乙内酰脲衍生物的固体分散体的制作方法
本发明涉及包含1-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-5,5-二甲基咪唑烷-2,4-二酮(在下文中可以称为“化合物i”)的固体分散体。具体地,本发明涉及包含化合物i、药学上可接受的聚合物和药学上可接受的阳离子物质的固体分散体。另外,本发明涉及含有固体分散体的药物组合物、用于制备固体分散体的方法以及使用药物组合物来治疗疾病的方法。
背景技术:
已知具有特定结构的螺咪唑啉酮衍生物具有有用的甲状旁腺激素(pth)样作用(专利文献(ptl)1)。例如,1-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-5,5-二甲基咪唑烷-2,4-二酮(化合物i)是具有强pth样作用和高代谢稳定性的化合物,并且其可以治疗可以通过pth样作用治疗的病况(包括甲状旁腺功能减退(hypoparathyroidism))(专利文献2)。此外,对该化合物的非侵入性全身或局部暴露诱导骨/软骨合成代谢,使得能够实现用于骨质疏松(osteoporosis)、牙周病中的骨质流失(bonelossinperiodontaldisease)、拔牙后的牙槽骨缺损(alveolarbonedefectaftertoothextraction)、骨关节炎(osteoarthritis)、关节软骨缺损(articularcartilagedefects)、无力性骨病(adynamicbonedisease)、软骨发育不全(achondroplasia)、低软骨形成症(hypochondroplasia)、骨软化症(osteomalacia)、骨折(bonefracture)等的预防、治疗、恢复和加速愈合的方法(专利文献3)。
期望将这样的药物开发成例如口服有效的剂型。然而,是否可以将药物开发成口服制剂取决于药物的生物利用度有多高。影响生物利用度的因素之一是药物的水溶性。通常,水溶性差或不溶的化合物在口服施用时显示出低生物利用度。改善活性成分的生物利用度和口服吸收度对于活性成分稳定地发挥其药用效果也是重要的。另外,在临床开发中,通常设想以高剂量使用药物以提高药物的血药浓度以改善治疗效果;因此,需要这样的制剂,即使当高剂量施用时其也是如此可溶的以致于其在消化道中充分溶解。
为了改善药物溶解度,已经制备了含有难溶性化合物和聚合物化合物的固体分散体(非专利文献(npl)1至4)。
此外,已知公开了含有可电离的药物、阳离子物质和分散体聚合物的固体分散体的文献(专利文献4)。以上文献中描述的发明所要解决的问题是使得本身难以形成固体分散体的一类化合物或更具体地具有低水溶性以及在用于形成喷雾溶液的挥发性溶剂中溶解度极差的一类化合物能够容易地形成固体分散体。
引用文献清单
专利文献
专利文献1wo2010/126030
专利文献2wo2014/092061
专利文献3wo2015/189901
专利文献4wo2008/047201
非专利文献
非专利文献1jpharmsci.1997年1月;86(1):1-12
非专利文献2aapspharmscitech,2018年4月,第19卷,第3期,第978-990页
非专利文献3mol.pharmaceutics2017,14,658-673
非专利文献4aapspharmscitech,2018年1月,第19卷,第1期,第326-337页
技术实现要素:
技术问题
如上所述,化合物i可用作药物化合物;然而,它在水中的溶解度差,并且需要改善的水溶性。
此外,如上所述,为了增加药物的血药浓度并且增强其治疗效果,可以以高剂量使用药物。因此,即使当以高剂量施用时,药物也必须具有足够高的溶解度以充分溶解在消化道中。
本申请的发明人发现,与当以其游离形的晶体使用时相比,当以其盐(例如苯磺酸盐)的晶体使用时,化合物i的水溶性可以提高数十倍。然而,已经发现尽管化合物i的苯磺酸盐显示出高水溶性,但是当在体内使用时,药物的生物利用度(即血药浓度)并未充分提高。
因此,对于包含化合物i的药物组合物,必须提供即使在体内也能够以高剂量使用药物的药物组合物(其具有较高的水溶性和高吸收度)。
更具体地,本发明的目的是响应于化合物i的水溶性差并且需要以高剂量使用的问题,提供具有无法通过应用常规技术实现的高溶解度并且在消化道中具有伴随的高吸收度的制剂。
问题的解决方案
作为在这种情况下的专门研究的结果,本发明人通过向上述化合物i和聚合物中添加阳离子物质来制备固体分散体,并且发现与含有各种聚合物和各种盐晶体的常规固体分散体的那些相比,该分散体在消化道中显示出较高的溶解度和伴随的吸收度。
具体地,本发明包括以下内容:
[1]一种固体分散体,所述固体分散体包含具有以下结构式的化合物i:
药学上可接受的聚合物和药学上可接受的阳离子物质;
[2]根据[1]所述的固体分散体,所述固体分散体包含以非晶形形式存在的化合物i;
[3]根据[1]或[2]所述的固体分散体,其中药学上可接受的聚合物是选自由以下组成的组的至少一种聚合物:聚乙烯吡咯烷酮、共聚维酮、聚乙烯醇、纤维素聚合物和甲基丙烯酸共聚物;
[4]根据[1]或[2]所述的固体分散体,其中药学上可接受的聚合物是选自由以下组成的组的至少一种聚合物:聚乙烯吡咯烷酮、共聚维酮、聚乙烯醇、羟丙基纤维素、乙酸琥珀酸羟丙甲纤维素和甲基丙烯酸共聚物ld;
[5]根据[1]至[4]中任一项所述的固体分散体,其中药学上可接受的阳离子物质是由pka值为11以上的碱提供的至少一种阳离子物质;
[6]根据[1]至[5]中任一项所述的固体分散体,其中药学上可接受的阳离子物质是选自由以下组成的组的至少一种:钠阳离子、钾阳离子和精氨酸阳离子;
[7]根据[1]至[6]中任一项所述的固体分散体,其中组合物中化合物i与聚合物的重量比为约1:9至约1:1;
[8]根据[1]至[7]中任一项所述的固体分散体,其中组合物中化合物i与聚合物的重量比为约1:2至约1:1;
[9]根据[1]至[8]中任一项所述的固体分散体,其中组合物中阳离子物质与化合物i的摩尔比为0.8以上;
[10]根据[1]至[9]中任一项所述的固体分散体,所述固体分散体还包含表面活性剂,其中表面活性剂是药学上可接受的表面活性剂;
[11]根据[1]至[10]中任一项所述的固体分散体,其中化合物i来源于:
游离形式的i型晶体:其粉末x射线衍射图包含以2θ表示的14.4°、15.3°、16.6°和18.7°(±0.2°)的衍射角的峰的化合物i晶体;和/或
游离形式的ii型晶体:其粉末x射线衍射图包含以2θ表示的7.9°、13.5°、15.9°和21.8°(±0.2°)的衍射角的峰的化合物i晶体;
[12]一种药物组合物,所述药物组合物包含根据[1]至[11]中任一项所述的固体分散体;
[13]一种用于制备固体分散体的方法,所述方法包括以下步骤:
(i)提供具有以下结构式的化合物i:
药学上可接受的聚合物和产生药学上可接受的阳离子物质的碱;
(ii)将化合物i、药学上可接受的聚合物和产生药学上可接受的阳离子物质的碱混合;以及
(iii)获得包含化合物i、药学上可接受的聚合物和药学上可接受的阳离子物质的固体分散体;
[14]根据[13]所述的方法,其中步骤(ii)和(iii)分别包括以下步骤:
(ii-a)通过将化合物i、药学上可接受的聚合物和产生药学上可接受的阳离子物质的碱在溶剂中混合来制备混合溶液;以及
(iii-a)从步骤(ii-a)中获得的混合溶液中移除溶剂;
[15]根据[14]所述的方法,其中步骤(iii-a)包括移除溶剂和通过喷雾干燥进行干燥的步骤;
[16]根据[13]至[15]中任一项所述的方法,其中在步骤(i)中,化合物i来源于:
游离形式的i型晶体:其粉末x射线衍射图包含以2θ表示的14.4°、15.3°、16.6°和18.7°(±0.2°)的衍射角的峰的化合物i晶体;和/或
游离形式的ii型晶体:其粉末x射线衍射图包含以2θ表示的7.9°、13.5°、15.9°和21.8°(±0.2°)的衍射角的峰的化合物i晶体;
[17]根据[12]所述的药物组合物,其中药物组合物用于预防或治疗甲状旁腺功能减退、预防或治疗骨质疏松、改善牙周病中的骨量减少、促进拔牙后的牙槽骨缺损的恢复、预防或治疗骨关节炎、促进关节软骨缺损的恢复、预防或治疗无力性骨病、预防或治疗软骨发育不全、预防或治疗低软骨形成症、预防或治疗骨软化症或者促进骨折的恢复;
[18]一种用于预防或治疗甲状旁腺功能减退、预防或治疗骨质疏松、改善牙周病中的骨量减少、促进拔牙后的牙槽骨缺损的恢复、预防或治疗骨关节炎、促进关节软骨缺损的恢复、预防或治疗无力性骨病、预防或治疗软骨发育不全、预防或治疗低软骨形成症、预防或治疗骨软化症或者促进骨折的恢复的方法,其中所述方法包括向需要其的患者施用根据[1]至[11]中任一项所述的固体分散体或根据[12]所述的药物组合物;
[19]根据[1]至[11]中任一项所述的固体分散体或根据[12]所述的药物组合物用于制备药物组合物的用途,所述药物组合物用于预防或治疗甲状旁腺功能减退、预防或治疗骨质疏松、改善牙周病中的骨量减少、促进拔牙后的牙槽骨缺损的恢复、预防或治疗骨关节炎、促进关节软骨缺损的恢复、预防或治疗无力性骨病、预防或治疗软骨发育不全、预防或治疗低软骨形成症、预防或治疗骨软化症或者促进骨折的恢复;以及
[20]根据[1]至[11]中任一项所述的固体分散体或根据[12]所述的药物组合物,其用于预防或治疗甲状旁腺功能减退、预防或治疗骨质疏松、改善牙周病中的骨量减少、促进拔牙后的牙槽骨缺损的恢复、预防或治疗骨关节炎、促进关节软骨缺损的恢复、预防或治疗无力性骨病、预防或治疗软骨发育不全、预防或治疗低软骨形成症、预防或治疗骨软化症或者促进骨折的恢复。
发明效果
本发明能够使得具有强pth样作用和高代谢稳定性的水溶性差的化合物1-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-5,5-二甲基咪唑烷-2,4-二酮的溶解度得到很大改善并且在消化道中的吸收度得到伴随的改善。
附图简述
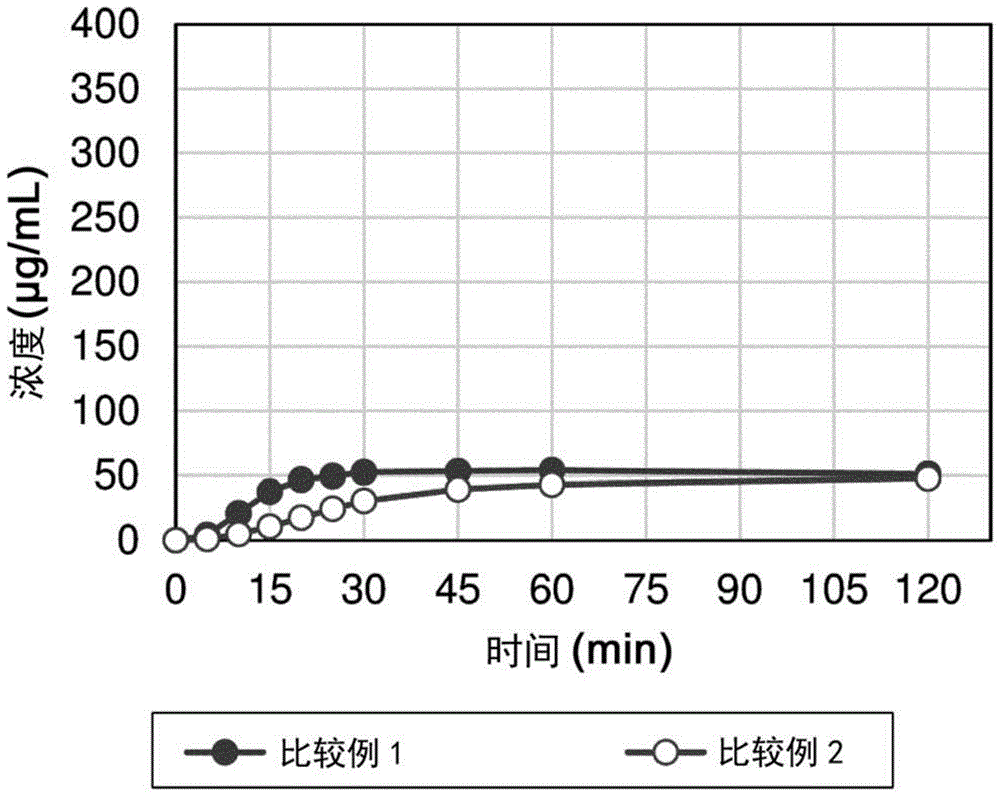
图1是显示出如在比较例1和2中测量的对固体分散体进行溶出测试的结果的图。纵轴表示化合物i的溶出浓度(μg/ml),并且横轴表示时间(分钟)。
图2是显示出如在比较例3至5中测量的对固体分散体进行溶出测试的结果的图。纵轴表示化合物i的溶出浓度(μg/ml),并且横轴表示时间(分钟)。
图3是显示出如在实施例1至3和比较例6和7中测量的对固体分散体进行溶出测试的结果的图。纵轴表示化合物i的溶出浓度(μg/ml),并且横轴表示时间(分钟)。
图4是显示出如在实施例4至6和比较例8中测量的对固体分散体进行溶出测试的结果的图。纵轴表示化合物i的溶出浓度(μg/ml),并且横轴表示时间(分钟)。
图5是显示出如在实施例1、7和8中测量的对固体分散体进行溶出测试的结果的图。纵轴表示化合物i的溶出浓度(μg/ml),并且横轴表示时间(分钟)。
图6是显示出如在实施例7、9和10中测量的对固体分散体进行溶出测试的结果的图。纵轴表示化合物i的溶出浓度(μg/ml),并且横轴表示时间(分钟)。
图7是显示出如在实施例11和12中测量的对固体分散体进行溶出测试的结果的图。纵轴表示化合物i的溶出浓度(μg/ml),并且横轴表示时间(分钟)。
图8是显示出如在实施例13和14中测量的对固体分散体进行溶出测试的结果的图。纵轴表示化合物i的溶出浓度(μg/ml),并且横轴表示时间(分钟)。
图9显示了游离形式的化合物i的i型晶体的粉末x-射线图(x-射线源:cukα1)。纵轴表示强度,并且横轴表示角度(2θ)。
图10显示了游离形式的化合物i的ii型晶体的粉末x-射线图(x-射线源:cukα1)。纵轴表示强度,并且横轴表示角度(2θ)。
实施方案描述
化合物i
本发明的固体分散体中含有的化合物是具有以下结构式的1-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-5,5-二甲基咪唑烷-2,4-二酮(化合物i)。
化合物i可以例如通过wo2014/092061的实施例3(化合物7)中描述的方法来制备。另外,游离形式的化合物i的i型晶体(制备实施例1)在ph6至7下的水溶性小于1μg/ml(37℃),并且例如在thf中在室温下的有机溶剂溶解度为80mg/ml以上,并且其是pka值为8和10的化合物。
换言之,用于本发明的化合物i具有极低的水溶性,但是在有机溶剂中具有相对高的溶解度。因此,化合物i在使用有机溶剂制备固体分散体的过程中不会引起任何特别的问题。
可以用于本发明的化合物i包括化合物i的任何同位素。化合物i的同位素是其中至少一个原子已被具有相同原子数(质子数)和不同质量数(质子和中子的数量的总和)的原子替代的化合物。化合物i中含有的同位素的实例包括氢原子、碳原子、氮原子、氧原子、硫原子和氟原子,并且它们的实例分别是2h和3h、13c和14c、15n、17o和18o、35s以及18f。特别地,通过发射放射性而衰变的放射性同位素(诸如3h或14c)可用于检查药物或化合物的体内组织分布。由于稳定的同位素不会衰减,因此存在量几乎不变,并且由于不存在放射性,因此可以安全地使用。化合物i的同位素可以按照常规方法通过用含有相应同位素的试剂代替用于合成的试剂来转化。
聚合物
可以用于本发明的固体分散体的聚合物应是药学上可接受的,并且优选地在生理相关的ph(例如1至8)下在水溶液中具有至少一定溶解度。几乎任何在1至8的ph范围的至少一部分内具有至少约0.1mg/ml的水溶性的聚合物可以是合适的。可以用于本发明的聚合物的实例包括以下非离子聚合物(中性聚合物)或离子聚合物。
在本文中,短语“药学上可接受的聚合物”中的短语“药学上可接受的”以本领域通常使用的含义使用;例如,其意指在作为接受药物施用的受试者的药物的常规用途范围内大体上不存在副作用或毒性。这同样适用于“药学上可接受的阳离子物质”和“药学上可接受的表面活性剂”。
非离子聚合物(中性聚合物)
在一个实施方案中,可以用于本发明的聚合物的实例包括非离子聚合物(中性聚合物)。换言之,聚合物优选地大体上不具有离子官能团。表述“大体上不具有离子官能团”意指共价连接至聚合物的离子基团的数量小于约0.05毫当量/克聚合物。优选地,所述数量小于约0.02毫当量/克中性聚合物。表述“离子官能团”意指在1至8的生理相关的ph范围的至少一部分内至少约10%会电离的官能团。这样的基团的pka值为约0至9。
可以用于本发明的中性聚合物的实例包括中性非纤维素聚合物。这样的中性非纤维素聚合物的实例包括以下:具有选自包括羟基、烷基酰氧基和环状酰胺的组的至少一种取代基的乙烯基聚合物和共聚物;至少一种亲水性的含有羟基的重复单元和至少一种疏水性的含有烷基或芳基的重复单元的乙烯基共聚物;聚乙烯醇;其重复单元中的至少一部分为未水解(乙酸乙烯酯)形式的聚乙烯醇;聚乙烯醇聚乙酸乙烯酯共聚物;聚乙烯吡咯烷酮;共聚维酮;丙烯酸酯和甲基丙烯酸酯共聚物;聚乙烯聚乙烯醇共聚物;和聚氧乙烯-聚氧丙烯嵌段共聚物(也称为泊洛沙姆)。
可以用于本发明的其他类型的中性聚合物的实例包括中性纤维素聚合物。表述“纤维素”意指已经通过使糖重复单元上的至少一部分羟基与化合物反应以形成酯或醚取代基而改性的纤维素聚合物。这样的中性纤维素聚合物的实例包括乙酸羟丙基甲基纤维素(hpmca)、羟丙基甲基纤维素(hpmc)、羟丙基纤维素(hpc)、甲基纤维素、羟乙基甲基纤维素、羟乙基纤维素、乙酸羟乙基纤维素和羟乙基乙基纤维素。
在另一个实施方案中,可以用于本发明的聚合物的实例包括中和的酸性聚合物。在2002年6月17日提交并且标题为“药物和中和的酸性聚合物的药物组合物”的美国公开专利申请us2003-0054038中更详细地描述了中和的酸性聚合物,所述申请的相关公开内容通过引用结合于此。表述“酸性聚合物”意指具有大量的酸性部分的任何聚合物。通常,大量的酸性部分将为每克聚合物约0.05毫当量以上的酸性部分。“酸性部分”包括足够酸性的任何官能团,其在与水接触或溶解在水中时能够至少部分地向水中贡献氢阳离子并且因此增加氢离子浓度。该定义包括pka小于约10的任何官能团或“取代基”(当官能团共价连接至聚合物时则被称为“取代基”)。
表述“中和的酸性聚合物”意指其中“酸性部分”或“酸性取代基”中的很大一部分的已被“中和”(换言之,以其去质子化形式存在)的任何酸性聚合物。被单质子酸(例如羧酸)取代的聚合物的“中和度”α被定义为聚合物上已被中和(即用碱去质子化)的酸性部分的分数。
通常,为了酸性聚合物被认为是“中和的酸性聚合物”,α必须是至少约0.01(或1%)、更优选地至少约0.1(10%)、仍更优选地至少约0.5(50%)并且最优选地至少0.9(90%)(意味着至少90%的酸性部分已被中和)。
可以以中和形式用于本发明的酸性聚合物的实例包括乙酸邻苯二甲酸纤维素、乙酸偏苯三酸纤维素、乙酸琥珀酸纤维素、邻苯二甲酸甲基纤维素、乙基邻苯二甲酸羟甲基纤维素(hydroxymethylcelluloseethylphthalate)、邻苯二甲酸羟丙基甲基纤维素、乙酸琥珀酸羟丙基甲基纤维素(也称为乙酸琥珀酸羟丙甲纤维素)(hpmcas)、乙酸马来酸羟丙基甲酯、偏苯三酸羟丙基甲酯、羧甲基乙基纤维素、聚丁酸邻苯二甲酸乙烯酯、聚乙酸邻苯二甲酸乙烯醇酯、甲基丙烯酸/丙烯酸乙酯共聚物(优选的质量比为1:99至99:1)、甲基丙烯酸/甲基丙烯酸甲酯共聚物(优选的质量比为1:99至99:1)和甲基丙烯酸共聚物。
中和的酸性聚合物可以通过本领域已知的任何常规方法形成,得到所需的中和度。通常,通过向含有酸性聚合物的溶液或组合物中添加足够量的碱来中和酸性聚合物。例如,可以将碱添加到酸性聚合物的溶液中,从而中和聚合物的酸性官能团。可以用于中和酸性聚合物的适当的碱包括上面关于本发明的固体分散体中存在的阳离子物质列出的那些。
在一个实施方案中,用于中和聚合物的碱的实例包括用于提供本发明的固体分散体中存在的阳离子物质的相同的碱。
离子聚合物
在一个实施方案中,可以用于本发明的聚合物的实例包括离子聚合物。换言之,聚合物大体上具有离子官能团,并且它们中的至少约10%在1至8的生理相关的ph范围的至少一部分内会电离。在它们会电离的ph范围内,离子聚合物通常分为酸性聚合物和碱性聚合物。酸性聚合物(或肠溶聚合物)是具有可溶于中性或碱性溶液的性质的聚合物,并且碱性聚合物是具有可溶于酸性或中性溶液的性质的聚合物。酸性聚合物的具体实例包括乙酸邻苯二甲酸纤维素、乙酸偏苯三酸纤维素、乙酸琥珀酸纤维素、邻苯二甲酸甲基纤维素、乙基邻苯二甲酸羟甲基纤维素、邻苯二甲酸羟丙基甲基纤维素、乙酸琥珀酸羟丙基甲基纤维素(hpmcas)、乙酸马来酸羟丙基甲酯、偏苯三酸羟丙基甲酯、羧甲基乙基纤维素、聚丁酸邻苯二甲酸乙烯酯、聚乙酸邻苯二甲酸乙烯醇酯、甲基丙烯酸/丙烯酸乙酯共聚物(优选的质量比为1:99至99:1)、甲基丙烯酸/甲基丙烯酸甲酯共聚物(优选的质量比为1:99至99:1)和甲基丙烯酸共聚物;并且它们被认为具有在行至从胃到小肠上段至中段之后开始快速溶解的性质。具体地,优选的聚合物展现出在ph6.8的日本药典级磷酸盐缓冲液中在120分钟内溶解的性质。碱性聚合物的具体实例包括甲基丙烯酸氨基烷基酯共聚物e和聚乙烯基缩醛二乙基氨基乙酸酯,它们被认为具有在胃中在酸性条件下开始快速溶解的性质。具体地,优选的聚合物展现出在ph1.2的日本药典溶出测试第一液体中在120分钟内溶解的性质。
对于本发明的固体分散体,也可以使用上述聚合物的共混物。换言之,除了单一类型的聚合物以外,术语“聚合物”也可以包括聚合物的共混物。
因此,在一个实施方案中,在本发明的固体分散体中,聚合物可以选自由以下组成的组:中性聚合物、离子聚合物或其混合物。
此外,在另一个实施方案中,可以用于本发明的聚合物包括选自由以下组成的组的聚合物:具有选自包括羟基、烷基酰氧基和环状酰胺的组的至少一种取代基的乙烯基聚合物和共聚物;至少一种亲水性的含有羟基的重复单元和至少一种疏水性的含有烷基或芳基的重复单元的乙烯基共聚物;聚乙烯醇;其重复单元中的至少一部分为未水解(乙酸乙烯酯)形式的聚乙烯醇;聚乙烯醇聚乙酸乙烯酯共聚物;聚乙烯吡咯烷酮;共聚维酮;丙烯酸酯和甲基丙烯酸酯共聚物;聚乙烯聚乙烯醇共聚物;聚氧乙烯-聚氧丙烯嵌段共聚物(也称为泊洛沙姆);乙酸羟丙基甲基纤维素(hpmca);羟丙基甲基纤维素(hpmc);羟丙基纤维素(hpc);甲基纤维素;羟乙基甲基纤维素;羟乙基纤维素、乙酸羟乙基纤维素;和羟乙基乙基纤维素(其为中性聚合物)、乙酸邻苯二甲酸纤维素;乙酸偏苯三酸纤维素;乙酸琥珀酸纤维素;邻苯二甲酸甲基纤维素;乙基邻苯二甲酸羟甲基纤维素;邻苯二甲酸羟丙基甲基纤维素;乙酸琥珀酸羟丙基甲基纤维素(hpmcas);乙酸马来酸羟丙基甲酯;偏苯三酸羟丙基甲酯;羧甲基乙基纤维素;聚丁酸邻苯二甲酸乙烯基酯;聚乙酸邻苯二甲酸乙烯醇酯;甲基丙烯酸/丙烯酸乙酯共聚物(优选的质量比为1:99至99:1);甲基丙烯酸/甲基丙烯酸甲酯共聚物(优选的质量比为1:99至99:1);和甲基丙烯酸共聚物(其为酸性聚合物),上述酸性聚合物的中和形式,及其混合物。
在一个实施方案中,可以用于本发明的优选聚合物包括选自由以下组成的组的聚合物:聚乙烯吡咯烷酮、共聚维酮、聚乙烯醇、纤维素聚合物和甲基丙烯酸甲基丙烯酸共聚物。更优选地,聚合物包括选自由以下组成的组的那些:聚乙烯吡咯烷酮、共聚维酮、聚乙烯醇、羟丙基纤维素、乙酸琥珀酸羟丙甲纤维素、和甲基丙烯酸共聚物ld。甚至更优选地,聚合物包括选自由以下组成的组的那些:聚乙烯吡咯烷酮、共聚维酮、羟丙基纤维素、和甲基丙烯酸共聚物ld。特别优选地,聚合物包括聚乙烯吡咯烷酮。
其中,作为聚乙烯吡咯烷酮,具体地,例如可以使用可以商品名kollidon30和90f商购获得的聚乙烯吡咯烷酮等。
作为共聚维酮,具体地,例如可以使用可以商品名kollidonva64商购获得的共聚维酮等。
作为聚乙烯醇,具体地,例如可以使用可以商品名parteckmxp商购获得的聚乙烯醇等。
作为羟丙基纤维素,具体地,例如可以使用可以商品名nissohpc-sl商购获得的羟丙基纤维素等。
作为甲基丙烯酸共聚物ld,具体地,例如,可以使用可以商品名eudragitl100-55商购获得的甲基丙烯酸共聚物ld等。
作为乙酸琥珀酸羟丙基甲基纤维素(乙酸琥珀酸羟丙甲纤维素)(hpmcas),具体地,例如,可以使用可以商品名aqoatas-lf商购获得的乙酸琥珀酸羟丙基甲基纤维素(hpmcas)等。
阳离子物质
本发明的固体分散体含有药学上可接受的阳离子物质。阳离子物质可以由与化合物i共溶解的碱提供。
在本发明中,“碱”的pka值优选地大于化合物i的pka值。由于化合物i的较低的pka值为约8,因此可以用于本发明的碱理想地具有优选地大于约9、甚至更优选地大于约10并且最优选地大于约11的pka值。在本文中,术语pka以其常规形式使用。换言之,pka是酸的电离常数的负对数。除非另有说明,否则pka假定为在25℃的蒸馏水中测量。
可以用于本发明的碱的实例包括:氢氧化物,诸如氢氧化钠、氢氧化钙、氢氧化钾、氢氧化镁、氢氧化铵和氢氧化胆碱;氧化物,诸如氧化镁和氧化钙;胺,诸如三(羟甲基)氨基甲烷、乙醇胺、二乙醇胺、n-葡甲胺、葡糖胺、乙二胺、n,n’-二苄基乙二胺、n-苄基-2-苯乙胺、环己胺、环戊胺、二乙胺、异丙胺、二异丙胺、十二胺和三乙胺;蛋白质,诸如明胶;和氨基酸,诸如赖氨酸、精氨酸、鸟嘌呤、甘氨酸和腺嘌呤。
药学上可接受的阳离子物质没有特别限制,只要它们用作药物即可,并且例如阳离子物质优选地选自由以下各项的阳离子组成的组:钾、钠、钙、镁、铝、铵、苯乍生(benzathine)(n,n’-二苄基乙二胺)、胆碱、二乙醇胺、乙二胺、葡甲胺(n-甲基葡糖胺)、苯乙苄胺(n-苄基苯乙胺)、二乙胺、哌嗪、氨丁三醇(2-氨基-2-羟甲基-1,3-丙二醇)、普鲁卡因及其混合物。更优选地,阳离子物质选自由以下组成的组:钾、钠、钙、镁、铝和铵的阳离子及其混合物。
在一个实施方案中,阳离子物质优选地是选自由pka值为11以上的碱提供的阳离子物质中的一种或多种。甚至更优选地,阳离子物质是选自由以下组成的组的一种或多种:钠、钾和精氨酸的阳离子。最优选地,阳离子物质是钠阳离子。
在又一个实施方案中,在本发明的固体分散体中,药学上可接受的聚合物和阳离子物质的优选组合的实例包括这样的情况,其中聚合物是聚乙烯吡咯烷酮,并且阳离子物质选自由钠、钾和精氨酸的阳离子及其混合物组成的组;并且更优选的实例包括以下:
聚合物是聚乙烯吡咯烷酮并且阳离子物质是钠阳离子;
在另一个实施方案中,聚合物是聚乙烯吡咯烷酮并且阳离子物质是钾阳离子;或者
在另一个实施方案中,聚合物是聚乙烯吡咯烷酮并且阳离子物质是精氨酸阳离子。
其他优选组合包括这样的情况,其中聚合物是共聚维酮,并且阳离子物质选自由钠、钾和精氨酸的阳离子及其混合物组成的组;并且更优选的实例包括以下:
聚合物是共聚维酮并且阳离子物质是钠阳离子;
在另一个实施方案中,聚合物是共聚维酮并且阳离子物质是钾阳离子;或者
在另一个实施方案中,聚合物是共聚维酮并且阳离子物质是精氨酸阳离子。
其他优选组合包括这样的情况,其中聚合物是聚乙烯醇,并且阳离子物质选自由钠、钾和精氨酸的阳离子及其混合物组成的组;并且更优选的实例包括以下:
聚合物是聚乙烯醇并且阳离子物质是钠阳离子;
在另一个实施方案中,聚合物是聚乙烯醇并且阳离子物质是钾阳离子;或者
在另一个实施方案中,聚合物是聚乙烯醇并且阳离子物质是精氨酸阳离子。
其他优选组合包括这样的情况,其中聚合物是羟丙基纤维素,并且阳离子物质选自由钠、钾和精氨酸的阳离子及其混合物组成的组;并且更优选的实例包括以下:
聚合物是羟丙基纤维素并且阳离子物质是钠阳离子;
在另一个实施方案中,聚合物是羟丙基纤维素并且阳离子物质是钾阳离子;或者
在另一个实施方案中,聚合物是羟丙基纤维素并且阳离子物质是精氨酸阳离子。
其他优选组合包括这样的情况,其中聚合物是乙酸琥珀酸羟丙甲纤维素,并且阳离子物质选自由钠、钾和精氨酸的阳离子及其混合物组成的组;并且更优选的实例包括以下:
聚合物是乙酸琥珀酸羟丙甲纤维素并且阳离子物质是钠阳离子;
在另一个实施方案中,聚合物是乙酸琥珀酸羟丙甲纤维素并且阳离子物质是钾阳离子;或者
在另一个实施方案中,聚合物是乙酸琥珀酸羟丙甲纤维素并且阳离子物质是精氨酸阳离子。
其他优选组合包括这样的情况,其中聚合物是甲基丙烯酸共聚物ld,并且阳离子物质选自由钠、钾和精氨酸的阳离子及其混合物组成的组;并且更优选的实例包括以下:
聚合物是甲基丙烯酸共聚物ld并且阳离子物质是钠阳离子;
在另一个实施方案中,聚合物是甲基丙烯酸共聚物ld并且阳离子物质是钾阳离子;或者
在另一个实施方案中,聚合物是甲基丙烯酸共聚物ld并且阳离子物质是精氨酸阳离子。
表面活性剂
本发明的固体分散体和含有其的药物组合物还可以含有表面活性剂。表面活性剂可以共分散在本发明的固体分散体中,或者其可以在固体分散体之外以与其他添加剂相同的方式混合(合并)。可以用于本发明的表面活性剂是药学上可接受的表面活性剂。
在本发明中,“表面活性剂”意指在分子中具有亲水基团和疏水基团二者的物质,并且包括离子表面活性剂和非离子表面活性剂。
离子表面活性剂是指当溶解在水中时电解并且形成离子(带电荷的原子或原子团)的离子表面活性剂。根据生成的离子的电荷,将离子表面活性剂进一步分为阴离子表面活性剂、阳离子表面活性剂和两性表面活性剂。
非离子表面活性剂的实例包括:糖酯型表面活性剂,诸如脱水山梨糖醇脂肪酸酯(c12-18)、聚氧乙烯(poe)脱水山梨糖醇脂肪酸酯(c12-18)和蔗糖脂肪酸酯;脂肪酸酯型,诸如poe脂肪酸酯(c12-18)、poe树脂酸酯和poe脂肪酸二酯(c12-18);醇型,诸如poe烷基醚(c12-18);烷基酚型表面活性剂,诸如poe烷基(c8-12)苯基醚、poe二烷基(c8-12)苯基醚和poe烷基(c8-12)苯基醚福尔马林缩合物;聚氧乙烯-聚氧丙烯嵌段聚合物型表面活性剂,诸如聚氧乙烯-聚氧丙烯嵌段聚合物和烷基(c12-18)聚氧乙烯-聚氧丙烯嵌段聚合物醚;烷基胺型,诸如poe烷基烷基胺(c12-18)和poe脂肪酸酰胺(c12-18);双酚型表面活性剂,诸如poe脂肪酸双苯基醚;聚芳环型表面活性剂,诸如聚氧化烯(poa)苄基苯基(或苯基苯基)醚和poa苯乙烯基苯基(或苯基苯基)醚;poe醚和酯型硅和氟表面活性剂;和植物油型表面活性剂,诸如poe蓖麻油和poe氢化蓖麻油。
非离子表面活性剂优选地包括聚乙二醇40硬脂酸酯、脱水山梨糖醇三油酸酯、聚氧乙烯(105)聚氧丙烯(5)二醇、聚氧乙烯氢化蓖麻油60、聚乙二醇35蓖麻油、聚桂醇和生育酚聚乙二醇琥珀酸酯(tpgs)。
阴离子表面活性剂的实例包括:硫酸盐型表面活性剂,诸如烷基硫酸盐(c12-18)、poe烷基醚硫酸盐(c12-18)、poe烷基苯基醚硫酸盐(c12-18)、poe苄基(或苯乙烯基)苯基(或苯基苯基)醚硫酸盐、聚氧乙烯和聚氧丙烯嵌段聚合物硫酸盐;磺酸盐型表面活性剂,诸如石蜡(烷烃)磺酸盐(c12-22)、α-烯烃磺酸盐(c14-16)、二烷基磺基琥珀酸盐(c8-12)、烷基苯磺酸盐(c12)、单或二烷基(c3-6)萘磺酸盐、萘磺酸盐-福尔马林缩合物、烷基(c8-12)二苯基醚二磺酸盐、木素磺酸盐、poe烷基(c8-12)苯基醚磺酸盐和poe烷基(c12-18)醚磺基琥珀酸半酯;羧酸型表面活性剂,诸如脂肪酸盐(c12-18)、n-酰基氨基酸盐(c12-18)和树脂酸盐;和磷酸盐型表面活性剂,诸如poe烷基(c12-18)醚磷酸盐、poe单或二烷基(c8-12)苯基醚磷酸盐、poe苄基(或苯乙烯基)苯基(或苯基苯基)醚磷酸盐、聚氧乙烯-聚氧丙烯嵌段聚合物和烷基(c8-12)磷酸盐。
阴离子表面活性剂优选地包括:单烷基硫酸盐,诸如月桂基硫酸钠、十四烷基硫酸钠、十六烷基硫酸钠和十八烷基硫酸钠;磺基琥珀酸二辛酯钠;月桂酰肌氨酸钠;和十二烷基苯磺酸钠。
在本发明中,在以适当的比率合并它们时,可以使用两种以上表面活性剂。
更优选的表面活性剂包括选自由以下组成的组的那些:单烷基硫酸盐、脱水山梨糖醇三油酸酯、聚氧乙烯(105)聚氧丙烯(5)二醇、聚氧乙烯氢化蓖麻油60、聚乙二醇35蓖麻油、磺基琥珀酸二辛基酯钠、月桂酰肌氨酸钠、十二烷基苯磺酸钠、生育酚聚乙二醇琥珀酸酯(tpgs)及其混合物。
甚至更优选的表面活性剂选自由以下组成的组:月桂基硫酸钠、聚氧乙烯氢化蓖麻油60、生育酚聚乙二醇琥珀酸酯(tpgs)及其混合物,并且表面活性剂的特别优选的实例是月桂基硫酸钠。
固体分散体
本发明的固体分散体包含化合物i、药学上可接受的聚合物和药学上可接受的阳离子物质。
在本文中,“固体分散体”意指药物的至少一部分分散在聚合物中。这样的固体分散体在本领域中有时被称为药物在聚合物中的“分子分散体”或“固溶体”。
在本文中,“晶体”意指化合物的特别固体形式,其在三维中显示出长程有序。术语“非晶形”是指不具有长程三维有序的材料,其不仅包括基本上无序的材料,而且还包括可以具有一定有序(小于三维的有序和/或在极短距离内的有序)的材料。材料的非晶形形式的另一个术语是材料的“非结晶”形式。
化合物i可以采取结晶形式或非晶形形式。在固体分散体中,化合物i的总重量的优选地至少约90重量%并且更优选地至少约95重量%是非晶形的。换言之,在固体分散体中,理想地,结晶形式的化合物i的量优选地不超过化合物i的总重量的约10重量%,并且更优选地不超过化合物i的总重量的约5重量%。
结晶的和非晶形的化合物i的量可以通过本领域已知的技术确定,诸如粉末x-射线衍射(xrpd)、扫描电子显微镜(sem)分析、固态nmr或包括差示扫描量热法(dsc)的热技术或者任何其他标准定量测量方法。
固体分散体中的非晶形化合物i可以以几种相存在。例如,其可以以化合物i的单相形式存在,或者以均匀地分散在整个聚合物中的固溶体形式存在,或者以这些状态的任何组合或介于它们之间的状态存在。优选地,化合物i和聚合物以固溶体形式存在。固溶体是热力学稳定的,并且在该溶体中化合物i以分子水平分散在聚合物中,即化合物i溶解。
当非晶形化合物i和聚合物的玻璃化转变温度(tg)相差多于约20℃时,可以通过测量固体分散体的tg来确定化合物i是否以固溶体的形式存在于聚合物中。如本文中使用的,tg是一种特征温度,在逐渐加热时玻璃状材料在该温度下经历从玻璃态到橡胶态的相对快速(即,在10-100秒内)的物理变化。非晶形材料(诸如聚合物或固体分散体)的tg可以通过多种技术诸如动态机械分析仪(dma)、膨胀仪、介电分析仪和dsc进行测量。尽管通过每种技术测量的精确值可以稍有不同,但是通常彼此之间落在10℃至30℃的范围内。当固体分散体显示出单一的tg时,分散体中单一非晶形相的化合物i的量通常小于约10重量%,这支持了分散体是大体上均匀的。
优选地,固体分散体显示出与使用的化合物i和聚合物各自的tg不同的至少一个tg,并且化合物i和聚合物的至少一部分以固溶体形式存在。
此外,从物理稳定性的方面来看,期望固体分散体具有高tg。根据friesen,5,mol.pharm.(2008)等,tg每增加10℃,固体分散体进行5%相分离所需的时间增加约十倍,并且当固体分散体的tg比储存温度高约30℃以上时,在该储存温度下在至少两年内将不会发生5%相分离(其可以被认为是物理稳定的)。例如,假设储存温度为30℃,如果固体分散体的tg为至少60℃以上、优选地80℃以上并且更优选地100℃以上,则可以预期在储存期间长期保持物理稳定性。
在本发明的固体分散体中,化合物i和聚合物的含量比取决于聚合物的性质并且没有特别限制,并且化合物i:聚合物(重量比)优选地在约1:100至约3:1之间宽泛地变化(例如,当仅化合物i和聚合物是组分时,化合物i的含量变为1重量%至75重量%)。化合物i:聚合物(重量比)的范围更优选为约1:9至约2:1、仍更优选约1:9至约1:1、甚至更优选约1:5至约1:1并且特别优选约1:2至约1:1。
在本文中,“约”意指优选地所描述的量±10%的范围,更优选地所描述的量±5%的范围,并且甚至更优选地所描述的量±1%的范围。
在本发明的固体分散体中,化合物i和阳离子物质之间的含量比没有特别限制,并且例如组合物中上述阳离子物质相对于化合物i的摩尔比优选地为0.8以上,并且化合物i:阳离子物质的摩尔比更优选为约1:0.8至1:10、甚至更优选约1:1至1:5并且特别优选约1:1至1:2。换言之,优选地,就摩尔比而言,阳离子物质相对于化合物i以略小、相等或过量的量存在。
相对于固体分散体的总重量,本发明的固体分散体优选地含有至少约1重量%的化合物i。在另一个方面,更优选地,相对于固体分散体的总重量,固体分散体理想地含有至少约5重量%、至少约10重量%、至少约15重量%、至少约20重量%、至少约25重量%、至少约30重量%、至少约35重量%、至少约40重量%、至少约45重量%或至少约50重量%的化合物i。
当本发明的固体分散体还含有表面活性剂时,化合物i和表面活性剂的含量比取决于表面活性剂的性质并且没有特别限制,并且化合物i:表面活性剂(重量比)优选地在约1:5至约5:1之间宽泛地变化。化合物i:表面活性剂(重量比)更优选地在约1:3至约3:1的范围内,并且甚至更优选地在约1:2至约2:1的范围内。
在又一个实施方案中,本发明的固体分散体可以由多个颗粒构成。上述颗粒中的每一个包含化合物i、药学上可接受的聚合物和药学上可接受的阳离子物质。这不同于化合物i颗粒和聚合物颗粒的简单的物理混合物。在优选的实施方案中,固体分散体由多个颗粒构成,并且每个颗粒包含化合物i、药学上可接受的阳离子物质和药学上可接受的聚合物。在这种情况下,优选地,化合物i、阳离子物质和聚合物为固溶体的形式。
用于制备固体分散体的方法
用于制备本发明的固体分散体的方法包括以下步骤:
(i)提供具有以下结构式的化合物i:
药学上可接受的聚合物和产生药学上可接受的阳离子物质的碱;
(ii)将化合物i、药学上可接受的聚合物和产生药学上可接受的阳离子物质的碱混合;以及
(iii)获得包含化合物i、药学上可接受的聚合物和药学上可接受的阳离子物质的固体分散体。
上述步骤(ii)和(iii)优选地分别包括以下步骤:
(ii-a)通过将化合物i、药学上可接受的聚合物和产生药学上可接受的阳离子物质的碱在溶剂中混合来制备混合溶液;以及
(iii-a)从步骤(ii-a)中获得的混合溶液中移除溶剂。
上述步骤(iii-a)优选地包括移除溶剂和通过喷雾干燥进行干燥的步骤。
在本发明的固体分散体中,固体分散体仅需要包括至少三种组分,即化合物i、药学上可接受的聚合物和药学上可接受的阳离子物质,并且在制备固体分散体的方法的上述步骤(i)中,提供化合物i的形式没有特别限制。
例如,化合物i可以以其游离形式提供到固体分散体中。备选地,化合物i可以以其药学上可接受的盐或水合物或溶剂化物的形式提供。
盐的实例包括无机酸盐、有机酸盐、无机碱盐、有机碱盐以及酸性或碱性氨基酸盐。
无机酸盐的优选实例包括盐酸盐、氢溴酸盐、硫酸盐、硝酸盐和磷酸盐。有机酸盐的优选实例包括乙酸盐、琥珀盐、富马酸盐、马来酸盐、酒石酸盐、柠檬酸盐、乳酸盐、硬脂酸盐、苯甲酸盐、甲磺酸盐、苯磺酸盐和对甲苯磺酸盐。无机碱盐的优选实例包括碱金属盐(诸如钠盐和钾盐)、碱土金属盐(诸如钙盐和镁盐)、铝盐和铵盐。有机碱盐的优选实例包括二乙胺盐、二乙醇胺盐、葡甲胺盐和n,n-二苄基乙二胺盐。酸性氨基酸盐的优选实例包括天冬氨酸盐和谷氨酸盐,并且碱性氨基酸盐的优选实例包括精氨酸盐、赖氨酸盐和鸟氨酸盐。
用于本发明的化合物i可以通过留在大气中而吸收水分并且成为水合物,并且这样的水合物也可以用作化合物i的原料。
另外,用于本发明的化合物i可以吸收某种其他溶剂以形成溶剂化物,并且这样的溶剂化物也可以用作化合物i的原料。
其中,化合物i优选地以其游离形式提供。
这样的化合物i可以通过wo2014/092061中描述的方法等获得。
此外,在本发明中,化合物i可以以结晶或非晶形形式提供。在结晶的情况下,化合物i的晶体包括多晶型物;然而,可以单独提供晶体形式,或者可以提供晶体形式的混合物。
如本文中的实施例中描述的,本发明人发现,用于本发明的化合物i的游离形式具有一些多晶型现象。结晶的多晶型物的实例包括由在14.4°、15.3°、16.6°和18.7°(±0.2°)的衍射角(2θ)处具有峰的粉末x-射线衍射图表征的晶体(在下文中称为“i型晶体”)和由在7.9°、13.5°、15.9°和21.8°(±0.2°)的衍射角(2θ)处具有峰的粉末x-射线衍射图表征的晶体(在下文中称为“ii型晶体”)。
作为药学上可接受的阳离子物质,可以使用上述阳离子物质,并且在上述步骤(i)中产生药学上可接受的阳离子物质的碱可以以固体形式提供或者以含有碱的溶液形式提供。作为优选的碱和阳离子物质,可以优选地使用与上述碱和阳离子物质类似的那些。
上述聚合物可以用作药学上可接受的聚合物,并且聚合物可以直接以固体形式或者通过将其预先溶解在溶剂中来提供到上述步骤(i)中。作为优选的聚合物,可以优选地使用与上述聚合物类似的那些。
当本发明的固体分散体还含有表面活性剂时,可以使用上述表面活性剂,并且在上述步骤(i)中,表面活性剂可以直接以固体形式或者通过将其预先溶解在溶剂中来提供。作为优选的表面活性剂,可以优选地使用与上述表面活性剂类似的那些。
在用于制备本发明的固体分散体的方法中,优选地,可以使用其中化合物i的至少一部分处于非晶形状态的任何常规方法。例如,当固体分散体由化合物i、药学上可接受的聚合物和产生药学上可接受的阳离子物质的碱形成时,作为溶剂工艺,可以使用非溶剂沉淀、冷冻干燥、喷涂、喷雾干燥等。作为熔融工艺,可以使用热熔挤出(hme)等。作为机械工艺,可以使用混合和粉碎等。
通常,溶剂工艺包括将至少一部分的药物、至少一部分的一种或多种聚合物组分和至少一部分的提供一种或多种阳离子物质的碱溶解在普通溶剂中的步骤。术语“溶剂”广义地使用并且包括溶剂的混合物。在本文中,“普通”意指溶剂(其可以是化合物的混合物)溶解至少一部分的药物和一种或多种聚合物。优选地,溶剂基本上溶解所有药物、所有聚合物和所有碱。
适用于溶剂处理的溶剂可以是药物、聚合物和碱相互可溶于其中的任何化合物。优选地,溶剂也是挥发性的,并且沸点为150℃以下。此外,溶剂应当具有相对较低的毒性,并且应当根据国际人用药品技术要求协调会(internationalcouncilforharmonizationoftechnicalrequirementsforpharmaceuticalsforhumanuse,ich)指南从固体分散体中移除至可接受的水平。将溶剂移除至该水平可能需要后续的处理步骤(诸如托盘干燥)。优选的溶剂包括:醇,诸如甲醇、乙醇、正丙醇、异丙醇和丁醇;酮,诸如丙酮、甲基乙基酮和甲基异丁基酮;酯,诸如乙酸乙酯和乙酸丙酯;以及各种其他溶剂,诸如乙腈、二氯甲烷、甲苯、1,1,1-三氯乙烷和四氢呋喃。在与挥发性溶剂的混合物中也可以少量使用低挥发性溶剂诸如二甲基乙酰胺或二甲亚砜。也可以以与水的混合物相同的方式使用溶剂的混合物(诸如50%甲醇和50%丙酮)。优选的溶剂是乙醇、甲醇、丙酮、四氢呋喃、乙酸乙酯、这些与水的混合物以及它们的混合物。此外,本发明的化合物i充分溶解在丙酮和四氢呋喃中,例如其在丙酮和水(体积比95:5)的混合溶液中的溶解度为50mg/ml以上,在四氢呋喃中的溶解度为80mg/ml以上,并且在四氢呋喃和甲醇(体积比95:5)的混合溶液中的溶解度为100mg/ml以上。特别地,四氢呋喃及其混合液可以被认为是优选的溶剂。
当化合物i、聚合物和提供阳离子物质的碱中的每一种的至少一部分溶解时,通过蒸发或通过与非溶剂混合来移除溶剂。示例性方法包括喷雾干燥、喷涂(锅包衣、流化床包衣等)、冷冻干燥以及通过将药物、聚合物和碱的溶液与co2、己烷、庚烷、适当ph的水或任何其他非溶剂快速混合进行沉淀。优选的是在移除溶剂之后获得大体上均匀的固体分散体。为了实现该目标,通常期望从溶液中快速移除溶剂,诸如通过其中溶液雾化并且药物和聚合物快速固化的过程。
喷雾干燥方法
可以通过喷雾干燥移除溶剂。术语“喷雾干燥”常规地使用,并且广泛地是指这样的方法,其涉及使液体混合物碎成小液滴(雾化)并且在喷雾干燥装置中将溶剂从混合物中快速移除,在所述喷雾干燥装置中存在用于将溶剂从液滴中蒸发的强驱动力。在perry’schemicalengineers’handbook(佩里化学工程师手册)第20-54至20-57页(第6版,1984)中一般地描述了喷雾干燥方法和喷雾干燥装置。在marshall,“atomizationandspray-drying(雾化和喷雾干燥)”,50chem.eng.prog.monogr.series2(1954)和masters,spraydryinghandbook(喷雾干燥手册)(第4版,1985)中概述了与喷雾干燥方法和装置有关的更多的细节。通常通过在干燥液滴的温度下保持喷雾干燥装置中溶剂的分压远低于溶剂的蒸气压来提供用于溶剂蒸发的强驱动力。这通过以下方式实现:(1)保持喷雾干燥装置中的压力处于部分真空(例如,0.01至0.50atm);或者(2)将液滴与温热干燥气体混合;或者(3)(1)和(2)二者。此外,可以通过加热喷雾溶液来提供溶剂蒸发所需的至少一部分热量。
可以在各种条件下将带有溶剂的进料喷雾干燥,但是仍产生具有可接受的性质的固体分散体。例如,可以使用各种类型的喷嘴将喷雾溶液雾化,由此将喷雾溶液作为小液滴的集合引入喷雾干燥室中。基本上任何类型的喷嘴都可以用于喷洒溶液,只要形成的液滴足够小,以使其足够干燥(通过溶剂蒸发),使得它们不粘附或覆盖喷雾干燥室壁即可。
最大液滴尺寸取决于喷雾干燥器中的尺寸、形状和流动模式而变化很大,但是当液滴离开喷嘴时,液滴的直径通常应小于约500μm。可以用于形成固体分散体的喷嘴类型的实例包括双流体喷嘴、喷泉型喷嘴、平扇型喷嘴、压力喷嘴和旋转雾化器。在优选的实施方案中,使用压力喷嘴。在2003年1月24日提交的美国公开专利申请号us2003/0185893中公开了细节,所述申请的公开内容通过引用结合于此。
可以在宽范围的温度和流速下将喷雾溶液递送至喷雾喷嘴或多个喷嘴。通常,喷雾溶液温度可以在刚好高于溶剂的凝固点至高于其环境压力沸点约20℃(通过对溶液加压)的任何范围内。在一些情况下,它可以是甚至更高的。到喷雾喷嘴的喷雾溶液流速取决于喷嘴类型、喷雾干燥器尺寸和喷雾干燥条件(例如,入口温度和干燥气体流速)可以在宽范围内变化。通常,在喷雾干燥方法中用于将溶剂从喷雾溶液中蒸发的能量主要来自干燥气体。
原则上,干燥气体基本上可以是任何气体,但是出于安全原因并且为了使固体分散体中药物或其他物质的不希望的氧化最小化,使用惰性气体诸如氮气、富含氮气的空气或氩气。通常将干燥气体在约60℃至约240℃的温度下引入干燥室中。
由于液滴的表面积与体积之比较大,并且用于溶剂蒸发的驱动力较大,因此液滴的固化时间很快。固化时间应小于约20秒、优选地小于约十秒并且更优选地小于一秒。这种快速固化对于使颗粒保持为均匀和均一的分散体而不是分离成富含药物的相和富含聚合物的相通常很重要。
固化后,固体粉末通常在喷雾干燥室中停留约5至60秒,以将溶剂从固体粉末中进一步蒸发。固体分散体在其离开干燥器时的最终溶剂含量应是低的,因为这降低了药物分子在固体分散体中的迁移率,从而改善其稳定性。通常,在其离开喷雾干燥室时,固体分散体的溶剂含量应小于10重量%、优选地小于2重量%并且更优选地小于1重量%。
成形后,可以使用合适的干燥方法干燥固体分散体以移除残留溶剂。实例包括托盘干燥、真空干燥、流化床干燥、微波干燥、带式干燥、旋转干燥和本领域已知的其他干燥方法。优选的二次干燥方法是真空干燥、托盘干燥等。为了使干燥期间化学降解最小化,可以在惰性气体(诸如氮气)下或在真空下进行干燥。
固体分散体通常为小颗粒的形式。颗粒的体积平均直径可以是小于500μm(直径)、或小于100μm(直径)、小于50μm(直径)或小于25μm(直径)。当通过喷雾干燥形成固体分散体时,所得分散体为这样的小颗粒的形式。
喷涂方法
在另一个实施方案中,通过将带有溶剂的进料溶液喷雾到种芯上来移除溶剂。种芯可以通过任何已知的方法(诸如熔融或喷雾凝结、挤出/滚圆、造粒和喷雾干燥)由任何合适的材料(诸如淀粉、微晶纤维素、糖或蜡)制造。可以使用药物领域中已知的包衣装置或造粒装置将进料溶液喷雾到这样的种芯上。实例包括锅包衣机(例如,可从日本东京的freundcorp.获得的hi-coater和可从英国利物浦的manesty获得的accela-cota)、流化床包衣机(例如,wurster包衣机或顶部喷雾器,可获自新泽西州拉姆齐的glattairtechnologies,以及瑞士布本多夫的niropharmasystems)和旋转造粒机(例如,可从freundcorp获得的cf-造粒机)。在该方法期间,种芯被进料溶液包被并且溶剂蒸发,得到含有固体分散体的包衣。如此形成的颗粒将具有与种芯密度类似的密度,并且可以改善组合物的加工和处理。
热熔挤出(hme)方法
熔融工艺可以使用常规的具有热源的搅拌器、捏合机等进行。此外,具有能够对其内部加压的结构的那些是更优选的。例如,可以使用在筒体中具有螺杆的挤出机(例如,单螺杆挤出机、双螺杆挤出机等)和注射成型机的注射装置(例如,双螺杆型挤出机)。其中,双螺杆型挤出机的注射装置是优选的(例如,可从美国马萨诸塞州的thermofisherscientific获得的pharma16双螺杆挤出机)。
在这种情况下,如有必要,将药物、聚合物和其他组分(诸如增塑剂和表面活性剂)从这些装置的料斗中装入保持在适当加热和熔融温度下的装置内部,并且旋转螺杆以将固体物质熔融并均匀混合。备选地,如有需要,可以在将其装入装置之前进行预混合。在本发明的制造方法中,压力、温度、粉末供给速率、模具直径、螺杆形状、螺杆转数等的设定条件根据药物、聚合物等的类型或者使用的装置的型号而不同,并且重要的是将它们组合以使温度保持在低于每种组分的分解温度,并且期望根据感兴趣的产品的特性改变这些设置。在熔融和捏合之后可以进行诸如冷却、固化和进一步的粉碎(如有需要)的处理,以获得本发明的固体分散体。
当本发明的固体分散体还包含表面活性剂时,表面活性剂可以共分散在本发明的固体分散体中,或者其可以在固体分散体之外以与其他添加剂相同的方式混合(合并)。当例如在上述溶剂工艺中共分散表面活性剂时,表面活性剂与化合物i、聚合物和碱一起溶解在溶剂中,并且通过喷雾干燥移除溶剂,这可以得到所需的固体分散体。另外,当通过在固体分散体外部涂覆来混合表面活性剂时,例如,使用常规掺合机/混合机等将表面活性剂与获得的固体分散体进行物理混合以制备期望的物理混合物。
制剂
如上所述获得的固体分散体可以直接作为药物组合物使用,或者在与药物制剂领域中使用的任何其他组分混合之后使用。
其他组分没有特别限制,只要它们是药学上可接受的即可,并且包括例如赋形剂以及如有必要的粘合剂、崩解剂、润滑剂、着色剂、调味剂、稳定剂、乳化剂、吸收促进剂、表面活性剂、ph调节剂、防腐剂和抗氧化剂。
包含本发明的固体分散体的药物组合物可以通过常用方法配制成片剂、散剂、细粒剂、颗粒剂、包衣片剂、胶囊剂、锭剂、栓剂、软膏剂、眼用软膏剂、泥罨剂等。通常,其优选地配制成可以口服施用的口服制剂。对于制剂,可以使用常用的赋形剂、粘合剂、润滑剂、着色剂、矫味剂以及如有需要的稳定剂、乳化剂、吸收增强剂、表面活性剂、ph调节剂、防腐剂、抗氧化剂等。通过将通常用作用于药物制剂的原料的组分组合通过常规方法来配制制剂。
例如,为了制备口服制剂,赋形剂以及其他的如有需要的粘合剂、崩解剂、润滑剂、着色剂、矫味剂、稳定剂、乳化剂、吸收增强剂、表面活性剂、ph调节剂、防腐剂、抗氧化剂等添加到本发明的固体分散体中,然后通过常规方法将其制成散剂、细粒剂、颗粒剂、片剂、包衣片剂、胶囊剂等。
赋形剂的实例包括乳糖、玉米淀粉、白糖、葡萄糖、甘露醇、山梨糖醇、淀粉、结晶纤维素、二氧化硅和偏硅酸铝镁。
粘合剂的实例包括聚乙烯醇、聚乙烯醚、甲基纤维素、乙基纤维素、阿拉伯胶、黄蓍胶、明胶、虫胶、羟丙基甲基纤维素、羟丙基纤维素、聚乙烯吡咯烷酮和聚丙二醇-聚氧乙烯嵌段聚合物。
崩解剂的实例包括淀粉、预胶化淀粉、琼脂、明胶粉、结晶纤维素、碳酸钙、氯化钠、碳酸氢钠、柠檬酸钙、硅酸酐、糊精、果胶、羧甲纤维素、羧甲纤维素钙和交联羧甲纤维素钠、低取代的羟丙基纤维素和羧甲基淀粉钠。
润滑剂的实例包括硬脂酸镁、硬脂酸钙、滑石、聚乙二醇、二氧化硅和氢化植物油。
作为着色剂,使用批准用作药物添加剂的那些。作为矫味剂,使用可可粉、薄荷脑、芳香散(empasm)、薄荷油、冰片和桂皮粉。
显然,这些片剂和颗粒剂可以是糖衣的或者如有需要以其他方式适当包衣的。
如在wo2014/092061和wo2015/189901中描述的,化合物i具有强pth样作用和高代谢稳定性,并且其使得能够治疗可以通过pth样作用治疗的病理病况(包括甲状旁腺功能减退)。此外,通过非侵入性全身暴露或局部暴露,诱导了骨/软骨合成代谢,并且其还可以提供用于预防以下各项、治疗以下各项、从以下各项中恢复和促进以下各项的愈合的方法:骨质疏松、牙周病中的骨量减少、拔牙后的牙槽骨缺损、骨关节炎、关节软骨缺损、再生障碍性骨病、软骨发育不全、低软骨形成症、骨软化症、骨折等。
本发明的药物组合物的剂量可以根据症状程度、年龄、性别、体重、施用形式、盐的类型、疾病的具体类型等进行适当选择。
剂量根据患者的疾病类型、症状水平、患者的年龄、性别和药物敏感性等而显著不同,但是用于成人的剂量通常为约0.03至1000mg/天、优选地0.1至500mg/天或更优选地0.1至100mg/天,一天施用一次或一天分几份施用。
溶出测试
在优选的实施方案中,本发明的固体分散体在体外溶出测试中提供了良好的溶出度。特别地,已知在使用人工模拟肠液(禁食状态的模拟肠液:fassif)作为测试溶液的体外溶出测试中改善的溶解度与良好的体内生物利用度(换言之,血药浓度增加)相关。例如,takano,23,pharm.res.(2006)等已经报道了口服施用时的消化道吸收率(fa),其根据使用上述fassif的体外溶出测试中对十二种模型药物的溶出曲线计算,显示出与由人体体内测试获得的fa的良好相关性。
合适的fassif是例如含有28.7mm磷酸二氢钾(kh2po4)、103.3mmkcl、3mm牛磺胆酸钠和0.8mml-α-磷脂酰胆碱并且使用naoh调节至ph6.5的水溶液。
体外溶出测试可以例如通过以下方式进行:将过量的本发明的组合物施加至加热至37℃的fassif溶液中,在使用桨式溶出测试器(例如,可从美国加利福尼亚的varianmedicalsystems获得的vk7010)进行搅拌的同时连续地取样,用0.45μmpvdf过滤器过滤样品,然后通过hplc对所得滤液中的化合物浓度进行定量。
在一个实施方案中,本发明的组合物在体外溶出测试中提供了良好的药物溶出度。这是在体外溶出测试的测试时间(例如至多120分钟)期间由对照组合物提供的溶出度的至少两倍。例如,当由对照组合物提供的溶出度在120分钟内为至多50μg/ml时,本发明的组合物在至少120分钟内提供至多100μg/ml的溶出度。更优选地,本发明的组合物提供了由对照组合物提供的溶出度的至少三倍或在一些情况下至少五倍以上的药物溶出度。本发明的组合物是包含药物、聚合物和阳离子物质的固体分散体,并且对照组合物包括结晶或非晶形形式的单一药物、仅含有药物和聚合物的固体分散体、仅含有药物和阳离子物质的分散体等。另外,当本发明的组合物包含影响药物溶解度的表面活性剂等时,包括含有相同量的相同组分的上述对照组合物。此外,除了体外溶出测试的测试时间内的最大药物溶出浓度以外,比较测试结束时(例如,120分钟)的药物浓度也是优选的。这是因为在假定具有良好的体内生物利用度时,考虑到在消化道中的保留等,甚至在相对较长的时间内保持高溶出度是优选的。
实施例
在下文中,将参照实施例更详细地描述本公开。然而,本公开可以在各种实施方案中实现,并且不被解释为限制于本文中描述的实施例。在下述实施例和比较例中,用作起始材料的化合物i是其游离形式的i型晶体或ii型晶体。在实施例1至12和比较例1至8中使用ii型晶体,并且在实施例13和14中使用i型晶体。
实施例1
该实施例描述了含有化合物i、作为聚合物的聚乙烯吡咯烷酮和作为阳离子物质的钠阳离子的固体分散体的形成。聚乙烯吡咯烷酮是n-乙烯基-2-吡咯烷酮的聚合物并且是非离子聚合物。
实施例1
将重量比为1:2的化合物i和聚乙烯吡咯烷酮(kollidon30,basf)(化合物33.3%和聚合物66.7%)在室温下以约12%固体含量浓度(就化合物i浓度而言为4%)溶解在乙醇(junseichemical)和2mnaoh水溶液(wakopurechemicals)(体积比为95:5)的混合物中。接下来,将获得的溶液用小型喷雾干燥器b-290(buchi)喷雾干燥以获得固体分散体(化合物i:钠摩尔比为1:1.6)。
例如,当制备2.4-g批料时,将19ml的乙醇和1ml的2mnaoh水溶液在玻璃烧杯中混合,并且在搅拌的同时添加0.8g的化合物i以将其溶解,然后在搅拌的同时添加1.6g的聚乙烯吡咯烷酮以将其溶解。将该溶液用小型喷雾干燥器b-290喷雾干燥,其中入口温度为50至70℃(出口温度为35至55℃),吸气器输出为100%,蠕动泵输出为20%,并且喷雾空气流量计高度为25至30mm。将喷雾干燥后回收的粉末在真空干燥器中在60℃干燥过夜,并且将所得粉末用作固体分散体。
表1示出了如下评价获得的固体分散体的理化性质的结果。使用用于测量x-射线粉末衍射(xrpd)的装置评价结晶状态,使用高效液相色谱(hplc)评价药物含量,使用用于测量热重(tg)的装置评价水含量,使用核磁共振(nmr)波谱仪评价残留溶剂,并且使用用于差示扫描量热(dsc)的装置评价玻璃化转变点。
xrpd结果显示出非晶形材料的光晕图案特征,表明化合物i以非晶形形式存在于固体分散体中。dsc结果显示出单一且非常高的玻璃化转变点(184℃)。其他性质即药物含量、水含量和残留溶剂如下所示。注意到当将与该实施例中相同的方式制备的固体分散体在40℃和30%rh的条件下储存六个月并且然后进行xrpd测量时,并未发现结晶峰,并且分散体具有足够的物理稳定性。
表1
实施例2和3
实施例2和3描述了这样的情况,其中钠阳离子用作阳离子物质并且羟丙基纤维素或甲基丙烯酸共聚物ld用作聚合物以通过与实施例1中相同的制备方法形成固体分散体。
实施例2
制备含有化合物i、作为聚合物的羟丙基纤维素(nissohpc-sl,nipponsoda)和作为阳离子物质的钠阳离子的固体分散体。羟丙基纤维素是具有纤维素主链的非离子聚合物,并且具有羟丙基基团作为官能团。制备条件与实施例1中相同。(化合物i:聚合物重量比为1:2,并且化合物i:钠摩尔比为1:1.6)
实施例3
制备含有化合物i、作为聚合物的甲基丙烯酸共聚物ld(eudragitl100-55,evonik)和作为阳离子物质的钠阳离子的固体分散体。甲基丙烯酸共聚物ld是离子(酸性)聚合物,并且是甲基丙烯酸和丙烯酸乙酯的共聚物。作为喷雾溶剂,使用甲醇(junseichemical)和2mnaoh水溶液(体积比为94.5:5.5)的混合物,并且其他条件与实施例1中相同。(化合物i:聚合物重量比为1:2,并且化合物i:钠摩尔比为1:1.8)
比较例1和2
比较例1和2描述了这样的情况,其中改变了作为活性药物成分的化合物i的形式。众所周知盐形成(游离形式晶体到盐晶体)和非晶化(游离形式晶体到非晶形游离形式)是用于改善溶解度的手段,并且尝试使用这些形式改善溶解度。
比较例1
该比较例描述了化合物i的苯磺酸盐。在80℃,将化合物i(4.0g)溶解在乙酸(40ml)和2mol/l苯磺酸水溶液(3.78ml)的混合物中。向该溶液中添加甲基乙基酮(20ml),并且将混合物搅拌十分钟,冷却至40℃,然后搅拌十分钟。在冷却至室温后,添加甲基乙基酮(20ml)并且将混合物搅拌十分钟。进一步添加甲基乙基酮(40ml),之后将沉淀的固体过滤并干燥。将干燥的固体(4.17g)悬浮在甲基乙基酮(60ml)中,并且在40℃搅拌两小时。此外,添加乙酸(2ml)和水(2ml)并且在40℃搅拌17小时,得到晶体,将其过滤并干燥以获得化合物i的苯磺酸盐晶体(3.75g)。
比较例2
该比较例描述了非晶形化合物i(非晶形游离形式)。将化合物i在室温下以约4%的浓度溶解在四氢呋喃(junseichemical)和水(体积比85:15)的混合物中。将获得的溶液用小型喷雾干燥器b-290喷雾干燥,其中入口温度为50至70℃(出口温度为35至55℃),吸气器输出为100%,蠕动泵输出为20%,并且喷雾空气流量计高度为25至30mm。将喷雾干燥后回收的粉末在真空干燥器中在室温下干燥过夜以获得非晶形游离形式。通过xrpd确认获得的粉末是非晶形的。
比较例3至5
比较例3至5描述了用化合物i和聚合物形成的固体分散体的形成。众所周知其中药物以非晶形状态分散在聚合物基质中的固体分散体是除了上述手段以外用于改善溶解度的手段,并且尝试使用该形式改善溶解度。
比较例3
该比较例描述了含有化合物i和作为聚合物的聚乙烯吡咯烷酮的固体分散体的形成。将重量比为1:2的化合物i和聚乙烯吡咯烷酮(33.3%化合物和66.7%聚合物)在室温下以约12%固体含量浓度(化合物i浓度为4%)溶解在四氢呋喃和甲醇(体积比70:30)的混合物中。接下来,将获得的溶液用小型喷雾干燥器b-290喷雾干燥。喷雾干燥条件与比较例2中相同。将喷雾干燥后回收的粉末在真空干燥器中在60℃干燥过夜以获得固体分散体。通过xrpd确认获得的粉末是非晶形的。
比较例4
该比较例描述了含有化合物i和作为聚合物的羟丙基纤维素的固体分散体的形成。制备条件与比较例3中相同(化合物i:聚合物重量比为1:2)。通过xrpd确认获得的粉末是非晶形的。
比较例5
该比较例描述了含有化合物i和作为聚合物的乙酸琥珀酸羟丙甲纤维素(shin-etsuaqoat-lf,shin-etsuchemical)的固体分散体的形成。乙酸琥珀酸羟丙甲纤维素是具有纤维素骨架的离子(酸性)聚合物,并且具有乙酰基和琥珀酰基基团作为官能团。使用四氢呋喃和水(体积比为85:15)的混合物作为喷雾溶剂,并且其他条件与比较例3中相同(化合物i:聚合物重量比为1:2)。通过xrpd确认获得的粉末是非晶形的。
比较例6
比较例6描述了其中在比较例2所示的化合物i的非晶化期间添加阳离子物质的情况,换言之,其中与实施例1至3相比未添加聚合物的情况。
比较例6
该比较例描述了含有化合物i和作为阳离子物质的钠阳离子的非晶形物质的形成。将化合物i在室温下以约4%的化合物i浓度溶解在乙醇和2mnaoh水溶液(体积比为95:5)的混合物中。将获得的溶液用小型喷雾干燥器b-290喷雾干燥,其中入口温度为50至70℃(出口温度为35至55℃),吸气器输出为100%,蠕动泵输出为20%,并且喷雾空气流量计高度为25至30mm(化合物i:钠摩尔比为1:1.6)。将喷雾干燥后回收的粉末在真空干燥器中在室温下干燥过夜。通过xrpd确认获得的粉末是非晶形的。
比较例7
比较例7描述了其中将聚乙烯吡咯烷酮聚合物与与比较例6中相同的组分物理混合的情况,或者更具体地,其中组合物与实施例1中相同但是化合物i和聚合物不形成固体分散体的情况。
比较例7
称量与比较例6中相同的组分(含有钠的非晶形化合物i(化合物i:钠摩尔比为1:1.6))和聚乙烯吡咯烷酮以将化合物i和聚合物的比率调整至1:2的重量比(33.3%化合物和66.7%聚合物),并且使用研钵和研杵将其混合。
对比较例1至7中制备的盐晶体、非晶形材料和固体分散体和实施例1至3中制备的本申请的固体分散体进行溶出测试的结果在图1至3中示出。向装入样品,使得20mg的化合物i将添加至温热至37℃的50ml的人工模拟肠液(禁食状态的模拟肠液(fassif))中(化合物浓度为400μg/ml)。在使用小型溶出测试器vk7010(varian)以50rpm的桨叶转速搅拌该混合物的同时,随时间(5、10、15、20、25、30、45、60和120分钟)取样,将这些用0.45-μmpvdf过滤器过滤,并且通过hplc对获得的滤液中的化合物i浓度进行定量。注意到在n=2至4的情况下测量相同样品中的每一个,并且在图中示出平均值(平均浓度)±标准偏差。
此外,为了评价游离形式的化合物i的i型晶体在fassif中的溶解度,将过量的游离形式的化合物i的i型晶体放入fassif中,并且在37℃浸泡和搅拌24小时后,对其进行离心。当通过hplc对分离的上清液中的化合物i浓度进行定量时,发现溶解度为1μg/ml。另外,以相同方式评价的游离形式的化合物i的ii型晶体的溶解度为2μg/ml。
根据溶出测试结果,在比较例1和2(图1)和比较例3至5(图2)中的任一个中,120分钟时的溶解度为至多约50μg/ml。这意味着与游离形式的i型晶体相比,确认溶解度提高约50倍。同时,实施例1至3的固体分散体的结果(图3)显示出高得多的溶解度。在120分钟时,与比较例3至5中的那些(相当于常规固体分散体)相比,它们显示出至多8倍高的溶解度;另外,与游离形式的i型晶体的溶解度相比,它们显示出至多236倍的溶解度改善效果。此外,尽管比较例3和实施例1使用相同的聚合物组分,并且比较例4和实施例2使用相同的聚合物组分,但是本实施例的固体分散体具有4至8倍高的溶解度。此外,在图3中,比较例6(其是仅含有钠的固体分散体的情况)和比较例7(其涉及将聚合物物理混合)中的溶解度结果与比较例1至5的那些相当。在这些情况下,用聚合物或阳离子物质形成的两组分体系不能显示出本实施例中观察到的高效果,并且仅当将组成要求中的三组分合并到固体分散体中时才观察到高溶出度改善效果。
实施例4至6和比较例8
实施例4至6和比较例8描述了这样的情况,其中将表面活性剂添加至固体分散体中。使用钠阳离子作为阳离子物质,并且使用羟丙基纤维素、共聚维酮和乙酸琥珀酸羟丙甲纤维素作为聚合物,使用与实施例1中相同的制备方法形成固体分散体,然后将用作表面活性剂的月桂基硫酸钠合并。
实施例4
如实施例1中那样制备含有化合物i、作为聚合物的羟丙基纤维素和作为阳离子物质的钠阳离子的固体分散体(化合物i:聚合物重量比为1:2,化合物i:钠摩尔比为1:1.6)。称量获得的固体分散体和月桂基硫酸钠(nikkolsls,nikkochemicals)称量,并且用研钵和研杵混合,使得月桂基硫酸钠的混合比变为化合物i的量的一半(化合物i:聚合物:月桂基硫酸钠=1:2:0.5)(重量比))。
实施例5
如实施例1中那样制备含有化合物i、作为聚合物的共聚维酮(kollidonva64,basf)和作为阳离子物质的钠阳离子的固体分散体(化合物i:聚合物重量比为1:2,化合物i:钠摩尔比为1:1.6)。共聚维酮是n-乙烯基-2-吡咯烷酮和乙酸乙烯酯的共聚物,并且是非离子聚合物。称量月桂基硫酸钠,并且添加至获得的固体分散体中,使得其混合比将是化合物i的量的一半(化合物i:聚合物:月桂基硫酸钠=1:2:0.5)(重量比)),并且使用研钵和研杵将其混合。
实施例6
制备含有化合物i、作为聚合物的乙酸琥珀酸羟丙甲纤维素和作为阳离子物质的钠阳离子的固体分散体。将其在室温下以约6%的固体含量浓度(就化合物i浓度而言为2%)溶解在四氢呋喃、乙醇、水和0.5mnaoh水溶液(wakopurechemicals)(重量比为50:20:20:10)的混合物中。应用的其他条件与实施例1中相同(化合物i:聚合物重量比为1:2,化合物i:钠摩尔比为1:1.6)。称量月桂基硫酸钠,并且添加至获得的固体分散体中,使得其混合比将是化合物i的量的一半(化合物i:聚合物:月桂基硫酸钠=1:2:0.5)(重量比)),并且使用研钵和研杵将其混合。
比较例8
如比较例6中那样制备含有化合物i和作为阳离子物质的钠阳离子的固体分散体(化合物i:钠摩尔比为1:1.6)。称量月桂基硫酸钠,并且添加至获得的固体分散体中,使得其混合比将是化合物i的量的一半(化合物i:月桂基硫酸钠=1:0.5)(重量比)),并且使用研钵和研杵将其混合。
溶出测试结果
对实施例4至6和比较例8中制备的含有表面活性剂的固体分散体进行溶出测试的结果在图4中示出。注意到在所有样品中,通过xrpd仅观察到来源于月桂基硫酸钠的峰,并且确认化合物i是非晶形的。从溶出度的结果来看,在120分钟时间点时,与其中将月桂基硫酸钠添加至不含有聚合物的固体分散体的比较例8相比,在其中将月桂基硫酸钠添加至含有聚合物的固体分散体的实施例4至6的情况中显示出约2至5倍高的溶解度。因此,表明可以将表面活性剂添加至上述三组分固体分散体以改善粉末的湿润性等(而不损害高溶解度)。
实施例7和8
实施例7和8描述了这样的情况,其中以不同于实施例1的化合物:聚合物比率,作为阳离子物质的钠阳离子和作为聚合物的聚乙烯吡咯烷酮形成固体分散体。
实施例7
制备含有化合物i、作为聚合物的聚乙烯吡咯烷酮和作为阳离子物质的钠阳离子的固体分散体。将重量比为1:1.5的化合物i和聚合物(40%化合物和60%聚合物)在室温下以约10%固体含量浓度(就化合物i浓度而言为4%)溶解在乙醇和2mnaoh水溶液(体积比为95:5)的混合物中。其他条件与实施例1中相同(化合物i:钠摩尔比为1:1.6)。
实施例8
制备含有化合物i、作为聚合物的聚乙烯吡咯烷酮和作为阳离子物质的钠阳离子的固体分散体。将重量比为1:1的化合物i和聚合物(50%化合物和50%聚合物)在室温下以约8%固体含量浓度(就化合物i浓度而言为4%)溶解在乙醇和2mnaoh水溶液(体积比为95:5)的混合物中。其他条件与实施例1中相同(化合物i:钠摩尔比为1:1.6)。
溶出测试结果
对实施例7和8中制备的固体分散体进行溶出测试的结果和用作比较的实施例1的结果在图5中示出。通过xrpd确认所有样品都是非晶形的。即使当改变化合物:聚合物重量比时,结果也显示出如实施例1中那样的高溶解度。
实施例9和10
实施例9和10描述了这样的情况,其中以与实施例7不同的钠摩尔比,作为阳离子物质的钠阳离子和作为聚合物的聚乙烯吡咯烷酮形成固体分散体。除了其中钠摩尔比为1.8的实施例3以外,在实施例1至8中以相对于化合物i的1.6的钠摩尔比制备喷雾溶液,并且尝试形成具有较低钠比例的固体分散体。
实施例9
制备含有化合物i、作为聚合物的聚乙烯吡咯烷酮和作为阳离子物质的钠阳离子的固体分散体。将重量比为1:1.5的化合物i和聚合物在室温下以约2.5%固体含量浓度(就化合物i浓度而言为1%)溶解在乙醇和5mnaoh水溶液(wakopurechemicals)(体积比为99.7:0.3)的混合物中(在喷雾溶液中,钠与化合物i的摩尔比为1.1)。其他条件与实施例1中相同。
实施例10
制备含有化合物i、作为聚合物的聚乙烯吡咯烷酮和作为阳离子物质的钠阳离子的固体分散体。将重量比为1:1.5的化合物i和聚合物在室温下以约10%固体含量浓度(就化合物i浓度而言为4%)溶解在乙醇和5mnaoh水溶液(体积比为98.4:1.6)的混合物中(在喷雾溶液中,钠与化合物i的摩尔比为1.3)。其他条件与实施例1中相同。
对于实施例7、9和10中制备的固体分散体,通过离子色谱法进行钠定量,并且通过hplc进行化合物定量。结果,确认所有固体分散体均以对于制备设定的摩尔比在其中含有钠。此外,对这些固体分散体进行溶出测试的结果在图6中示出。结果表明即使当改变钠的摩尔比时也具有与先前实施例中相同的高溶解度。注意到通过xrpd确认获得的粉末是非晶形的。
实施例11至14
实施例11至14描述了这样的情况,其中使用除了钠阳离子以外的各种阳离子物质形成固体分散体。作为阳离子物质,检查了钾阳离子和精氨酸阳离子。
实施例11
制备含有化合物i、作为聚合物的聚乙烯吡咯烷酮和作为阳离子物质的钾阳离子的固体分散体。将重量比为1:2的化合物i和聚合物在室温下以约12%固体含量浓度(就化合物i浓度而言为4%)溶解在乙醇和1mkoh水溶液(wakopurechemicals)(体积比为90:10)的混合物中。其他条件与实施例1中相同(化合物i:钾摩尔比为1:1.6)。
实施例12
制备含有化合物i、作为聚合物的羟丙基纤维素和作为阳离子物质的钾阳离子的固体分散体。将重量比为1:2的化合物i和聚合物在室温下以约12%固体含量浓度(就化合物i浓度而言为4%)溶解在乙醇和1mkoh水溶液(体积比为90:10)的混合物中。其他条件与实施例1中相同(化合物i:钾摩尔比为1:1.6)。
对实施例11和12中制备的固体分散体进行溶出测试的结果在图7中示出。结果表明即使当使用钾阳离子作为阳离子物质时,溶解度也高于不含有阳离子物质的两组分固体分散体(诸如比较例3至6中的那些)的溶解度。
实施例13和14
实施例13和14描述了这样的情况,其中使用精氨酸阳离子作为阳离子物质来形成固体分散体。在这些实施方案中,尝试通过热熔挤出进行制备,而不是通过上述喷雾干燥方法进行制备。
实施例13
制备含有化合物i、作为聚合物的共聚维酮和作为阳离子物质的精氨酸阳离子的固体分散体。将在研钵中以1:1:8的重量比进行物理混合的化合物i、l-精氨酸(wakopurechemicals)和聚合物放入小型双螺杆热熔挤出机ze-9(three-tec)中。在螺杆旋转速度20rpm和机筒温度(h1/h2/h3=150/220/210℃)的条件下对混合物进行热熔挤出。将获得的棒状样品用样品磨粗略地粉碎,并且将获得的粒状粉末制成固体分散体(化合物i:精氨酸摩尔比为1:3.6)。
实施例14
制备含有化合物i、作为聚合物的聚乙烯醇(parteckmxp,merck)和作为阳离子物质的精氨酸阳离子的固体分散体。聚乙烯醇是乙烯醇的非离子聚合物。其他条件与实施例13中相同(化合物i:聚合物重量比为1:8,化合物i:精氨酸摩尔比为1:3.6)。
对实施例13和14中制备的固体分散体进行溶出测试的结果在图8中示出。结果表明即使当使用精氨酸阳离子作为阳离子物质时,溶解度也高于不含有阳离子物质的两组分固体分散体(诸如比较例3至6中的那些)的溶解度。另外,结果表明不仅可以通过喷雾干燥方法制备固体分散体,而且可以通过热熔挤出制备固体分散体。
实施例15体内吸收度测试
该实施例描述了对在体外溶出测试中显示出高溶出度的实施例进行的体内(比格犬)测试的结果。评价口服施用组合物(如下面描述的那些)时化合物i的暴露(cmax和auc)。
组合物a:使用填充有与比较例1相同的苯磺酸盐晶体和不帮助溶出的各种添加剂的明胶胶囊(尺寸#00)。
组合物b:使用填充有与实施例1相同的固体分散体的明胶胶囊(尺寸#00)。
组合物c:使用填充有与实施例2相同的固体分散体的明胶胶囊(尺寸#00)。
组合物d:使用填充有与月桂基硫酸钠混合的与实施例5相同的固体分散体的明胶胶囊(尺寸#00)。
所使用的动物物种是12至18月龄的比格犬(雄性),并且作为预处理,在开始口服施用前一小时,在后肢中以6μg/kg的剂量肌内施用0.25ml/kg的五肽胃泌素(sigma-aldrich)。将含有组合物的胶囊口服施用至在施用前一天的17:00移除残留食物的禁食动物,然后使用导管对动物口服施用自来水(以50ml/动物)并且用10ml空气冲洗。在施用之前和在施用后0.083、0.5、1、2、4、7和24小时时从前臂头静脉中收集血液(0.6ml)。将收集的血液立即离心(12,000rpm),并且通过hplc-ms/ms对获得的血浆中的化合物i浓度进行定量。
表2显示了体内测试的剂量(剂量)、n数量(使用的狗的数量)和结果(cmax、aucinf和生物利用度)。换言之,表2显示了使用实施例1、2和5以及比较例1进行的体内(比格犬)测试的结果,并且显示了当口服施用每种组合物时的化合物i的暴露(cmax、auc和生物利用度)。
在本文中,cmax是观察到的血浆中化合物i的最大浓度,并且aucinf是血浆中化合物i浓度相对于时间的曲线下面积。这些值是相对于进行施用的狗的数量的平均值。通过aucinf除以剂量(相对于通过给予不同狗组的2mg/kg静脉内施用获得的剂量)的百分比(%)计算得到生物利用度。
表2
根据测试结果,施用实施例的固体分散体的组展现出比施用含有化合物i的苯磺酸盐晶体的组合物的组更高的吸收度(暴露),并且观察到对于cmax的约1.4至2.6倍的改善和对于aucinf的1.9至3.7倍的改善。这表明本发明的固体分散体不仅改善了体外溶出度,而且还改善了体内生物利用度。
制备实施例1:游离形式的化合物i的i型晶体的制备
将化合物i(1g)悬浮在乙醇(8ml)和水(2ml)的混合物中,并且在75℃搅拌三小时。在将悬浮液冷却至45℃后,将其再搅拌一天。将获得的晶体过滤并且干燥以获得化合物i的i型晶体(0.9g)。
i型晶体具有优异的储存稳定性。
制备实施例2:游离形式的化合物i的ii型晶体的制备
将化合物i溶解在其量10倍(v/w)的二甲亚砜中,分配到小瓶中,然后冷冻干燥。向通过冷冻干燥获得的固体中添加其量五倍(v/w)的1-丁醇,并且将混合物搅拌一周,以获得化合物i的ii型晶体。
制备实施例1和2中获得的晶体的xrpd在以下条件下测量:
测量设备:smartlab(由rigaku制造)
对阴极:cu
管电压:45kv
管电流:200ma
扫描范围:5°至35°
取样宽度:0.02°
图9显示出i型晶体的测量结果,并且图10显示出ii型晶体的测量结果。
工业适用性
本发明的固体分散体能够使得1-(3,5-二甲基-4-(2-((4-氧代-2-(4-(三氟甲氧基)苯基)-1,3,8-三氮杂螺[4.5]癸-1-烯-8-基)磺酰基)乙基)苯基)-5,5-二甲基咪唑烷-2,4-二酮(一种具有强pth样作用和高代谢稳定性的水溶性差的化合物)的溶解度得到很大改善并且在消化道中的吸收度得到伴随的改善。
- 还没有人留言评论。精彩留言会获得点赞!