eu或更少、或约0.01eu或更少。
[0278]
本公开的组合物还可包含至少约109个微细胞或被杀死的细菌细胞,例如至少约1
×
109、至少约2
×
109、至少约5
×
109或至少8
×
109个。在一些实施方式中,组合物包含不超过约10
11
个微细胞或被杀死的细菌细胞,例如不超过约1
×
10
11
或不超过约9
×
10
10
,或不超过约8
×
10
10
个。
[0279]
iv.将活性剂负载到微细胞或被杀死的细菌细胞中
[0280]
活性剂或抗肿瘤剂,如小分子药物、蛋白质和功能性核酸,可以直接通过将多个完
整的微细胞与活性剂在缓冲剂中共同培育而被包装到微细胞中。缓冲剂组成可以根据本领域公知的条件来改变,以优化活性剂在完整微细胞中的负载。缓冲剂还可以根据试剂而变化(例如,在核酸有效载荷的情况下,根据要负载在微细胞中的核酸的核苷酸序列或长度)。适合负载的示例性缓冲剂包括但不限于磷酸盐缓冲盐水(pbs)。在包装后,活性剂将保留在微细胞内部,并受到保护以免降解。关于在无菌盐水中培育的包装sirna的微细胞进行的长时间培育研究表明,例如,sirna没有泄漏。
[0281]
活性剂如功能性核酸或可以被核酸编码的蛋白质可通过将编码该活性剂的载体如质粒转化到亲本细菌细胞中而被引入微细胞。当由亲本细菌细胞形成微细胞时,微细胞保留质粒和/或表达产物抗肿瘤剂的某些拷贝。包装和表达产物到微细胞中的更多细节被提供在wo 03/033519中,其内容整体通过引用并入本公开中。
[0282]
wo 03/033519中提供的数据证明,例如,携载哺乳动物基因表达质粒的重组微细胞可被递送至吞噬细胞和非吞噬细胞。wo 03/033519还描述了用游离复制质粒dna上携载的异源核酸对产生微细胞的亲本细菌菌株的遗传转化。在分离亲本细菌和微细胞后,一些游离dna被分离到微细胞中。所得重组微细胞容易被哺乳动物吞噬细胞吞食,并在细胞内吞噬溶酶体内降解。另外,一些重组dna逃脱吞噬酶体膜,并被转运到表达重组基因的哺乳动物细胞核中。
[0283]
在其他实施方式中,可以将针对不同mrna靶标的多种核酸包装在相同微细胞中。这种方法可用于对抗药物抗性和凋亡抗性。例如,癌症患者通常呈现对化疗药物的抗性。这种抗性可以通过基因的过表达来介导,如多药抗性(mdr)泵和抗凋亡基因等。为了抵抗这种抗性,可以使微细胞包装治疗显著浓度的针对mdr相关基因的功能性核酸,并在化疗前给予患者。此外,由于大多数分子靶标易受突变并具有多个等位基因,将针对不同mrna靶标的多种功能性核酸包装到相同微细胞中可提高治疗成功率。在wo 2009/027830中提供了将核酸直接包装到微细胞中的更多细节,其内容整体通过引用并入本公开中。
[0284]
可以通过在包含微细胞的细胞外介质和微细胞胞质之间产生药物的浓度梯度,将小分子药物(无论亲水性还是疏水性)包装在微细胞中。当细胞外介质的药物浓度高于微细胞胞质时,药物自然会沿该浓度梯度移动,进入微细胞胞质。但是,当浓度梯度颠倒时,药物不会移动到微细胞外。药物负载过程及其惊人性质的更多细节被发现于例如美国专利申请公开号2008/0051469,其内容通过引用具体并入本文。
[0285]
为了给微细胞负载通常不溶于水的药物,可以首先将药物溶解在适当的溶剂中。例如,可以将紫杉醇溶解在乙醇和cremophore el(聚乙氧基化蓖麻油)的1:1混合物中,然后在pbs中稀释以获得紫杉醇溶液,该溶液在水性介质中部分稀释,并携载最少量的有机溶剂以确保药物保持处于溶液。可以在该最终培养基中培育微细胞以进行药物负载。因此,发明人发现,甚至疏水性药物也可以扩散到微细胞的胞质或膜中,以达到高度的和治疗上显著的细胞质药物载荷。这是出乎意料的,因为微细胞膜是由疏水性磷脂双层构成的,预见其阻止疏水性分子扩散到细胞质中。各种代表性小分子药物在微细胞中的负载已被已被证明,示例了不同的尺寸和化学性质:多柔比星、紫杉醇、氟紫杉醇、顺铂、长春碱、孟沙特罗(monsatrol)、胸苷酸合酶(ts)抑制剂osi
‑
7904、伊立替康、5
‑
氟尿嘧啶、吉西他滨和卡铂。另外,全面地,所得的包装小分子药物的微细胞在体外和体内显示出显著的抗肿瘤功效。
[0286]
v.将微细胞靶向特定的哺乳动物细胞和肿瘤
[0287]
发明人发现肿瘤细胞周围的血管显示出完整性的丧失。也就是说,甚至在血脑屏障(bbb)环境中,血管也具有大开窗并且是“泄漏的”。在癌细胞建立时,其会分泌促进新血管形成的物质——被称为血管生成的过程。这些血管生长迅速,并且与正常血管不同,其是泄漏的,具有从50nm到1.2μm范围的“孔”(开窗)(超透性脉管系统)。目前认为诸如脂质体的药物递送粒子通过被动过程实现肿瘤靶向,所述被动过程涉及从支持肿瘤微环境的渗漏脉管系统渗出。hobbs等,1998。尽管已经表明异常肿瘤微环境的特征在于间质性高血压,并且这种现象可限制抗癌抗体治疗剂的到达(access),但并非呈现为绝对障碍,以免疫脂质体(nielsen等,2002)和量子点缀合的抗体(gao等,2004)为例。这种现象对于edv也一样,edv具有携载特异性定向肿瘤抗体的额外优势。在静脉注射后,edv渗出到肿瘤微环境中,然后通过癌细胞表面受体的参与和内吞作用进行主动靶向。因此,与常规理解相比,大如微细胞(即远大于上述bbb的共识孔径限制)的粒子却仍小于泄漏血管壁的开窗;因此,其可以通过这些开窗被动地渗入并进入肿瘤微环境。
[0288]
在进入肿瘤微环境后,微细胞能够触发受体介导的宿主肿瘤细胞内化并被其摄取。因此,包装有抗肿瘤剂的微细胞将该剂释放到肿瘤细胞的细胞质中,将其杀死。
[0289]
根据本公开的另一方面,如上所述,组合物的微细胞或被杀死的细菌细胞通过配体被导向靶标哺乳动物肿瘤细胞。在一些实施方式中,配体是“双特异性的”。也就是说,配体对微细胞和哺乳动物(肿瘤)细胞组分均显示特异性,从而其使给定的囊泡结合至靶标细胞,从而使靶标细胞吞食前者。双特异性配体将微细胞靶向肿瘤细胞的应用被进一步描述于wo 05/056749和wo 05/079854,并且双特异性配体将被杀死的细菌细胞靶向肿瘤细胞的应用被进一步描述于美国专利号8,591,862,其对应内容通过引用整体并入本文。在这种配体附接于囊泡后,配体的空位特异性(“单特异性”)适用,直到其与靶标(肿瘤)哺乳动物细胞相互作用。多个肿瘤靶向配体是本领域已知的(hong等,2011;hoelder等,2012;galluzzi等,2013)。几种肽,例如生长抑素(sst)肽、血管活性肠肽(vip)、arg
‑
gly
‑
asp(rgd)肽和铃蟾肽/胃泌素释放肽(bbn/grp),已被成功地表征肿瘤受体成像(de jong等,2009;tweedle,2009;schottelius和wester 2009;igarashi等,2011;laverman等,2012)。
[0290]
靶向肿瘤的肽序列可以主要通过三种不同的方式进行选择:(1)从天然蛋白衍生(nagpal等,2011);(2)化学合成和基于结构的合理改造(andersson等,2000;merrifield,2006);(3)肽库筛选(gray和brown,2013)。在这些方法中,噬菌体展示技术是一种常规的但使用最广泛的方法,其具有许多优点,如易于处理以及可以有效筛选大量不同的肽(deutscher,2010)。
[0291]
在肿瘤细胞上而非正常细胞上过表达的受体是体内肿瘤成像的极佳候选者。迄今为止,已鉴定了多种肿瘤靶向肽及其类似物,如下所述。
[0292]
arg
‑
gly
‑
asp(rgd)肽——rgd特异性结合至整联蛋白受体(ruoslahti,1996)。整联蛋白构成两个亚基(α和β亚基)。整联蛋白家族,尤其是αvβ3,与肿瘤血管生成和转移相关。其是在血管生成过程中在内皮细胞上过度表达,但在大多数正常器官中几乎检测不到。因此,其被广泛用于诊断成像。
[0293]
铃蟾肽(bbn)/胃泌素释放肽(grp)——两栖类bbn及其相关肽由一系列神经肽组成,这些神经肽呈现各种生理效果,如外分泌和内分泌、热调控、蔗糖调控以及细胞生长(ohki
‑
hamazaki等,2005)。铃蟾肽样肽受体具有4个亚型:神经调节素b受体、铃蟾肽3受体、
grp受体和铃蟾肽4受体。这些受体在多种肿瘤如乳腺癌、卵巢癌和胃肠道间质肿瘤中过表达。
[0294]
胆囊收缩素(cck)/胃泌素肽——cck和胃泌素是在胃肠道以及中枢神经系统中发挥多种生理作用的结构和功能上相似的肽(matsuno等,1997)。已经鉴定出三种类型的cck受体(cck1、cck2和cck2i4sv),其均属于gpcr的超家族。其中,cck2/胃泌素受体在人类癌症如间质性卵巢癌和星形细胞瘤等中经常被发现。
[0295]
α
‑
黑素细胞刺激激素(α
‑
msh)——α
‑
msh是线性十三肽,主要负责皮肤色素沉着调控(singh和mukhopadhyay,2014)。α
‑
msh及其类似物呈现对黑素皮质素
‑
1受体(mc
‑
1r)的结合亲和力,该受体在超过80%的人类黑素瘤转移中表达,因此被广泛用作黑素瘤靶向成像和放射疗法的媒介。
[0296]
神经肽y(npy)——npy是一种36个氨基酸的肽,属于胰腺多肽家族(tatemoto,2004)。npy受体在包括神经母细胞瘤、肉瘤和乳腺癌在内的各种肿瘤中过表达。
[0297]
神经降压素(nt)——nt是一种13个氨基酸的肽,靶向已在各种肿瘤(如导管胰腺腺癌、小细胞肺癌和甲状腺髓样癌)中鉴定出来的nt受体(tyler
‑
mcmahon等,2000)。因此,其是用于癌症成像的有吸引力的候选者。
[0298]
前列腺特异性膜抗原(psma)——前列腺癌细胞在细胞表面上过表达psma(silver等,2007;ghosh和heston,2004;mhawech
‑
fauceglia等,2007;santoni等,2014)。有几种可得的靶向psma的放射性药物,包括[
68
ga]ga
‑
psma
‑
hbed
‑
cc(也称为[
68
ga]ga
‑
psma
‑
11[pet])、单克隆抗体[
177
lu]lu/[
90
y]y
‑
j591(治疗)、[
123
i]i
‑
mip
‑
1072(平面/spect)、[
131
i]i
‑
mip
‑
1095(治疗)和治疗剂psma
‑
i&t和dkfz
‑
psma
‑
617(psma
‑
617),其用
68
ga标记进行pet或用
177
lu标记用于治疗。
[0299]
生长抑素(sst)肽——sst是天然存在的具有14或28个氨基酸的环肽激素(weckbecker等,2003)。其可以抑制胰岛素、胰高血糖素和其他一些激素的分泌。生长抑素受体(sstr;五个亚型sstr1
‑
sstr5)在多种肿瘤(包括神经胶质瘤、神经内分泌肿瘤和乳腺肿瘤)中过表达。gep系统的神经内分泌肿瘤(nen)最常起源于胰腺、空肠、回肠、盲肠、直肠、阑尾和结肠。所有gep
‑
nen的共同特征是内分泌和神经细胞的复合特征。分化良好的nen过表达生长抑素受体(sstr),尤其是sstr
‑
2亚型。
[0300]
物质p——物质p是属于被称为速激肽的神经肽家族的十一肽(strand,1999)。物质p是已被发现在各种癌细胞上表达的神经激肽1受体(nk1r)已知特定内源配体。
[0301]
t140——t140是具有一个二硫桥的14个氨基酸的肽,并且是4型趋化因子受体(cxcr4)的反向激动剂(burger等,2005)。其衍生物被广泛用作cxcr4成像剂。
[0302]
肿瘤分子靶向肽1(tmtp1)——tmtp1是已被发现特异性结合高度转移性癌细胞,尤其是来自一般肝微转移的癌细胞的5个氨基酸的肽(yang等,2008)。
[0303]
血管活性肠肽(vip)——vip是具有28个氨基酸的神经肽(igarashi等,2011)。其促进血管扩张、细胞生长和增殖。其作用主要受两种受体亚型(vpac1和vpac2)控制。大量vip受体在包括胰腺腺癌和神经内分泌肿瘤在内的许多肿瘤上表达。
[0304]
借助配体和细胞膜上组分如多糖、糖蛋白或多肽之间的相互作用,可以将配体附接到囊泡的细胞膜上。表达的配体被锚定在囊泡的表面上,使得配体的肿瘤表面组分结合部分暴露,从而该部分可以在囊泡和哺乳动物肿瘤细胞接触时结合靶标哺乳动物细胞表面
受体。
[0305]
可选地,配体可以由细菌源囊泡的活体对应物(例如由微细胞的亲本细胞或成为被杀死细胞之前的细菌细胞)表达和展示。在这种情况下,配体不需要针对囊泡的特异性,而仅对哺乳动物细胞的特征性组分显示特异性。也就是说,这种组分就无需是肿瘤细胞本身独有的,甚至不需要是被治疗的特定种类的肿瘤细胞所独有的,只要肿瘤细胞在其表面上呈递该组分。
[0306]
在静脉内给予后,囊泡在肿瘤微环境中迅速积累。根据上述渗漏的肿瘤脉管系统而发生的这种积累引起囊泡包装的治疗有效载荷递送至肿瘤细胞,然后肿瘤细胞内化包装的囊泡。
[0307]
发明人已经发现,这种递送方法适用于多种哺乳动物肿瘤细胞,包括通常对微细胞的特异性粘附和内吞作用难治的细胞。例如,包含针对抗her2受体或抗egf受体的抗体的配体可以使微细胞与一系列被靶向的非吞噬细胞(例如肺癌、卵巢癌、脑癌、乳腺癌、前列腺癌和皮肤癌细胞)上的对应受体结合。
[0308]
因此实现的结合是在每种类型的非吞噬细胞摄取囊泡之前。也就是说,在本发明的环境中,合适的靶标细胞呈递细胞表面受体,该细胞表面受体通过囊泡上的配体的结合引起该囊泡的内吞作用。
[0309]
更具体地,本发明人发现,(a)微细胞或被杀死的细菌细胞上的配体与(b)哺乳动物细胞表面受体之间的相互作用可以激活摄取途径,在此称为“受体介导型内吞”(rme)途径进入靶标宿主细胞(如肿瘤细胞)的晚期内体/溶酶体区室。通过该rme途径,发明人发现,细菌源囊泡通过早期的内体、晚期的内体和溶酶体进行加工,导致其有效载荷释放到哺乳动物宿主细胞的细胞质中。而且,作为核酸的有效载荷不仅逃脱了在晚期内体/溶酶体区室中的完全降解,而且被宿主细胞表达。
[0310]
用于这种递送方法的肿瘤靶向配体可以是“双特异性的”,如上所述,因为其分别结合携载有效载荷的囊泡和靶标细胞上的表面成分,并且其与后者组分的相互作用导致囊泡摄取到rme途径中。在任何情况下,根据本发明,给定的靶标细胞表面受体可以是配体结合的候选物——如果与组分的相互作用实际上进入了内吞途径,内吞途径需要从靶标细胞表面进行胞质内化。这种候选物在本发明中的适用性容易通过如下测定评估:其中在其表面上呈递候选组分的细胞类型与携载配体的微细胞在体外共同培育,该配体结合该候选物并且还与荧光染料或其他适合检测的标志物连接,例如通过共聚焦显微镜进行目视评估(这种类型的体外测定被描述于macdiarmid等,2007b,第436页图3的说明。因此,通过这样的测定,观察到的标记物的内化构成所测试的靶标细胞表面受体适于本发明的阳性指示。
[0311]
根据本发明,配体可以是呈现所期望的一种或多种特异性的任何多肽或多糖。优选的配体是抗体。在其当前使用中,术语“抗体”涵盖通过体外或体内产生免疫原性响应而获得的免疫球蛋白分子。因此,“抗体”类别包括单克隆抗体和人源化抗体,如单链抗体片段(scfv)、双特异性抗体等。大量的不同双特异性蛋白质和基于抗体的配体是已知的,如caravella and lugovskoy,curr.opin.chem.biol.,14:520
‑
28(2010)的评论文章所证,通过引用将其整体并入本文。可以通过已知的重组dna技术获得根据本公开有用的抗体。
[0312]
因此,作为非限制性实例,携载对表面组分如肿瘤抗原的特异性的抗体可用于将微细胞靶向待治疗的肿瘤中的细胞。在这方面,示例性的细胞表面受体包括rtk表皮生长因
子受体(egfr)、血管内皮生长因子受体(vegfr)、血小板源生长因子受体(pdgfr)和胰岛素样生长因子受体(igfr)中的任意种,其中每一种均在若干实体肿瘤(包括脑瘤)和叶酸受体中高度表达,叶酸受体在一些垂体腺瘤中过表达。这样的双特异性配体也可以靶向突变体或变体受体,例如il
‑
13rα2受体,其在人成胶质细胞瘤多形性肿瘤的50%至80%中表达,参见wykosky等,2008;和jarboe等,2007;debinski等,2000;和okada等,1994),但是其区别于在正常组织中表达的其生理对应物il4r/il13r。参见hershey,2003。因此,正常脑细胞实际上不存在il13rα2。参见debinski和gibo,2000。此外,转移到大脑的肿瘤可能过度表达某些受体,该受体也可以是合适的靶标。例如,da silva等(2010)表明,乳腺癌的脑转移灶表达了her家族的rtk的所有成员。her2在20%的脑转移中被扩增和过表达,egfr在21%的脑转移中被过表达,her3在60%的脑转移中被过表达,并且her4在22%的脑转移中被过表达。值得注意,her3表达在居于脑中的乳腺癌细胞中是增加的。
[0313]
候选靶标细胞表面受体的示例是受体酪氨酸激酶或“rkt”(以与其他整合膜蛋白相似的速率经历组成性内化(内吞)的跨膜蛋白质家族)的成员。参见goh和sorkin,2013。rkt家族描述于lemmon and schlessinger,cell,141(7):1117
‑
134(2010)。示例性rtk是erbb egfr、erbb2、erbb3、erbb4 ins insr、igf1r、insrr pdgf pdgfr.α.、pdgfr.β.、csf1r/fms、kit/scfr、fit3/flk2 vegf vegfr1/fit1、vegfr2/kdr、vegfr3/fit4 fgf fgfr1、fgfr2、fgfr3、fgfr4 ptk7 ptk7/cck4 trk trka、trkb、trkc ror ror1、ror2 musk met、ron axl、mer、tyro3 tie tie1、tie2 eph epha1
‑
8、epha10、ephb1
‑
4、ephb6 ret ryk ddr ddr1、ddr2 ros lmr lmr1、lmr2、lmr3 alk、ltk styk1 surtk106/styk1。
[0314]
合适的靶标细胞表面受体的另一个候选物是结合叶酸和还原的叶酸衍生物并介导四氢叶酸向细胞内部的传递的膜缔合高亲和力叶酸结合蛋白(叶酸受体)家族;在同类细胞因子(如il13)的内化中发挥作用的膜结合细胞因子受体家族;在某些癌细胞上表达并介导同类单克隆抗体(例如cd20情况下的利妥昔单抗(rituximab))的内化的表面抗原,如cd20、cd33、间皮素和hm1.24;以及粘附受体(整合素)家族,其是通过内体途径运输的跨膜糖蛋白并且是癌细胞粘附的主要介质。在本发明的一个实施方式中,肿瘤细胞表面受体包括整联蛋白、神经调节素b受体,铃蟾肽3受体、grp受体、铃蟾肽4受体、cck2/胃泌素、黑皮质素
‑
1受体(mc
‑
1r)、神经肽y(npy)受体、神经降压素(nt)受体、前列腺特异性膜抗原(psma)、生长抑素(sst)受体、神经激肽1受体(nk1r)、4型趋化因子受体(cxcr4)、血管活性肠肽(vip)、表皮生长因子受体(egfr)、血管内皮生长因子受体(vegfr)、血小板源生长因子受体(pdgfr)、胰岛素样生长因子受体(igfr)或其任意组合。
[0315]
根据本发明的另一实施方式,细胞表面受体是在疾病状态下的靶标细胞上独特表达,但是在健康状态下保持不表达、低水平表达或不可及的抗原。可以被本发明的靶向配体特异性结合的这种靶标抗原的实例可以有利地选自epcam、ccr5、cd19、her
‑
2neu、her
‑
3、her
‑
4、egfr、psma、cea、muc
‑
1(粘蛋白)、muc2、muc3、muc4、muc5、muc5、muc7、bhcg、lewis
‑
y.cd20、cd33、cd30、神经节苷脂gd3、9
‑
o
‑
乙酰基
‑
gd3、gm2、globo h、岩藻糖基gm1、聚sa、gd2、碳酸酐酶ix(mn/ca ix)、cd44v6、sonic hedgehog(shh)、wue
‑
1、血浆细胞抗原、(膜结合)ige、黑色素瘤硫酸软骨素蛋白聚糖(mcsp)、ccr8、tnf
‑
α前体、steap、间皮素、a33抗原、前列腺干细胞抗原(psca)、ly
‑
6;桥粒芯蛋白4(desmoglein 4)、e
‑
钙粘着蛋白新表位、胎儿乙酰胆碱受体、cd25、ca19
‑
9标志物、ca
‑
125标志物和muellerian抑制物(mis)受体ii型、
preparation)的形式。这种长效制剂可以通过植入(例如,皮下或肌内)或肌内注射来给予。在一些实施方式中,给予包括肠内或肠胃外给予。在一些实施方式中,给予包括选自口服、颊部、舌下、鼻内、直肠、阴道、静脉内、肌内和皮下注射的给予。
[0325]
在一些方面,提供了含微细胞的组合物,其包含治疗有效量的抗肿瘤剂。根据本公开,抗肿瘤剂的“治疗有效”量是在论试剂的剂量,例如在被给予于对象时引起药理响应的sirna或超细胞毒性药物。
[0326]
因此,在本公开的环境中,可以在携载有治疗有效载荷的微细胞被给予时,参考动物模型或人对象中的肿瘤或肿瘤症状的预防或改善来判断治疗有效量,如下文进一步描述。对于具体对象证明在给定情况下“治疗有效量”的量可能不是对于100%的类似地接受肿瘤治疗的对象有效,即使这种剂量被从业技术人员视作“治疗有效量”。在这方面,合适的剂量还将根据例如肿瘤的类型、阶段和严重程度而变化。
[0327]
当“治疗有效”用于指代药物组合物中的微细胞数目时,该数目可以基于包装在微细胞中的抗肿瘤剂和该药剂治疗肿瘤的功效来确定。在这方面,可以用临床或病理参数如肿瘤质量(tumor mass)来测量治疗效果。因此,肿瘤质量的减少或肿瘤质量增大的减少可用于测量治疗效果。
[0328]
a.给予途径
[0329]
本发明的制剂可以通过各种途径和向哺乳动物体内的各种位点给予,以局部或全身地实现期望的治疗效果。递送可以例如通过以下完成:通过口服给予,通过将制剂施用于体腔,通过吸入或吹入,或者通过肠胃外、肌内、静脉内、门内(intraportal)、肝内、腹膜、皮下、肿瘤内或皮内给予。给予方式和位点取决于靶标细胞的位置。例如,可通过靶向性微细胞的静脉内递送而更有效地治疗肿瘤转移。原发性卵巢癌可通过靶向性微细胞的腹膜内递送来治疗。也可以采用组合途径。例如,在转移性膀胱癌中,可以在膀胱内以及静脉内给予负载有细胞毒性药物和靶向受体的微细胞,并可以将包装佐剂的(受体靶向的或非靶向的)微细胞与包装靶向药物的微细胞一起静脉内给予。靶向性药物包装微细胞的原位给予可以靶向膀胱表面暴露的肿瘤,而静脉内给予的微细胞的充分组合可以靶向组织定位肿瘤,并且还引发抗肿瘤免疫响应。
[0330]
b.纯度
[0331]
本发明的微细胞基本上不含污染性亲本细菌细胞。因此,含微细胞的制剂优选包含每107个微细胞少于约1个污染性亲本细菌细胞、每108个微细胞少于约1个污染性亲本细菌细胞、每109个微细胞少于约1个污染性亲本细菌细胞、每10
10
个微细胞少于约1个污染性亲本细菌细胞、或每10
11
个微细胞少于约1个污染性亲本细菌细胞。
[0332]
纯化微细胞的方法是本领域已知的,并被描述于pct/ib02/04632。一种这样的方法结合了错流过滤(进料流平行于膜表面;forbes,1987)和死端过滤(进料流垂直于膜表面)。任选地,在过滤组合之前可以在低离心力下进行差速离心,以去除细菌细胞的一部分,从而富集上清液的微细胞。
[0333]
另一种纯化方法采用在生物相容性介质中进行的密度梯度离心。离心后,从梯度中收集微细胞带,并且任选地,对微细胞进行更多轮密度梯度离心以使纯度最大化。该方法可以进一步包括对包含微细胞的样品进行差速离心的初步步骤。当在低离心力下进行时,差速离心将去除一部分亲本细菌细胞,从而富集上清液的微细胞。
[0334]
特别有效的纯化方法利用细菌细丝化来增加微细胞纯度。因此,微细胞纯化方法可以包括以下步骤:(a)使包含微细胞的样品经历诱导亲本细菌细胞采取细丝形式的条件,然后(b)过滤样品以获得纯化的微细胞制剂。
[0335]
已知的微细胞纯化方法也可以被组合。一种高度有效的方法组合如下:
[0336]
步骤a:产生微细胞的细菌细胞培养物的差速离心。该步骤,可在2,000g下进行约20分钟,去除大多数亲本细菌细胞,而在上清液中留下微细胞;
[0337]
步骤b:使用等渗且无毒的密度梯度介质进行密度梯度离心。此步骤将微细胞与多种污染物(包括亲本细菌细胞)分开,并使微细胞的损失最低。优选地,该步骤在纯化方法中重复进行;
[0338]
步骤c:通过0.45μm滤器的错流过滤,以进一步减少亲本细菌细胞污染;
[0339]
步骤d:应激诱导的残留亲本细菌细胞细丝化。这可以通过使微细胞悬浮液经历几种应激诱导环境条件中的任一种来完成;
[0340]
步骤e:抗生素处理以杀死亲本细菌细胞;
[0341]
步骤f:错流过滤以除去小污染物,如膜泡、膜碎片、细菌碎片、核酸、介质组分等和浓缩微细胞。可以使用0.2μm的滤器将微细胞与小污染物分离,可以使用0.1μm的滤器浓缩微细胞;
[0342]
步骤g:死端过滤以消除细丝状死亡细菌细胞。此步骤可使用0.45μm的滤器;和
[0343]
步骤h:从微细胞制剂去除内毒素。抗脂质a包覆的磁珠可用于该步骤。
[0344]
c.给予时间表
[0345]
总体上,本文公开的制剂可以以常规测试限定的合适剂量使用,以获得最佳的生理效果,同时使任何潜在的毒性最小化。剂量方案可以根据多种因素选择,所述因素包括患者的年龄、体重、性别、医疗状况;待治疗疾病的严重程度、给予途径以及患者的肾和肝功能。
[0346]
实现以最小副作用产生最大功效的范围内的微细胞和药物浓度的最佳精确度可需要基于微细胞的动力学以及药物对于靶标位点和靶标细胞的可利用性的方案。在确定治疗方案的最佳浓度时,可以考虑微细胞或药物的分布、平衡和消除。当组合使用时,可以调节微细胞和药物的剂量,以达到期望的效果。
[0347]
此外,可以使用药代动力学/药效学建模系统来优化制剂的剂量给予。例如,可以选择一种或多种剂量方案,并且可以使用药代动力学/药效学模型来确定一种或多种剂量方案的药代动力学/药效学特征。接下来,可以基于具体的药代动力学/药效学特征选择其中一种实现期望的药代动力学/药效学响应的剂量方案以给予。参见,例如,wo 00/67776。
[0348]
具体地,可以经几周的过程至少一周一次给予所述制剂。在一个实施方式中,经几周至几个月至少一周一次给予所述制剂。
[0349]
更具体地,所述制剂可以至少一天一次给予,给予约2、约3、约4、约5、约6、约7、约8、约9、约10、约11、约12、约13、约14、约15、约16、约17、约18、约19、约20、约21、约22、约23、约24、约25、约26、约27、约28、约29、约30、约31天。可选地,所述制剂可以每天约一次、每约2、约3、约4、约5、约6、约7、约8、约9、约10、约11、约12、约13、约14、约15、约16、约17、约18、约19、约20、约21、约22、约23、约24、约25、约26、约27、约28、约29、约30或约31天或更多天约一次给予。
[0350]
可选地,所述制剂可以每周约一次、每约2、约3、约4、约5、约6、约7、约8、约9、约10、约11、约12、约13、约14、约15、约16、约17、约18、约19或约20周或更多周约一次给予。可选地,所述制剂可以至少一周一次给予,给予约2、约3、约4、约5、约6、约7、约8、约9、约10、约11、约12、约13、约14、约15、约16、约17、约18、约19或约20周或更多周。
[0351]
可选地,所述制剂可以每周约两次、每约2、约3、约4、约5、约6、约7、约8、约9、约10、约11、约12、约13、约14、约15、约16、约17、约18、约19或约20周或多周约两次给予。可选地,所述制剂可以至少一周一次给予,给予约2、约3、约4、约5、约6、约7、约8、约9、约10、约11、约12、约13、约14、约15、约16、约17、约18、约19或约20周或更多周。
[0352]
可选地,所述制剂可每月约一次、每约2、约3、约4、约5、约6、约7、约8、约9、约10、约11或约12个月或更多月约一次给予。
[0353]
所述制剂可以单日剂量给予,或者总日剂量可以一日两次、三次、四次的分剂量给予。
[0354]
在将微细胞在药物之前给予的方法中,药物的给予可以在给予微细胞后几分钟至几小时的任何时间发生。可选地,药物可以在微细胞之后数小时至数天,可能数周至数月的任何时间给予。
[0355]
更具体地,微细胞可以在药物前至少约1、约2、约3、约4、约5、约6、约7、约8、约9、约10、约11、约12、约13、14小时、约15、约16、约17、约18、约19、约20、约21、约22、约23或约24小时给予。此外,微细胞可以在药物给予前至少约1、约2、约3、约4、约5、约6、约7、约8、约9、约10、约11、约12、约13、约14、约15、约16、约17、约18、约19、约20、约21、约22、约23、约24、约25、约26、约27、约28、约29、约30或约31天给予。在又一个实施方式中,微细胞可以在药物前至少约1、约2、约3、约4、约5、约6、约7、约8、约9、约10、约11、约12、约13、约14、约15、约16、约17、约18、约19或约20周或更多周给予。在进一步实施方式中,微细胞可以在药物前至少约1、2、3、4、5、6、7、8、9、10、11或12个月给予。
[0356]
在另一个实施方式中,在药物之后给予微细胞。微细胞的给予可在药物给予后几分钟到几小时的任何时间发生。可选地,微细胞可以在药物后数小时至数天,可能数周至数月的任何时间给予。
[0357]
vii.治疗癌症的方法
[0358]
本文所述的组合物可用于治疗患有癌症的对象。本文公开的方法包括向对象给予有效量的根据本发明的组合物,所述组合物包含至少一种抗肿瘤剂、i型干扰素激动剂、ii型干扰素激动剂或i型干扰素激动剂和ii型干扰素激动剂的组合。抗肿瘤剂、i型干扰素激动剂、ii型干扰素激动剂或i型干扰素激动剂和ii型干扰素激动剂的组合被包含在一个或多个微细胞中。
[0359]
在另一方面,用于治疗患有癌症的对象的组合物还包含药学上可接受的载体。
[0360]
在另一方面,本文公开的方法可用于治疗患有癌症的对象,其中所述对象是人类、非人灵长类、犬、猫、牛、绵羊、马、兔、小鼠或大鼠。
[0361]
在另一方面,本文公开的方法可用于治疗癌症。在一些实施方式中,癌症包括肺癌、乳腺癌、脑癌、肝癌、结肠癌、胰腺癌或膀胱癌。
[0362]
在一些实施方式中,癌症包括急性成淋巴细胞性白血病;急性髓性白血病;肾上腺皮质癌;aids相关癌症;aids相关淋巴瘤;肛门癌;阑尾癌;星形细胞瘤;非典型畸胎样/横纹
肌样肿瘤;基底细胞癌;膀胱癌;脑干神经胶质瘤;脑肿瘤(包括脑干神经胶质瘤、中枢神经系统非典型畸胎样/横纹肌样肿瘤、中枢神经系统胚胎肿瘤、星形细胞瘤、颅咽管瘤、成室管膜细胞瘤、室管膜瘤、髓母细胞瘤、髓上皮瘤、中间分化的松果体实质瘤、幕上原发性神经外胚层肿瘤);乳腺癌;支气管肿瘤;burkitt淋巴瘤;原发位点未知的癌症;类癌肿瘤;原发位点未知的癌;中枢神经系统非典型畸胎样/横纹肌样肿瘤;中枢神经系统胚胎肿瘤;宫颈癌;儿童期癌症;脊索瘤;慢性淋巴细胞性白血病;慢性髓性白血病;慢性骨髓增生性障碍;结肠癌;结肠直肠癌;颅咽管瘤;皮肤t细胞淋巴瘤;内分泌胰腺胰岛细胞肿瘤;子宫内膜癌;成室管膜细胞瘤;室管膜瘤;食道癌;成感觉神经细胞瘤;ewing肉瘤;颅外生殖细胞肿瘤;性腺外生殖细胞肿瘤;肝外胆管癌;胆囊癌;胃癌;胃肠类癌肿瘤;胃肠道间质细胞肿瘤;胃肠道间质肿瘤(gist);妊娠滋养层肿瘤;神经胶质瘤;毛细胞白血病;头颈癌;心脏癌;hodgkin淋巴瘤;下咽癌;眼内黑色素瘤;胰岛细胞肿瘤;kaposi肉瘤;肾癌;langerhans细胞组织细胞增生症;喉癌;唇癌;肝癌;恶性纤维组织细胞瘤骨癌;髓母细胞瘤;髓上皮瘤;黑色素瘤;merkel细胞癌;merkel细胞皮肤癌;间皮瘤;原发隐匿的转移性鳞状颈癌;口癌;多发性内分泌肿瘤综合征;多发性骨髓瘤;多发性骨髓瘤/浆细胞瘤;蕈样真菌病;骨髓增生异常综合征;骨髓增生性肿瘤;鼻腔癌;鼻咽癌;成神经细胞瘤;非hodgkin淋巴瘤;非黑素瘤皮肤癌;非小细胞肺癌;口唇癌;口腔癌;口咽癌;骨肉瘤;其他脑和脊髓肿瘤;卵巢癌;卵巢上皮癌;卵巢生殖细胞肿瘤;卵巢低恶性潜在肿瘤;胰腺癌;乳头状瘤病;鼻旁窦癌;甲状旁腺癌;骨盆癌;阴茎癌;咽癌;中间分化的松果体实质性肿瘤;成松果体细胞瘤;垂体瘤;浆细胞肿瘤/多发性骨髓瘤;胸膜肺母细胞瘤;原发性中枢神经系统(cns)淋巴瘤;原发性肝细胞肝癌;前列腺癌;直肠癌;肾癌;肾细胞(肾)癌;肾细胞癌;呼吸道癌;视网膜母细胞瘤;横纹肌肉瘤;唾液腺癌;sezary综合征;小细胞肺癌;小肠癌;软组织肉瘤;鳞状细胞癌;鳞状颈癌;胃癌;幕上原发性神经外胚层肿瘤;t细胞淋巴瘤;睾丸癌;咽喉癌;胸腺癌;胸腺瘤;甲状腺癌;过渡细胞癌;肾盂和输尿管过渡细胞癌;滋养层肿瘤;输尿管癌;尿道癌;子宫癌;子宫肉瘤;阴道癌;外阴癌;waldenstrom巨球蛋白血症;或wilm肿瘤。
[0363]
在一些实施方式中,脑癌或肿瘤选自脑干神经胶质瘤、中枢神经系统非典型畸胎样/横纹肌样肿瘤、中枢神经系统胚胎肿瘤、星形细胞瘤、颅咽管瘤、成室管膜细胞瘤、室管膜瘤、髓母细胞瘤、髓上皮细胞瘤、中间分化的松果体实质性肿瘤、幕上原发性神经外胚层肿瘤和成松果体细胞瘤。
[0364]
viii.定义
[0365]
除非另有定义,本文中使用的技术和科学术语具有本发明所属领域的普通技术人员通常理解的含义。除非另有说明,在以下描述和实施例中提及的材料、试剂等可从商业来源获得。
[0366]
为了方便起见,下面提供在说明书、实施例和所附权利要求中使用的某些术语和短语的含义。其他术语和短语遍及说明书中被定义。
[0367]
除非上下文另外明确指出,单数形式“一个”、“一种”和“所述”包括复数指代。
[0368]
术语“约”是指所理解的数字不限于在此阐述的确切数值,并且旨在在不背离本发明的范围的情况下指代实质上在所述数值左右的数值。如本文中所用,“约”将被本领域普通技术人员理解,并且将在其使用的环境中以某种程度变化。如果在给定其使用环境下本领域普通技术人员不清楚的该术语的使用,则“约”表示具体项目的上至正负10%。
[0369]
本文可互换使用的“个体”、“对象”、“宿主”和“患者”是指期望诊断、处理或治疗的任何哺乳动物对象。在一个优选的实施方式中,所述个体、对象、宿主或患者是人。其他对象可以包括但不限于牛、马、犬、猫、豚鼠、兔、大鼠、灵长类和小鼠。
[0370]
本文可互换使用的“癌症”、“肿瘤”、“瘤”、“恶性病”和“癌”是指呈现异常生长表型的细胞或组织,其特征在于细胞增殖控制明显丧失。有几种主要类型的癌症。癌是始于皮肤或始于界定或覆盖内部器官的组织的癌症。肉瘤是始于骨、软骨、脂肪、肌肉、血管或其他结缔组织或支撑组织的癌症。白血病是始于血液形成组织如骨髓并导致大量异常血细胞生成并进入血液的癌症。淋巴瘤和多发性骨髓瘤是始于免疫系统细胞的癌症。中枢神经系统癌症是始于脑和脊髓组织的癌症。本发明的方法和组合物特别适用于癌前、恶性、转移前、转移和性非转移性细胞。
[0371]
术语“治疗”(“treatment”、“treating”、“treat”)等是指在肿瘤患者中获得期望的药理和/或生理效果。该效果可以就完全或部分预防肿瘤或其症状而言是预防性的,和/或可以就部分或完全稳定或治愈和/或可归因于肿瘤的不利效应而言是治疗性的。治疗包括对哺乳动物,特别是人类的肿瘤的任何治疗。期望的效果具体地是肿瘤响应,其可以通过肿瘤质量减小或肿瘤质量增加抑制来测量。除肿瘤响应外,总生存期、无进展生存期或肿瘤复发前时间增加或不利效应的减少也可在临床上用作期望的治疗效果。
[0372]
如本文所用,术语“给予”包括直接给予至另一者、自我给予、以及处方或指导给予本文所公开的药剂。
[0373]
如本文所用,短语“有效量”和“治疗有效量”分别是指提供在需要这种治疗的对象中给予该活性剂的特定药理效果的对象中的活性剂剂量或血浆浓度。要强调的是,活性剂量的有效量不总是有效地治疗本文所述的状况/疾病,即使这种剂量被本领域技术人员认为是有效量。
[0374]
如本文所用,术语“活性剂”是可用于治疗对象的任何小分子药物、蛋白质、功能性核酸或编码功能性核酸的多核酸。活性剂可以是本文所述的抗肿瘤剂、功能性酸、i型干扰素激动剂或ii型激动剂中的任意种。
[0375]
本文所用的短语“药学上可接受的”是指在合理的医学判断范围内,适合于体内使用而没有过度毒性、刺激性、过敏反应或其他问题或并发症,与合理的收益/风险比相对应的那些化合物、材料、组合物和/或剂型。
[0376]
术语“内吞作用”包括(1)吞噬作用和(2)胞饮作用,其本身是包括下列在内的类别:(2a)不需要受体结合的巨胞饮作用以及(2b)网格蛋白(clathrin)介导的内吞作用和(2c)小窝(细胞膜穴样内陷,caveolae)介导的内吞作用和(2d)网格蛋白/小窝非依赖性内吞作用,这些全部趋向于进入晚期内体/溶酶体途径。本发明人发现,微细胞上的配体与哺乳动物细胞表面受体之间的相互作用激活了特定的内吞途径,其涉及到受体介导的晚期内体/溶酶体区室的内吞作用(rme)。借助于这种内吞作用途径,本发明人进一步发现微细胞能够将其有效载荷释放到靶标哺乳动物细胞的细胞质中。在有效载荷是编码核酸的情况下,该核酸不仅在内体/溶酶体区室中不被完全降解,而且在靶标哺乳动物细胞中表达。
[0377]
以下实施例仅是示例性的,而非限制性的,并且提供了对本发明的更完整的理解。实施例表明,可以通过(1)给予携载rnai序列的靶向性重组微细胞,该rnai序列被设计用于减少或消除抗药性编码基因(一个或多个)的表达,以及(2)给予靶向性的包装药物的微细
胞,该微细胞携载癌细胞敏感的药物。
[0378]
提供以下实施例以示例本发明。然而,应当理解,本发明不限于这些实施例中描述的具体条件或细节。在整个说明书中,对包括美国专利在内的公众可得的文件的任何和所有参考均通过引用具体地地并入。
[0379]
工作实施例
[0380]
实施例1:小鼠的临床前研究
[0381]
此实施例表明,微细胞(edv)在小鼠异种移植模型中实现化疗药物的有效递送并抑制肿瘤生长。edv靶向技术已在多种癌症的小鼠异种移植模型上进行了测试,该癌症包括结肠癌、乳腺癌、卵巢癌、白血病、肺癌、间皮瘤和子宫癌。此外,由于edv靶向肿瘤细胞表面受体(包括egfr、人表皮生长因子受体2(her2)、间皮素(msln)和cd33),利用了多种靶向部分。最后,经测试的靶向edv包括多种细胞毒性药物,包括多柔比星、紫杉醇、monastrol、伊立替康、超细胞毒性药物(如pnu
‑
159682)和新型胸苷酸合酶抑制剂(osi
‑
7904)。pnu
‑
159682是一种蒽环类类似物,其细胞毒性是多柔比星的数千倍。osi
‑
7904是一种具有抗肿瘤活性的叶酸苯并喹唑啉类似物。作为胸苷酸合酶抑制剂,osi
‑
7904非竞争性结合至胸苷酸合酶,导致胸腺嘧啶核苷酸合成和dna复制的抑制。
[0382]
在所有情况下,即使使是大型(>1000mm3)肿瘤,在使用特异靶向的和包装药物的edv时也观察到肿瘤稳定化或消退(参见表5)。用游离药物(未包装在edv中)治疗的对照小鼠呈现预期的静脉炎毒性以及最终体重减轻和死亡。重复给予edv包装的药物后未观察到这些毒性。
[0383]
惊人地,即使通过edv递送的细胞毒性药物的浓度比全身递送少上至8,000倍,通过edv递送的细胞毒性药物浓度仍足以最大化抗肿瘤功效。
[0384]
[0385][0386]
实施例2:犬类的临床前研究
[0387]
此实施例表明,负载有药物的微细胞(edv)可以被安全地给予犬。
[0388]
在患有一系列内源性肿瘤(n=41)的犬中进行的犬毒理学研究表明,上至98个剂量的靶向的和药物包装的edv可以经超过2年的过程被安全地给予单只动物。一些犬中用药后,温度有轻度升高(上至l℃的升高),伴随白介素
‑
6(il
‑
6)、白介素
‑
10(il
‑
10)和肿瘤坏死因子α(tnf
‑
α)升高,这与任何重大不利事件无关。
[0389]
此外,在犬中进行的临床前研究表明,以靶向cd3的负载有多柔比星的edv可以抑制肿瘤的生长。对两只患有晚期非hodgkin淋巴瘤的犬用靶向cd3的包装多柔比星的edv的治疗,二者均显示出明显的肿瘤消退,如淋巴结尺寸的显著减少所证。macdiarmid等,2007b。用靶向cd33的和包装多柔比星的edv治疗时,超过60%的具有血管肉瘤的犬显示肿瘤稳定化或消退。
[0390]
在另一项对患有晚期脑癌的犬(n=17)的研究中,用负载有多柔比星的靶向egfr的edv治疗该动物。macdiarmid等,2016。对单只犬给予浓度为1x10
10egfr
微细胞
dox
的98个重复剂量(11只犬接受>20个剂量),未观察到毒性迹象。客观响应率为23.53%(17只犬中的4只;95%置信区间,6.8
‑
49.8%)。在评价肿瘤响应的15只犬中,2只对治疗有完全响应(cr),2只对治疗有部分响应(pr)(肿瘤体积减少90
‑
98.95%),10只具有稳定的疾病(sd),1只显示进展性疾病(pd)。
[0391]
使用
123
碘放射性标记的靶向egfr的edv在两只患有脑癌的犬中进行的生物分布研究显示,靶向型edv定位于脑肿瘤,表明edv可以规避血脑屏障以进入肿瘤环境。胃肠道中的一些定位表明通过粪便排泄。
[0392]
实施例3:猴类的临床前研究
[0393]
此实施例表明,微细胞(edv)技术被猴子良好地耐受。
[0394]
进行了三项恒河猴试验,以评价空白edv(每剂量上至2x10
10
个)、靶向egfr的负载多柔比星的edv(每剂量上至2x10
10
个)和靶向egfr的负载紫杉醇的edv(每剂量上至1x10
11
个)的毒性。每周一次用edv治疗猴子,治疗5周(35天重复剂量测试)。
[0395]
如关于犬类所见,用药后会出现短暂的温度升高(上至1℃的升高),并伴随il
‑
6升高。在这些时候,炎性标志物c反应蛋白也增加,但是没有观察到显著的毒性或不利事件。在仅用靶向egfr的负载多柔比星的edv的治疗过程中,观察到tnf
‑
α轻度升高。总共72只猴已安全地通过edv技术被给药。
[0396]
实施例4:临床前研究中的炎性和免疫响应。
[0397]
此实施例表明,在临床前小鼠研究(实施例1)、临床前犬类研究(实施例2)和临床前猴类研究(实施例3),中仅观察到较小的炎性响应。这些响应快到在用药后4小时被解决。没有观察到血液学或生化参数的其他显著变化。在整个治疗过程中,动物的外观和行为保持健康。
[0398]
在犬类研究和猴类试验中评价了抗产物抗体的形成。关于给予靶向型edv考虑的免疫响应为:
[0399]
·
对edv表面暴露的免疫显性抗原的血清抗体响应,该抗原是lps的o多糖组分(igg或igm响应)。抗o
‑
多糖抗体响应是t细胞非依赖性的,并且不呈现记忆响应。
[0400]
·
血清抗体对小鼠igg单克隆抗体的响应,该小鼠igg单克隆抗体用于将edv靶向肿瘤细胞表面受体(例如egfr)的bsab的构建。
[0401]
在患有血管肉瘤或脑癌的犬中,通过第3
‑
4个剂量的靶向性的和包装多柔比星的edv的,血清抗lps igg滴度上升到平均约10,000。后续用药未导致滴度进一步升高。在3只猴试验(健康动物)中的抗lps igg滴度总体上显示出在平稳之前的前2
‑
3个剂量过程轻度升高。响应在很大程度上是剂量依赖性的,对于最高剂量水平的
egfr
edv
dox
,上升到刚好超过100的最大滴度。这些被认为是微弱的抗体响应,因为抗革兰氏阴性菌疫苗中预期的抗o多糖抗体滴度总体上在百万级。
[0402]
猴类试验中的抗lps igm滴度响应也是轻度的,在
egfr
edv
dox
治疗时上升到刚好超过100,而用非靶向edv治疗时上升到1,000。第3
‑
4个剂量后,滴度不再增加。
[0403]
在猴类研究中还测量了对bsabs构建中使用的单克隆抗体的免疫原性响应,在以靶向egfr的edv(小鼠igg)治疗的猴子中观察到响应egfr抗体的滴度轻度升高。这些结果表明,靶向bsab的包装药物的edv的给予可能不引起显著的抗lps免疫响应,抗lps免疫响应可阻止后续剂量的有效性。这对于免疫系统可能受到损害的癌症患者尤其相关,因为这表明重复给予可能是可行的治疗选择。
[0404]
实施例5:在晚期实体肿瘤中评价靶向erbitux的包装紫杉醇的edv(
egfr(erb)
edvs
pac
)的首次人体1期临床试验
[0405]
此实施例显示了使用微细胞(edv)将紫杉醇递送至晚期实体肿瘤的有前
景的结果。
[0406]
在该试验中显示,对靶向erbitetx(西妥昔单抗)的包装紫杉醇的edv在人类患者中被良好地耐受,但是由于临床试验中的不利事件或剂量限制性毒性,相当多的患者不得不终止研究。此外,尽管这种治疗策略可以实现疾病稳定化,但是本研究中没有任何患者对治疗呈现部分或完全响应。此试验数据的结果发表在solomon等,2015中。
[0407]
首次人体试验被被设计为剂量递增研究,以确定靶向egfr的负载紫杉醇的edv(
egfr(erb)
edvs
pac
)的安全性、耐受性和最大耐受剂量或推荐2期剂量。注意,用于将edv靶向egfr的抗体是基于erbitux序列的。其他目标是评价对iv注射
egfr(erb)
edvs
pac
的免疫和炎性响应,和根据recist标准评价对治疗的响应。
[0408]
该研究在澳大利亚墨尔本的3家肿瘤诊所进行,并在australian new zealand clinical trials registry进行了注册(编号actrn12609000672257)。最终研究报告是可获得的,并且该研究已发表在solomon等,2015。
[0409]
患者是至少18岁的成年人,患有上皮恶性病,尚无其标准治疗性处理。
[0410]
egfr(erb)
edvs
pac
被以20分钟iv输注每周给予,周期由5周治疗组成。其然后是无治疗的一周,其中患者接受通过mri、ct和/或fdg
‑
pet进行的放射学评估。如果肿瘤保持稳定或对治疗有响应,或者如果其从治疗中获得临床收益,并且其未经历任何剂量限制性毒性(dlt)或其他需要中断治疗的不利事件(ae),则患者可以继续接受更多周期的治疗。
[0411]
在7种剂量水平下递送了共236个剂量:每剂量1x108、1x109、3x109、1x10
10
、1.5x10
10
、2x10
10
和5x10
10
个靶向egfr(erb)的包含紫杉醇的edv。28名患者中22名完成了至少1个完整周期的治疗(5个每周一次剂量),1名患者经9个完整周期(约14个月)接受了45个剂量。没有发生与治疗相关的死亡。最大耐受剂量被确定为1x10
10
个
egfr(erb)
edvs
pac
,在该剂量水平以上观察到显著毒性,具体地是长期发烧和肝功能测试(lft)短暂升高的形式。在所示群体中,该治疗总体上被良好耐受,具有可接受的安全性。
[0412]
下表6列出了临床研究和发现的总结。
[0413]
表6:临床数据总结
‑
egfr(erb)
edvs
pac
[0414]
[0415]1一个完整周期由5个每周一次剂量组成。
[0416]
2 1个完整周期治疗完成后评价响应。
[0417]
至少可能与研究治疗相关的最常见不利事件是低度发热(发烧)和僵直(rigor)(寒战),上至60%的患者经历(1
‑
2级严重程度)。用药后4小时,大多数患者经历了细胞因子il
‑
6、il
‑
8和il
‑
10的轻度短暂升高。总体上在接受剂量后的24小时内水平恢复到基线。这与对治疗的轻微炎性响应一致。
[0418]
5名患者经历了被认为严重的治疗相关不利事件,并且8名患者经历了剂量限制性毒性或需要剂量减少的不利事件。这些事件被总结在表7中,并在下面的叙述中进行描述。
[0419]
表7:严重不利事件或剂量限制毒性
[0420][0421]1×
108剂量水平下的1个患者经历了符合dlt标准的lft升高。该事件未被认为是严重的,并且针对随后的剂量水平修改了dlt的定义。mtd以上的剂量水平下的四名患者经历了不符合修改后的dlt标准的lft升高,但是这些患者还经历了严重的治疗相关临床症状(发热、僵直、恶心、呕吐),并且安全委员会决定降低剂量。
[0422]
三名患者被招募至第一剂量水平,1x10
8egfr(erb)
edvs
pac
。一名患者在其5个剂量中的3个剂量后经历了磷酸水平的3级下降。在每种情况下,水平在用药后24小前恢复正常,并且没有临床症状。另一名患者在第3个剂量后4小时经历了肝酶丙氨酸转氨酶(alt)和天冬氨酸转氨酶(ast)的无症状3级升高,其在下一剂量前恢复至基线。这些事件符合方案的最初剂量限制毒性(dlt)定义,并且正在进行的患者剂量减少至5x10
7egfr(erb)
edvs
pac
,并且一名患者在研究过程中接受了共45个剂量。另外3名患者被招募接受1x10
8egfr(erb)
edvs
pac
,这3名患者未报告药物相关的不利事件。修改了方案的dlt定义,以排除在治疗7天内解决的生化异常。
[0423]
剂量增加到1x10
9egfr(erb)
edvs
pac
。第二个剂量后两天,一名患者经历了严重的关节疼痛。这伴随着细胞因子干扰素
‑
α显著升高,表明病毒感染。该患者入院观察,后来被诊断为反应性关节炎,这被归类为严重不利事件(sae)。经过一番考虑,安全委员会决定谨慎进行,并将此事件定义为dlt。因此,该组也扩大至6名患者。试验中没有其他患者经历类似事件。在第一个周期完成时,安全数据支持进一步增加剂量。
[0424]
三名患者组被招募至下一个剂量水平,3x10
9egfr(erb)
edvs
pac
。一名患者在接受仅1
个剂量后由于迅速进展性疾病而退出,随后因疾病死亡。第四名患者被招募至相同剂量水平。所有患者均耐受该剂量,而无任何主要顾虑,并且安全性数据支持进一步增加剂量。该组中的一名患者在第一个周期后实现疾病稳定化,并完成了两个周期共10个剂量。一名患者由于其骨髓的疾病浸润仅接受了五个剂量中的四个剂量。
[0425]
三名患者组被招募至下一个剂量水平,即1x10
10egfr(erb)
edvs
pac
。患者耐受这个剂量水平,而无任何主要顾虑,并且在第1周期完成时,安全性数据支持进一步增加剂量。三名患者中有两名实现了疾病稳定化,并且分别完成了三个和五个周期,15个和25个剂量。
[0426]
两名患者被招募至下一个剂量水平,即5x10
10egfr(erb)
edvs
pac
。他们接受了此剂量水平下的一个剂量,并且均经历了肝酶alt和ast的3
‑
4级升高。这些变化是短暂的,因此不符合方案的dlt的修改后定义,但是,由于患者经历了其他ae如发烧、僵直和恶心(在一种情况下导致因sae住院),因此做出了将剂量水平降低至1x10
10egfr(erb)
edvs
pac
的决定。这些患者还经历了炎性标志物il
‑
6、il
‑
8、l
‑
10和tnf
‑
α的显著升高。这两名患者中的一名继续实现疾病稳定化,并在剂量降低后完成了两个完整的周期。
[0427]
为了确定最大耐受剂量(mtd),一名患者被招募至中间剂量水平2x10
10egfr(erb)
edvs
pac
。患者接受此水平下的一个剂量,并且类似地经历了alt和ast的3
‑
4级短暂升高,伴有发烧、僵直、恶心和呕吐以及炎性标志物升高。还观察到临床显著的乳酸脱氢酶(ldh)和γ
‑
谷氨酰转移酶(ggt)升高。同样,尽管这些参数不符合方案的dlt的修改后定义,升高的肝酶仍被认为是sae,并且判断将剂量降低至1x10
10egfr(erb)
edvs
pac
在临床上是适当的。该患者实现疾病稳定化,并且完成了四个周期,接受个19剂量。
[0428]
在确定mtd的进一步尝试中,三名患者被招募至中间剂量水平,1.5x10
10egfr(erb)
edvs
pac
。这些患者中的一名接受了其第一个剂量治疗而没有任何不利反应,但其由于疾病快速进展而没有继续治疗。另一名患者经历了3级低血压,这被认为是剂量限制性严重不利事件,因此其剂量水平降至5x10
9egfr(erb)
edvs
pac
。该组中的最后一名患者在其第一个剂量后经历了ast的3级升高,并伴有治疗相关症状(发烧、僵直、呕吐)以及炎性标志物升高,并且随后的剂量降低至1x10
10egfr(erb)
edvs
pac
。
[0429]
因此,可以得出结论,
egfr(erb)
edvs
pac
的mtd是剂量水平1x10
10
,并且另外3名患者被招募至该剂量水平。一名患者在一个剂量后由于推测的细胞因子释放综合征而退出研究。该患者具有先存的咳嗽,并且由于锁骨上物质压在头臂静脉上而经历偶尔的晕厥事件。用药后2
‑
4小时之间,患者发热,开始咳嗽,并在工作人员的见证下经历了3
‑
4次晕厥。该患者仍留院观察并诊断为细胞因子释放综合征,尽管未检测到ifn
‑
γ升高。该患者经历了il
‑
6、il
‑
8和il
‑
10升高,类似于其他患者在此剂量水平或以上的观察结果,这很可能代表对药物产品的细菌组分的炎性响应,而不是一般的细胞因子释放综合征。尽管此事件确实代表了sae,但安全委员会认为其由于涉及先存疾病而不符合dlt标准。其余两名患者完成了第一周期的治疗,无任何主要顾虑。
[0430]
没有因治疗引起的不利事件而导致的死亡。总体而言,治疗被良好耐受,并且对于预期的目标群体没有特别的安全顾虑。
[0431]
所有患者在筛选时的鼠伤寒沙门氏菌(抗lps)和erbitux抗体均为阴性。除一名患者外,所有患者在用
egfr(erb)
edvs
pac
治疗后均产生阳性沙门氏菌抗体滴度(27/28=96%)。抗lps抗体滴度在第3个剂量(第15天)达到峰值并且保持该水平——尽管重复用药。没有患者
产生阳性erbitux抗体滴度。
[0432]
评价了完成第1周期的22名患者的肿瘤响应。观察到的最佳响应是稳定疾病(sd)(没有患者实现根据recist标准的部分或完全响应)。在第1周期结束时,在10/22(45.5%)的患者中观察到稳定的疾病,并且12/22(55.5%)显示进展性疾病(pd)。剂量水平1下的一名患者完成了9个完整的治疗周期,其疾病从第4周期结束开始交替稳定和进展。其是产生pd的最长时间,197天。
[0433]
总之,首次人体研究表明,包装有紫杉醇的edv被良好地耐受,并且45.5%的患者呈现疾病稳定化。该实施例还表明,期望改善癌症治疗策略以改善生存(情况)和疾病响应。
[0434]
实施例6:评价靶向egfr的包装多柔比星的edv(
egfr(v)
edvs
dox
)在复发性成胶质细胞瘤中的1期临床试验
[0435]
此实施例表明,在患有复发性成胶质细胞瘤的患者中,用靶向egfr的包装多柔比星的微细胞(edv)进行的治疗被良好耐受。50%的患者呈现疾病的稳定化,但是没有患者经历部分或完全的响应。whittle et al.,j.clin.neurosci.,22(12):1889
‑
1894(2015)。
[0436]
复发性成胶质细胞瘤试验被设计为剂量递增研究以确定靶向egfr的负载多柔比星的edv(
v
edvs
dox
)的安全性、耐受性和最大耐受剂量或推荐2期剂量。注意,用于将edv靶向egfr的抗体与当前方案的抗体(基于vectibix的序列)相同。其他目标是评估对iv给予的
v
edvs
dox
的免疫和炎性响应,和根据神经肿瘤响应评估(rano)标准评估对治疗的响应。
[0437]
该研究于2013年2月5日开始,于2014年6月26日结束。其在澳大利亚悉尼和墨尔本的4家肿瘤诊所进行,并在australian new zealand clinical trials registry注册(编号actrn12613000297729)。最终临床研究报告是可获得的。该研究基于草案列举发表于whittle et al.,j.clin.neurosci.,22(12):1889
‑
1894(2015)。
[0438]
患者是至少18岁的成年人,有病理记录的和明确诊断的复发性世界卫生组织(who)iv级成胶质细胞瘤,其已在接受标准护理治疗(包括最大程度的安全手术切除、标准辅助放射/替莫唑胺和维护替莫唑胺治疗)后经历了疾病复发或进展。
[0439]
v
edvs
dox
每周以20分钟iv输注被给予,周期由8周治疗组成。在每个周期结束时,患者进行通过磁共振成像(mri)放射学评估其肿瘤。如果肿瘤保持稳定或对治疗有响应,或者如果其从治疗获得临床收益,并且其没有经历任何需要中断治疗的dlt或其他ae,则患者可以继续接受更多周期的治疗。
[0440]
经3个剂量水平共递送197个剂量:每剂量2x109、5x109和8x10
9v
edvs
dox
。14名患者中8名完成了至少1个完整周期的治疗(8个每周一次剂量),1个患者经几乎6个完整疗程(约12个月)接受了47个剂量。没有发生与治疗相关的死亡,并且没有患者经历剂量限制性毒性或需要中止治疗的其他不利事件。下表8展示了临床研究和发现的总结。
[0441][0442]
1 一个完整周期由8个每周一次剂量组成。
[0443]2对于完成至少1个治疗周期的患者。
[0444]3复发性成胶质细胞瘤的历史生存中位数为约5个月。
[0445]
4 2年存活的患者数量和百分比。
[0446]
至少可能与研究治疗相关的最常见不利事件为低度发热(发烧)、恶心和僵直(寒战),上至50%的患者(总体上1
‑
2级严重程度)经历。大多数患者在用药后3小时经历了轻度的短暂的细胞因子il
‑
6、il
‑
8、il
‑
10和tnf
‑
α升高。水平总体上在接受用药后24小时内恢复到基线。这与对治疗的轻度炎性响应一致。
[0447]
根据national cancer institute’s common terminology criteria for adverse events(nci
‑
ctcae),五名患者经历了3级或更高严重程度的治疗相关ae。这些事件被总结在下表9中,并在下面的叙述中被描述。
[0448][0449]
两名患者经历了严重程度≥3级严重程度的不利事件,其被认为是严重的。剂量服用后的晚上,5x109剂量下的一名患者在第1周期的第2个剂量后晚上因3级身体虚弱并伴有1级发烧的严重不利事件住院。这不被认为是剂量限制性毒性,因为患者在研究入选时对抗产物抗体(edv的沙门氏菌lps组分的抗体)呈阳性。该患者继续接受了再2个剂量的治疗,没有重复发生该事件。
[0450]
8x109剂量水平下的另一名患者在第1周期的第1个剂量后4小时经历了严重不利事件,3级症状性低血压,其导致住院进行静脉补水。这不被认为是dlt,因为其很可能归因于被治疗患者的大肠杆菌尿路感染。该患者接受了后续另外3个剂量研究药物。
[0451]
3名患者经历了不被认为严重的≥3级严重程度的不利事件。一名患者在一些剂量后经历了肝酶3级升高。这些不被认为是剂量限制性毒性或严重不利事件,因为其是无症状且短暂的,在两个剂量之间恢复到基线。两名患者经历了3级低磷血症,并且在一例中其伴有4级淋巴细胞减少症。这些也不被认为是剂量限制性毒性或严重不利事件,因为其是短暂的并且不需要治疗干预。
[0452]
总体而言,治疗被良好耐受,并且对于预期的目标群体没有特别的安全顾虑。
[0453]
为了评价对edv治疗的免疫响应,在筛选时评估了沙门氏菌的抗体。14名患者中的13名(93%)在筛选时呈沙门氏菌抗体阴性。分配至剂量水平2的一名患者在筛选时呈阳性。所有患者显示抗体滴度初始升高直至第3个剂量。在后续剂量中滴度保持而没有进一步增加,尽管一名患者接受了总共47个剂量。没有患者产生针对vectibix的抗体。
[0454]
为了评价功效,对完成第1周期的8名患者评价了肿瘤响应。没有患者经历完全或部分减轻,但是50%的患者显示疾病稳定,没有患者经历疾病进展。一名患者被报告在整个研究过程中具有稳定疾病,接受了几乎6个完整的治疗周期(约12个月)。
[0455]
追踪完成至少1个完整治疗周期的8名患者的生存(情况)。8名患者全部生存超过5
‑
7个月的中位历史生存期,并且中位os为15.1个月(范围9.1到>18.4)。四名对象在最后一次接触时还存活,并在此时进行了审查。这些患者中的两名(14.3%)完成了至少1个治疗周期,生存>18个月。
[0456]
总之,靶向egfr的包装多柔比星的edv对复发性成胶质细胞瘤的治疗显示了有前
景的结果,几乎没有不利事件,并且50%的患者经历了疾病稳定化。然而,需要改善的治疗策略。
[0457]
实施例7:在间皮瘤中评价包装有微小rna
‑
16模拟物的靶向egfr的edv(
egfr(v)
edvs
mirna16a
)的1期临床试验
[0458]
此实施例表明,包装有微小rna
‑
16的靶向egfr的edv在16名进行功效测试患者中的1名间皮瘤患者中给出了部分响应;而62.5%的患者经历了稳定化的疾病,并且31.3%的患者经历了进展性疾病。试验数据发表在zandwijk et al.,lancetoncol.,18(10):1386
‑
1396(2017)和kao et al.,am.j.respir.crit.care med.,191(12):1467
‑
1469(2015)。这种治疗策略总体上被良好耐受。
[0459]
在开放标签、多中心、探索性1期研究中,在复发性恶性胸膜间皮瘤(mpm)患者中评价了
v
edvs
mirna16a
。该试验的主要目的是确立
v
edvs
mirna16a
的最大耐受剂量和dlt以评价多次用药的效果,和检测
v
edvs
mirna16a
的早期功效迹象。注意,用于将edv靶向egfr的抗体与当前方案的抗体(基于vectibix的序列)相同。该试验的次要目的是评估接受
v
edvs
mirna16a
的患者的生活质量,和监测治疗过程中eastern cooperative oncology group(ecog)的表现状态和肺功能参数的变化。探索性目的评价了治疗过程中免疫和细胞因子标志物的变化。
[0460]
为符合资格,患者必须具有mpm的组织学或细胞学证明,并在其肿瘤组织中有egfr表达的证据。患者包括18岁以上的男性和女性,并且ecog表现状态为0或1,预期寿命为至少3个月。患者必须在标准1线或2线治疗方案过程中或之后呈现疾病进展,并且需要具有足够的骨髓、肝和肾功能。
[0461]
将
v
edvs
mirna16a
每周一次或每周两次以20分钟iv输注给予,周期由8周治疗组成。在每个周期结束时,患者进行其肿瘤的放射学评估。根据修订的实体肿瘤响应评价标准(recist)评估肿瘤响应。肺量测定法、fdg
‑
pet扫描和ct扫描用于评估疾病程度。
[0462]
该试验于2014年10月2日在澳大利亚悉尼的三家肿瘤诊所开始,并于2016年11月24日结束。该研究已在australian new zealand clinical trials registry(编号actrn 12614001248651)和clinicaltrials.gov(编号nct02369198)注册。5个组招募了总共27名患者,其中26名患者接受了共316个剂量的
v
edvs
mirna16a
(一名对象在接受任何治疗前死亡,被排除在进一步分析之外)。这项研究已发表在kao等,2015和van zandwijk等,2017。
[0463]
评价的剂量水平为5x109,每周一次或两次;和2.5x109,每周两次。为避免细胞因子反应增加,还评价了剂量从1x109逐渐增加到1期等效剂量的剂量适应性。还评价了为后续剂量的预先用药逐渐减少dex的地塞米松(dexamethasone)(dex)适应性。每周一次以5x10
9v
edvs
mir16a
确定mtd。在所示群体中,该治疗总体上被良好耐受,并具有可接受的安全性发现。
[0464]
下表10中展示了临床研究和发现的总结。
[0465][0466]1入选的27名患者中的1名在接受治疗前死亡,并被排除在进一步分析之外。
[0467]2一个完整的周期由8周治疗组成(8个每周一次剂量或16个每两周一次剂量)。
[0468]3对于完成至少1个治疗周期的患者。
[0469]4输注相关的反应包括以下任一种:寒战、僵直、发热、心动过速、盗汗或高血压。
[0470]
24名患者中的16名完成了至少1个完整治疗周期,其中2名患者接受了总共至少40个剂量(≥5个完整治疗周期)。观察到的最佳响应是1名患者的部分响应。如下所述,该患者呈现响应
v
edvs
mir16a
治疗的近完全缓解。10名患者(62.5%)呈现稳定化的疾病,并且5名患者(31.3%)呈现进展性疾病。中位生存期为36.5周,或8.4个月(范围9.3
‑
>119.6周),其中9名(60.0%)生存≥6个月,其中5名在自治疗开始>12个月时存活并且良好。参见图4。
[0471]
特别注意,一名患者(组1的患者#5)在第一个周期结束时呈现剧烈临床响应(参见kao et al.,am.j.respir.crit.care med.,191(12):1467
‑
1469(2015))。在8周时间结束时,其pet
‑
ct扫描显示“完全”代谢响应,并且胸部ct扫描显示部分响应并在4周后确认。目标成像响应伴随着呼吸功能测试参数的显著改善。
[0472]
最常见的治疗相关不利事件是输注相关的反应(96.2%),包括寒战、僵直、发热、心动过速或高血压(盗汗也包括在这一类别中)。其中大多数是轻度至中度的严重程度。输注后14名患者(53.8%)还经历了肿瘤位点的非心脏性胸痛。这些反应通过对方案的修改而解决,其中所有对象均在第1周期接受了适应性的递增剂量方案,导致输注相关的反应较少和其严重程度较低。实验室检查表明,在大多数患者中,在
v
edvs
mirna16a
输注后不久,炎性细胞因子和嗜中性粒细胞短暂升高并且淋巴细胞短暂减少,与轻度炎性响应一致。
[0473]
8名患者经历了9个严重不利事件,该不利事件至少可能与治疗相关。非心脏性胸痛和输注相关反应均发生在2名患者中。三名患者经历了剂量限制性毒性,并且另外两名对象经历了被认为剂量限制性但不符合剂量限制性毒性标准的毒性,因为其发生在剂量限制性毒性窗之外。没有发生治疗相关的死亡。这些事件被总结在下表11中,并在下面的叙述中
进行描述。
[0474][0475]
三名患者被招募至组1(每周一次,5x109)。其中一名经历了肿瘤位点周围的剂量限制性毒性非心脏性胸痛。该对象前往降低的剂量,随后递增回到完全强度。该组扩大以纳入总共6名对象,没有进一步的dlt。
[0476]
组2的2名对象中的2名(每周两次,5x109)经历了导致剂量减少或研究退出的毒性。第一是输注相关的反应,其被归类为剂量限制性毒性。该对象前往降低的剂量,随后递增回到完全强度。第二事件是持续性ecg变化伴有同时冠状动脉缺血,其未被归类为剂量限制性毒性,因为其未在第4周用药之内发生(在下文进一步讨论)。该对象被从研究剔除,并且没有其他对象入选该组(最大给予剂量)。
[0477]
六名对象入选了另一组研究(组3,每周一次5x109,剂量递增),其中所有对象均以减少的剂量开始,随后递增至完全强度,以最小化研究药物的输注反应。在该组中没有对象经历剂量限制性毒性。
[0478]
两名对象入选了组4(每周两次2.5x109+剂量递增),并且没有经历剂量限制性毒性。然而,由于与每周两次用药相关的显著临床负担,决定终止该组的招募。
[0479]
9名对象入选了另一组研究(组5,每周一次5x109+剂量递增+dex适应性),以评价包括剂量递增以及地塞米松预先用药逐渐减少的用药方案。参见图4。在该组中一名对象未接受任何治疗。在其余8名对象中,2名经历了毒性,导致剂量减少或研究退出。第一是takotsubo(应激相关的)心肌病,其被归类为剂量限制性毒性(在下文进一步讨论)。该对象退出研究。第二是类过敏反应,其未被归类为剂量限制性毒性,因为其在第7周用药内发生。但是,该对象被从研究剔除,并且该组的招募中止。最大耐受剂量被确定为每周一次5x10
9v
edvs
mirna16a
。
[0480]
在这项研究中注意到了多个以前在其他适应症的不同edv产品的试验中未观察到的心脏事件。两名对象经历了心脏事件的严重不利事件,导致剂量降低或退出(局部缺血和takotsubo心肌病)。缺血被认为不太可能是由于研究治疗所致,因为该事件发生于用药后7天,并且该对象有在前冠心病病史。该对象在较早剂量期间经历了ecg变化。心肌病事件后于输注反应,可能是dex递减(tapering)的结果。该对象也有冠状动脉疾病病史。另外三名对象在用药后经历了短暂的ecg变化(t波异常),其没有被归类为严重。这些不与肌钙蛋白升高、缺血或lvef变化相关。所有事件均被解决,并且对象继续接受其他剂量治疗,而无进一步的ecg异常。然而,鉴于恶性胸膜间皮瘤患者的高龄以及与疾病相关的发病,这些观察
结果共同致使安全委员会建议对于研究的其余部分以及任何未来试验采取更严格的心脏排除标准和额外的心脏监测。
[0481]
用
v
edvs
mirna16a
进行的总体治疗总体上被良好耐受,上至5x109edv的最大耐受剂量,并且在给予足够的监测和适当的递增剂量方案的情况下,此特定患者群体无重大顾虑。
[0482]
因此,此实施例显示了通过使用靶向egfr的edv将mirna16a递送至癌细胞来治疗间皮瘤的有前景的结果。然而,此实施例结果还表明需要改善的间皮瘤治疗策略。
[0483]
实施例8:体外细胞毒性测定表明超毒性药物pnu
‑
159682比其他化疗剂更大程度地抑制肿瘤细胞系的增殖。
[0484]
此实施例证明,pnu
‑
159682——一种超毒性细胞毒性化疗剂——与宽范围的其他化疗药物相比,是更有效的癌细胞生长抑制剂。
[0485]
pnu
‑
159682是蒽环奈莫柔比星(mmdx)的高效代谢产物,并且细胞毒性比其母体化合物(mmdx和多柔比星)高超过3,000倍;因此,pnu
‑
159682被认为是“超毒性”化疗药物。由于超毒性药物的毒性水平会导致严重的不利事件,包括死亡,一般化疗通常不可能使用这种药物。
[0486]
将pnu
‑
159682和其他所示化疗剂以图5
‑
10所示的浓度添加至肿瘤细胞系。
[0487]
将所有细胞再培育72小时,然后根据制造商的说明,利用celltiter 96aqu
eous one solution cell proliferation assay(promega corp.,madison,wi,usa)进行比色mts细胞增殖测定(cory et al.,cancer commun.,3(7):207
‑
12(1991))。比色测量结果在490nm下读取。
[0488]
更高的细胞毒性:这些体外细胞毒性测定的结果表明,与通过一系列已知的化疗剂观察到的细胞毒性相比,pnu
‑
159682对人肺癌细胞系a549呈现更高的细胞毒性,如图5a所示。具体地,pnu
‑
159682抑制a549细胞的程度远高于多柔比星。图5b。
[0489]
已知对常规化疗剂呈现抗性的抗癌细胞有效性:进一步,图6显示pnu
‑
159682和多卡霉素对源自iv期患者的两种肾上腺皮质癌细胞系呈现有效的细胞毒性效果,该肾上腺皮质癌细胞系对多柔比星、米托坦、紫杉醇、奥沙利铂和米托蒽醌具有高度抗性。此外,图7显示pnu
‑
159682抑制对多柔比星、紫杉醇和多西他赛具有抗性的人乳腺癌细胞系mda
‑
mb
‑
468的增殖。图8显示pnu
‑
159682抑制人结肠直肠癌细胞系caco
‑
2(图8a)和hct116(图8b)的增殖,该细胞系对多柔比星、顺铂具有抗性。图9显示pnu
‑
159682可以抑制对多柔比星和紫杉醇ivax具有抗性的成胶质细胞瘤细胞系u87
‑
mg的增殖。图10a显示pnu
‑
159682可以抑制对吉西他滨敏感的人胰腺细胞系的增殖(miapaca
‑
2细胞,图10a),即使这些细胞对多柔比星、gemzar、甲氨蝶呤、奥沙利铂、伊立替康和5
‑
氟尿嘧啶(5
‑
fu)具有抗性。图10b显示了pnu
‑
159682可以抑制作为吉西他滨抗性细胞的人胰腺细胞系(miapaca
‑
2gemr细胞,图10b)的增殖,即使这些细胞对gemzar具有抗性。
[0490]
这些数据表明,高度期望在癌症治疗中使用超毒性化疗剂如pnu
‑
159682,因为该药物可有效用于治疗对常规非超毒性化疗药物呈现抗性的癌症。
[0491]
实施例9:用靶向egfr的edv递送的pnu
‑
159682可以克服小鼠异种移植模型中人肺癌细胞的抗性。
[0492]
此实施例表明,利用微细胞(edv)递送超毒性化疗剂如pnu
‑
159682有效地抑制肺癌异种移植模型中的肿瘤生长。
[0493]
通过在存在多柔比星(dox)的情况下连续培养和选择dox抗性克隆,使a549(肺癌)细胞形成多柔比星抗性。然后将这些细胞作为异种移植物植入balb/c nu/nu小鼠。当肿瘤体积达到~150mm3时,将4个不同组的小鼠(每组n=7)在图11中实心箭头指示的时间点用下列进行静脉内(iv)治疗:(i)盐水,(ii)靶向表皮生长因子受体(egfr)的负载有多柔比星的edv(
egfr
edvs
tmdox
),(iii)靶向egfr的负载有pnu
‑
159682的edv(
egfr
edvs
tm682
),以及(iv)负载有pnu
‑
159682的非靶向型edv(edvs
tm682
)。靶向egfr并负载有pnu
‑
159682的edv的组成被图形描绘在图1中。
[0494]
图11所示的结果显示,
egfr
edvs
tmdox
不具有抗肿瘤功效,因此肿瘤呈现dox抗性。相比之下,用
egfr
edvs
tm682
治疗的小鼠显示完全肿瘤稳定化。当盐水治疗的肿瘤达到500mm3至700mm3范围内的肿瘤体积时,在图6中用星号(*)指示的时间点将治疗变为
egfr
edvs
tm682
。惊人地,结果显示高度显著的抗肿瘤功效,甚至已达到500mm3至700mm3范围内的体积的肿瘤中。
[0495]
实施例10:用靶向egfr的edv递送功能性dna试剂有效抑制间皮瘤(msto)和肾上腺皮质癌(acc)癌细胞生长。
[0496]
此实施例表明,利用微细胞(edv)递送靶向polo样激酶1(plk1)的sirna、靶向核糖核苷酸还原酶1(rrm1)的sirna或mirna16a可以有效地抑制癌细胞的生长。
[0497]
已显示polo样激酶1(plk1)和核糖核苷酸还原酶1(rrm1)在几种非小细胞肺癌(nsclc)细胞系中过表达,该细胞系包括a549、a549mdr(dox抗性a549细胞系,其过表达多药抗性膜泵(mdr)、h2122、h358和h441。图12显示了gapdh(g)、ksp(k)、plk1(p)和rrm1的表达,并且该表达相对于gapdh在所示的nsclc细胞系中的表达被显示。
[0498]
为了测试plk1或rrm1是否是对于癌症治疗有用的靶标,合成了靶向rrm1(sirrm1)和plk1(siplk1)的抑制性非编码小干扰rna(sirna),并将其包装在edv中以递送至癌细胞系。
[0499]
发现靶向rrm1的sirna抑制间皮瘤和肾上腺皮质癌细胞的增殖。将靶向egfr的包装sirrm1的edv转染到msto(间皮瘤细胞系)或h295r(肾上腺皮质癌细胞系)中。转染后五天,测量细胞增殖,并且结果示于图13中,并且显示与用非靶向的包装sirrm1的edv或靶向egfr的包装sinonsense的edv进行的对照转染相比细胞增殖抑制高度显著。
[0500]
在balb/c nu/nu小鼠的间皮瘤(msto)异种移植研究中,与盐水或靶向egfr的包装si加扰(siscrambled)的edv相比,用靶向egfr的包装sirrm1的edv进行静脉内(iv)治疗显示出高度显著的抗肿瘤功效,如图14所示。分离自接受包装sirrm1的edv的小鼠中的肿瘤显著小于来自接受阴性对照的小鼠的肿瘤。图15。
[0501]
发现mirna16a抑制间皮瘤癌细胞的增殖。在balb/c nu/nu的间皮瘤(msto)异种移植研究中,与盐水或靶向egfr的包装si加扰的edv相比,用靶向egfr的包装mirna16a的edv进行的静脉内(iv)治疗显示出高度显著的抗肿瘤功效,如图14所示。分离自接受包装mirna16a的edv的小鼠的肿瘤显著小于接受阴性对照的小鼠的肿瘤。图15。
[0502]
靶向egfr的负载siplk的edv(
egfr
edv
tmsiplk
)诱导癌症患者源肿瘤球形体(acc001)的凋亡,如图16所示。无任何有效载荷的edv(
egfr
edv
tm
)或带有靶向无关荧光素酶序列的rna干扰分子的edv(
egfr
edv
tm荧光素酶
)用作阴性对照。与这些阴性对照相比,通过测量细胞碎片(图16a)和测量annexin/碘化丙锭(pi)比(图16b)确定
egfr
edv
tmsiplk
诱导了acc001球形体的凋
亡。与阴性对照相比,
egfr
edv
tmsiplk
治疗还导致亚g1细胞周期的大量细胞停滞,如图17所示。因此,用sirna抑制plk1表达是诱导acc001肾上腺皮质癌细胞的凋亡和细胞周期停滞的有效策略。
[0503]
实施例11:在异种移植模型中用靶向egfr的edv递送i型干扰素激动剂增强了负载有细胞毒性药物的edv的抗肿瘤功效。
[0504]
此实施例表明,利用微细胞(edv)递送与i型干扰素激动剂组合的化疗剂是治疗癌症如肺癌异种移植的有效策略。i型干扰素激动剂可以与化疗剂位于相同或不同的微细胞中。在本实施例中,化疗剂是超毒性药物pnu
‑
159682,而i型干扰素激动剂是40mer双链dna。
[0505]
将balb/c nu/nu小鼠中的a549(肺癌)异种移植用各种edv组合通过在尾静脉中静脉内注射进行治疗,如图18所示。小鼠用下列治疗:(i)实心三角形=
egfr
edvs
pnu
‑
159682
+edvs
40mer
,(ii)实心圆形=
egfr
edvs
pnu
‑
159682
,(iii)空心正方形=
egfr
edvs
pnu
‑
159682
+edv,(iv)空心三角形=
egfr
edvs
pnu
‑
159682
+edvs
50mer
,(v)实心正方形=盐水。该是i型干扰素激动剂。
[0506]
在异种移植物植入后第24、27、29、31、34、36和38天用这些edvs组合治疗小鼠,如图18中的向上箭头所示。如图18所示,测试的所有edv组合均导致肿瘤生长稳定化。相比之下,盐水处理的对照组呈现上至体积~650mm3的肿瘤生长。在第36天和第38天,用
egfr
edvs
pnu
‑
159682
+edvs
50mer
治疗肿瘤体积为~650mm3的盐水组小鼠,如图18中的下箭头所示。
[0507]
用包含pnu
‑
159682和靶向egfr的微细胞(edv)(
egfr
edvs
pnu
‑
159682
)组合包含40mer双链dna的edv(edvs
40mer
)治疗具有~650mm3大体积肿瘤的小鼠导致肿瘤急剧消退。具体而言,在仅5天之内,肿瘤体积就从~650mm3减小至~250mm3,或在5天内肿瘤尺寸减少62%。结果总结在下表中。
[0508][0509]
此外,包含40mer双链dna的edv(edvs
40mer
)与包含pnu
‑
159682和靶向egfr的微细胞(edv)(
egfr
edvs
pnu
‑
159682
)组合在肺癌的小鼠异种移植模型中诱导了与单独
egfr
edvs
pnu
‑
159682
治疗的肿瘤细胞相比更显著的肿瘤消退,如图19所示。在图19中,balb/c nu/nu小鼠用下列治疗:(i)实心圆形=
egfr
edvs
pnu
‑
159682
,(ii)实心三角形=
egfr
edvs
pnu
‑
159682
+edvs
40mer
,或(iii)实心正方形=盐水。结果总结在下表中。
[0510][0511][0512]
总之,包装在微细胞中的i型ifn激动剂增强
egfr
edvs
pnu
‑
159682
治疗的抗肿瘤效果。
[0513]
实施例12:连同佐剂i型ifn激动剂(edv
40mer
或edvs
60mer
)和ii型ifn激动剂(imukin)的edvs
pnu
‑
159682
的临床评价。
[0514]
此实施例表明,i型和ii型ifn激动剂在患有晚期实体肿瘤的人类患者中增强了
egfr
edvs
pnu
‑
159682
的抗癌效果。显著地,甚至在晚期胰腺癌患者中,这种治疗在仅3个剂量后也产生肿瘤标志物水平下降90%、并且患者呈现生活质量显著提高。
[0515]
用包含药物、i型ifn激动剂和ii型ifn激动剂的微细胞进行治疗:本公开的发明人进行了临床案例研究,其中对象接受靶向且负载型edv与负载有1型ifn激动剂40mer双链dna(edvs
40mer
)(1型ifn激动剂)或60mer双链dna(edvs
60mer
)(1型ifn激动剂)的微细胞的组合。
[0516]
三名患有晚期实体肿瘤的对象接受了
egfr(v)
edvs
pnu
‑
159682
与佐剂edvs
40mer
(1型ifn激动剂)的组合治疗作为designer edv研究(澳大利亚,墨尔本)的一部分。根据同情使用法规,另外两名患者接受了负载且靶向型的edv与edvs
60mer
(1型ifn激动剂)组合治疗。一名被诊断为iv期胰腺癌的同情使用患者接受了
egfr(v)
edvs
pnu
‑
159682
和edvs
40mer
(1型ifn激动剂)或edvs
60mer
(1型ifn激动剂)的治疗,并且第二名诊断为复发性肾上腺皮质癌的同情使用患者接受了
egfr(v)
edvs
pnu
and edvs
60mer
+imukin(ii型ifn)。
[0517]
临床研究
[0518]
包含药物和i型ifn激动剂的微细胞:在具有晚期实体肿瘤的对象中的一项开放标签的、单中心的、探索性1期研究(designer edv研究)中,在3名患者中评价了
egfr(v)
edvs
pnu
和佐剂edvs
40mer
(1型ifn激动剂)。为符合条件,患者应具有晚期实体肿瘤的组织学或细胞学证明,并且有证据显示其肿瘤组织中egfr表达以促进靶向。患者必须已在给予标准的线、二线或三线治疗方案期间或之后显示疾病进展。该试验的主要目的是确立
egfr(v)
edvs
pnu
‑
159682
与佐剂edvs
40mer
(1型ifn激动剂)的安全性和耐受性。
[0519]
该试验在澳大利亚墨尔本的一个中心启动,并已在australian new zealand clinical trials registry注册(编号actrn 12617000037303)。
[0520]
试验中的3名患者接受了共13个剂量的2.5x109的
egfr(v)
edvs
pnu
‑
159682
和5x108的edvs
40mer
(1型ifn激动剂)。治疗被每周一次以20分钟iv输注给予,周期由8周治疗组成。在每个周期结束时,患者进行其肿瘤的放射学评估。
[0521]
可得的安全性数据是有限的,但治疗总体上被良好耐受,无意外的ip相关不利反应。如给予其他edv产品所见,患者总体上经历炎性细胞因子il
‑
6、il
‑
8和tnf
‑
α的短暂增加,在治疗剂与治疗剂之间恢复到基线。观察到的血液学参数变化在很大程度上反映了在
先用edv治疗剂进行的试验中观察到的变化,包括白血细胞(wbc)的轻度自我限制性升高、用药后3小时嗜中性粒细胞的升高以及伴随的淋巴细胞和单核细胞减少。在下一个时间点,下一次用药之前,参数恢复正常。一些对象经历了血清磷酸盐水平的轻度降低,这不需要干预并且在剂量与剂量之间恢复到基线。
[0522]
两名患者经历了涉及输注相关反应的不利事件,僵直和发烧在用药后约1小时开始。这些患者入院过夜观察,并且该事件在第二天被解决。由于剂量限制性毒性,一名患者退出研究。第二名患者继续研究并接受了其他剂量。
[0523]
同情使用研究
[0524]
第一个同情使用案例研究在悉尼royal north shore hospital进行。患者是一名68岁女性,其被诊断患有iv期胰腺癌。其已进行whipple手术(胰十二指肠切除术(pancreaticoduodenectomy)),并且吉西他滨作为一线治疗。其还接受了folfirinox治疗方案,但产生了转移性肝病。其肿瘤细胞被体外测试,发现对pnu
‑
159682敏感。
[0525]
该患者接受每两周一次剂量的edv产品,包括负载pnu的和靶向egfr的edv与不同免疫调节佐剂的组合。这些以20ml输注被iv递送。其如用于本方案预期接受了包括pnu的靶向egfr(v)的edv和靶向itg(609)的edv,以及edvs
40mer
(1型ifn激动剂)或edvs
60mer
(1型ifn激动剂)。该患者总共接受了45个剂量的
egfr(v)
edvs
pnu
‑
159682
+edvs
40mer
(或相关产品edvs
60mer
)。
egfr(v)
edvs
pnu
‑
159682
和
itg(609)
edvs
pnu
‑
159682
的剂量分别递增上至最大值2x109和4x109,并且edvs
40mer/60mer
以5x108的设定剂量被给予。
[0526]
患者对治疗耐受很好,无ip相关的严重不利事件。初步结果表明在用药后炎性细胞因子il
‑
6、il
‑
8和tnf
‑
α短暂增加,类似于给予其他edv产品时所见。这些响应总体上经过后续剂量减少。在用药后,抗炎性细胞因子il
‑
10也短暂增加。值得注意,ifn
‑
α在贯穿研究的不同时间点增加,这可能是edvs
40mer/60mer
刺激的结果。用药后2小时未检测到ifn
‑
γ升高。
[0527]
显著地,在前3个剂量后,相当于仅10天的治疗,患者的肿瘤标志物(ca 19
‑
9)的水平下降了超过90%。10个剂量后,其再进一步下降,肿瘤标志物水平降低几乎95%。她还显示显著的体重增加——相比之下大多数患有iv期胰腺癌的患者经历的恶病质状态,并报告了生活质量显著改善。因此,该病例研究的初步安全性和功效结果因此非常有前景,特别是在晚期胰腺癌相关的预后不良的情况下。
[0528]
包含药物、i型ifn激动剂和ii型激动剂的微细胞:在具有非常严重肿瘤负荷的末期肾上腺皮质癌患者中的第二例同情使用案例研究在royal north shore hospital(悉尼)被治疗。
[0529]
该患者每接受10个每两周一次剂量的
egfr(v)
edvs
pnu
‑
159682
(包含抗肿瘤剂的微细胞,其中抗肿瘤剂的剂量范围为1x109至4x109)、edvs
60mer
(包含i型ifn激动剂的微细胞,其中1型ifn激动剂的剂量范围为5x108至2x109[edvs])和imukin(ii型ifn,其中ii型ifn的剂量范围为5μg(1x105iu)至30μg(6x105iu))。
[0530]
患者对治疗耐受非常好,在用药后直至60分钟仅经历轻度温度升高。这在edv产品的给予上是期待的。不幸的是,患者有很高的疾病负担,包括高水平的皮质醇——已知这是一种严重的免疫系统抑制剂,并且在第7周期间的ct扫描显示疾病进展,因此该患者退出了研究。
[0531]
总之,5名患者接受了共69个剂量的
egfr(v)
edvs
pnu/dox
或
egfr(v)
edvs
pnu/pnu
+
edvs
40mer/60mer
(i型ifn激动剂)
±
imukin(ii型ifn)。这些治疗被耐受良好,并且添加免疫调节佐剂看起来并没有改变单药负载和靶向型edv的安全性。
[0532]
实施例13:添加ifn
‑
γ(ii型ifn激动剂)增强了负载有多柔比星的靶向表皮生长因子受体的edv的抗肿瘤功效,并且在各种癌症的异种移植模型中引起肿瘤消退。
[0533]
此实施例表明,在小鼠异种移植模型中,使用微细胞(edv)递送多柔比星与ifn
‑
γ组合提高了抗肿瘤效果。
[0534]
肺癌:为在肺癌模型中研究组合
egfr
edvs
dox
和ifn
‑
γ(ii型ifn)的抗肿瘤效果,建立balb/c nu/nu小鼠中的a549(肺癌)异种移植物,并将其分成通过静脉内尾静脉注射接受不同治疗组合的四组。第1组接受无菌生理盐水(图20,空心菱形)。第2组每剂量接受ifn
‑
γ(0.5x104iu)(图20,实心三角形)。第3组接受
egfr
edvs
dox
(图20,实心正方形)。第4组每剂量接受
egfr
edvs
dox
和ifn
‑
γ(0.5x104iu)(图20,实心圆形)。
[0535]
用
egfr
edvs
dox
治疗的小鼠实现了a549肺癌异种移植物的肿瘤稳定化(图20,实心正方形)。相比之下,用
egfr
edvs
dox
和ifn
‑
γ治疗的小鼠在共6个剂量后到第43天显示了高度显著的肿瘤消退(图20,实心圆形)。用单独ifn
‑
γ治疗的小鼠不显示抗肿瘤功效(图20,实心三角形),并且肿瘤如盐水治疗组中一般生长(图20,空心菱形)。因此,将
egfr
edvs
dox
和ifn
‑
γ(ii型ifn)组合在肺癌小鼠异种移植模型中导致肿瘤消退,如下表14总结。
[0536][0537]
乳腺癌:为在乳腺癌模型中研究组合
egfr
edvs
dox
和ifn
‑
γ的抗肿瘤效果,建立了在balb/c nu/nu小鼠中的mda
‑
mb 468异种移植物,并将其分成通过静脉内尾静脉注射接受不同治疗组合的四组。第1组接受无菌生理盐水(图21,空心菱形)。第2组每剂量接受ifn
‑
γ(0.5x104iu)(图21,实心三角形)。第3组接受
egfr
edvs
dox
(图21,实心正方形)。第4组每剂量接受
egfr
edvs
dox
和ifn
‑
γ(0.5x104iu)(图21,实心圆形)。
[0538]
用
egfr
edvs
dox
治疗的小鼠实现了mda
‑
mb 468乳腺癌异种移植物的肿瘤稳定化,但是到~第25天,肿瘤又开始生长,这可能是由于对多柔比星的抗性产生所致(图21,实心正方形)。相比之下,用
egfr
edvs
dox
和ifn
‑
γ进行的小鼠治疗显示出高度显著的肿瘤消退,并且到第30天,在总共6个剂量之后,这些肿瘤更像是疤痕组织(图21,实心圆形)。用单独ifn
‑
γ治疗的小鼠不显示抗肿瘤功效(图21,实心三角形),并且肿瘤如盐水治疗组中一般生长(图21,空心菱形)。在图22中描绘的另外的实验中,用
egfr
edvs
dox
治疗的小鼠再次实现了mda
‑
mb 468乳腺癌异种移植物的肿瘤消退,但是到~第23天,肿瘤又开始生长,这可能是由于对多柔比星的抗性产生所致(图22,实心正方形)。相比之下,用
egfr
edvs
dox
和ifn
‑
γ治疗的小鼠显示出高度显著的肿瘤消退,并且到第28天,在总共3个剂量后,这些肿瘤更像是疤痕组织(图22,实心圆形)。用单独ifn
‑
γ治疗的小鼠不显示抗肿瘤功效(图22,实心三角形),并且肿瘤如盐水治疗组中一般生长(图22,空心菱形)。因此,将
egfr
edvs
dox
和ifn
‑
γ(ii型ifn)组
合导致乳腺癌小鼠异种移植模型中肿瘤消退,如下表15总结。
[0539][0540]
肺癌:为研究
egfr
edvs
dox
和ifn
‑
γ(ii型干扰素)在多柔比星抗性肺癌模型中的抗肿瘤效果,首先使a549肺癌细胞在存在多柔比星(dox)的情况下在培养中生长,并且建立dox抗性衍生细胞系。然后,建立balb/c nu/nu小鼠中的dox抗性a549异种移植物,并将其分成通过静脉内尾静脉注射接受不同治疗组合的四组。第1组每周两次接受无菌生理盐水(图23,空心菱形)。第2组接受
egfr
edvs
dox
(图23,实心三角形)。第3组每剂量接受
egfr
edvs
dox
和ifn
‑
γ(0.75x104iu)(图23,实心正方形)。第4组每剂量接受
egfr
edvs
dox
和ifn
‑
γ(0.5x104iu)(图23,实心圆形)。如图23所示,第1、2和3组是每周两次的指示剂量(通过图23中的x轴上用实心三角形表示);而第4组小鼠每周三次被给予指示剂量(通过图23中的x轴上的空心三角形表示)。每周用
egfr
edvs
dox
和ifn
‑
γ(0.5或0.75x104iu)治疗两次或三次的小鼠达到了抗性a549肺癌异种移植物的肿瘤稳定化(图23)。相比之下,用
egfr
edvs
dox
治疗的小鼠不显示抗肿瘤功效,并且肿瘤如盐水治疗组中一般生长。该结果表明,将ifn
‑
γ(0.5或0.75
×
104iu)连同
egfr
edvs
dox
一起包括在通常对单独后者具有抗性的肿瘤的治疗中对于实现肿瘤稳定化是至关重要的。因此,将
egfr
edvs
dox
和ifn
‑
γ组合可以克服肺癌异种移植模型中的抗性,如下表16总结。
[0541][0542]
实施例14:用
egfr
edvs
pnu
或
itg
edvs
pnu
‑
159682
+edvs
40mer
(1型ifn激动剂)+imukin(ii型
ifn)治疗具有晚期内源性肿瘤的犬
[0543]
此实施例表明,在犬研究中,用微细胞技术以及另外与干扰素γ(imukin)(ii型ifn)一起递送超毒性药物pnu
‑
159682和i型ifn激动剂(40mer双链dna)被良好耐受。
[0544]
对患有内源性晚期癌症的犬进行了毒理学研究。犬是呈现晚期肿瘤的伴侣动物。从各宠物主人处获得知情同意。
[0545]
用负载pnu的靶向egfr或itg(整联蛋白)的edv与免疫调节佐剂(edvs
60mer/50mer
和/或imukin)组合治疗13只患有脑癌、肉瘤或黑色素瘤的犬。对患有脑癌或肉瘤的犬用靶向egfr的edv治疗(n=9),并且对患有黑素瘤的犬用靶向itg的edv(n=4)治疗。在有或无佐剂的情况下,以不同的组合给予共185个每周一次或每两周一次剂量,单只犬接受上至73个剂量。给予的edv剂量是上至5x109的负载pnu且靶向性的edv,上至2x109的edvs
40mer/50mer
和每剂量25μg/m2的imukin。所有组合都总体上被良好耐受,其中最常见的不利事件与给予其他edv产品时所见(轻度嗜睡、发烧、恶心、呕吐)相似。免疫调节佐剂的添加未呈现改变总谱或增加edv用药下所见的ae频率。
[0546]
对血液学参数的显著效果包括在用药后3小时的白细胞亚群(wbc、嗜中性粒细胞、淋巴细胞、单核细胞和嗜酸性粒细胞)轻度短暂减少。在添加或不添加免疫调节佐剂的情况下,观察到相似的变化。值得注意,在其他犬类和人类临床研究中,嗜中性粒细胞在用药后3小时总体上是增加的而不是减少。嗜中性粒细胞的这种轻度减少看起来是用负载pnu的edv治疗犬特异性的。
[0547]
在用药后3小时,犬经历了il
‑
6、il
‑
8、il
‑
10、il
‑
12p40和tnf
‑
α的短暂升高。总体上,包含免疫调节佐剂的剂量导致的这些细胞因子的诱导略高于单独包含负载pnu的edv的剂量。其还导致用药后il
‑
2水平降低。这些数据为与负载抗肿瘤剂且靶向性的edv组合使用的免疫调节性edv佐剂的安全性和耐受性提供了支持。
[0548]
观察到的最佳响应是7只可评价动物中6只(85.7%)的病情稳定化,尽管1只犬达到了近乎部分响应(肿瘤尺寸减少了29.8%)。一只犬经研究过程显示了稳定化的疾病,经超过11个月的治疗接受了73个剂量。这只犬由于肿瘤质量压在视神经上而呈现视力损失;然而,经治疗过程视力得以恢复,表明临床症状改善。
[0549]
实施例15:在人类临床研究中靶向和负载型edv不激活ifn
‑
γ
[0550]
此实施例表明,包装有紫杉醇或多柔比星的edv在人类患者中诱导了细胞因子响应,与toll样受体激活一致,但是这种edv治疗并未诱导ii型干扰素响应。
[0551]
为了了解通过给予靶向和负载型edv激活的途径,在人中首次人类试验(上文实施例5)和复发性成胶质细胞瘤人类临床试验(上文实施例6)中评价了一组细胞因子。
[0552]
图24显示了首次人类研究(实施例5)中对
egfr(erb)
edvs
pac
治疗的细胞因子响应,并且图25显示了在复发性成胶质细胞瘤研究中对
egfr(v)
edvs
dox
的细胞因子响应。如图24和25所示,两种治疗均为il
‑
6、il
‑
8、il
‑
10和程度较小的tnf
‑
α在用药后3
‑
4小时瞬时升高,并到24小时或下一剂量之前恢复至基线。对负载有紫杉醇或多柔比星的edv的细胞因子响应与toll样受体途径的激活一致,特别是tlr4,已知其被细菌lps刺激。il
‑
12仅在首次人类研究中的2名患者中随机升高(图24),尽管升高与用药不一致。
[0553]
值得注意,i
‑
型干扰素ifn
‑
α贯穿研究在随机阶段下在首次人类研究中的3/22名患者中(参见图24)和在复发性成胶质细胞瘤研究中的1/14患者中(参见图25)升高。ifn
‑
α
的诱导与用药不一致。干扰素途径总体上是通过病毒刺激而不是如同tlr途径通过细菌刺激而激活的,因此在研究期间这些选定的患者可能同时具有未报告的病毒感染(例如感冒)。但是,测试的所有其他细胞因子,包括ifn
‑
γ,均不受靶向和负载型edv用药的影响。这表明ii型ifn
‑
γ的激活(通过添加imukin)可以是通过刺激不同的抗肿瘤途径来增强靶向和负载型edv的功效的可行方法。
[0554]
实施例16:用于癌症的细胞免疫疗法:靶向肿瘤的包装超毒性药物的细菌源纳米细胞引出的新途径
[0555]
此实施例表明,靶向肿瘤细胞表面受体的edv纳米细胞充当癌症免疫疗法,能够通过递送超级细胞毒素pnu
‑
159682(682)结合先天性和适应性免疫系统的激活而对肿瘤进行双重攻击。
[0556]
此实施例显示了靶向型edv纳米细胞递送能够杀死微环境中的肿瘤细胞并伴有主要的th1细胞因子响应的682激活的m1巨噬细胞和nk细胞。这然后是树突状细胞成熟,导致抗原呈递和肿瘤特异性cd8
+
t细胞的生成。超级细胞毒素递送与先天性和适应性抗肿瘤免疫响应激活的组合产生有效的癌细胞免疫疗法,在临床肿瘤学中具有潜力。
[0557]
此实施例首次证明了edv纳米细胞的全新细胞免疫治疗功能,其中其不仅能够在肿瘤细胞内递送细胞毒性药物,而且同时引发针对肿瘤的先天性和适应性免疫响应。临床上,在同情使用案例患者中,显示了负载682的edv克服了抗药性,同时刺激适应性免疫。临床前研究表明,edv治疗产生的免疫治疗途径涵盖了解决通过免疫系统产生有效抗肿瘤响应所需的所有必要步骤的方法。此实施例示例了edv激活先天性免疫系统的细胞的能力,包括巨噬细胞、nk细胞和树突状细胞。这然后是树突状细胞成熟和抗原呈递,导致适应性t细胞响应,其中肿瘤特异性细胞毒性t细胞产生并导致其他免疫细胞进一步募集到肿瘤微环境。该方法通过创建免疫原性肿瘤微环境以及还作用于多个免疫细胞亚群,从而避免患者中可能出现的原发性和/或适应性抗性,而规避了免疫疗法的当前一些陷阱。
[0558]
结果:响应于edv治疗的m1巨噬细胞极化和树突状细胞成熟:由于edv源于鼠伤寒沙门氏菌(salmonella typhimurium)细菌的事实,edv外膜含有大量的脂多糖(lps)含量(macdiarmid等,2007b)。公知高剂量的lps与巨噬细胞的相互作用导致巨噬细胞激活和m1极化。为了确定edv是否能够引发相似的表型响应,将raw264.7细胞与ep
‑
edv
‑
682和ep
‑
edv一起培育,并考察巨噬细胞表型和细胞因子产生中的变化。已知共刺激性cd86表达的表达是巨噬细胞极化的表型指示剂以及抗肿瘤m1肿瘤相关巨噬细胞(tam)的标志(dong等,2016)。
[0559]
ep
‑
edv
‑
682和ep
‑
edv都能够引起cd86表达水平的显著增加,最有可能是响应edv表面上lps的存在,而单独682并没有引起相同的响应(图27a)。此外,经edv处理的raw细胞显示出促炎性细胞因子tnf
‑
α和il
‑
6的显著增加,其已被认为是造成th1巨噬细胞极化的原因(yuan等,2015)(图35a和35b)。值得注意,已用ep
‑
edv
‑
682处理然后与raw264.7细胞共培养的小鼠肿瘤细胞(4t1和ct26ep12.1)也能够产生raw264.7细胞上的cd86表达显著增加(图27b)以及促炎性细胞因子tnf
‑
α和il
‑
6的产生的显著增加(图27c
‑
27d)。然而,单独ep
‑
edv或682处理不能产生cd86表达的任何显著变化,并且682处理显示th1细胞因子的产生无增加,表明响应于edv负载的682的细胞死亡对于充分引起随后的m1巨噬细胞极化是必要的。
[0560]
还考察了响应于ep
‑
edv、ep
‑
edv
‑
682和682处理的肿瘤细胞死亡对树突状细胞成熟的影响(图27e
‑
27i)。将骨髓源树突状细胞(bmdc)与处理过的肿瘤细胞(4t1和ct26ep12.1)共培养48小时,然后评估1型干扰素ifnα和ifnβ的产生。树突状细胞的1型干扰素产生增加被充分确立作为树突状细胞成熟和抗原呈递增强的机制,并且对于其与其他免疫细胞亚群(包括nk细胞和t细胞)的相互作用至关重要(fitzgerald
‑
bocarsly和feng,2007;simmons等,2012)。bmdc与ep
‑
edv
‑
682处理的4t1细胞的共培养显示两种1型干扰素均显著增加,其中ifnαmrna的产生增加~100倍,并且ifnβmrna的产生增加~70倍(图27e)。类似地,bmdc与ep
‑
edv
‑
682处理的ct26ep12.1的共培养显示ifnαmrna的产生增加~300倍并且ifnβmrna的产生增加~60倍(图27f)。
[0561]
另外,为了评估负载到edv中的药物的差异是否对1型干扰素产生有任何影响。ct26ep12.1用ep
‑
edv
‑
dox处理,并显示ifnβmrna的产生~20倍显著增加,但ifnαmrna的产生仅略微非显著增加。ep
‑
edv和682处理不能与任一细胞系共培养中引发1型干扰素mrna的产生增加。但是,ct26ep12.1的多柔比星(dox)处理确实事实上显示出ifnβmrna的产生~5倍显著增加(图27f)。已知对肿瘤细胞的多柔比星处理会导致免疫原性细胞死亡,因此能够促进树突状细胞成熟。但是,随单独该药物发生的这种类型的死亡总体上需要远高于ic50的药物剂量(showalter等,2017)。bmdc与edv以及药物处理的4t1和ct26ep12.1细胞的共培育导致cd86、mhc ii类和cd80在24h内上调(图27g
‑
27i),在小鼠jaws ii细胞中观察到相似的结果(图35c
‑
35e)。与edv处理过的肿瘤细胞共培养的bmdc还呈现tnfα、il
‑
12p40和il
‑
6的产生极大和显著增加(图27j
‑
27l)。这些结果组合表明负载有682的edv能够使巨噬细胞朝向m1抗肿瘤表型极化,以及促进树突状细胞成熟,而单独682不能引起类似的响应。
[0562]
实施例17:超级细胞毒素pnu
‑
159682的有效递送产生了杀肿瘤的cd11b
+
先天免疫细胞
[0563]
由于682是对于甚至抗药性癌细胞的ic50也在pm范围内的超级细胞毒素(quintieri等,2005),其因这种化合物相关的严重全身毒性而不能在临床上使用(staudacher和brown,2017)。但是,当被包封在edv中时,超级细胞毒素(例如682)可以被有效地递送到肿瘤,而副作用很少,如在ep
‑
edv
‑
682治疗的小鼠中最低限度的减重(<5%)、很少甚至没有皮毛起伏(fur ruffling)、和没有嗜睡或驼背姿势表现所证,ep
‑
edv
‑
682治疗的小鼠关联在前的涉及用携载多种治疗有效载荷的edv治疗的小鼠的研究(macdiarmid等,2009;macdiarmid等,2007a;macdiarmid等,2007b;sagnella等,2008)(图35a
‑
35d)。
[0564]
在此,在ep
‑
edv
‑
682治疗的携载乳腺脂肪垫中的4t1肿瘤或皮下ct26ep12.1肿瘤的balb/c小鼠中观察到显著的肿瘤消退(图28a
‑
28b)。ep
‑
edv
‑
682还能够促进携载t84人异种移植物和抗药性a549/mdr异种移植物的无胸腺balb/c裸鼠的显著肿瘤减少,以及引起大(~600mm3)a549/mdr肿瘤的显著肿瘤减少(图28c
‑
28d)。
[0565]
虽然已经充分确立了edv可以有效向肿瘤递送化疗剂,但最初的体外实验表明edv还可以以多种方式充当免疫治疗剂,包括但不限于驱动m1巨噬细胞极化。为了确立这些结果是否扩展到体内对肿瘤微环境内先天免疫系统的刺激,从经盐水、ep
‑
edv或ep
‑
edv
‑
682处理并与其相应的肿瘤细胞在xcelligence实时细胞分析仪(rtca)中以5:1(效应体:靶标)的比例离体共培养的4t1或ct26ep12.1小鼠肿瘤中分离cd11b
+
免疫细胞(图28e
‑
28g)。与粘附4t1细胞共培养的cd11b
+
细胞显示初始粘附和沉降阶段导致细胞指数增加,然后活性阶
段——其中细胞指数在cd11b
+
细胞有效杀死粘附肿瘤细胞的情况下稳定减少,或在肿瘤细胞未被有效溶解并继续生长的情况下增加。
[0566]
如示,分离自已被ep
‑
edv
‑
682治疗的小鼠肿瘤的cd11b
+
细胞高度有效杀死4t1,而分离自ep
‑
edv或盐水治疗的肿瘤的cd11b
+
细胞未杀死4t1肿瘤细胞(图28e)。巨噬细胞在4t1肿瘤(cd45
+
cd11b
+
ly6g ly6c
+
)内的分型显示m1/m2比例增加,如表达cd86:cd206的巨噬细胞的比例的增加所证(图28f)。此外,来自ep
‑
edv
‑
682 4t1肿瘤负载小鼠的cd11b
+
在与4t1细胞离体共培养时呈现,相对于盐水和ep
‑
edv处理的小鼠,巨噬细胞炎性蛋白1α(ccl3/mip
‑
1α)的产生2倍增加(图36i)。mip
‑
1α的局部化产生已经被认为是负责吸引免疫效应细胞浸润到肿瘤微环境中并增强效应细胞介导的抗肿瘤免疫响应的主要因素(allen等,2018;zibert等,2004)。
[0567]
与粘附ct26ep12.1细胞共培养的cd11b
+
细胞类似地呈现从用ep
‑
edv
‑
682治疗的小鼠的肿瘤分离的cd11b
+
细胞的细胞溶解活性增强(图28g)。总体而言,来自ct26ep12.1肿瘤的cd11b
+
细胞比来自4t1肿瘤的那些更具活性,如xcelligence rtca上所有三个治疗组的细胞指数稳定下降所示。然而,在ep
‑
edv
‑
682处理组中细胞溶解更明显,并且在cd11b
+
细胞添加后1小时内开始,在cd11b
+
细胞添加后10h下降至42%活力。相比之下,在ep
‑
edv处理的样品中细胞溶解开始用了近5小时,并且盐水处理组中用了7小时,并且在cd11b
+
细胞添加后10h的活力分别为76%和86%。从ct26ep12.1肿瘤中分离的cd11b
+
细胞的流式细胞术分析显示,在ep
‑
edv
‑
682治疗的肿瘤中,cd86:cd206比例(m1/m2比例)显著增加(图28h)。
[0568]
与具有免疫功能的小鼠品系一样,在无胸腺裸鼠的抗药性人肺癌异种移植物(a549/mdr)中也观察到巨噬细胞极化的转变,其中观察到在egfr
‑
edv
‑
682治疗的小鼠脾脏中的m1/m2比与盐水治疗的小鼠或用无效减少肿瘤生长的edv治疗的小鼠相比增加了~2
‑
3倍(图35e)。另外,在经egfr
‑
edv
‑
682治疗的t84肿瘤中也检测到m1/m2比的小幅而显著的增加(图35f)。与这两种具有免疫功能的小鼠癌症模型相似,从经egfr
‑
edv
‑
682处理的带有a549/mdr肿瘤的裸鼠肿瘤分离的cd11b
+
细胞呈现更优的肿瘤细胞溶解,其中在cd11b
+
细胞添加至a549/mdr肿瘤细胞后6.5h与用盐水处理的那些相比有28%细胞溶解,如通过xcelligence rtca确定(图35g和35h)。
[0569]
实施例18:nk细胞在ep
‑
edv
‑
682处理后在体内采取抗肿瘤表型
[0570]
此实施例的目的是确定包含抗肿瘤剂的细菌微细胞对nk细胞功能的影响。
[0571]
为了探究携载682的edv对nk细胞功能的影响,在用ep
‑
edv
‑
682、ep
‑
edv或盐水进行2周治疗后,从携载4t1或ct26ep12.1肿瘤的edv治疗的balb/c小鼠和对照balb/c小鼠的脾脏分离nk细胞。在xcelligence rtca中将脾脏nk细胞与其相应的肿瘤细胞以20:1的e:t比共培养,并经3
‑
4天时间分析肿瘤细胞死亡(图29a
‑
29d)。
[0572]
两种肿瘤模型中的ep
‑
edv
‑
682治疗的小鼠的nk细胞均通过对靶标肿瘤细胞的显著而有效的细胞溶解而被证实抗肿瘤特性,而用盐水或ep
‑
edv治疗的那些显示对其靶标肿瘤细胞极少甚至没有细胞溶解潜力。来自携载ct26ep12.1的小鼠的nk细胞显示出对靶标ct26ep12.1细胞的快速细胞溶解,在共培养的最初几个小时内下降至近60%靶标细胞活力,并在50小时后维持仅18%靶标细胞活力。来自ep
‑
edv治疗的ct26ep12.1的nk细胞具有低水平的溶细胞能力,在50h后具有70
‑
80%的靶标细胞活力,而来自盐水治疗的小鼠的nk细胞在相同时期内显示~82%的靶标细胞活力(图29d)。来自ep
‑
edv
‑
682治疗的携载4t1肿
瘤的小鼠的nk细胞虽然仍具有高度活性,但在70小时后显示出下降至~36%靶标细胞活力的更渐进的细胞溶解特性,而来自盐水或ep
‑
edv治疗的小鼠的nk细胞保持≥90%的靶标细胞活力(图29a和29b)。此外,从ep
‑
edv
‑
682治疗的小鼠分离的nk细胞与nk/4t1共培养呈现,与来自盐水治疗的小鼠的那些相比,tnfα和rantes的产生均增加超过2倍(图29f和29g)。
[0573]
从携载a549/mdr或t84肿瘤的无胸腺小鼠的脾脏分离的nk细胞显示出与具有免疫能力的小鼠肿瘤模型相似的细胞溶解特征(图36a和36c)。egfr
‑
edv
‑
682治疗的小鼠的nk细胞,在与其靶标肿瘤细胞共培养时,导致a549/mdr和t84肿瘤中的靶标细胞活力均低于40%,而两者的盐水对照均保持靶标细胞活力≥70%。在t84/nk细胞共培养物中粒酶b产生的考察表明,在包含来自ep
‑
edv
‑
682治疗的小鼠的nk细胞的共培养物中粒酶b的水平显著较高(图36b)。
[0574]
通过流式细胞术对4t1肿瘤内的nk细胞的考察表明,ep
‑
edv
‑
682治疗的肿瘤中的nk细胞(cd45+、cd11b+、dx5+)具有显著增加的nkg2d表达,其已知是肿瘤免疫监视中重要的nk激活受体(图29e)。肿瘤细胞表面上nkg2d配体的上调足以抵消nk抑制信号,从而能够实现肿瘤细胞细胞溶解(morvan和lanier,2016)。针对nkg2d结合nk配体rae
‑
1、h60a和mult
‑
1的小鼠肿瘤细胞系(包括ct26ep12.1和4t1)筛选显示,4t1和ct26ep12.1均具有在4个细胞系中最高水平的h60a表达,并且这是在这两个细胞系中总体表达最高的配体。此外,小鼠乳腺癌细胞系4t1和emt
‑
6细胞在筛选的所有细胞系中均显示最高的rae
‑
1表达,但水平远低于h60a,而ct26ep12.1仅具有非常低水平的该配体。最后,除4t1细胞外,所有其他细胞系均未显示mult
‑
1的表达(图29h)。
[0575]
为了进一步考察这些配体和nkg2d受体在分离的nk细胞的细胞溶解活性中的作用,从ep
‑
edv
‑
682治疗的携载4t1肿瘤的小鼠的脾脏中分离了nk细胞。在xcelligence rtca系统中与4t1细胞共培养之前,将nk细胞与被设计以阻断nkg2d受体与其特定配体结合的抗体一起培育(图29i)。rae
‑
1抗体导致4t1细胞的nk细胞溶解的~13%抑制,而h60a抗体导致~21%抑制,并且两种抗体的组合抑制了nk细胞的~25%的细胞溶解能力(图29j)。
[0576]
实施例19:肿瘤微环境内的主要th1细胞因子响应在ep
‑
edv
‑
682治疗后呈现
[0577]
此实施例的目的是探究edv治疗后肿瘤微环境内的细胞因子环境。
[0578]
4t1和ct26ep12.1肿瘤在最终治疗后24小时被收获,并以非酶促方式轻轻地解离,确保无细胞溶解,从而可以评估间质性肿瘤细胞因子水平(图30a
‑
30b)。由于肿瘤尺寸的显著差异,对肿瘤进行称重和测量,并计算每克组织的细胞因子水平,以解决这些尺寸差异。两种肿瘤均响应ep
‑
edv
‑
682治疗呈现肿瘤微环境内tnfα显著增加,尽管在ct26ep12.1肿瘤中这种增加更为明显,其中在ep
‑
edv
‑
682治疗后与盐水治疗相比有>10倍增加。类似地,ep
‑
edv
‑
682治疗导致间质ifnα浓度在两种肿瘤中均显著增加,其中在4t1肿瘤中ifnα水平~2倍增加,并且ct26ep12.1肿瘤中15倍增加。
[0579]
ep
‑
edv
‑
682治疗的细胞因子水平最显著的变化出现在ct26ep12.1肿瘤中,其中出现ifnγ水平500倍增加,而在4t1肿瘤中出现ifnγ水平小幅而显著的2倍增加。ep
‑
edv
‑
682治疗导致的il
‑
lβ水平在4t1肿瘤中显著降低,但在ct26ep12.1肿瘤中显示升高,尽管不显著。在4t1肿瘤中出现il
‑
2(~4倍)和il
‑
4(~3倍)显著增加,而ep
‑
edv
‑
682治疗后il
‑
6水平无显著变化。在ep
‑
edv
‑
682治疗的小鼠的ct26ep12.1肿瘤中,观察到il
‑
2水平显著增加超过150倍,而il
‑
6水平显著增加3倍,il
‑
4水平非显著增加2倍。还考察了mip
‑
1α(ccl3)和
rantes(ccl5),其已被证明是免疫细胞如抗原呈递细胞、nk细胞和t细胞浸润的主要决定因素(allen等,2018)。在ep
‑
edv
‑
682治疗的肿瘤中,4t1和ct26ep12.1均呈现这两种趋化因子的水平升高,其中用ep
‑
edv
‑
682治疗的ct26ep12.1肿瘤中的mip
‑
1α水平和4t1肿瘤中的rantes水平升高~3倍。此外,在ct
‑
26ep12.1和4t1 ep
‑
edv分别治疗肿瘤中均出现rantes和mip
‑
1α水平增加2
‑
2.5倍,尽管这并不显著。总体上,ep
‑
edv治疗导致间质肿瘤细胞因子和趋化因子水平与盐水治疗组相似。
[0580]
除了考察间质肿瘤细胞因子外,还评估了来自被治疗动物的脾细胞的细胞因子产生(tnfα、ifnγ、il
‑
1β、il
‑
2和il
‑
10)。脾细胞被单独培养或与来自其相应小鼠的分散(dispersed)肿瘤细胞共培养。用盐水、ep
‑
edv和ep
‑
edv
‑
682进行的全身治疗对来自4t1或ct26ep12.1肿瘤模型的脾细胞的细胞因子产生没有显著影响(图30c
‑
30g)。然而,当脾细胞被与其相应的被治疗肿瘤一起培养时,情况就不再如此。与仅脾细胞以及来自两种肿瘤模型的盐水和ep
‑
edv治疗的小鼠的共培养物相比,来自ep
‑
edv
‑
682治疗的小鼠的共培养物中的tnfα产生显著增加(图30c)。在4t1模型中,与盐水治疗的小鼠相比,来自ep
‑
edv
‑
682治疗的小鼠的共培养物的il
‑
2产生显著增加(图30d)。此外,当将来自盐水处理的小鼠的脾细胞与其相应的肿瘤细胞共培养时,il
‑
2的产生显著减少。与从携载ct26ep12.1肿瘤的小鼠分离出的单独脾细胞相比,所有共培养物中的il
‑
2产生均显著增加。il
‑
1β产生的唯一显著变化发生在来自4t1模型的共培养物中,其中与ep
‑
edv
‑
682治疗的小鼠相比,来自盐水治疗的小鼠的样品呈现显著增加,相应于体内观察到的差异(图30e)。单独脾细胞与从携载4t1肿瘤的盐水治疗小鼠分离的共培养物之间的ifnγ产生显著降低,并且在ep
‑
edv
‑
682治疗的脾细胞/肿瘤细胞共培养物中显著高于来自盐水或ep
‑
edv治疗的携载4t1肿瘤携载小鼠的共培养物的那些(图30f)。在ct26ep12.1肿瘤模型中,与盐水和ep
‑
edv共培养物相比,在ep
‑
edv
‑
682治疗的脾细胞/肿瘤细胞共培养物中ifnγ的产生减少,但是这仅对ep
‑
edv有意义。最后,在来自ct26ep12.1治疗小鼠的盐水处理的脾细胞/肿瘤细胞共培养物中,与单独脾细胞和edv治疗组相比,il
‑
10产生显著增加(图30g)。
[0581]
实施例20:ep
‑
edv
‑
682治疗导致肿瘤特异性cd8+t细胞产生
[0582]
最初的体外实验表明,edv处理可通过直接相互作用或因响应负载有有效化疗剂的靶向型edv的细胞死亡而导致树突状细胞成熟。因此,该实验旨在考察该结果是否可以平移到体内dc成熟和抗原呈递,导致产生肿瘤特异性cd8
+
细胞毒性t细胞。
[0583]
2周治疗后,从已用盐水、ep
‑
edv或ep
‑
edv
‑
682治疗的4t1或ct26ep12.1肿瘤携载小鼠取出脾脏,并分离cd8
+
t细胞。然后将cd8
+
t细胞添加到相应的肿瘤细胞,并利用xcelligence rtca考察其特异性识别和杀死那些细胞的能力(图31a和31c)。从携载4t1的小鼠分离并经盐水或ep
‑
edv处理的cd8
+
t细胞对4t1细胞不呈现细胞毒性,而从用ep
‑
edv
‑
682治疗的小鼠分离的cd8
+
t细胞在30小时后诱导靶标细胞的50%细胞溶解(图31b)。
[0584]
从携载ct26ep12.1的小鼠中分离并用ep
‑
edv
‑
682处理的cd8
+
t细胞在杀死靶标ct26ep12.1细胞方面高度有效,其中在将效应细胞添加到靶标细胞后20小时观察到81%的细胞毒性(图31d)。值得注意,甚至ep
‑
edv处理也能够引起ct26ep12.1模型中肿瘤特异性cd8
+
t细胞的产生,其中在20h后出现40%细胞毒性,而来自盐水治疗小鼠的cd8
+
t细胞对ct26ep12.1细胞不显示特异性。4t1肿瘤内cd8
+
t细胞的流量分析显示,经ep
‑
edv
‑
682治疗的小鼠的肿瘤(cd45
+
、cd3
+
、cd8
+
)内的cd8
+
t细胞的百分比小幅而显著增加(图31e)。另外,
在ep
‑
edv
‑
682治疗的小鼠的肿瘤内,观察到调节性t细胞(cd45
+
、cd3
+
、cd4
+
、cd25
+
)的百分比显著降低2倍(图31f)。还通过流动考察了4t1肿瘤携载小鼠的肿瘤引流淋巴结的t细胞数量。与盐水和ep
‑
edv治疗的小鼠相比,ep
‑
edv
‑
682治疗的小鼠的淋巴结中观察到总t细胞数(cd3
+
)显著增加以及cd4
+
和cd8
+
t细胞数均显著增加。edv处理的小鼠(图31g)。还检测到经ep
‑
edv
‑
682治疗的携载4t1肿瘤的小鼠的淋巴结中成熟树突状细胞的显著增加(图31h)。来自ep
‑
edv
‑
682治疗的小鼠的分离的cd8
+
t细胞与4t1细胞之间的相互作用的可视化显示,这些t细胞能够附接至肿瘤细胞并将穿孔素(绿色)排入肿瘤细胞(图31i)。
[0585]
实施例21:iv期胰腺导管腺癌案例中患者对egfr
‑
edv
‑
682的响应
[0586]
此实验涉及在患有iv期胰导管腺癌(pdac)的患者(ceb)中egfr
‑
edv
‑
682治疗的同情案例使用的临床观察。
[0587]
ceb的诊断评价包括腹部的计算机轴向断层扫描(ct)(2017年5月),其揭示了多个低密度肝病灶。肿瘤并不热衷于(适于,avid on)正电子发射断层扫描(pet)。标准生物化学和血液学测试总体上是平常的。还评估了血清ca19
‑
9和c反应蛋白(crp)。低血清水平的ca 19
‑
9(在一些胃肠道恶性肿瘤(尤其是胰腺癌)中表达的碳水化合物抗原)已被证明是总体生存率和治疗响应的预后指标。同样,crp水平升高也已被证明与不良的临床结果显著相关(szkandera等,2014)。甚至在吉西他滨和folfirinox后,ceb也呈现ca19
‑
9水平>l20,000ku/l,比正常水平高3,000倍,以及显著升高的crp水平64mg/l。
[0588]
考察从胰头部和尾部切除的肿瘤组织获得的pdac细胞的药物敏感性。头部和尾部pdac细胞均呈现低敏感性,对一线和二线药物有部分或没有响应(图37a)。相比之下,两者均显示对682的极端敏感性,其中ic50在pm范围内(图37a)。此外,通过流式细胞术发现表皮生长因子受体(egfr)以每细胞>200,000个拷贝过表达(图37),因此是靶向负载有682的edv进行治疗的理想受体。
[0589]
ceb很好地耐受携载682且靶向egfr的edv(e
‑
edv
‑
d682),并贯穿周期报告了健康状况(well
‑
being)显著提高。在此期间,她的ecog表现状态从2降至0。用药后3小时有tnfα和il
‑
6短暂升高,其在第1个剂量之后最高并且在后续剂量中显著较低,这可能表明耐受性增强。贯穿周期,血液学参数没有变化并且白细胞计数(wcc)保持正常。甚至在14个剂量后,生化结果也平常。值得注意的是在第13个剂量时,ca19
‑
9标记物从>120,000ku/l稳定下降至5,310ku/ml,并且crp水平从64mg/l下降至7mg/l(图32a和32b)。
[0590]
在第12个剂量后,来自外周血单核细胞(pbmc)的主要免疫细胞亚群的免疫分型揭示了可以支持有利的抗肿瘤响应的多种细胞类型内的变化(门控策略
‑
图38)。在与筛选剂量(d1)相比时,总体cd14
+
单核细胞,巨噬细胞和树突状细胞的前体,在d12时从15.28%增加到31.39%(增加105%)(图32c),包括中间体(cd14+cd16++)单核细胞亚群(增加69%;图32d)。中间单核细胞显示了最高的将抗原呈递给t细胞的能力,并具有优越的il
‑
12和ifn
‑
γ的抗原特异性诱导能力(ziegler
‑
heitbrock和hofer,2013)。
[0591]
还观察到髓样树突状细胞(mdc)增加28%和浆细胞样dc(pdc)减少60%(图32f)。负责驱动cd8
+
效应t响应的clec9a+髓样树突状细胞(mdc)也增加(在d12为98%)(图32e)。该增加与d12时的总细胞毒性cd8
+
t细胞(60%)的增加一致,包括效应cd8
+
t细胞(增加50%)(图32g)。效应cd8
+
t细胞库是最近与单核细胞、巨噬细胞或树突状细胞呈递的抗原相互作用的cd8
+
t细胞,并含有肿瘤和/或edv抗原特异性t细胞。在d12时,表达衰竭的程序性死亡
‑
1(pd
‑1+
)表型的细胞毒性cd8
+
t细胞(指示延长的细胞激活和表达pd
‑
l1的肿瘤细胞的pd
‑
1连接的易感性)与d1相比降低了17%。该过程通常发生在肿瘤和肿瘤引流淋巴结内。在本案例研究中观察到的结果与在临床前小鼠模型中观察到那些有相似趋势。
[0592]
讨论:此数据表明,负载有超细胞毒素682的靶向型edv不仅能够有效地将该药物递送至肿瘤位点,而且还可以通过刺激多个免疫细胞亚群而充当免疫治疗剂。已经证明了ep
‑
edv
‑
682治疗将包括巨噬细胞、nk细胞和cd8
+
t细胞的免疫细胞亚群推向能够有效消除肿瘤细胞的抗肿瘤表型的能力。当与药物的有效性相结合时,这会导致对肿瘤的双重攻击。
[0593]
在静脉内给予后,edv通过其渗漏的脉管系统外渗到肿瘤中,其中携载其毒性有效载荷的靶向型edv的给予剂量的≥30%在2hr时间内直接沉积到肿瘤微环境中(macdiarmid等,2007b)。epcam靶向型edv与肿瘤细胞(在此例中为4t1和ct26ep12.1)表面表达的epcam结合,然后被内化,有效地将其有效载荷(682)直接递送到肿瘤细胞内。682是高度有效的超级细胞毒素,在递送至肿瘤细胞的24h内导致快速凋亡(图33a)。然后,通过ep
‑
edv
‑
682治疗产生的凋亡细胞和damp信号可以与先天性免疫细胞(如肿瘤相关的巨噬细胞(tam)相互作用,并刺激cd86的上调和th1促炎性细胞因子(如tnfα和il
‑
6)的产生(图33b)。
[0594]
此外,edv本身也可以直接与tam相互作用,产生相似的mi极化的,尽管预期这在当前系统中将以非常低的水平发生。在此,已经证明了ep
‑
edv
‑
682治疗在4种不同的肿瘤模型中的肿瘤微环境内转移m1:m2平衡的能力。尽管在不同的肿瘤模型中这种转移的程度不同,但是显示m1极化的增加转化为由从已被ep
‑
edv
‑
682治疗的小鼠的肿瘤分离的tam造成的肿瘤细胞溶解的增加。除了表型向m1转移之外,来自ep
‑
edv
‑
682治疗的小鼠的肿瘤的tam还分泌了增加量的mip
‑
1α(图33c),一种已被确定在促进免疫细胞募集并且尤其是nk细胞、cd4
+
t细胞和cd8
+
t细胞的肿瘤浸润中起作用的趋化因子(allen等,2018)。
[0595]
edv治疗允许实现在体内响应于垂死肿瘤细胞的肿瘤微环境内的dc形成(priming)和成熟(图33d)。在成熟过程中,dc迁移至肿瘤引流淋巴结以抗原呈递给t细胞,从而增加cd4
+
t辅助细胞和肿瘤特异性cd8
+
ctl的产生,启动对肿瘤的适应性免疫响应(图33e)。
[0596]
与增强巨噬细胞和dc抗肿瘤功能结合,edv治疗能够引起nk细胞激活,导致细胞毒性增加(图33f)。在用ep
‑
edv
‑
682治疗的小鼠肿瘤内的nk细胞上观察到nkg2d受体上调,并且从受体被证明显著促进从ep
‑
edv
‑
682治疗的小鼠分离的nk细胞的细胞溶解能力。此外,未成熟的、中间的和成熟的小鼠nk细胞表达可与趋化因子miplα和rantes结合的ccr1和ccr5趋化因子受体,二者均在ep
‑
edv
‑
682治疗的肿瘤中以及通过ep
‑
edv
‑
682治疗的小鼠的巨噬细胞和nk细胞被上调(bernardini等,2016)。
[0597]
趋化因子,如mip1α和rantes,负责将辅助和效应免疫细胞(包括nk细胞、巨噬细胞和t细胞)进一步募集到肿瘤微环境(图33g)(allen等,2018;bernardini等,2016;zibert等,2004)。在包括巨噬细胞、nk细胞和dc在内的edv治疗导致的初始先天免疫响应之后,产生适应性免疫响应,其中肿瘤特异性ctl和t辅助细胞产生,然后被募集到肿瘤位点(图33h)。然后,肿瘤特异性ctl靶向并溶解肿瘤细胞,进一步促进已被其他免疫细胞亚群与靶向型药物负载型edv组合产生主要抗肿瘤环境。药物负载型edv治疗可引起主要th1的响应,如肿瘤微环境中th1细胞因子(tnfα、ifnα、ifnγ、il
‑
2和il
‑
6)的增加所证明。如前所述,先天免疫细胞亚群在被激活时成为这些具体细胞因子中一种或多种的主要来源。t细胞同样
能够产生所有上述细胞因子(belardelli和ferrantini,2002;lee和margolin,2011)。先天免疫细胞或t细胞释放这些细胞因子造成其他免疫细胞的共同刺激、激活、生长和增加的抗原呈递,形成反馈环路,其进一步增强免疫系统的抗肿瘤活性(图33i)(lee和margolin,2011)。
[0598]
方法与材料
[0599]
engeneic dream vector(edv):edv是从血清伤寒肠炎沙门氏菌(salmonella enterica serovar typhimurium)(s.typhimurium)mincde
‑
菌株制备和纯化的,如前所述(macdiarmid等,2007b)。先前已经描述了药物负载、抗体靶向、冻干和剂量制备(macdiarmid等,2007b;sagnella等,2018)。edv制剂经过严格的质量控制,其中使用zetasizer nano系列和nanosight lm20(malvern仪器)利用动态光散射评估edv尺寸和数量。使用endosafe便携式测试系统(charles river)评估内毒素水平。从edv
tm
制剂提取药物并通过hplc进行定量,如前所述(macdiarmid等,2007b)。
[0600]
流式细胞术:所有流式细胞术均在beckman coulter gallios 6c上进行,并使用kaluza软件(beckman coulter)进行分析。
[0601]
细胞培养:使raw264.7细胞(atcc)在含有10%fcs的dulbecco改良eagle培养基(dmem)(sigma)中生长,并使用细胞刮刀进行传代。使小鼠肿瘤细胞系(4t1和ct26)在包含10%fcs的rpmi
‑
1640培养基(sigma)中单层生长,并使用磷酸盐缓冲盐水(pbs)/胰蛋白酶edta每周2
‑
3次传代。将所有细胞保持在37℃、在含有5%co2的潮湿气氛中的培养中,并常规地进行筛选,并发现不含支原体。使用quantum simply cellular抗大鼠igg微球(bangs laboratory),使用apc抗小鼠cd326(biolegend)利用流式细胞术,对小鼠细胞系中的epcam表达和受体数量进行定量。由于显示ct26是epcam阴性的,使用lipofectamine 2000(thermo fisher)用含有小鼠epcam orf克隆(nm_008532.2)(genescript)的pcdna3.1+c dyk转染细胞。利用g418选择获得表达epcam的ct26克隆的纯种群,并如上所述关于epcam表达筛选细胞。考察克隆的生长速率、药物敏感性和体内致瘤性,并选择具有高epcam表达且上述3个参数与亲本ct26细胞系相似的一种用于所有后续研究(ct26ep12.1)。
[0602]
骨髓源dc(bmdc):从balb/c小鼠的股骨和胫骨分离骨髓。在红细胞溶解和洗涤后,将细胞重悬于aimv+5%fbs+2
‑
巯基乙醇+青霉素/链霉素+20ng/ml gm
‑
csf(miltenyi biotec)中,并使其生长7天。
[0603]
用edv处理raw264.7细胞:将raw264.7细胞以每孔3x105个细胞接种在6孔板中,并培育过夜。然后,将培养基换成含有以下中的一种的新制培养基:1pg/ml lps(sigma);100pmol pnu
‑
159682(national levena);ep
‑
edv
‑
682(500:1和1000:1edv:细胞);ep
‑
edv(5000:1edv:细胞);或不予处理。在处理后6小时和24小时利用细胞刮刀收获细胞,并将样品用dapi(sigma)、抗cd45 brilliant violet 510(biolegend)、抗cd86apc
‑
cy7(biolegend)和抗cd206 af488(r&d systems)染色并通过流式细胞术来评估。
[0604]
巨噬细胞和dc/肿瘤细胞共培养:用versene(gibco)收获ct26ep12.1和4t1细胞,并将细胞收集在1ml eppendorf管中。将细胞重悬于补充有10%fbs(bovogen)的1ml dmem(sigma)中,其中包含:ep
‑
edv(1000:1和5000:1
‑
edv:细胞);ep
‑
edv
‑
682(500:1和1000:1
‑
edv:细胞);ep
‑
edv
‑
dox(10,000:1
–
edv:细胞)、100pmol pnu
‑
159682、5mm多柔比星或单独培养基。通过mts和xcelligence实时实验确定药物和edv量,以使选定的浓度导致治疗后24
小时内细胞死亡开始。然后用pbs彻底清洗细胞,以去除任何非粘附的edv或过量的药物。将处理后的肿瘤细胞与raw264.7或bmdc以1:1的肿瘤细胞:raw264.7/bmdc/jaws ii比例培养过夜。收集上清液用于elisa分析。使用细胞刮刀收集raw264.7/肿瘤细胞共培养物,并将样品用dapi(sigma)、抗cd45 brilliant violet 510(biolegend)、抗cd86 apc
‑
cy7和抗cd206 af488染色,并通过流式细胞术评估。将jaws ii/肿瘤细胞和bmdc/肿瘤细胞共培养物用versene收集,并用dapi(sigma)、cd11b af488(abeam)、cd11c pe(molecular probes)、抗cd45 brilliant violet 510或pecy5(biolegend)、抗cd86 apc
‑
cy7、mhc ii类pecy5(thermo fisher)、mhc ii类brilliant violet 421(biolegend)、7
‑
aad(biolegend)和/或cd80 pe(thermo fisher)染色,并通过流式细胞术评估。使用rnaeasy plus mini试剂盒(qiagen),按照制造商的规程,从bmdc/肿瘤细胞共培养物提取rna。简而言之,将细胞在rlt缓冲剂中溶解并均质化,并通过gdna消除剂旋转柱。将70%乙醇添加到流通液(flow through)中,然后将样品通过rneasy旋转柱,洗涤并在无rnase的水中洗脱。在eppendorf生物光度计+(eppendorf biophotometer plus)上测定rna浓度。使用superscript
tm
vilo
tm
cdna合成试剂盒(thermo fisher),按照制造商的规程,将rna用于逆转录cdna。将转录的cdna以1:2稀释用于qpcr。各qpcr反应均包含5ul taqman快速高级母液(thermo fisher)、0.5ul 20x taqman引物/探针混合物(ifnαmm03030145_gh、ifnb1mm00439552_s1、gapdh mm99999915_g1;thermo fisher)和2.5ul水。将8μl的混合物加上2μl的cdna加入96孔板中。使用applied biosystems实时pcr系统进行qpcr。将数据导出到excel,并根据aact计算相对定量。
[0605]
体内肿瘤模型:所有动物工作均按照aec 1/2016、aec 14/2016、aec 15/2011和aec11/2017的engeneic动物伦理准则进行。对于4tl和ct26ep12.1模型,雌性balb/c小鼠从6
‑
8周龄的动物资源中心(animal resources centre)获得。对于t84和a549/mdr模型,5
‑
7周龄的balb/c fox1
nu
/arc从动物资源中心获得。在观察至少1周后,对小鼠在右手侧第3个乳腺脂肪垫中注射5x10
4 4t1细胞/50μl pbs,或在balb/c小鼠的右胁腹皮下注射2x10
5 ct26ep12.1/100μl pbs。对于人类异种移植,将5x10
6 a549/mdr或1x10
7 t84/100μl pbs/基质胶(sigma)皮下注射到右胁腹。4t1模型在肿瘤诱导后第7天开始治疗,此时平均肿瘤尺寸为~90mm3;ct26ep12.1模型在第9天开始治疗,此时平均肿瘤尺寸为~125mm3。通过2周每周3次静脉内尾静脉注射,以以下治疗中的一种治疗小鼠:盐水、l x 109靶向epcam的edv(ep
‑
edv)或1x109靶向epcam的负载有pnu
‑
159682的edv(ep
‑
edv
‑
682)。测量肿瘤3次/周,并将肿瘤体积计算为π/6(长度x宽度x高度)。在2周的时间结束时,对小鼠进行人道安乐死,并收集肿瘤和脾脏进行离体分析。当肿瘤分别达到100
‑
120mm3和120
‑
150mm3时,开始a549/mdr和t84肿瘤的治疗。用盐水、1x109靶向egfr的负载有多柔比星的edv(egfr
‑
edv
‑
dox)、1x109靶向egfr的负载有pnu
‑
159682的edv(egfr
‑
edv
‑
682)或1x109非靶向的负载有pnu
‑
159682的edv(edv
‑
682)。
[0606]
从4t1和ct26ep12.1肿瘤分离cd11b
+
细胞:利用tissue dissociation试剂盒(miltenyi biotec)在37℃下按照制造商的说明,使用gentlemacs
tm
解离器,将肿瘤解剖,称重并酶促消化。解离后,使用rbc溶解缓冲剂(sigma)去除红细胞。洗涤后,使细胞通过70μm细胞过滤器以除去任何团块。通过使用macs分离器(miltenyi biotec)上的ls柱上的cd11b macs珠(miltenyi biotec),通过阳性选择,来纯化cd11b
+
细胞。通过流式细胞术利用apc抗
小鼠cd11b(biolegend)评估分离的cd11b
+
细胞群体的纯度,并显示其为~80%纯(图39a)。
[0607]
从脾脏中分离nk和cd8:利用dounce均质器均质化脾脏,并通过70μm筛网过滤器过滤以获得单细胞悬浮液,然后使用rbc溶解缓冲剂进行红细胞溶解。然后洗涤脾细胞并进行细胞计数,然后再进行nk或cd8
+
t细胞分离。使用nk细胞分离ii试剂盒(miltenyi biotec)或cd8a
+
t细胞分离试剂盒(miltenyi biotec),按照制造商的说明,通过阴性选择从解离的脾细胞分离nk细胞和cd8
+
t细胞。通过利用macs分离器(miltenyi biotec)的ls柱分离细胞。通过流式细胞术评估nk细胞和cd8
+
t细胞制剂,并且nk细胞纯度始终高于90%(图39b),而cd8
+
t细胞纯度始终高于86%(图39c)。在nk细胞介导的细胞溶解测定之前,将nk细胞在补充有10%fbs的rpmi
‑
1640培养基中于37℃放置过夜。分离后立即将cd8
+
t细胞添加到肿瘤细胞中以评估cd8
+
t细胞的细胞溶解。
[0608]
xcelligence监测的肿瘤细胞cd11b
+
、cd8
+
和nk细胞的细胞溶解:通过xcelligence dp系统实时监测细胞生长特征和肿瘤细胞死亡。为此,覆盖组织培养孔底部的圆形电极检测电阻抗的变化,并将阻抗值转换为细胞指数(ci)。细胞指数测量结果直接对应于细胞粘附强度和细胞数量。将靶标细胞(4t1、ct26ep12.1、a549/mdr或t84)接种到e
‑
plate 16中。使细胞附接并增殖直至其已达到其对数生长期。将效应细胞(cd11b
+
细胞、nk细胞或cd8
+
t细胞)以以下效应细胞
‑
靶标细胞比添加至靶标细胞:5:1(cd11b
+
:肿瘤细胞)、20:1(nk)细胞:小鼠肿瘤细胞)、10:1(nk:人肿瘤细胞)和30:1(cd8
+
t细胞:肿瘤细胞)。在添加效应细胞后,系统进行定期测量(每5或15分钟),3
‑
4天,以监测免疫细胞介导的肿瘤细胞杀伤。
[0609]
nk细胞介导的细胞溶解抑制:首先通过流式细胞术,使用抗rae
‑
1α/β/γ
‑
pe(miltenyi biotec)、抗h60a
‑
pe(miltenyi biotec)和抗mult
‑
1pe(r&d systems),筛选nk细胞配体表达的小鼠肿瘤细胞系。对于基于这些配体表达水平的nk细胞介导的细胞溶解抑制,将效应nk细胞在3μg/ml以下nk细胞配体的阻断mab的存在下添加至靶标细胞:抗rae
‑
1αβγ(r&d systems)或抗h60(r&d systems),分开或混合。xcelligence数据在excel中被转换,并被导出到prism(graphpad software)进行图形和统计分析。
[0610]
肿瘤/脾脏流式细胞术:将肿瘤和脾脏如上所述解离。在红血细胞溶解后,将细胞与fc阻断剂(block)1:10在macs缓冲剂(miltenyi biotec)中培育10分钟。在10分钟培育后,将细胞洗涤一次,并与一级抗体组在黑暗中在冰上在macs缓冲剂中培育15分钟。将细胞洗涤2次,然后重悬浮在macs缓冲剂中用于流式细胞术分析。以下抗体用于t细胞、nk细胞和巨噬细胞染色组:抗cd45 pecy7(biolegend)、抗cd45 bv510(biolegend)、抗cd3e apc
‑
efluor780(ebioscience)、抗cd3 apc(molecular probes)、抗cd4 pe
‑
tr(abeam)、抗cd8a fitc(ebioscience)、抗cd8 bv510(biolegend)、抗cd25 pe(abeam)、抗cd314(nkg2d)pe
‑
efluor610(ebioscience)、抗cd335(nkp46)pecy7(biolegend)、抗cd27 bv421(biolegend)、抗cd183(cxcr3)bv510(biolegend)、抗nkg2a/c/e fitc(ebioscience)、抗cd11b apc(bd pharmingen)、抗ly6c fitc(biolegend)、抗ly6g bv510(biolegend)、抗f4/80pe dazzle594(biolegend)、抗cd206 pecy7(biolegend)和抗cd86 apc
‑
cy7(biolegend)。单个染色对照和/或versacomp(beckman coulter)珠用于荧光补偿。dapi(sigma)、碘化丙锭(sigma)、draq5(thermo fisher)或live/dead yellow(thermo fisher)用于活细胞检测。使用未染色和同型对照来确定自身荧光水平和确认抗体特异性。
[0611]
细胞因子和趋化因子检测(肿瘤和脾细胞):为了测量小鼠肿瘤中的间质细胞因子
zandwijk等,2017)。在每剂量前和每剂量后3小时评价血清生物化学、血液学和细胞因子表达。至少每两周一次监测ca19
‑
9和c反应蛋白的水平。在用药前和周期结束时通过流式细胞术考察外周血单核细胞(pbmc)的抗肿瘤免疫细胞数量变化。从最初的手术切除获得肿瘤组织,并培养pdac细胞并测试其药物敏感性和表面受体表达。
[0617]
统计:使用graphpad prism软件包执行所有统计分析。数据表示为平均值
±
标准偏差(sd)或平均值的标准误差(sem)。两组之间的统计学显著性通过student t检验确定。3种或更多组之间的统计学显著性通过单因素anova然后tukey多重比较检验来确定。肿瘤消退研究的重要性通过双因素anova然后tukey多重比较检验来确定。对于所有测试,p值如下:*p≤0.05,**p≤0.01,***p≤0.001,和****p≤0 0001
[0618]
实施例22:jawsii细胞的edv
αgc
处理以及αgc通过cd1d配体的后续表面呈递此实施例对比了针对癌细胞的αgc和游离αgc的edv递送。
[0619]
使用的细胞:小鼠未成熟单核细胞jawsii(crl
‑
11904
tm
)。
[0620]
制备perfecta3d 96孔悬滴板:使用p1000移液器对3d悬滴板的上侧和下侧托盘储液部填充融化的1%琼脂糖(1g琼脂糖溶解在100ml水中,在微波中溶解,并使其冷却至~50℃)。使板干燥并在室温下沉降至少30分钟。然后,对悬滴板的外侧孔填充50μl无菌细胞培养基(无细胞)/孔。
[0621]
用edv
αgc
处理jawsii球形体:用100ng/mlαgc(阳性对照)处理jawsii细胞;将空白微细胞和微细胞
αgc
与未处理的细胞相比,并在处理后8h、16h、24h和48h收集(图46a
‑
46d)。
[0622]
jawsii细胞解离成单细胞悬浮液:使jawsii细胞在t25或t75烧瓶中作为半悬浮培养物生长。通过使用移液器移液,将培养基小心地收集到无菌的50ml管中,并将烧瓶的培养表面用5ml的无菌pbs洗涤两次,并在每次洗涤后收集在相同无菌50ml管中。通过加入5ml的0.25%胰蛋白酶/edta收集粘附的细胞,并在37℃下培育3分钟或直到所有细胞从烧瓶表面提起。通过使用移液器轻轻移液,将提起的细胞小心地分解成单个细胞,并转移到先前步骤中使用的样品无菌50ml管中。然后将细胞悬浮液以300g离心7分钟,并轻轻倒出上清液。用手指轻拂管底部使细胞团解离,然后重悬于5ml预热的jawsii培养基中。通过使用移液器小心地移液,将细胞悬浮液进一步解离成单个细胞。为了确定细胞数,将10μl细胞悬浮液与10μl台盼蓝溶液混合,并使用eve自动细胞计数器进行分析。
[0623]
初始处理准备:各处理组各时间点使用6个悬滴悬浮液样品。将5
×
104个jawsii细胞和5
×
108个微细胞(1∶1000微细胞/细胞比)用于每个处理样品,并在jawsii细胞培养基中以50μl的总体积进行培养。准备了额外的未处理样品用于同型对照。通过在12,000g下离心7分钟使适量的微细胞成团,并通过移液小心除去上清液。将适量的活jaws细胞(基于上一节中的细胞计数)添加到成团的微细胞中。然后通过轻轻移液使微细胞解离为单微细胞的细胞悬浮液。然后通过添加无菌培养基补足各样品的最终体积。对于未处理的和αgc处理的样品,5x10
4 jawsii细胞用于各样品,并在jawsii细胞培养基中以50μl的总体积进行培养。将适量的活jawsii细胞转移到eppendorf管中。然后通过添加无菌培养基补足各样品的最终体积。将1000ng/ml的αgc直接添加到用1000ng/ml的αgc(阳性对照)处理组处理的jawsii细胞的细胞悬浮液中。然后将样品以50μl处理悬浮液/孔小心地接种到悬滴板的各孔中,并在5%co2下在37℃下培育直到收集。
[0624]
用抗αgalcer:mcd1d复合单克隆抗体对处理后的jawsii细胞染色:使用p200移液
管小心收集各悬滴孔的全部内容物,并将其转移到eppendorf管中。各处理组总共收集6个样品到1个管中。将1:1000pe缀合的抗小鼠αgalcer:mcd1d复合单克隆抗体和1:1000pe缀合的小鼠igg1同型对照加入适当的样品中,并通过轻轻涡旋混合。galcer:mcd1d单克隆抗体与galcer:mcd1d复合体的细胞表面暴露部分结合。然后将样品在黑暗中于室温培育20分钟。然后通过在350g下离心5分钟使样品成团。通过小心移液除去上清液,并将团块重悬浮,并在500μl facs缓冲剂中洗涤一次。然后通过在350g下离心5分钟来收集细胞,将其重悬浮于250pl facs缓冲剂中,并转移到facs管中。将1μl的dapi添加到各样品中,并通过轻轻旋转管将其混合。然后使用gallios流式细胞仪分析样品。
[0625]
结果:流式细胞术数据(图45)显示,与用单独和未处理的微细胞处理的jawsii细胞相比,用微细胞
α
‑
gc
和游离α
‑
gc处理的jawsii细胞在用抗galcer:mcd1d染色后有明显变化。这种阳性染色证实了成功地微细胞将α
‑
gc递送至jawsii细胞并且随后通过cd1d分子在细胞表面上抗原呈递,该cd1d分子在细胞表面呈递糖脂。α
‑
gc的呈递是至关重要的步骤,其通过不变的nkt细胞触发抗肿瘤活性所必需的ii型ifn级联而导致受体识别。
[0626]
实施例23:在同系小鼠模型中使用
ep
微细胞
dox
和微细胞
α
‑
gc
组合治疗的体内研究(balb/c小鼠中的
ep
ct26鼠结肠癌)
[0627]
此实施例示例了含有治疗剂的微细胞和含有ii型干扰素激动剂的微细胞抗肿瘤的功效。该结果表明,缺乏i型干扰素激动剂的组合物可用于有效治疗肿瘤。
[0628]
小鼠和治疗(实验1
‑
3):6
‑
7周龄的雌性balb/c小鼠得自animal resources company in western australia。在实验开始之前,使小鼠适应环境一周。用表达epcam抗原的质粒稳定转化ct26细胞(小鼠结肠癌),并建立稳定的克隆(epclone 12.1)。该克隆在细胞表面上表达epcam。所有动物实验均按照澳大利亚national health and medical research council关于实验动物的养护和使用的指南以及engeneic animal ethics committee的批准进行。
[0629]
通过在每只小鼠的左胁腹皮下注射2x105个细胞/100μl pbs,建立ct26(epclone 12.1)同种移植物。肿瘤在植入后8天内生长至~125mm3的起始体积。将小鼠随机分组,每个治疗组8只小鼠。用
ep
微细胞
dox
、微细胞
α
‑
gc
和
ep
微细胞
dox+
微细胞
α
‑
gc
(组合)治疗肿瘤,与单独盐水治疗对比。
[0630]
用药两周每周3次进行。在单一或组合治疗中,
ep
微细胞
dox
以1x109个微细胞/剂量用药。微细胞
α
‑
gc
在实验1(图40)和3(图42)中以1x107,并且在实验2(图40)中以1x108用药;其中在肿瘤体积达到800mm3时,盐水组也受到挑战。
[0631]
结果:与盐水和
ep
微细胞
dox
处理相比,接受
ep
微细胞
dox+
微细胞
α
‑
gc
的组合治疗组的所有3个实验均显示肿瘤进展明显停止。该结果支持通过向
ep
微细胞
dox
添加微细胞
α
‑
gc
处理来产生免疫辅助效果的理论。用单独微细胞
α
‑
gc
进行的治疗也显示所有3个实验的肿瘤进展停止,尽管没有达到组合治疗的程度,如实验2最佳显示。
[0632]
在实验2中,在治疗变为药物和α
‑
gc edv介导的组合疗法后,盐水治疗的对照肿瘤呈现显著的肿瘤消退(图41)。在实验终止前三天内,已达到800mm3的肿瘤降至600mm3以下。
[0633]
不同尺寸的肿瘤的剂量评价;小鼠和治疗(实验4):通过将2
×
105个细胞/100μ1pbs皮下注射入6
‑
7周龄balb/c雌性小鼠的左侧腹来建立ct26(epclone12.1)同系移植物。在治疗开始之前,使肿瘤长到~200
‑
250mm3或600
‑
800mm3。将小鼠随机分为6组,每组3只小
鼠。小鼠仅接受一个剂量。治疗组包括;盐水(图43c)、
ep
微细胞
dox
(1x109)(图43f)、微细胞
α
‑
gc
(1x106)(图43e)、微细胞
α
‑
gc
(1x107)(图43d)、
ep
微细胞
dox
1x109+微细胞
α
‑
gc
(l x 106)(图43b)、
ep
微细胞
dox
(1x109)+微细胞
α
‑
gc
(1x107)(图43a)。
[0634]
将小鼠在治疗后24小时处死以得到200
‑
250mm3肿瘤(图43),在16小时和24小时处死以得到600
‑
800mm3肿瘤(图44)。
[0635]
结果:通过如上所述单一剂量治疗不同尺寸的肿瘤,进一步研究了微细胞
α
‑
gc
单独或组合用药在携载ct26同系肿瘤的balb/c小鼠中的效果。值得注意,发现在携载200
‑
250mm3以及400
‑
600mm3肿瘤的小鼠中,肿瘤均在24小时用药内出现明显的坏死(黑色)。这种效果在较大的肿瘤中更为明显,并且在对照组中见不到。
[0636]
总之,此数据表明,包含治疗剂的微细胞和ii型干扰素激动剂的组合显示出抗肿瘤功效。该结果表明,缺乏i型干扰素激动剂的组合物可用于有效治疗肿瘤。
[0637]
对于本领域技术人员将显而易见,在不脱离本发明的精神或范围的情况下,可以对本发明的方法和组合物进行各种改动和变型。因此,意图本发明覆盖本发明的改动和变型,只要其落入所附权利要求及其等同形式的范围。
[0638]
被引用的出版物
[0639]
ablasser et al.,nat.immunol.,10(10):1065
‑
72(2009).
[0640]
ablasser et al.,nature,498:380
–
384(2013a).
[0641]
ablasser et al.,nature,503:530
–
534(2013b).
[0642]
adamus et al.,contemp.oncol(ponzn),22(1a):56
‑
60(mar.2018).
[0643]
aduro biotech inc.(2016),novartis pharmaceuticals.study of the safety and efficacy of miw815(adu
‑
s100)in patients with advanced/metastatic solid tumors or lymphomas.2020.clinicaltrials.gov[internet].bethesda(md):national library of medicine(us).identifier:nct02675439.available from:https://clinicaltrials.gov/show/nct02675439.(cited 01 july 2016).
[0644]
ahmadzadehfar et al.,“radioembolization of liver tumors with yttrium
‑
90microspheres,”semin nucl med.2010;40(2):105
–
121.
[0645]
ahmadzadehfar et al.,“therapeutic response and side effects of repeated radioligand therapy with 177lu
‑
psma
‑
dkfz
‑
617of castrate
‑
resistant metastatic prostate cancer,”oncotarget.2016;7(11):12477
–
12488.
[0646]
alexopoulou et al.,nature,413:732
‑
738(2001).
[0647]
alzahrani as,alshaikh o,tuli m,al
‑
sugair a,alamawi r,al
‑
rasheed mm.diagnostic value of recombinant human thyrotropin
‑
stimulated 123i whole
‑
body scintigraphy in the follow
‑
up of patients with differentiated thyroid cancer.clin nucl med.2012;37(3):229
–
234.
[0648]
andersson l,blomberg l,flegel m,lepsa l,nilsson b,verlander m.large
‑
scale synthesis of peptides.pept sci.2000;55:227
–
250.
[0649]
anguille et al.,pharmacological reviews 67,731
‑
753(2015).
[0650]
barber et al.,curr.opin.immunol.,23(1):10
‑
20(2011).
[0651]
belardelli et al.,trends in immunology 23,201
‑
208(2002).
tumors,”cancer research,aacr annual meeting 2017;april 1
‑
5,2017(published july 2017)).
[0711]
hansen et al.,embo j.,33(15):1654
‑
66(2014).
[0712]
harry,e.j.,mol.microbiol.,40(4):795
‑
803(2001).
[0713]
hershey,j.allergy clin.immunol.,111:677
‑
90(2003).
[0714]
hiraga et al.,j.bacteriol.,171:1496
‑
1505(1989).
[0715]
hobbs et al.,proc.natl.acad.sci.usa,95(8):4607
‑
4612(1998).
[0716]
holman bl,tumeh ss.single
‑
photon emission computed(spect):applications and potential.jama.1990;263(4):561
–
564.
[0717]
hornung et al.,nature,458:514
–
518(2009).
[0718]
hu&lutkenhaus,mol.microbio.,34(1):82
‑
90(1999).
[0719]
iftode et al.,crit.rev.biochem.mol.biol.,34:141
‑
80(1999).
[0720]
igarashi h,fujimori n,ito t,nakamura t,oono t,nakamura k,suzuki k,jensen rt,takayanagi r.vasoactive intestinal peptide(vip)and vip receptors—elucidation of structure and function for therapeutic applications.int j clin med.2011;2:500
–
508.
[0721]
immune design(2016),merck sharp&dohme corp.study of intratumoral g100 with or without pembrolizumab in patients with follicular non
‑
hodgkin’s lymphoma.2017.clinicaltrials.gov[internet].bethesda(md):national library of medicine(us).identifier:nct02501473.available from:https://clinicaltrials.gov/show/nct02501473.(cited 01 july 2016).
[0722]
jarboe et al.,cancer res.,67:7983
‑
86(2007).
[0723]
jenkins et al.,british journal of cancer 118,9
‑
16(2018).
[0724]
jung et al.,translational oncology 11,686
‑
690(2018).
[0725]
kao et al.,am.j.respir.crit.care med.,191(12):1467
‑
1469(2015).
[0726]
kao et al.,american journal of respiratory and critical care medicine 191,1467
‑
1469(2015).
[0727]
kawai and akira,nat.immunol.,11:373
–
384(2010).
[0728]
khalil et al.,proc nat’l acad.usa,106:11667
‑
72(2009).
[0729]
kim et al.,proc.natl.acad.sci.usa,107:15181
–
15186(2010).
[0730]
kota et al.,cell,137:1005
‑
17(2009).
[0731]
kramer
‑
marek g,capala j.the role of nuclear medicine in modern therapy of cancer.tumour biol.2012;33(3):629
–
640.
[0732]
kranzusch et al.,cell rep.,3:1362
–
1368(2013).
[0733]
krieg et al.,nature,374:546
‑
549(1995).
[0734]
kwekkeboom dj,de herder ww,kam bl,et al.treatment with the radiolabeled somatostatin analog[177 lu
‑
dota 0,tyr3]octreotate:toxicity,efficacy,and survival.j clin oncol.2008;26(13):2124
–
2130.
[0735]
landskron et al.,journal of immunology research 2014,149185(2014).
[0736]
lee et al.,cancers 3,3856
‑
3893(2011).
[0737]
lemmon and schlessinger,cell,141(7):1117
‑
134(2010).
[0738]
leung and amarasinghe,curr.opin.struct.biol.,36:133
–
141(2016).
[0739]
li et al.,science,341:1390
–
1394(2013b).
[0740]
liu et al.,science,347(6227):aaa2630(2015).
[0741]
lu et al.,structure,18:1032
–
1043(2010).
[0742]
ma et al.,mol.microbiol.,54:99
‑
108(2004).
[0743]
macdiarmid et al.,plos one,11(4)(2016).
[0744]
macdiarmidet al.nature biotechnology 27,643
‑
651(2009).
[0745]
macdiarmid et al.,cell cycle 6,2099
‑
2105(2007a).
[0746]
macdiarmidet al.,cancer cell 11,431
‑
445(2007b).
[0747]
majkowska et al.,“complexes of low energy beta emitters 47sc and 177lu with zoledronic acid for bone pain therapy,”appl radiat isot.2009;67(1):11
–
13.
[0748]
mankan et al.,embo j.,33:2937
–
2946(2014).
[0749]
mcwhirter et al.,j.exp.med.,206:1899
–
1911(2009).
[0750]
marq et al.,j.biol.chem.,286:6108
–
6116(2011).
[0751]
matsuno m,matsui t,iwasaki a,arakawa y.role of acetylcholine and gastrin
‑
releasing peptide(grp)in gastrin secretion.j gastroenterol.1997;32:579
–
586.
[0752]
medimmune llc(2016).a study of medi9197 administered in subjects with a solid tumor cancer.2018.clinicaltrials.gov[internet].bethesda(md):national library of medicine(us).identifier:nct02556463.available from:https://clinicaltrials.gov/show/nct02556463.(cited 01 jul 2016).
[0753]
mellman et al.,nature 480,480
‑
489(2011).
[0754]
merrifield r.solid
‑
phase peptide synthesis.adv enzymol relat areas mol biol.2006;32:221
–
296.
[0755]
meulen and brady,hum.vaccin.immunother.,13(1):15
‑
16(2017).
[0756]
mhawech
‑
fauceglia p,zhang s,terracciano l,et al.prostate
‑
specific membrane antigen(psma)protein expression in normal and neoplastic tissues and its sensitivity and specificity in prostate adenocarcinoma:an immunohistochemical study using mutiple tumour tissue microarray technique.histopathology.2007;50(4):472
–
483.
[0757]
morvan et al.,nature reviews cancer 16,7
‑
19(2016).
[0758]
muller et al.,frontiers in immunology 8,304(2017).
[0759]
m
ü
ller c,zhernosekov k,u,et al.a unique matchedof terbium radioisotopes for pet and spect and for alpha
‑
and beta
‑
radionuclide therapy:an in vivo proof
‑
of
‑
concept study with a new receptor
‑
targeted folate derivative.j nucl med.2012;53(12):1951
–
1959.
rev cell dev biol.1996;12:697
–
715.
[0781]
sagnella et al.,molecular cancer therapeutics 17,1012
‑
1023(2018).
[0782]
santoni m,scarpelli m,mazzucchelli r,et al.targeting prostate
‑
specific membrane antigen for personalized therapies in prostate cancer:morphologic and molecular backgrounds and future promises.j biol regul homeost agents.2014;28(4):555
–
563.
[0783]
sawa
‑
wejksza et al.,archivum immunologiae et therapiae experimentalis 66,97
‑
111(2018).
[0784]
sazar,“activating the natural host defense;hiltonol(poly
‑
iclc)and malignant brain tumors,oncovir,inc.,www.oncovir.com/id2(accessed july 11,2018).
[0785]
sharma et al.cell 168,707
‑
723(2017).
[0786]
sharpe,immunological reviews 276,5
‑
8(2017).
[0787]
showalter,cytokine 97,123
‑
132(2017).
[0788]
silver da,pellicer i,fair wr,heston wd,cordon
‑
cardo c.prostate
‑
specific membrane antigen expression in normal and malignant human tissues.clin cancer res.1997;3(1):81
–
85.
[0789]
simmons et al.the journal of immunology 188,3116
‑
3126(2012).
[0790]
singh m,mukhopadhyay k.alpha
‑
melanocyte stimulating hormone:an emerging anti
‑
inflammatory antimicrobial peptide.biomed res int.2014;2014:874610.
[0791]
sioud,m.,trends pharmacol.sci.,25:22
‑
8(2004).
[0792]
schoggins et al.,nature,505:691
–
695(2014).
[0793]
solomon et al.,plos one,10:1
‑
17(2015).
[0794]
staudacher et al.,british journal of cancer 117,1736
‑
1742(2017).
[0795]
strand,fl.neuropeptides:regulators of physiological processes.mit press;1999.
[0796]
stewart and d’ari,j.bacteriol.,174:4513
‑
6(1992).
[0797]
sun et al.,science,339(6121):786
–
791(2013).
[0798]
sun et al.,biochem.biophys.res.commun.,280:788(2001).
[0799]
sun x,li y,liu t,li z,zhang x,chen x.peptide based imaging agents for cancer detection.adv drug deliv rev.110
‑
111:38
‑
51(2017).
[0800]
szkandera et al.,british journal of cancer 110,183
‑
188(2014).
[0801]
takahashi h,emoto k,dubey m,castner dg,grainger dw.imaging surface immobilization chemistry:correlation with cell patterning on non
‑
adhesive hydrogel thin films.adv funct mater.2008;18:2079
–
2088.
[0802]
takaoka et al.,nature,448:501
–
505(2007).
[0803]
takeshita et al.,molec.ther.,18:181
‑
87(2010).
[0804]
tanpure et al.,bioorg.med.chem.,21:8019
‑
32(2013).
[0805]
tatemoto,k.neuropeptide y:history and overview,neuropeptide y and related peptides.springer;2004.p.1
‑
21.
[0806]
teunissen et al.,“endocrine tumours of the gastrointestinal tract.peptide receptor radionuclide therapy,”best pract res clin gastroenterol.2005;19(4):595
–
616.
[0807]
tyler
‑
mcmahon bm,boules m,richelson e.neurotensin:peptide for the next millennium.regul pept.2000;93:125
–
136.
[0808]
unterholzner et al.,nat.immunol.,11:997
–
1004(2010).
[0809]
unterholzner et al.,immunobiology,128(11):1312
‑
21(2013).
[0810]
van zandwijk et al.,lancet oncol.,18(10):1386
‑
1396(2017).
[0811]
van zandwijk et al.,the lancet oncology 18,1386
‑
1396(2017).
[0812]
ventola,pharmacy and therapeutics 42,452
‑
463(2017).
[0813]
wallace et al.,springer seminars in immunopathology 27,49
‑
64(2005).
[0814]
walrand s,hesse m,renaud l,jamar f.the impact of imagebias on pet/ct 90
y dosimetry after radioembolization.j nucl med.2015;56(3):494
–
495.
[0815]
wang et al.,nat.struct.mol.biol.,17:781
–
787(2010).
[0816]
wang et al.,immunity,41(6):919
‑
33(2014).
[0817]
weckbecker g,lewis i,albert r,schmid ha,hoyer d,bruns c.opportunities in somatostatin research:biological,chemical and therapeutic aspects.nat rev drug discov.2003;2:999
–
1017.
[0818]
white&mccubrey,leukemia,15:1011
‑
1021(2001).
[0819]
whittle et al.,j.clin.neurosci.,22(12):1889
‑
1894(2015).
[0820]
whittle et al.,journal of clinical neuroscience:official journal of the neurosurgical society of australasia 22,1889
‑
1894(2015).
[0821]
wu et al.,science,339:826
–
830(2013).
[0822]
wykosky et al.,clin cancer res.,14:199
‑
208(2008).
[0823]
xiaet al.,nat.immunol.,16:366
–
375(2015).
[0824]
yague et al.,gene ther.,11:1170
‑
74(2004).
[0825]
yang w,luo d,wang s,wang r,chen r,liu y,zhu t,ma x,liu r,xu g.tmtp1,a novel tumor
‑
homing peptide specifically targeting metastasis.clin cancer res.2008;14:5494
–
5502.
[0826]
yang et al.,nat.immunol.,11:487
–
494(2010).
[0827]
yi et al.,plos one,8(10):e77846(2013).
[0828]
yuan et al.,scientific reports 5,14273(2015).
[0829]
zhang et al.,j.immunol.,186:4541
–
4545(2011a).
[0830]
zhang et al.,nat.immunol.,12:959
–
965(2011b).
[0831]
zhang et al.,cell rep.,6:421
–
430(2014).
[0832]
zibert et al.,human gene therapy 15,21
‑
34(2004).
[0833]
ziegler
‑
heitbrock et al.,frontiers in immunology 4,23(2013).
[0834]
zitvogel et al.,nature reviews immunology 15,405
‑
414(2015).
[0835]
u.s.patent no.8,591,862.
[0836]
u.s.patent no.7,183,105.
[0837]
u.s.2008/0051469.
[0838]
wo 2000/067776.
[0839]
wo 2003/033519.
[0840]
wo 2004/113507.
[0841]
wo 2005/056749.
[0842]
wo 2005/079854.
[0843]
wo 2009/027830.
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1