用作神经保护剂的醌还原酶2抑制剂
用作神经保护剂的醌还原酶2抑制剂
背景技术:
1.以氯喹(cq)和羟基氯喹(hq)为原型的氨基喹啉类是最初被开发用于治疗疟疾的醌还原酶2(qr2)抑制剂,但后来被发现对其他适应症有治疗效果,包括,尤其是,自身免疫性疾病,如系统性红斑狼疮(sle)和类风湿性关节炎(ra)。singer等人,"update on immunosuppressive therapy," curr. opin. rheumatol. 1998, 10:169
‑
173;wallace, "the use of chloroquine and hydroxychloroquine for non
‑
infectious conditions other than rheumatoid arthritis or lupus: a crucial review," lupus 1996, 5 suppl 1:s59
‑
64。对于sle和ra,氨基喹啉类是一线疗法的主要药物,且常与其他药物组合使用。氨基喹啉类不仅改善sle和ra的病征和症状,而且对脂质代谢有有益的效果,并减少血栓的发生。在患有炎症性或糜烂性骨关节炎的患者中观察到类似的益处。当用作移植物抗宿主病、癌症和hiv的辅助疗法时也显示了功效。savarino等人,"effects of chloroquine on viral infections: an old drug against today's diseases" lancet infect. dis. 2003, 3(11):722
‑
7;savarino等人,"risks and benefits of chloroquine use in anticancer strategies," lancet oncol. 2006, 7(10):792
‑
3;sotelo等人,"adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double
‑
blind, placebo
‑
controlled trial," ann. intern. med. 2006, 144(5):337
‑
43。
2.氯喹(cq)在神经保护中的潜力以前已经在卒中、兴奋性毒性和外伤性损伤的临床前模型中进行过研究,但其治疗机制仍不清楚。在永久性mca闭塞模型中,cq显著限制了向脑中损伤部位的小胶质细胞和pmn迁移,减少了反应性星形细胞胶质化和新生血管形成,且减少了60%的卒中量。giulian等人,"the role of mononuclear phagocytes in wound healing after traumatic injury to adult mammalian brain," j. neurosci. 1989, 9:4416
‑
4429; ivanova等人,"cerebral ischemia enhances polyamine oxidation: identification of enzymatically formed 3
‑
aminopropanal as an endogenous mediator of neuronal and glial cell death," j. exp. med. 1998, 188:327
‑
340。cq 还响应各种刺激在体外通过小胶质细胞减少了细胞因子的生成。giulian,"microglia and the immune pathology of alzheimer disease," am. j. hum. genet. 1999, 65:13
‑
18。
3.由于一些疟疾对cq耐药,还考察了衍生化合物。例如,ware 等人的us 2006/0074105描述了某些喹啉和喹唑啉衍生物,据说其在治疗疟疾和自身免疫性疾病中有用。
4.虽然cq和hq在临床上通常被用作自身免疫性病症的一线疗法,但它们的功效受限于严重的副作用。最重要和最具特征的毒性是视网膜毒性,除非剂量有限,否则长期使用可能导致“牛眼黄斑病变”和失明。心脏毒性虽然罕见,但也可能发生,表现为传导紊乱(如束支传导阻滞)和/或与充血性心力衰竭相关的心肌病。长期cq或hq疗法后心脏和视网膜活检的电子显微镜检查显示特殊的细胞质包涵体,被理解为是溶酶体(以及视网膜和皮肤中的黑素小体)高度药物累积的直接后果。值得注意的是,在治疗剂量期间,cq能够在皮肤、视网
膜、肾和肝细胞中累积至mm浓度,而血浆浓度保持<1
µ
m。
5.仍然需要开发另外的氨基喹啉醌还原酶2(qr2)抑制剂,特别是还具有减少的溶酶体累积以便降低毒性的那些。
技术实现要素:
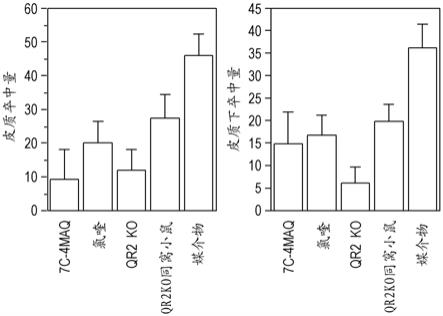
6.根据一些实施方案,本发明提供了一种在有需要的对象中治疗急性神经损伤的方法,包括向所述对象施用式i的化合物:其中:w为n或n
+
o
‑
;x为cr
14
或n;r1为h或三氟甲基;r2为nr7r8、or
11
、sr
12
或烷基;r3为h或or
13
;r4为h或甲氧基;r5为h、cl或三氟甲基;r6为h、nr9r
10
或三氟甲基;r7为h、c1‑5烷基、杂芳基烷基、环烷基、杂环烷基、杂环基、芳基、杂芳基、脲基、硫脲基、烯基、炔基、酰胺基、氨基、烷氧基、烷基氨基、烷基膦酸酯、烷基腈、卤代烷基或任选被下列基团取代的卤代烷基:c1‑5烷基、环烷基、环烯基、杂环烷基、杂芳基、烯基、炔基、酰胺基、烷氧基、烷基氨基、羟基烷基、卤素、羟基、羧酸酯、烷基羧酸酯、酰基叠氮、磺酰胺或卤代烷基;r8为h、c1‑5烷基、环烷基、杂环烷基、芳基、杂芳基、脲基、硫脲基、烯基、炔基、酰胺基、氨基、烷氧基、烷基氨基、烷基膦酸酯、烷基腈、卤代烷基或任选被下列基团取代的卤代烷基:c1‑5烷基、环烷基、环烯基、杂环烷基、杂芳基、烯基、炔基、酰胺基、烷氧基、羟基烷基、卤素、羟基、羧酸酯、烷基羧酸酯、酰基叠氮、磺酰胺或卤代烷基;r9为h、o、c1‑5烷基、环烷基、杂环烷基、芳基、杂芳基、烷基氨基,烷基腈或任选被下列基团取代的烷基膦酸酯:c1‑5烷基、环烷基、杂环烷基、芳基、杂芳基或烷基氨基;r
10
为h、o、c1‑5烷基、环烷基、杂环烷基、芳基、杂芳基、烷基氨基、烷基腈或任选被下列基团取代的烷基膦酸酯:c1‑5烷基、环烷基、杂环烷基、杂芳基或烷基氨基;r
11
为烷基、芳基或任选被下列基团取代的杂芳基:烷基、卤代烷基、芳基或杂芳基;r
12
为烷基、芳基或任选被下列基团取代的杂芳基:烷基、卤代烷基、芳基或杂芳基;
r
13
为烷基或任选被烷基或卤代烷基取代的芳基;以及r
14
为h或芳基;或其药学上可接受的盐或前药。
7.在一些实施方案中,w为n。在一些实施方案中,x为cr
14
。在一些实施方案中,r1为h。在一些实施方案中,r2为nr7r8。在一些实施方案中,r3为h。在一些实施方案中,r4为h。在一些实施方案中,r5为cl。在一些实施方案中,r6为h。在一些实施方案中,r7为h。在一些实施方案中,r8为被杂芳基取代的c1‑5烷基。在一些实施方案中,r
14
为h。
8.在一些实施方案中,所述化合物在大约ph4至ph5具有正的logd值。
9.在一些实施方案中,所述化合物为式i(a)化合物:其中r7和r8各自独立地为h或c1‑5烷基,其中所述c1‑5烷基任选被环烷基、杂环烷基、杂环基、芳基或杂芳基(其各自可进一步被任何合适的取代基取代,例如,c1‑5烷基、环烷基、环烯基、杂环烷基、杂芳基、烯基、炔基、酰胺基、烷氧基、烷基氨基、羟基烷基、卤素、羟基、羧酸酯、烷基羧酸酯、酰基叠氮、磺酰胺或卤代烷基)取代。
10.在一些实施方案中,r7和r8中的一个为h,且另一个为c1‑5烷基,其中所述c1‑5烷基任选被环烷基、杂环烷基、杂环基、芳基或杂芳基(其各自可进一步被任何合适的取代基取代,例如,c1‑5烷基、环烷基、环烯基、杂环烷基、杂芳基、烯基、炔基、酰胺基、烷氧基、烷基氨基、羟基烷基、卤素、羟基、羧酸酯、烷基羧酸酯、酰基叠氮、磺酰胺或卤代烷基)取代。
11.在一些实施方案中,所述化合物在大约ph4至ph5具有正的logd值。
12.还提供了一种在有需要的对象中治疗急性神经损伤的方法,包括向所述对象施用式ii的化合物:其中r'选自吡啶
‑2‑
基甲基、吡啶
‑3‑
基甲基、1
‑
苄基哌啶
‑4‑
基、4
‑
氰基
‑
2,2
‑
二乙基丁基、2
‑
氯环戊基、4
‑
(二乙基氨基)丁
‑2‑
基、1
‑
(呋喃
‑2‑
基)乙基、1
‑
环丙基乙基、1
‑
乙基哌啶
‑4‑
基、5
‑
氨基
‑
2,2
‑
二乙基戊基和2
‑
(二乙基磷酰基)
‑1‑
甲基乙基,或其药学上可接受的盐或前药。
13.在一些实施方案中,所述化合物在大约ph4至ph5具有正的logd值。
14.在一些实施方案中,急性神经损伤包括创伤性脑损伤。在一些实施方案中,急性神经损伤包括蛛网膜下腔出血。在一些实施方案中,急性神经损伤包括术后认知功能障碍。在一些实施方案中,急性神经损伤包括缺氧性脑损伤。在一些实施方案中,急性神经损伤包括缺血性脑损伤。
15.还提供如本文所教导的用于治疗急性神经损伤的方法中的活性化合物。进一步提供如本文所教导的活性化合物在制备用于治疗急性神经损伤的药物中的用途。在一些实施方案中,急性神经损伤包括创伤性脑损伤。在一些实施方案中,急性神经损伤包括蛛网膜下腔出血。在一些实施方案中,急性神经损伤包括术后认知功能障碍。在一些实施方案中,急性神经损伤包括缺氧性脑损伤。在一些实施方案中,急性神经损伤包括缺血性脑损伤。
16.进一步提供一种在有需要的对象中治疗血管性痴呆的方法,包括向所述对象施用式i的化合物:其中:w为n或n
+
o
‑
;x为cr
14
或n;r1为h或三氟甲基;r2为nr7r8、or
11
、sr
12
或烷基;r3为h或or
13
;r4为h或甲氧基;r5为h、cl或三氟甲基;r6为h、nr9r
10
或三氟甲基;r7为h、c1‑5烷基、杂芳基烷基、环烷基、杂环烷基、芳基、杂环基、杂芳基、脲基、硫脲基、烯基、炔基、酰胺基、氨基、烷氧基、烷基氨基、烷基膦酸酯、烷基腈、卤代烷基或任选被下列基团取代的卤代烷基:c1‑5烷基、环烷基、环烯基、杂环烷基、杂芳基、烯基、炔基、酰胺基、烷氧基、烷基氨基、羟基烷基、卤素、羟基、羧酸酯、烷基羧酸酯、酰基叠氮、磺酰胺或卤代烷基;r8为h、c1‑5烷基、环烷基、杂环烷基、芳基、杂芳基、脲基、硫脲基、烯基、炔基、酰胺基、氨基、烷氧基、烷基氨基、烷基膦酸酯、烷基腈、卤代烷基或任选被下列基团取代的卤代烷基:c1‑5烷基、环烷基、环烯基、杂环烷基、杂芳基、烯基、炔基、酰胺基、烷氧基、羟基烷基、卤素、羟基、羧酸酯、烷基羧酸酯、酰基叠氮、磺酰胺或卤代烷基;r9为h、o、c1‑5烷基、环烷基、杂环烷基、芳基、杂芳基、烷基氨基、烷基腈或任选被下
列基团取代的烷基膦酸酯:c1‑5烷基、环烷基、杂环烷基、芳基、杂芳基或烷基氨基;r
10
为h、o、c1‑5烷基、环烷基、杂环烷基、芳基、杂芳基、烷基氨基、烷基腈或任选被下列基团取代的烷基膦酸酯:c1‑5烷基、环烷基、杂环烷基、芳基、杂芳基或烷基氨基;r
11
为烷基、芳基或任选被下列基团取代的杂芳基:烷基、卤代烷基、芳基或杂芳基;r
12
为烷基、芳基或任选被下列基团取代的杂芳基:烷基、卤代烷基、芳基或杂芳基;r
13
为烷基或任选被烷基或卤代烷基取代的芳基;以及r
14
为h或芳基;或其药学上可接受的盐或前药。
17.在一些实施方案中,w为n。在一些实施方案中,x为cr
14
。在一些实施方案中,r1为h。在一些实施方案中,r2为nr7r8。在一些实施方案中,r3为h。在一些实施方案中,r4为h。在一些实施方案中,r5为cl。在一些实施方案中,r6为h。在一些实施方案中,r7为h。在一些实施方案中,r8为被杂芳基取代的c1‑5烷基。在一些实施方案中,r
14
为h。
18.在一些实施方案中,所述化合物在大约ph4至ph5具有正的logd值。
19.在一些实施方案中,所述化合物为式i(a)的化合物:其中r7和r8各自独立地为h或c1‑5烷基,其中所述c1‑5烷基任选被环烷基、杂环烷基、杂环基、芳基或杂芳基(其各自可进一步被任何合适的取代基取代,例如,c1‑5烷基、环烷基、环烯基、杂环烷基、杂芳基、烯基、炔基、酰胺基、烷氧基、烷基氨基、羟基烷基、卤素、羟基、羧酸酯、烷基羧酸酯、酰基叠氮、磺酰胺或卤代烷基)取代。
20.在一些实施例中,r7和r8中的一个为h,且另一个为c1‑5烷基,其中所述c1‑5烷基任选被环烷基、杂环烷基、杂环基、芳基或杂芳基(其各自可进一步被任何合适的取代基取代,例如,c1‑5烷基、环烷基、环烯基、杂环烷基、杂芳基、烯基、炔基、酰胺基、烷氧基、烷基氨基、羟基烷基、卤素、羟基、羧酸酯、烷基羧酸酯、酰基叠氮、磺酰胺或卤代烷基)取代。
21.在一些实施方案中,所述化合物在大约ph4至ph5具有正的logd值。
22.还提供了一种在有需要的对象中治疗血管性痴呆的方法,包括向所述对象施用式ii的化合物:
其中r'选自吡啶
‑2‑
基甲基、吡啶
‑3‑
基甲基、1
‑
苄基哌啶
‑4‑
基、4
‑
氰基
‑
2,2
‑
二乙基丁基、2
‑
氯环戊基、4
‑
(二乙基氨基)丁
‑2‑
基、1
‑
(呋喃
‑2‑
基)乙基、1
‑
环丙基乙基、1
‑
乙基哌啶
‑4‑
基、5
‑
氨基
‑
2,2
‑
二乙基戊基和2
‑
(二乙基磷酰基)
‑1‑
甲基乙基,或其药学上可接受的盐或前药。
23.在一些实施方案中,所述化合物在大约ph4至ph5具有正的logd值。
24.还提供如本文所教导的用于治疗血管性痴呆的方法中的活性化合物。进一步提供如本文所教导的活性化合物在制备用于治疗血管性痴呆的药物中的用途。
25.还进一步提供了一种在有需要的对象中治疗中枢神经系统狼疮(cns lupus)的方法,包括向所述对象施用式i的化合物:其中:w为n或n
+
o
‑
;x为cr
14
或n;r1为h或三氟甲基;r2为nr7r8、or
11
、sr
12
或烷基;r3为h或or
13
;r4为h或甲氧基;r5为h、cl或三氟甲基;r6为h、nr9r
10
或三氟甲基;r7为h、c1‑5烷基、杂芳基烷基、环烷基、杂环烷基、杂环基、芳基、杂芳基、脲基、硫脲基、烯基、炔基、酰胺基、氨基、烷氧基、烷基氨基、烷基膦酸酯、烷基腈、卤代烷基或任选被下列基团取代的卤代烷基:c1‑5烷基、环烷基、环烯基、杂环烷基、杂芳基、烯基、炔基、酰胺基、烷氧基、烷基氨基、羟基烷基、卤素、羟基、羧酸酯、烷基羧酸酯、酰基叠氮、磺酰胺或卤代烷基;r8为h、c1‑5烷基、环烷基、杂环烷基、芳基、杂芳基、脲基、硫脲基、烯基、炔基、酰胺基、氨基、烷氧基、烷基氨基、烷基膦酸酯、烷基腈、卤代烷基或任选被下列基团取代的卤代烷基:c1‑5烷基、环烷基、环烯基、杂环烷基、杂芳基、烯基、炔基、酰胺基、烷氧基、羟基烷基、卤素、羟基、羧酸酯、烷基羧酸酯、酰基叠氮、磺酰胺或卤代烷基;r9为h、o、c1‑5烷基、环烷基、杂环烷基、芳基、杂芳基、烷基氨基、烷基腈或任选被下列基团取代的烷基膦酸酯:c1‑5烷基、环烷基、杂环烷基、芳基、杂芳基或烷基氨基;r
10
为h、o、c1‑5烷基、环烷基、杂环烷基、芳基、杂芳基、烷基氨基、烷基腈或任选被下列基团取代的烷基膦酸酯:c1‑5烷基、环烷基、杂环烷基、杂芳基或烷基氨基;
r
11
为烷基、芳基、或任选被下列基团取代的杂芳基:烷基、卤代烷基、芳基或杂芳基;r
12
为烷基,芳基,或任选被下列基团取代的杂芳基:烷基、卤代烷基、芳基或杂芳基;r
13
为烷基或任选被烷基或卤代烷基取代的芳基;以及r
14
为h或芳基;或其药学上可接受的盐或前药。
26.在一些实施方案中,w为n。在一些实施方案中,x为cr
14
。在一些实施方案中,r1为h。在一些实施方案中,r2为nr7r8。在一些实施方案中,r3为h。在一些实施方案中,r4为h。在一些实施方案中,r6为h。在一些实施方案中,r7为h。在一些实施方案中,r8为被杂芳基取代的c1‑5烷基。在一些实施方案中,r
14
为h。
27.在一些实施方案中,所述化合物在大约ph4至ph5具有正的logd值。
28.在一些实施方案中,所述化合物为式i(a)的化合物:其中r7和r8各自独立地为h或c1‑5烷基,其中所述c1‑5烷基任选被环烷基、杂环烷基、杂环基、芳基或杂芳基(其各自可进一步被任何合适的取代基取代,例如,c1‑5烷基、环烷基、环烯基、杂环烷基、杂芳基、烯基、炔基、酰胺基、烷氧基、烷基氨基、羟基烷基、卤素、羟基、羧酸酯、烷基羧酸酯、酰基叠氮、磺酰胺或卤代烷基)取代。
29.在一些实施方案中,r7和r8中的一个为h,且另一个为c1‑5烷基,其中所述c1‑5烷基任选被环烷基、杂环烷基、杂环基、芳基或杂芳基(其各自可进一步被任何合适的取代基取代,例如,c1‑5烷基、环烷基、环烯基、杂环烷基、杂芳基、烯基、炔基、酰胺基、烷氧基、烷基氨基、羟基烷基、卤素、羟基、羧酸酯、烷基羧酸酯、酰基叠氮、磺酰胺或卤代烷基)取代。
30.在一些实施方案中,所述化合物在大约ph4至ph5具有正的logd值。
31.还提供了一种在有需要的对象中治疗中枢神经系统狼疮的方法,包括向所述对象施用式ii的化合物:
其中r'选自吡啶
‑2‑
基甲基、吡啶
‑3‑
基甲基、1
‑
苄基哌啶
‑4‑
基、4
‑
氰基
‑
2,2
‑
二乙基丁基、2
‑
氯环戊基、4
‑
(二乙基氨基)丁
‑2‑
基、1
‑
(呋喃
‑2‑
基)乙基、1
‑
环丙基乙基、1
‑
乙基哌啶
‑4‑
基、5
‑
氨基
‑
2,2
‑
二乙基戊基和2
‑
(二乙基磷酰基)
‑1‑
甲基乙基,或其药学上可接受的盐或前药。
32.在一些实施方案中,所述化合物在大约ph4至ph5具有正的logd值。
33.还提供如本文所教导的用于治疗中枢神经系统狼疮的方法中的活性化合物。进一步提供如本文所教导的活性化合物在制备用于治疗中枢神经系统狼疮的药物中的用途。
附图说明
34.图1a
‑
图1b:图1a示出氯喹的logd图,和图1b示出羟基氯喹的logd图。图中阴影区域代表在体内遇到的溶酶体ph的潜在范围。在溶酶体ph下,氯喹和羟基氯喹二者的logd都基本为负数,反映了这些分子累积的电荷和它们的膜通透性的丧失。
35.图2a
‑
图2e分别示出实施例化合物a
‑
e的logd图。请注意,对于每个实施例化合物,logd值在溶酶体ph(ph 4至5)保持为正数。
36.图3a
‑
图3c:图3a提供了在小鼠中在4小时(左)和24小时(右)短暂大脑中动脉闭塞(mcao)卒中演变的弥散加权核磁共振成像(dw
‑
mri)图像。图3b提供了在mcao后用氯喹(cq)与媒介物相比的神经学评分(左)和rotorod评价(右)。图3c示出了三天时皮质(左)和皮质下的卒中量,比较了实施例化合物f(7c
‑
4maq)、氯喹(cq)、qrii缺失小鼠、qrii缺失小鼠同窝小鼠和媒介物。
37.图4呈现了在tbi后rotorod(左)和morris水迷宫(右)表现结果,比较了实施例化合物f(7c
‑
4maq)与氯喹(cq)和媒介物。
38.图5示出了颅内出血模型中右侧基底节出血的t2和磁敏感加权成像(swi)。
39.图6示出了深低温停循环(dhca)后的神经元凋亡。在使用cq(25mg/kg,水平阴影条)、pbs(等体积,黑色条)、qr2抑制剂7c
‑
4maq(25mg/kg,垂直阴影条)或媒介物(50% dmso,开放条)治疗的大鼠中dhca后48小时皮质和海马中的tunel分析。
40.图7示出了dhca后的神经元坏死。在使用cq(25mg/kg,水平阴影条)、pbs(等体积,黑色条)、qr2抑制剂7c
‑
4maq(25mg/kg,垂直阴影条)或媒介物(50% dmso,开放条)治疗的大鼠中dhca后48小时皮质和海马中的酸性品红
‑
天青石蓝染色。
41.图8示出了在cpob/dhca前2小时使用qr2抑制剂7c
‑
4maq(25mg/kg,开放条)或50% dmso(实心条)治疗的大鼠中,术后(pod)第1天和第2天通过neuroscore分析的神经学结果。
42.图9示出了通过mri adc
‑
灌注测量的全脑灌注。a,在第3天和第32天,将假手术小鼠的adc
‑
灌注强度与具有bcas的所有小鼠的集合组进行比较。100%灌注定义为假手术组平均adc
‑
灌注强度。在第3天bcas小鼠(n=17)的灌注显著少于假手术小鼠(n=5)(**p<0.01),但在第32天灌注回升到正常水平。b,bcas术后三天,代表性的上色adc
‑
灌注mr序列叠加在灰度冠状t2加权序列上。注意,与其他治疗组相比,假手术大脑中的灌注增加(adc
‑
灌注信号强度增加)。c,治疗组于第3天和第32天进行adc
‑
灌注。第3天,假手术组(n=5)的灌注显著高于所有其他组(p<0.05,组效应;假手术
×
n
‑
mcq,n=4,p<0.01;假手术
×
cq,n=5,p<0.01;假手术
×
媒介物,n=9,p<0.05)。数值代表平均值
±
sem。*p<0.05,**p<0.01。
43.图10示出morris水迷宫(mwm)上的学习表现。a,逃逸潜伏期。与媒介物对照(媒介
物,n=10;p<0.05,组效应;p<0.01,n
‑
mcq
×
媒介物)相比,施用7c
‑
4maq(“n
‑
mcq”)(n=10)的小鼠表现出减少的逃逸潜伏期,且与假手术小鼠(假手术,n=14)难以区别。mwm测试的第5天后,使水下平台可见,且所有的组间差异消失。b,逃逸潜伏期。施用cq(n=10)的动物的表现概况与它们的n
‑
mcq对应动物(p<0.05,组效应;p<0.001,cq
×
媒介物)相似。c,探针试验。与所有其他治疗组(p<0.05,组效应)相比,施用n
‑
mcq的小鼠在目标象限中花费显著更多的时间。d,游泳速度。对治疗组未观察到游泳速度的差异。数值代表平均值
±
sem。*p<0.05,**p<0.01,***p<0.001.5,p<0.01;假手术
×
媒介物,n=9,p<0.05),数值代表平均值
±
sem。*p<0.05,**p<0.01。
44.图11示出氨基喹啉类减少bcas小鼠的白质束(wm束)中的小胶质细胞增生和星形细胞增生。a,在第3天,中间的cc(前囟=0 mm)的代表性iba
‑
1和gfap染色。b,在第3天和第32天,多个白质束(wm束)中的iba
‑
1免疫阳性细胞密度。与其他治疗组(n=5)相比,媒介物对照(n=9)在第3天和第32天的多个白质束(wm束)中具有显著更高的iba
‑
1阳性细胞密度。c,gfap免疫阳性细胞密度。与所有其他治疗组相比,媒介物对照在第3天和第32天在cc中具有显著更高的gfap阳性细胞的密度。数值代表平均值
±
sem。*p<0.05,**p<0.01,***p<0.001。与所有其他治疗组相比,在第3天和第32天在cc中的阳性细胞。数值代表平均值
±
sem。*p<0.05,**p<0.01,***p<0.001。
45.图12示出抑制qr2减少了bcas小鼠白质束(wm束)中的氧化应激。在第32天在ic中,媒介物组(n=9)表现出比所有其他治疗组(cq,n=8;n
‑
mcq,n=7;媒介物,n=9)显著更高的8
‑
ohdg染色密度、以及在第3天在cc中比假手术组和n
‑
mcq组更高的密度。数值代表平均值
±
sem。*p<0.05,**p<0.01,***p<0.001。
具体实施方式
46.本文提供了治疗急性神经损伤、血管性痴呆或中枢神经系统狼疮的方法。在一些实施方案中,提供可用于抑制醌还原酶2(qr2)的喹啉和喹唑啉衍生物用于这样的治疗。
47.本文引用的所有专利参考文献的公开内容在与本文所公开内容一致的程度内通过引用并入本文。除非上下文另有明确指示,如在本发明说明书和所附权利要求书中所使用的,单数形式的“一个”、“一种”和“所述”也意欲包括复数形式。除非上下文另有明确指示,如在本发明说明书和所附权利要求书中所使用的,单数形式的“一个”、“一种”和“所述”也意欲包括复数形式。此外,当提及诸如化合物的量、剂量、时间、温度等的可测量值时,本文中使用的术语“约”和“大约”意欲包含规定量的20%、10%、5%、1%、0.5%或甚至0.1%的变化。此外,如本文所用,“和/或”和“/”是指并包含一个或多个相关联的所列项目的任何和所有可能的组合,以及当以替代(“或”)解释时则没有组合。
48.i. 定义本文使用以下定义。
49.如本领域所知,“h”是指氢原子。“c”是指碳原子。“n”是指氮原子。“o”是指氧原子。
[0050]“卤素”是指f、cl、br或i。“cl”是指氯,“i”是指碘,“f”是指氟,且“br”是指溴。
[0051]“酰基”为基团
‑
c(o)r,其中r为合适的取代基(例如,乙酰基、丙酰基、丁酰基、苯甲酰基或烷基苯甲酰基)。
[0052]
本文所使用的“烷基”是指含有1或2至10或20或更多个碳原子(例如,c2、c3、c4、
c5、c6、c7、c8、c9、c10、c11、c12、c13、c14、c15等)的直链或支链饱和烃基。烷基的代表性实例包括但不限于甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基、叔丁基、正戊基、异戊基、新戊基、正己基、3
‑
甲基己基、2,2
‑
二甲基戊基、2,3
‑
二甲基戊基、正庚基、正辛基、正壬基、正癸基等。在一些实施方案中,烷基是具有1至3、4或5个碳原子的“低级烷基”。
[0053]
本文所使用的“烯基”为具有一个或多个双键的直链或支链不饱和烃基。
[0054]
本文所使用的“炔基”为具有一个或多个三键的直链或支链不饱和烃基。
[0055]“氨基”是基团
‑
nh2。本文所用的“酰胺”或“酰胺基”是指具有与氮原子(n)相连的羰基(c=o)的有机官能团。“烷基氨基”是指如本文所定义的烷基通过氮原子(
‑
nh
‑
)附加到母体分子。
[0056]
本文所使用的“烷氧基”是指如本文所定义的烷基通过氧原子(
‑
o
‑
)附加到母体分子。烷氧基的代表性实例包括但不限于甲氧基、乙氧基、丙氧基、2
‑
丙氧基、丁氧基、叔丁氧基、戊氧基、己氧基等。
[0057]
本文所使用的“芳基”是指具有一个或多个芳香环的环系统。芳基的代表性实例包括薁基(azulenyl)、茚满基、茚基、萘基、苯基、四氢萘基等。本发明的芳基可被1、2、3、4或5个独立地选自以下的取代基取代:烯基、烯氧基、烷氧基、烷氧基烷氧基、烷氧羰基、烷基、烷基羰基、烷基羰基氧基、烷基亚磺酰基、烷基磺酰基、烷硫基、炔基、芳基、芳氧基、叠氮基、芳基烷氧基、芳基烷基、芳氧基、羧基、氰基、甲酰基、卤素、卤代烷基、卤代烷氧基、羟基、羟基烷基、巯基、硝基、氨磺酰基、磺基、磺酸酯、nr'r''(其中,r'和r''独立地选自氢、烷基、烷基羰基、芳基、芳基烷基和甲酰基)和
‑
c(o)nr'r'' (其中r'和r''独立地选自氢、烷基、烷基羰基、芳基、芳基烷基和甲酰基)。
[0058]“环烷基”是指单环或稠合多环c3至c10饱和烃基。“杂环烷基”是指其中一个或多个碳原子已被独立地选自o、n和s的原子替代的环烷基。
[0059]
本文所使用的“卤代烷基”是指含有1或2至10或20或更多个碳原子(例如,c2、c3、c4、c5、c6、c7、c8、c9、c10、c11、c12、c13、c14、c15等)的直链或支链的烃基,其中至少一个氢原子已被卤素(f、cl、br或i)替代。“卤代烷基”的代表性实例包括但不限于氟烷基(例如,氟甲基(
‑
ch2f)、二氟甲基(
‑
chf2)或三氟甲基(cf3))。
[0060]
本文所使用的“杂环基”是指包含至少一个选自o、n及s的杂原子的单环、双环或三环环系统。单环杂环系统示例为含有1、2、3或4个独立地选自o、n和s的杂原子的任何5或6员环。5员环具有0至2个双键,和6员环具有0至3个双键。单环环系统的代表性实例包括但不限于:氮杂环丁烷、氮杂
䓬
、氮杂环丙烷、二氮杂
䓬
、1,3
‑
二氧戊环、二噁烷、二噻烷、呋喃、咪唑、咪唑啉、咪唑烷、异噻唑、异噻唑啉、异噻唑烷、异噁唑、异噁唑啉、异噁唑烷、吗啉、噁二唑、噁二唑啉、恶二唑烷、噁唑、噁唑啉、噁唑烷、哌嗪、哌啶、吡喃、吡嗪、吡唑、吡唑啉、吡唑烷、吡啶、嘧啶、哒嗪、吡咯、吡咯啉、吡咯烷、吡咯烷、四氢呋喃、四氢噻吩、四嗪、四唑、噻二唑、噻二唑啉、噻二唑烷、噻唑、噻唑啉、噻唑烷、噻吩、硫代吗啉、硫代吗啉砜、亚砜、噻喃、三嗪、三唑、三噻烷等。双环环系统示例为上述任一单环环系统稠合到本文所定义的芳基、本文所定义的环烷基或本文所定义的另一单环环系统。双环环系统的代表性实例包括但不限于,例如,苯并咪唑、苯并噻唑、苯并噻二唑、苯并噻吩、苯并噁二唑、苯并噁唑、苯并呋喃、苯并吡喃、苯并噻喃、苯并二噁英、1,3
‑
苯并间二氧杂环戊烯、噌啉、吲唑、吲哚、吲哚啉、吲嗪、萘啶、异苯并呋喃、异苯并噻吩、异吲哚、异吲哚啉、异喹啉、酞嗪、吡喃并吡啶、喹啉,喹嗪、
喹喔啉、喹唑啉、四氢异喹啉、四氢喹啉、噻喃并吡啶等。含氮杂环基的实例包括但不限于,吡咯烷基、哌啶基、哌嗪基、吗啉基等。
[0061]“杂芳基”意指其中一个或多个碳原子已被独立地选自o、n和s的原子替代的环状芳香烃基。杂芳基的实例包括吡啶基、嘧啶基、咪唑基、噻吩基、呋喃基、吡嗪基、吡咯基、吡喃基、异苯并呋喃基、色烯基、呫吨基、吲哚基、异吲哚基、吲嗪基、三唑基、哒嗪基、吲唑基、嘌呤基、喹嗪基、异喹啉基、喹啉基、酞嗪基、萘啶基、喹喔啉基、异噻唑基和苯并[b]噻吩基。优选的杂芳基是五元环和六元环,并包含一到三个独立地选自o、n和s的杂原子。杂芳基,包括每个杂原子,可以不被取代或被1至4个合适的取代基取代,只要化学上是可行的。例如,杂原子s可被一个或两个氧基取代,其可显示为=o。含氮杂芳基的实例包括但不限于,吡啶基、嘧啶基、吡嗪基、哒嗪基、咪唑基、吡咯基、吡唑基、噻唑基、三唑基、异噻唑基、吲哚基、苯并咪唑基、苯并噁唑基、喹啉基、异喹啉基、喹唑啉基、吖啶基、咔唑基、氮杂
䓬
基、1,4
‑
二氮杂
䓬
基、嘌呤基、蝶啶基、酞嗪基等。
[0062]“羟基”是指基团
‑
oh。
[0063]“腈基”是指基团
‑
cn。
[0064]“硝基”是指基团
‑
no2。
[0065]“砜”是指磺酰基官能团,
‑
so2r,其中r是任何共价连接的一个或多个原子。
[0066]“亚砜”是指基团
‑
s(o)r,其中r是任何共价连接的一个或多个原子。
[0067]“硫醇”或“巯基”是指基团
‑
sh或其互变异构体=s。
[0068]“脲基”是指基团
‑
nhconh2。“硫脲基”是指基团
‑
nhcsnh2。
[0069]“药学上可接受的盐”是保留特定化合物的游离酸和碱的生物有效性且不是在生物学上或其它方面不期望的盐。药学上可接受的盐的实例包括但不限于,硫酸盐、焦硫酸盐、重硫酸盐、亚硫酸盐、亚硫酸氢盐、磷酸盐、单氢磷酸盐、二氢磷酸盐、偏磷酸盐、焦磷酸盐、氯化物、溴化物、碘化物、乙酸盐、丙酸盐、癸酸盐、辛酸盐、丙烯酸盐、甲酸盐、异丁酸盐、己酸盐、庚酸盐、丙酸盐、草酸盐、丙二酸盐、琥珀酸盐、辛二酸盐、癸二酸盐、富马酸盐、马来酸盐、丁炔
‑
1,4
‑
二酸盐、己炔
‑
1,6
‑
二酸盐、苯甲酸盐、氯苯甲酸盐、甲基苯甲酸盐、二硝基苯甲酸盐、羟基苯甲酸盐、甲氧基苯甲酸盐、邻苯二甲酸盐、磺酸盐、二甲苯磺酸盐、苯乙酸酯、苯丙酸盐、苯丁酸盐、柠檬酸盐、乳酸盐、γ
‑
羟基丁酸盐、乙醇酸盐、酒石酸盐、甲烷磺酸盐、丙磺酸盐、萘
‑1‑
磺酸盐、萘
‑2‑
磺酸盐和扁桃酸盐。
[0070]
本领域已知的“前药”是可在生理条件下或通过溶剂分解或代谢转化为具有药学活性的特定化合物的化合物。在t. higuchi和v. stella,prodrugs as novel delivery systems, a.c.s. symposium series的第14卷以及edward b. roche, 编辑, bioreversible carriers in drug design, american pharmaceutical association and pergamon press, 1987中,对此进行了深入的讨论,两者都通过引用以其整体并入本文。还参见美国专利no. 6680299。实例包括由对象在体内代谢成本文所述活性化合物的前药,其中,如果化合物中存在醇或羧酸基团,则前药为所述醇或羧酸基团的酯;如果化合物中存在醇基,则前药为所述醇基的缩醛或缩酮;如果化合物中存在胺基,则前药为所述胺基的n
‑
曼尼希碱或亚胺;或者如果化合物中存在羰基,则前药为所述羰基的席夫碱、肟、缩醛、烯醇酯、噁唑烷或噻唑烷,如美国专利no. 6680324和美国专利no. 6680322所描述的。
[0071]
如本领域所理解的,术语“任选取代的”表示指定基团或未被取代或被一个或多个
合适的取代基取代。“取代”的“取代基”是代替母体有机分子上氢原子的基团。
[0072]
ii、活性化合物根据一些实施方案在本文提供的活性化合物为式i的化合物:其中:w为n或n
+
o
‑
;x为cr
14
或n;r1为h或三氟甲基;r2为nr7r8、or
11
、sr
12
或烷基;r3为h或or
13
;r4为h或甲氧基;r5为h、cl或三氟甲基;r6为h、nr9r
10
或三氟甲基;r7为h、c1‑5烷基、杂芳基烷基、环烷基、杂环烷基、杂环基、芳基、杂芳基、脲基、硫脲基、烯基、炔基、酰胺基、氨基、烷氧基、烷基氨基、烷基膦酸酯、烷基腈、卤代烷基或任选被下列基团取代的卤代烷基:c1‑5烷基、环烷基、环烯基、杂环烷基、杂芳基、烯基、炔基、酰胺基、烷氧基、烷基氨基、羟基烷基、卤素、羟基、羧酸酯、烷基羧酸酯、酰基叠氮、磺酰胺或卤代烷基;r8为h、c1‑5烷基、环烷基、杂环烷基、芳基、杂芳基、脲基、硫脲基、烯基、炔基、酰胺基、氨基、烷氧基、烷基氨基、烷基膦酸酯、烷基腈、卤代烷基或任选被下列基团取代的卤代烷基:c1‑5烷基、环烷基、环烯基、杂环烷基、杂芳基、烯基、炔基、酰胺基、烷氧基、羟基烷基、卤素、羟基、羧酸酯、烷基羧酸酯、酰基叠氮、磺酰胺或卤代烷基;r9为h、o、c1‑5烷基、环烷基、杂环烷基、芳基、杂芳基、烷基氨基、烷基腈或任选被下列基团取代的烷基膦酸酯:c1‑5烷基、环烷基、杂环烷基、芳基、杂芳基或烷基氨基;r
10
为h、o、c1‑5烷基、环烷基、杂环烷基、芳基、杂芳基、烷基氨基、烷基腈或任选被下列基团取代的烷基膦酸酯:c1‑5烷基、环烷基、杂环烷基、杂芳基或烷基氨基;r
11
为烷基、芳基或任选被下列基团取代的杂芳基:烷基、卤代烷基、芳基或杂芳基;r
12
为烷基、芳基或任选被下列基团取代的杂芳基:烷基、卤代烷基、芳基或杂芳基;r
13
为烷基或任选被烷基或卤代烷基取代的芳基;以及r
14
为h或芳基;或其药学上可接受的盐或前药。
[0073]
在式i的一些实施方案中,w为n。在式i的一些实施方案中,x为cr
14
。在式i的一些实
施方案中,r1为h。在式i的一些实施方案中,r2为nr7r8。在式i的一些实施方案中,r3为h。在式i的一些实施方案中,r4为h。在式i的一些实施方案中,r5为cl。在式i的一些实施方案中,r6为h。在式i的一些实施方案中,r7为h。在式i的一些实施方案中,r8为被杂芳基取代的c1‑5烷基。在式i的一些实施方案中,r
14
为h。
[0074]
在式i的一些实施方案中,所述化合物为式i(a)的化合物:其中r7和r8各自独立地为h或c1‑5烷基,其中所述c1‑5烷基任选被环烷基、杂环烷基、杂环基、芳基或杂芳基(其各自可进一步被任何合适的取代基取代,例如,c1‑5烷基、环烷基、环烯基、杂环烷基、杂芳基、烯基、炔基、酰胺基、烷氧基、烷基氨基、羟基烷基、卤素、羟基、羧酸酯、烷基羧酸酯、酰基叠氮、磺酰胺或卤代烷基)取代。
[0075]
在式i(a)的一些实施方案中,r7和r8中的一个为h,且另一个为c1‑5烷基,其中所述c1‑5烷基任选被环烷基、杂环烷基、杂环基、芳基或杂芳基(其各自可进一步被任何合适的取代基取代,例如,c1‑5烷基、环烷基、环烯基、杂环烷基、杂芳基、烯基、炔基、酰胺基、烷氧基、烷基氨基、羟基烷基、卤素、羟基、羧酸酯、烷基羧酸酯、酰基叠氮、磺酰胺或卤代烷基)取代。
[0076]
在式i的一些实施方案中,所述化合物为:4
‑
{2
‑
[(7
‑
氯喹啉
‑4‑
基)氨基]乙基}苯酚,
7
‑
氯
‑
n
‑
(吡啶
‑2‑
基)喹啉
‑4‑
胺,7
‑
氯
‑
n
‑
(吡啶
‑3‑
基)喹啉
‑4‑
胺,或7
‑
氯
‑
n
‑
甲基喹啉
‑4‑
胺,或其药学上可接受的盐。
[0077]
在式i的一些实施方案中,所述化合物为:7
‑
氯
‑
n
‑
甲基喹啉
‑4‑
胺或其药学上可接受的盐。
[0078]
本文进一步提供的活性化合物为式ii的化合物:其中r'选自吡啶
‑2‑
基甲基、吡啶
‑3‑
基甲基、1
‑
苄基哌啶
‑4‑
基、4
‑
氰基
‑
2,2
‑
二乙基丁基、2
‑
氯环戊基、4
‑
(二乙基氨基)丁
‑2‑
基、1
‑
(呋喃
‑2‑
基)乙基、1
‑
环丙基乙基、1
‑
乙基
哌啶
‑4‑
基、5
‑
氨基
‑
2,2
‑
二乙基戊基和2
‑
(二乙基磷酰基)
‑1‑
甲基乙基,或其药学上可接受的盐或前药。
[0079]
在式ii的一些实施方案中,所述化合物为:6
‑
甲氧基
‑
n
‑
(吡啶
‑2‑
基甲基)喹啉
‑8‑
胺,或n
‑
[1
‑
(呋喃
‑2‑
基)乙基]
‑6‑
甲氧基喹啉
‑8‑
胺,或其药学上可接受的盐。
[0080]
进一步的活性化合物可在ware, jr.等的美国专利申请公开no. 2006/0074105中找到,该申请通过引用并入本文。化合物可根据已知的方法制备,如egan等人, j. med. chem. 2000, 43:283
‑
291; stocks等人, j. med. chem. 2002, 45:4975
‑
4983中所描述的;或者通过本文在下面提供的实施例中描述的方法制备。
[0081]
在上述式i或式ii化合物的一些实施方案中,所述化合物在约ph 4至ph 5具有正的logd值。
[0082]
除非另有说明,本文所描述的结构还意欲包括所述结构的所有对映异构体、非对映异构体和几何(或构象)形式;例如,每个不对称中心的r和s构型,(z)和(e)双键异构体,以及(z)和(e)构象异构体。因此,本发明化合物的单一立体化学异构体以及对映异构体、非对映异构体和几何(或构象)混合物在本发明的范围内。除非另有说明,本发明化合物的所有互变异构形式都在本发明的范围内。互变异构形式包括化合物的酮
‑
烯醇互变异构体。此外,除非另有说明,否则本发明化合物的所有旋转异构体形式都在本发明的范围内。除非另有说明,本文所描述的结构还意欲包括不同之处仅在于存在一个或多个同位素富集原子的化合物。例如,除了用氘或氚替代氢或者用富含
13
c
‑
或
14
c
‑
的碳替换碳之外,具有本发明结构的化合物都在本发明的范围内。这样的化合物可用作,例如,生物测定中的分析工具或探
针。
[0083]
iii. 使用方法如上文所指出,本文所教导的活性化合物可用于治疗急性神经损伤、血管性痴呆或中枢神经系统狼疮。
[0084]
急性神经损伤包括,但不限于,创伤性脑损伤和非创伤性急性脑损伤。创伤性脑损伤,如本领域所知,是由单一或重复的外部机械力(如来自突然加速或减速的钝力或剪切力)引起的大脑损伤和/或功能障碍。创伤性脑损伤包括,但不限于,脑震荡、挫伤和出血,包括脑实质、硬膜下、硬膜外和蛛网膜下腔出血。其他急性神经损伤包括缺氧或缺血性脑损伤(例如动脉卒中(局灶性、全身性)、静脉梗死、感染、围手术期脑损伤等)引起的损害。
[0085]
血管性痴呆是由急性脑血管损害引起的痴呆或认知功能障碍(cognitive deficit),常与多发性脑血管事件(如卒中)有关。
[0086]
中枢神经系统狼疮(cns lupus)是指系统性红斑狼疮(sle)患者的神经和/或行为的临床综合征。中枢神经系统狼疮在临床上可表现为突发性意识模糊(acute confusion)、疲劳、头痛、轻度认知障碍、谵妄、昏迷、痴呆、感觉/运动/自主神经缺失和/或癫痫(后者在狼疮患者中比一般人群更常见)。中枢神经系统狼疮也可表现为心理障碍,如抑郁、躁狂和/或精神病。也可能有更多的局灶性神经功能缺失且可继发于狼疮相关的脑和脊柱的栓塞性、血栓性或血管炎性梗死以及颅神经病变。中枢神经系统狼疮的病理生理机制可能包括脑炎、横贯性脊髓炎、神经炎和脑或脊柱的卒中(栓塞性、血栓性或血管炎性)。
[0087]
本文所使用的术语“治疗”是指向患有损伤、疾病或病症或处于损伤、疾病或病症的风险的患者赋予益处(例如,改善或降低发展一个或多个症状的风险,所述症状如认知功能障碍和/或运动功能障碍)、使损伤或症状的进展延迟等的任何类型的治疗。
[0088]
本发明主要涉及人类对象的治疗,但本发明也可在动物对象上实施,特别是哺乳动物对象,如用于兽医目的和/或用于药物筛选和/或药物开发目的的小鼠、大鼠、狗、猫、家畜和马。
[0089]
iv. 制剂在一些实施方案中,一种或多种活性化合物可提供于药学上可接受的载体中。载体应是可接受的,即它们与制剂的任何其他成分相容,并且不会对其接受者有害。在一些实施方案中,药学上可接受的载体是无菌的(例如,无内毒素或无热原的水,或无内毒素或无热原的盐水)。
[0090]
本发明的制剂可包括短期、快速起效、快速补偿(rapid
‑
offset)、控释、缓释、延迟释放和脉冲释放制剂,前提是所述制剂实现施用如本文所述的化合物。参见remington's pharmaceutical sciences (第18版;mack publishing company, eaton, pa., 1990),其整体通过引用并入本文。
[0091]
根据本发明的药物制剂可适于各种递送模式,包括口服、肠胃外(包括静脉内、肌肉内、皮下、皮内和经皮)、局部(包括皮肤、口腔和舌下)和直肠施用。
[0092]
根据本发明的合适剂量单位形式的实例为片剂、胶囊、在合适的液体媒介物中的经口施用的液体制剂、用于肌肉内和静脉内施用的在合适的液体媒介物中的无菌制剂、栓剂和在合适的药学上可接受的载体中的用于即时制备无菌可注射制剂的无菌干制剂。用于固体口服药物剂量单位形式的合适的固体稀释剂或载体可选自脂类、碳水化合物、蛋白质
和矿物固体;例如,淀粉、蔗糖、高岭土、磷酸氢钙、明胶、阿拉伯胶、玉米淀粉、滑石粉等。硬胶囊和软胶囊二者都可以用合适的稀释剂和赋形剂配制;例如,食用油、滑石粉、碳酸钙等,以及硬脂酸钙。用于口服施用的液体制剂可在水中或含有悬浮剂的水溶液中制备;例如,羧甲基纤维素钠、甲基纤维素、阿拉伯胶、聚乙烯吡咯烷酮、聚乙烯醇等。在一些实施方案中,可包括防腐剂,例如对羟基苯甲酸酯、氯丁醇、苄基醇、苯酚等。参见mccall的美国专利no. 4159331。
[0093]
用于治疗性治疗的活性化合物的施用量可能取决于医生确定的患者的年龄、体重和状况。在一些实施方案中,施用和/或药物剂量单位形式可提供每剂量约0.05 mg至约100 mg的活性化合物。在一些实施方案中,活性化合物的提供量为约1μg/kg接收者体重至约1g/kg接收者体重,或每千克(kg)体重10μg至100mg,或每千克体重0.1mg至50mg。
[0094]
在以下非限制性实施例中更详细地解释本发明。
实施例
[0095]
实施例1:qr2的非亲溶酶体氨基喹啉抑制剂的开发氯喹和羟基氯喹是优先在细胞溶酶体中累积的亲溶酶体药物。它们的结构是:对于氯喹,叔胺氮的pka为10.32,且喹啉氮的pka为7.29。在酸性溶酶体ph4至5.5,几乎100%的氯喹因此被双重质子化,使分子带有2
+
电荷,使得分子具有强亲水性、膜不通透性,并因此被捕获在酸性细胞器中。
[0096]
通过检测药物的辛醇
‑
水分配系数logd,可以定量处理该捕获现象,logd描述了在不同ph下化合物的所有形式的相对分配性质。对于给定的ph,具有正的logd的化合物是相对亲脂性的且膜通透性更强,而具有负的logd的化合物是亲水性的且膜通透性更弱。
[0097]
图1a示出了氯喹的logd,且图1b示出了羟基氯喹的logd。图中阴影区域代表在体内遇到的溶酶体ph的潜在范围。在溶酶体ph值下,氯喹和羟基氯喹二者的logd基本为负数,反映了这些分子的累积的电荷和它们膜通透性的丧失。
[0098]
采用化学蛋白体学(chemoproteomics)策略生成对qr2有选择性的4
‑
氨基喹啉支架化学库。利用一系列化学信息学工具,我们用计算机对该库进行筛选(mine),并已识别出对qr2具有纳摩尔到微摩尔抑制的氨基喹啉衍生物,这些衍生物还具有避免溶酶体累积的化学性质,从而解决了导致cq/hq的最常见毒性的机制。
[0099]
下面提供的实施例化合物及其各自的logd预测如图2a
‑
2e所示。请注意,无论酸性如何,每种化合物的logd值保持为正数(> 0.5),表明这些分子在溶酶体的ph(ph 4至5)将保持亲脂性(并因此膜通透性)。
[0100]
实施例化合物a(logd示于图2a中):4
‑
{2
‑
[(7
‑
氯喹啉
‑4‑
基)氨基]乙基}苯酚实施例化合物b(logd示于图2b中):6
‑
甲氧基
‑
n
‑
(吡啶
‑2‑
基甲基)喹啉
‑8‑
胺实施例化合物c(logd示于图2c中):n
‑
[1
‑
(呋喃
‑2‑
基)乙基]
‑6‑
甲氧基喹啉
‑8‑
胺实施例化合物d(logd示于图2d中):
7
‑
氯
‑
n
‑
(吡啶
‑2‑
基)喹啉
‑4‑
胺实施例化合物e(logd示于图2e中):7
‑
氯
‑
n
‑
甲基喹啉
‑4‑
胺实施例化合物f:7
‑
氯
‑
n
‑
(吡啶
‑3‑
基)喹啉
‑4‑
胺下面表1呈现了与氯喹(cq)相比,这些非亲溶酶体4
‑
氨基喹啉类的药物相似性的另外的评估。亲脂性效率,lipe(又称配体亲脂性效率),是一个将效力与亲脂性相关联的药物设计和发现参数。lipe被定义为pic50(
‑
log ic50)减去计算的log p,clog p:lipe = pic
50 –ꢀ
clogplipe用于评估体内药物特异性,更高的值预示效力增加以及不希望的或偏离目标的相互作用的可能性降低。许多公开的4
‑
氨基喹啉qr2抑制剂的lipe高于cq,因此,预示比cq更好的毒性曲线,与通过消除溶酶体捕获(lysosomotropism )预期的显著降低的毒性无关。
[0101]
表1:qr2 的4
‑
氨基喹啉抑制剂药物相似性的经验和计算参数。
[0102]
实施例2:7
‑
氯代化合物的合成与表征将在甲胺水溶液(40% 20 ml 260 mmol,26 eq.)中的4,7
‑
二氯喹啉(2.0 g,10.2 mmol)的悬浮液在微波容器中于90℃(初始功率设定为150w)加热2小时。通过tlc(2% 在ch2cl2中的meoh)对反应混合物进行分析,表明原料已完全消耗。用h2o(100 ml)稀释反应混合物,并在真空收集不溶物。滤饼用h2o洗涤并真空中干燥,给出纯产物,为白色微晶固体(1.8g,92%)。1h nmr (dmso
‑
d6, 300 mhz) δ 8.40 (d, j = 5.1 hz, 1h), 8.16 (d, j = 9.0 hz, 1h), 7.77 (s, 1h), 6.38 (d, j = 5.4 hz, 1h), 2.86 (d, j = 5.4 hz, 3h). esims: m/z = 193 [(m+h)
+
]。
[0103]7‑
取代
‑4‑
(吡啶
‑3‑
基)
‑
甲基氨基喹啉类的一般方法。将7
‑
取代
‑4‑
氯喹啉(5.1 mmol)、3
‑
氨基甲基吡啶(0.70 g,6.2 mmol,1.2eq.)和1
‑
丁醇(5 ml)的混合物在130℃(浴温)的密封厚壁压力容器(12 ml)中加热24小时。将容器冷却至室温,并将内容物用et2o(150 ml)稀释。真空除去不溶物。将滤饼溶解在最小量的meoh中,并将所得溶液添加到硅胶中(~3g)。在减压下将混合物浓缩至干。快速柱层析(rediseprf sio2(40 g),100% ch2cl2→
75%(90:10,ch2cl2:meoh,含10% nh3)给出所需产物。
[0104]
x=cl(白色固体,0.92 g,67%)。
[0105]1h nmr (dmso
‑
d6, 400 mhz) δ 8.61 (s, 1h), 8.42 (s, 1h), 8.27 (m, 2h), 8.00 (s, 1h), 7.75 (m, 2h), 7.46 (d, j = 8.8 hz, 1h), 7.31 (m, 3h), 6.39 (d, j = 5.4 hz, 1h), 4.55 (d, j = 5.4 hz, 2h). esims: m/z = 270 [(m+h)
+
]。
[0106]
实施例3:qr2抑制在脑梗死中的保护作用及非亲溶酶体qr2抑制剂治疗效果的证据。
[0107]
氯喹(cq)的神经保护功效在短暂性大脑中动脉(mca)闭塞小鼠模型中得到了证明。在72小时的尸体解剖组织学评估显示,缺血发作90分钟后单次腹腔注射(i.p.)施用cq(25 mg/kg)可导致总卒中量减少55%,通过弥散加权核磁共振成像(dw
‑
mri)测量,在4到24小时内,卒中发展相应减少,且神经学评分和运动功能提高,如图3a
‑
3c所示。
[0108]
在同一模型中还测试了非亲溶酶体qr2选择性4
‑
氨基喹啉、7
‑
氯
‑
n
‑
甲基喹啉
‑4‑
胺(7c
‑
4maq,上文所示实施例化合物f)的神经保护功效。在该动物模型中,7
‑
氯
‑
n
‑
甲基喹啉
‑4‑
胺实现突出显著的神经保护,以同等的单次急性期剂量(25 mg/kg)相比较,比cq施用后所见到的的皮质卒中量减少近2x(图3c)。
[0109]
值得注意的是,25mg/kg的剂量仍然比对该化合物测定的ld
50
低20多倍。最后,我们在同一mca闭塞/再灌注模型中将qr2缺失小鼠与其同窝对照进行比较,也如图3c所示。
[0110]
实施例4:创伤性脑损伤(tbi)在弥漫性闭合脑损伤的小鼠tbi模型中研究了qr2抑制的神经保护可能性(laskowitz 等人, "neuroprotective pentapeptide cn
‑
105 is associated with reduced sterile inflammation and improved functional outcomes in a traumatic brain injury murine model." sci. rep. 2017 apr 21;7:46461)。如图4所示,tbi后在神经认知和神经运动功能评估中均发现明显的提高。单次25 mg/kg施用7
‑
氯
‑
n
‑
甲基喹啉
‑4‑
胺(7c
‑
4maq,上文所示的实施例化合物f)相比媒介物可使rotorod潜伏期提高20%(n=12/gp),且更显著的是,即使在受伤后1个月后,在morris水迷宫表现中也有62%的提高(n=12/gp)。
[0111]
与在卒中模型中一样,当将7c
‑
4maq与cq相比较,以及将qr2缺失小鼠与它们的同窝对照相比较时,观察到几乎相同的趋势。
[0112]
实施例5:颅内出血(ich)。
[0113]
在小鼠颅内出血性损伤模型中研究qr2抑制 (lei等人,"neuroprotective pentapeptide cn
‑
105 improves functional and histological outcomes in a murine model of intracerebral hemorrhage." sci. rep. 2016 oct 7;6:34834)。在该研究中,我们研究了7c
‑
4maq、氯喹(cq)和8
‑
氨基喹啉、伯氨喹,目的是测试实施例化合物的功效,同时也进一步探究治疗作用的分子机制。
[0114]
此前有报道称,氨基喹啉类如cq和羟基氯喹抑制qr2反应的后半部分,而其他喹啉类如伯氨喹则抑制前半部分。在该特定的ich模型(图5)中,注意到以前的神经保护干预仅在组织学和分子水平、而不是行为水平显示出统计学上可辨别的治疗效果。在我们的实验中,cq疗法也导致运动功能非显著的14%的提高(p=0.3)(rotorod评估)。然而,7c
‑
4maq施用在单次25 mg/kg的腹腔注射(i.p.)剂量后导致行为结果在统计学上显著的21%的提高(p=
0.013,双尾t,n=21)(数据未显示)。同样值得注意的是,仅选择性地抑制qr2反应的第一阶段的伯氨喹,在等量的腹腔注射(i.p.)剂量后,导致运动功能35%的恶化(p=0.0001)(数据未显示)。
[0115]
实施例6:术后认知功能障碍使用心肺旁路(cpb)和深低温停循环(dhca)的主要心血管手术后的围手术期脑损伤(pci)仍然是导致不良脑预后的显著原因。在最初在杜克大学podgoreanu博士实验室开发的一个确立已久的大鼠模型中,我们比较了cq相比7c
‑
4maq 的qr2抑制对心肺旁路(cpb)/深低温停循环(dhca)后脑预后的影响 (de lange 等人, "a novel survival model of cardioplegic arrest and cardiopulmonary bypass in rats: a methodology paper," j cardiothorac surg. 2008 aug 19;3:51)。结果如图6、7和8所示。对于这个cpb/dhca模型,禁食的成年雄性sprague
‑
dawley大鼠(10
‑
12周龄)被用吸入的2
‑
2.5%的异氟烷麻醉,插管并机械通气。插管放置在尾动脉和右颈外静脉。然后动物在cpb上冷却30分钟,并在16
‑
18℃的颅周温度进行dhca。dhca 60分钟后,重新启动cpb,动物被重新温暖30分钟,并在≥35.5℃的温度与cpb分离。术后第1天进行mri,术后第1天和第2天进行神经学评估,然后第2天后处死动物。
[0116]
初步结果的mri分析显示,与它们各自的对照组相比,使用氯喹(cq)或实施例化合物(7c
‑
4maq)治疗的动物中的钆螯合物测量的术后血脑屏障通透性降低了3%(p<0.05)。用cq或7c
‑
4maq治疗的动物在皮质和海马中也显示出更少的凋亡和坏死神经元(图6,图7)。最后,7c
‑
4maq治疗的大鼠在术后第1天和第2天表现出显著提高的神经学评分(图8)。
[0117]
实施例7:痴呆,血管亚型血管性痴呆是由慢性脑灌注不足引起的,其临床特征是mri上的白质病变和执行功能下降。近来的研究显示,痴呆大鼠模型以及痴呆人类患者中海马的醌氧化还原酶2(qr2)的表达显著增加,表明qr2可能是治疗靶点。我们检验了氯喹和7c
‑
4maq对血管性痴呆小鼠模型的神经保护作用。使用定量免疫化学、mri和行为任务(包括morris水迷宫和rotorod)的组合来确定生理、细胞和功能结果。
[0118]
如图9
‑
图12所示,两种qr2抑制剂都提高了morris水迷宫的表现,同时减少星形细胞增生、小胶质细胞增生和氧化应激标记物。请注意,在图9
‑
图12中,7c
‑
4maq被称为使用先前的名称“n
‑
mcg”。尽管功能结果和细胞炎症响应有所提高,但治疗组和对照组之间白质损伤的结构标志物没有变化。这些结果为qr2在痴呆中的病理作用及其作为治疗靶点的潜力提供了证据。此外,结果表明,功能相关的神经保护是通过独立于那些导致痴呆相关的白质病变(通常在mri上表征)的机制的机制而发生的。
[0119]
上述内容是对本发明的阐述,但不应被解释为限制本发明。本发明由以下权利要求限定,其中包括权利要求的等价物。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1