P53WT肿瘤的治疗方法与流程
相关申请和支持
本申请要求于2018年10月30日提交的美国临时申请62/752382的优先权和权益,该临时申请的全部内容通过引用特此并入。
本发明是在美国国立卫生研究院(nationalinstitutesofhealth)授予的授权号r01ca063113r01ca173023和p01ca203655的政府支持下完成的。政府拥有本发明的某些权利。
本发明涉及治疗p53野生型(wt)肿瘤的方法。特别地,本发明提供了基于鼠双微体2(mdm2)抑制剂以及酪蛋白激酶1α(ck1α)降解剂和/或mdm4抑制剂的组合的针对p53wt肿瘤的新疗法。
背景技术:
默克尔细胞癌(mcc)是一种侵袭性皮肤神经内分泌癌,在美国的发病率在过去的20年中增加了三倍[1,2]。2008年,feng等人在10个mcc肿瘤中的8个里发现了默克尔细胞多瘤病毒(mcv,mcpyv)的克隆整合[3]。mcv阳性mcc包含整合的mcv基因组拷贝,并表达小t抗原(st)和截短形式的大t抗原(lt)[4]。mcc肿瘤相关的截短的lt保留了n末端lxcxe(rb结合基序),但删除了病毒复制所需的c末端dna结合和解旋酶结构域[3]。mcvst和截短的lt的表达可以促进几种细胞类型的增殖和转化,这与其在mcc中的致癌作用一致[5]。
原型多瘤病毒猿猴空泡病毒40(sv40)lt与视网膜母细胞瘤相关蛋白rb(rb1)和细胞肿瘤抗原p53(tp53)结合并使其抑癌功能失活[6]。相反,mcvlt与rb结合,但不与p53结合[6]。mcc的下一代测序表明,病毒阴性mcc通常带有p53和rb突变以及uv损伤特征[7,8]。相反,病毒阳性mcc通常包含野生型rb和p53,且没有紫外线损伤的证据[7,8]。考虑到病毒阳性mcc中存在野生型p53,诸位发明人怀疑mcvt抗原可在功能上使p53活性失活。
p53在多种癌症中均发生突变。可替代地,野生型p53可以通过过表达mdm2(靶向p53的泛素连接酶)或mdm4(mdmx)而功能失活[9,10]。mdm2和mdm4二者具有相似的结构,具有n末端p53结合结构域和c末端ring结构域[11]。尽管mdm4不直接泛素化p53,但它的ring结构域促进了泛素向mdm2的募集[11]。mdm4还具有自抑制结构域,可减少与p53的结合[12]。酪蛋白激酶1α(ck1α,csnk1a1)可以减轻mdm4的自抑制相互作用[13]。
技术实现要素:
本发明提供了包含mdm2抑制剂和酪蛋白激酶1α(ck1α)降解剂和/或mdm4抑制剂的新组合,用于在受试者的p53wt肿瘤的治疗中使用。
在根据本发明的组合中,mdm2抑制剂可以选自由以下组成的组:努林(nutlin)-3,伊达努林(idasanutlin)(rg7388、ro5503781,罗氏公司(roche)),rg7775(ro6839921,罗氏公司),ro5045337(罗氏公司),amg232(安进公司(amgen)),ds3032(ds3032b,第一三共株式会社(daiichisankyo)),alrn-6924(艾勒朗公司(aileron)),krt-232(卡托斯公司(kartos)),atsp-7041,cgm097(诺华公司(novartis))和hdm201(诺华公司),优选地是hdm201,即(s)-5-(5-氯-1-甲基-2-氧代-1,2-二氢-吡啶-3-基)-6-(4-氯-苯基)-2-(2,4-二甲氧基-嘧啶-5-基)-1-异丙基-5,6-二氢-1h-吡咯并[3,4-d]咪唑-4-酮。
在根据本发明的组合中,酪蛋白激酶1α(ck1α)降解剂和/或mdm4抑制剂选自由以下组成的组:沙利度胺,泊马度胺,来那度胺和sc-24-ur99(诺华公司)即4-(3-氨基-1-(4-氯-5-甲基-6-(甲基氨基)吡啶-3-基)-5-氟-1h-吲唑-6-基)萘-1-醇,优选地是来那度胺即(rs)-3-(7-氨基-3-氧代-1h-异吲哚-2-基)哌啶-2,6-二酮、也称为revlimid。
本发明提供了特别是与mdm2抑制剂hdm201和ck1α降解剂来那度胺的此类组合。
根据本发明的组合可以包含另外的抗癌剂。
根据本发明的另外的抗癌剂可以选自:
flt3抑制剂(例如吉特瑞尼、奎扎替尼、米哚妥林),
bcl2抑制剂(例如那维托克莱克斯、维尼托克莱克斯),
其他mdm2抑制剂(例如努林-3、伊达努林、amg232、ds-3032b、alrn6924/atsp7041),
去甲基化剂(hma)(例如委丹扎[氮胞苷、5-氮胞苷]、达珂[地西他滨]、胍地西他滨),
蒽环(例如伊达比星、道诺霉素、多柔比星、泛艾霉素);
抗cd33抗体(例如灭髓瘤[吉妥珠单抗]、瓦达土昔单抗)
及其他试剂(例如arac[阿糖胞苷])。
可以通过根据本发明的组合治疗的p53wt肿瘤可以是实体瘤或血液肿瘤。实体瘤可以是肉瘤例如脂肪肉瘤或软组织肉瘤,淋巴瘤例如非霍奇金淋巴瘤(nhl)、特别是套细胞淋巴瘤(mcl),黑素瘤例如皮肤黑素瘤或葡萄膜黑素瘤,母细胞瘤(例如神经母细胞瘤),结肠肿瘤,结直肠肿瘤,肾脏肿瘤,和肝肿瘤或皮肤癌例如默克尔细胞癌(mcc),尤其是默克尔细胞多瘤病毒(mcv)阳性mcc。血液肿瘤可以是急性髓性白血病(aml)、多发性骨髓瘤(mm)、骨髓增生异常综合征(mds)或急性淋巴细胞白血病(all)。
具体地,本发明提供了以下实施例:
1.一种组合,其包含(a)鼠双微体2(mdm2)抑制剂以及(b)酪蛋白激酶1α(ck1α)降解剂和/或鼠双微体4(mdm4)抑制剂。
2.一种组合,其包含(a)mdm2抑制剂以及(b)ck1α降解剂和/或mdm4抑制剂,用于在受试者的p53野生型(wt)肿瘤的治疗中使用。
3.一种治疗受试者的p53wt肿瘤的方法,所述方法包括向所述受试者施用(a)mdm2抑制剂以及(b)ck1α降解剂和/或mdm4抑制剂的组合。
4.如实施例1所述的组合、如实施例2所述的用于使用的组合、或如实施例3所述的方法,其中所述mdm2抑制剂选自由以下组成的组:努林-3,伊达努林(rg7388、ro5503781),rg7775(ro6839921),amg232,ds3032(ds3032b),alrn-6924,atsp-7041,cgm097,和hdm201即(s)-5-(5-氯-1-甲基-2-氧代-1,2-二氢-吡啶-3-基)-6-(4-氯-苯基)-2-(2,4-二甲氧基-嘧啶-5-基)-1-异丙基-5,6-二氢-1h-吡咯并[3,4-d]咪唑-4-酮。
5.如实施例1所述的组合、如实施例2所述的用于使用的组合、或如实施例3所述的方法,其中所述mdm2抑制剂选自努林-3、伊达努林和hdm201组成的组。
6.如实施例1所述的组合、如实施例2所述的用于使用的组合、或如实施例3所述的方法,其中所述mdm2抑制剂是hdm201。
7.如实施例1、4-6中任一项的所述的组合,如实施例2、4-6中任一项所述的用于使用的组合,或如实施例3-6中任一项所述的方法,其中所述ck1α降解剂和/或mdm4抑制剂选自由以下组成的组:沙利度胺,泊马度胺,来那度胺即(rs)-3-(7-氨基-3-氧代-1h-异吲哚-2-基)哌啶-2,6-二酮、也称为revlimid,和sc-24-ur99即4-(3-氨基-1-(4-氯-5-甲基-6-(甲基氨基)吡啶-3-基)-5-氟-1h-吲唑-6-基)萘-1-醇。
8.如实施例1、4-6中任一项的所述的组合,如实施例2、4-6中任一项所述的用于使用的组合,或如实施例3-6中任一项所述的方法,其中所述ck1α降解剂和/或mdm4抑制剂选自来那度胺和sc-24-ur99组成的组。
9.如实施例1所述的组合、如实施例2所述的用于使用的组合、或如实施例3所述的方法,其中所述mdm2抑制剂选自努林-3、伊达努林和hdm201组成的组,并且其中所述ck1α降解剂和/或mdm4抑制剂选自来那度胺和sc-24-ur99组成的组。
10.如实施例1所述的组合、如实施例2所述的用于使用的组合、或如实施例3所述的方法,其中所述mdm2抑制剂是hdm201,并且所述ck1α降解剂和/或mdm4抑制剂是来那度胺。
11.如实施例2、4-10中任一项所述的用于使用的组合,或如实施例3-10中任一项所述的方法,其中所述p53wt肿瘤是实体瘤。
12.如实施例11所述的用于使用的组合、或如实施例11中任一项所述的方法,其中所述实体瘤选自由以下组成的组:肉瘤例如脂肪肉瘤或软组织肉瘤,淋巴瘤例如非霍奇金淋巴瘤(nhl)、特别是套细胞淋巴瘤(mcl),黑素瘤例如皮肤黑素瘤或葡萄膜黑素瘤,母细胞瘤(例如神经母细胞瘤),结肠肿瘤,结直肠肿瘤,肾脏肿瘤,肝肿瘤和皮肤癌例如默克尔细胞癌(mcc)。
13.如实施例11所述的用于使用的组合、或如实施例11所述的方法,其中所述实体瘤是默克尔细胞癌(mcc)。
14.如实施例13所述的用于使用的组合、或如实施例13所述的方法,其中所述默克尔细胞癌(mcc)是默克尔细胞多瘤病毒(mcv)-阳性mcc。
15.如实施例2、4-10中任一项所述的用于使用的组合,或如实施例3-10中任一项所述的方法,其中所述p53wt肿瘤是血液肿瘤(或血液恶性肿瘤)。
16.如实施例15所述的用于使用的组合、或如实施例15所述的方法,其中所述血液肿瘤选自由以下组成的组:急性髓性白血病(aml)、多发性骨髓瘤(mm)、骨髓增生异常综合征(mds)和急性淋巴细胞白血病(all)。
17.如实施例15所述的用于使用的组合、或如实施例15所述的方法,其中所述血液肿瘤选自多发性骨髓瘤(mm)和骨髓增生异常综合征(mds)组成的组。
18.如实施例15所述的用于使用的组合、或如实施例15所述的方法,其中所述血液肿瘤是骨髓增生异常综合征(mds)。
19.如实施例2所述的用于使用的组合、或如实施例3所述的方法,其中所述mdm2抑制剂是hdm201,其中所述ck1α降解剂和/或mdm4抑制剂是来那度胺,并且其中所述p53wt肿瘤是mcv阳性mcc。
20.如实施例2所述的用于使用的组合、或如实施例3所述的方法,其中所述mdm2抑制剂是hdm201,其中所述ck1α降解剂和/或mdm4抑制剂是来那度胺,并且其中所述p53wt肿瘤是mds。
21.如前述实施例中任一项所述的组合/用于使用的组合/方法,进一步包括一种或多种另外的抗癌剂。
本文所述的组合可以提供有益的抗癌作用,例如,增强的抗癌作用、降低的毒性、和/或减少的副作用。例如,第一治疗剂(例如本文披露的任何治疗剂),和第二治疗剂(例如一种或多种另外的治疗剂),或全部,可以按与单一疗法剂量相比达到相同的治疗效果所需的更低的剂量施用。因此,披露了用于使用前述组合疗法来治疗增殖性障碍的组合物和方法。
在一些实施例中,一种用本文所述的组合治疗受试者,例如患有本文所述的癌症的受试者的方法包括施用所述组合作为治疗方案的一部分。在一个实施例中,治疗方案包含一种或多种,例如两种、三种、或四种本文所述的组合。在一些实施例中,按至少一个阶段、并且任选地两个阶段,例如第一阶段和第二阶段,将治疗方案施用至受试者。在一些实施例中,所述第一阶段包括剂量递增阶段。在一些实施例中,所述第一阶段包括一个或多个剂量递增阶段,例如,第一、第二、或第三剂量递增阶段。在一些实施例中,所述剂量递增阶段包括施用包含例如,如本文所述的两种、三种、四种、或更多种治疗剂的组合。在一些实施例中,所述第二阶段包括剂量扩展阶段。在一些实施例中,所述剂量扩展阶段包括施用包含例如如本文所述的两种、三种、四种、或更多种治疗剂的组合。在一些实施例中,所述剂量扩展阶段包含与剂量递增阶段相同的两种、三种、四种、或更多种治疗剂。
在一些实施例中,所述第一剂量递增阶段包括施用包含两种治疗剂、例如两种本文所述的治疗剂的组合,其中一种或两种治疗剂的最大耐受剂量(mtd)或推荐的扩展剂量(rde)已经确定。在一些实施例中,在所述第一剂量递增阶段之前,向受试者施用在所述第一剂量递增阶段中作为单一药剂施用的治疗剂。
在一些实施例中,所述第二剂量递增阶段包括施用包含三种治疗剂、例如三种本文所述的治疗剂的组合,其中一种、两种、或全部治疗剂的最大耐受剂量(mtd)或推荐的扩展剂量(rde)已经确定。在一些实施例中,所述第二剂量递增阶段在所述第一剂量递增阶段结束之后开始。在一些实施例中,所述第二剂量递增阶段包括施用一种或多种在所述第一剂量递增阶段中施用的治疗剂。在一些实施例中,在不进行所述第一剂量递增阶段的情况下进行所述第二剂量递增阶段。
在一些实施例中,所述第三剂量递增阶段包括施用包含四种治疗剂,例如四种本文所述的治疗剂的组合,其中一种、两种、三种、或全部治疗剂的最大耐受剂量(mtd)或推荐的扩展剂量(rde)已经确定。在一些实施例中,所述第三剂量递增阶段在所述第一或第二剂量递增阶段结束之后开始。在一些实施例中,所述第三剂量递增阶段包括施用一种或多种(例如全部)在所述第二剂量递增阶段中施用的治疗剂。在一些实施例中,所述第三剂量递增阶段包括施用一种或多种在所述第一剂量递增阶段中施用的治疗剂。在一些实施例中,在不进行所述第一、第二、或这两个剂量递增阶段的情况下进行所述第三剂量递增阶段。
在一些实施例中,所述剂量扩展阶段在所述第一、第二或第三剂量递增阶段结束之后开始。在一些实施例中,所述剂量扩展阶段包括施用在所述剂量递增阶段,例如所述第一、第二、或第三剂量递增阶段中施用的组合。在一个实施例中,在所述剂量扩展阶段中,从受试者获得活检。
不希望受理论束缚,据信在一些实施例中,包括剂量递增阶段和剂量扩展阶段的治疗方案允许新的药剂或方案进入以用于组合、组合的快速产生、和/或评估可耐受的组合的安全性和活性。
在此,诸位发明人证明了mcvst充当转录激活因子以增加mdm2和ck1α的水平,进而与mdm4共同抑制p53在mcc中的功能。诸位发明人进一步证明了在mcc中靶向mdm2和mdm4两者的协同功效。
默克尔细胞癌(mcc)是一种侵袭性皮肤癌。病毒(默克尔细胞多瘤病毒,mcv)阴性mcc在rb和p53中包含失活突变,而mcv阳性mcc通常包含野生型rb和p53。诸位发明人证明了mcv大t抗原与rb结合导致p53激活,而mcv小t抗原通过增加mdm2和ck1α(mdm4的激活因子)的水平来减少p53激活。来那度胺或特异性mdm4抑制剂与mdm2抑制剂协同作用对ck1α进行靶向降解,以激活p53并诱导细胞凋亡。诸位发明人的工作揭示了mcv调控mcc中p53的机制,并证明了组合靶向mdm2和mdm4在p53野生型肿瘤中的效用。
默克尔细胞多瘤病毒(mcv)占所有默克尔细胞癌(mcc)(皮肤的高度侵袭性神经内分泌癌)的约80%。mcv阳性mcc表达小t抗原(st)和大t抗原(lt)的截短形式,且通常包含野生型p53(tp53)和rb(rb1)。相反,病毒阴性mcc包含tp53和rb1中的失活突变。尽管mcv截短的lt可以结合并抑制rb,但它不结合p53。诸位发明人在此披露了mcvlt与rb结合,导致arf(mdm2的抑制剂)水平升高和p53激活。但是,st的共表达减少了p53的激活。
mcvst将myc同源物mycl(l-myc)募集到ep400染色质重塑复合物上,并激活特异性靶基因。诸位发明人观察到mcv阳性mcc细胞系中ep400的耗竭导致增加的p53靶基因表达。诸位发明人怀疑mcvst-mycl-ep400复合物可以在功能上使p53失活,但是其潜在机制尚不清楚。ep400耗竭后,chip和rna-seq整合分析确定了mdm2和ck1α(mdm4的激活因子)是st-mycl-ep400复合物的靶基因。此外,mcv阳性mcc细胞表达高水平的mdm4。mdm2抑制剂与靶向ck1α的来那度胺或mdm4抑制剂的组合引起p53的协同激活,导致小鼠mcv阳性mcc细胞和mcc来源的异种移植物中发生凋亡反应。这些结果支持mdm2和mdm4在病毒阳性mcc和其他p53野生型肿瘤中的双重靶向。
附图说明
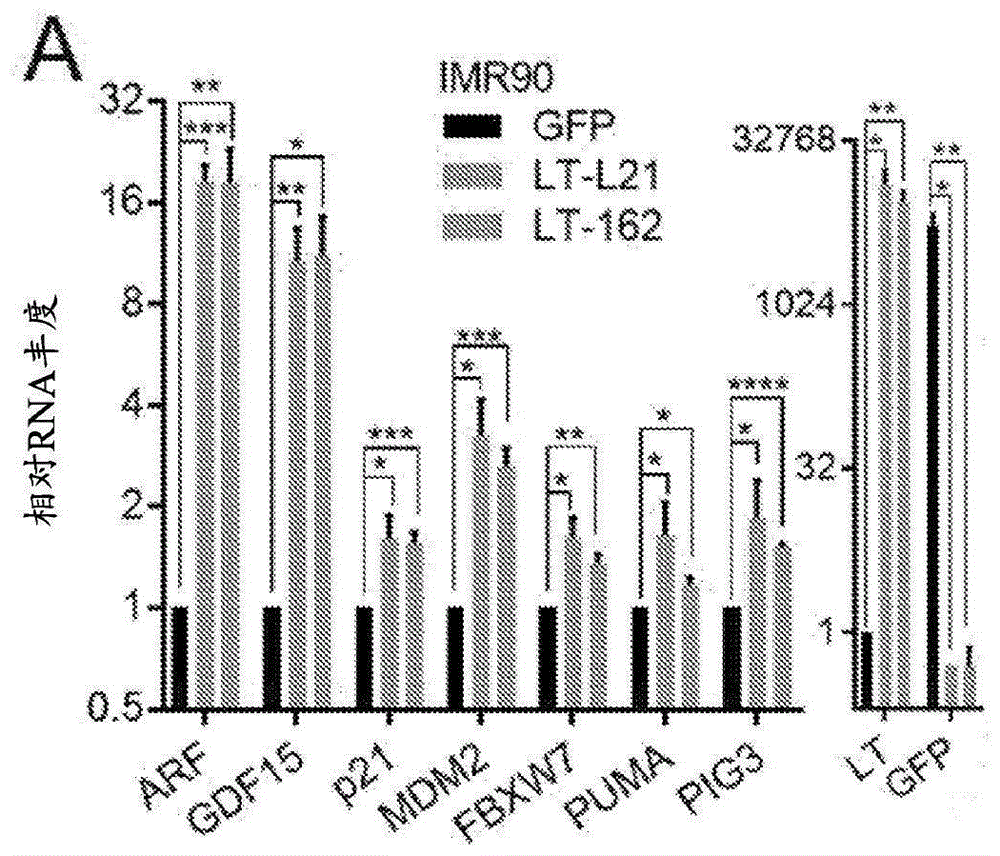
图1.默克尔细胞多瘤病毒大t抗原激活p53反应而小t抗原抑制p53反应。
a.mcvlt的截短的肿瘤亚型的诱导型表达增加imr90细胞中arf和p53靶基因。用强力霉素(dox)处理24小时诱导gfp或lt-l21和lt-162截短的lt的表达。lt、arf和p53靶基因rna水平相对于gfp诱导的细胞的那些归一化,而gfp水平相对于lt-l21样品归一化。数据显示为平均值±sd;*学生t检验p<0.05,**p<0.005,***p<0.0005,****p<0.00005。
b.诱导imr90细胞40小时以表达gfp、st、lt-l21或lt-l21与st。在dox之前(-)或之后(+)制备裂解物。p53反应的激活通过p53、磷酸丝氨酸15p53(p-p53)、乙酰赖氨酸382p53(ac-p53)、p21和裂解的parp的水平升高(**)来反映。
c.lxcxe基序(e216k)中l21的表达而不是lt突变体的表达通过抑制rb和诱导arf激活p53。
图2.mdm2和ck1α是st-mycl-ep400复合物的转录靶标。
a.火山图说明了相对于对照shrna,在用诱导型shrna耗竭ep400后,mkl-1mcc细胞系中差异表达的p53靶基因。针对统计显著性,将每个基因log2倍数变化相对于-log10p值作图。绿点表示满足2倍变化临界值的基因,而红点表示经调整的p值小于0.1。
b.在shrna被诱导8天后,用mkl-1细胞进行rt-qpcr。将读数相对于rplp0和未诱导的样品归一化。实验进行了三次并取平均值。数据显示为平均值±sd;*学生t检验p<0.05,**p<0.005,***p<0.0005,****p<0.00005。
c.用max、ep400、st和igg抗体进行chip,然后对mkl-1细胞中指定的启动子进行qpcr。chip-qpcr进行了3次,显示了平均输入百分比。
d.mcvt抗原的耗竭导致mdm2、ck1α和mdm4水平降低。用特异性shrna转导mkl-1细胞5天,并收获用于蛋白质印迹。
e.在mcvt抗原存在(+)或不存在(-)下,针对imr90细胞中mdm2启动子,max、ep400和st抗体的chip-qpcr。chip独立进行了五次。
f.努林-3处理不会引发p53反应,但是mcvt抗原会增加表达p53dd的imr90细胞中mdm2、mdm4和ck1α的水平。将mkl-1和imr90-p53dd用努林-3(1μm)处理24小时。
图3.mdm4在mcv阳性mcc中过表达。
a.收获来自mcc细胞系和人包皮成纤维细胞(hff)的rna,用于mdm4(完整)、mdm4-fl(全长变体)和mdm4-s(短剪接变体)的rt-qpcr。mdm4水平用rplp0、18srrna和β-肌动蛋白rna对照的几何平均值进行归一化。数据显示为平均值±sd;*对于mdm4-fl,学生t检验p<0.05。
b.用所示抗体对mcc细胞系和hff进行蛋白质印迹。由于失活性突变,ms-1和mcc13过表达p53,而mkl-2和mcc26未表达可检出水平的p53。
图4.mdm2和mdm4的抑制增强mcc细胞系中的p53激活。
a.来那度胺在mkl-1中通过努林-3增强p53激活。将mkl-1细胞用努林-3(5μm)、来那度胺(len,10μm)或两者处理40小时。
b.来那度胺消耗ck1α,但在uiso细胞中不会通过努林-3增强p53激活。加或不加来那度胺(10μm),将mkl-1(mcv+mcc)或uiso(mcv-mcc)细胞用努林-3(1μm)处理24小时。值得注意的是,uiso的mdm2和mdm4蛋白少于mkl-1。
c.来那度胺和泊马度胺(以较低程度)而非沙利度胺与努林-3共同激活p53。与来那度胺、泊马度胺(10μm)或沙利度胺(10μm)一起,用努林-3处理mkl-1细胞24小时。
d.来那度胺处理减少mdm4与p53和激活的mdm2的结合。将mkl-1细胞用努林-3(5μm)、来那度胺(10μm)或两者处理40小时,并收获其以用针对mdm4、p53和ck1α的抗体进行免疫沉淀,然后进行蛋白质印迹。
e.通过crispr转导耗竭ck1α增强了努林-3的p53激活。加或不加来那度胺,将稳定表达两种ck1αsgrna中的每一种的mkl-1细胞用努林-3(1μm)处理24小时。来那度胺进一步降低了ck1α,sgrna并未完全耗竭。
f.mdm4抑制剂sc-24-ur99(ur99)与mdm2抑制剂共同激活p53。加或不加来那度胺(1μm)或ur99(0.1μm),将mkl-1细胞用努林-3(1μm)或hdm201(0.1μm)处理。
图5.mdm2和ck1α-mdm4的抑制协同诱导通过细胞凋亡引起的细胞死亡。
a.用几种浓度的mdm2抑制剂、努林-3、rg7388或amg232处理mkl-1和ms-1细胞,处理96小时后进行xtt测定。**多重t检验p值<0.005。
b.bliss协同测试显示了努林-3和来那度胺或sc-24-ur99之间的强协同作用,但与沙利度胺无。
c.使用来那度胺、努林-3或两种药物处理16小时的mkl-1细胞进行bh3分析。实验进行了三次。数据显示为平均值±sd;*学生t检验p<0.05。
d.scid小鼠中的mkl-1mcc异种移植物对hdm201和来那度胺的组合处理有反应。当异种移植肿瘤为200mm3时,每天口服施用hdm201(40mg/kg)、来那度胺(50mg/kg)或两种药物。数据显示为平均值±sem;hdm201与组合处理之间的多重t检验*p<0.05#-由于肿瘤体积达到最大允许大小,研究终止。
e.模型:默克尔细胞多瘤病毒t抗原使默克尔细胞癌对于靶向p53-mdm2-mdm4途径敏感。
图6(本文中也称为图s1)。
a.mcvlt的全长或截短的肿瘤亚型的诱导型表达激活imr90细胞中的p53。用强力霉素(dox)处理40小时诱导gfp或wt(野生型)lt、fl(全长)lt、162、168和l21截短的lt的表达。在dox之前(-)或之后(+)制备裂解物。p53反应的激活通过p53、磷酸丝氨酸15p53(p-p53)、乙酰赖氨酸382p53(ac-p53)、p21和裂解的parp的水平升高(**)来反映。
b.rt-qpcr显示imr90细胞中lt-l21的诱导增加了已知p53靶基因p21的水平。单独或与作为剪接变体的st一起,将l21-lt诱导4、8、12、16、20、24、36、48、60和72小时,并收集rna用于rt-qpcr。显示的数据是三个独立实验的代表值。**双因素anovap值<0.005。
c.lxcxelt突变体可以结合vps39,但不结合rb。收集稳定表达gfp、lt-l21或lt-l21e216k突变体的hct116细胞,用于lt免疫沉淀。
图7(本文中也称为图s2)。
a.rna-seq的归一化计数表明,mkl-1中相对于对照shrna,ep400shrna的mdm2水平降低而p21水平增加。
b.使用两种独立的shrna使ep400耗竭后,与对照shrna相比,mdm2、ck1α(csnk1a1)和ep400的rna-seq倍数变化显示mdm2和ck1α与ep400的水平降低。显示了bonferroni校正的经调整的p值。
c.诱导mkl-1细胞8天以表达ep400或对照shrna。蛋白质印迹显示,使用ep400shrna,mdm2和ep400水平降低,而p53和p21水平升高。
d.用指示的抗体,对具有(+)或不具有(-)mcvt抗原(lt-l21和st)的、稳定表达htert、p53dd、mycl(imr90-p53dd)的imr90细胞进行印迹。
e.max、ep400和st的两个独立的chip-seq在mdm2和ck1α启动子处显示峰,也由h3k4me3标记出。
f.st的chip-qpcr显示mcvst与表达mcvt抗原的imr90-p53dd细胞中mdm2和ck1α启动子结合。实验进行了两次。数据显示为平均值±sd;*p<0.05,n.s>=0.05。
g.st和ep400在p53耗竭的mkl-1细胞中富集mdm2和ck1α启动子。使用稳定表达p53shrna的mkl-1。实验进行了两次。
图8(本文中也称为图s3)。
a.mdm4-fl(全长)与mdm4-s(短变体)的比率。
b.对101例mcc的cbioportal分析揭示,与无tp53或rb1突变的mcc(64例)相比,37例肿瘤带有tp53或rb1突变和更高水平的突变。mdm2和mdm4显示频繁的拷贝数增加(3-8拷贝)和扩增(>8拷贝)。
图9(本文中也称为图s4)。
a.来那度胺迅速降低mkl-1中的ck1α蛋白。加或不加mg132(10μm),将mkl-1细胞用来那度胺(len,10μm)和/或环己酰亚胺(cx,5μm)处理2、4或6小时。p53是环己酰亚胺脉冲追踪处理的阳性对照。
b.加或不加来那度胺(10μm),将mkl-1、waga、peta、broli和ms-1mcv阳性mcc细胞系用努林-3(5μm)处理40小时。
c.来那度胺(10μm)和泊马度胺(10μm)(以较低程度)而非沙利度胺(10μm)可降低ck1α蛋白水平。
d.将mkl-1细胞用努林-3、来那度胺或两者处理16小时,然后用环己酰亚胺处理2和4小时。使用licor定量蛋白质印迹条带,且每种处理相对于0小时时间点进行归一化。对于dmso和来那度胺处理的样品,p53的半衰期估计为1.5小时,而对于努林-3处理的样品,p53的半衰期估计为4小时。实验进行了3次,数据显示为平均值±sd;**双因素anova<0.005。
e.将mkl-1细胞用努林-3(5μm)、来那度胺(10μm)或两者处理24或40小时,并收获以使用mdm4、p53和ck1α进行ip蛋白质印迹。
图10(本文中也称为图s5)。
a-c.加或不加来那度胺(固定浓度5μm),将mkl-1细胞用努林-3(a)、rg7388(b)或amg232(c)处理。xtt在96小时处理后进行。
d.加或不加来那度胺,将mkl-1(具有野生型p53)和ms-1(具有p53突变)用努林-3处理96天,并进行xtt测定以测量相对于dmso对照归一化的相对细胞活力。
e.来那度胺和泊马度胺(以较低程度)而非沙利度胺与努林-3协同作用降低mkl-1活力。xtt测定在处理96小时后进行,三次生物学重复。数据显示为平均值±sd;*p<0.05,**p<0.005,***p<0.0005,****p<0.00005以及n.s>=0.05。
图11(本文中也称为图s6)。
a.compusyn协同测试表明努林-3和rg7388与来那度胺具有协同作用。在处理mkl-1细胞96小时后进行xtt测定。ci值小于1表示协同作用。
b-d.努林-3与沙利度胺(b)、来那度胺(c)或sc-24-ur99(d)的剂量反应矩阵。
e.zip协同测试
f.hsa协同测试。
具体实施方式
lt激活p53反应,而st抑制p53反应。
为了研究mcvt抗原对正常细胞中p53的影响,将表达gfp或肿瘤来源的截短或全长形式的lt的强力霉素诱导型载体引入imr90二倍体肺成纤维细胞(图1a,s1a)。lt的截短形式包括l21(编码残基1-292)、162(残基1-320)和168(残基1-275),每个均包含完整的lxcxe基序[6]。通过rt-qpcr评估,imr90中lt-l21或lt-162表达显著提高了arf和几个p53靶基因(包括gdf15和p21(cdkn1a))的水平(图1a)。rb的抑制激活e2f转录因子,导致arf水平升高[10]。arf是主要p53降解e3连接酶mdm2的有效抑制剂[14]。lt表达增加了p53以及磷酸丝氨酸15p53(p-p53)和p21的蛋白水平,表明p53活化(s1a)。lt-162的表达还导致裂解的parp水平升高(**),表明发生了凋亡反应。
lt和st在mcc肿瘤中作为整合的mcpyv病毒dna的剪接变体共表达。为了在imr90细胞系中模拟这一点,诸位发明人引入了共表达st的lt-l21的基因组形式。当st与lt-l21共表达时,与对仅lt-l21的反应相比,观察到较低水平的p53激活(p21、p-p53和乙酰赖氨酸382或ac-p53)(图1b,s1b)。结果表明,当共表达时,截短lt可以激活p53,而st可以减少这种反应。为了确定增加的arf水平是否需要lt与rb结合,诸位发明人在lt-l21的lxcxe基序(e216k)中引入了点取代突变。当在hct116细胞中稳定表达时,l21-e216k无法共沉淀rb而是保留与vps39的结合,而vps39与lt(s1c)的不同区域结合[5]。当在imr90细胞中表达时,l21-e216k不会增加arf、p21、p53或ac-p53的水平(图1c)。这些结果表明lt与rb的结合有助于提高arf水平和激活p53反应。值得注意的是,st与l21-e216k共表达时,检测到mdm2水平升高,但未检测到p21水平升高。
mdm2和ck1α是st-mycl-ep400复合物的转录靶标。
诸位发明人最近报道,mcvst将myc同源物mycl(l-myc)募集到ep400染色质重塑复合物上,以结合特异性基因启动子并激活其表达[15]。为了鉴定mkl-1细胞中由st-mycl-ep400复合物调控的基因,使用三种不同的shrna在ep400耗竭后进行rna-seq[15]。利用报道的rna-seq结果,诸位发明人评估了已知p53靶基因的基因表达变化[9]。如火山图所示,ep400耗竭导致包括p21(cdkn1a)在内的许多p53靶基因水平升高(图2a)。使用1.5的倍数变化临界值,总计198个p53靶基因中有59个基因被上调,而17个基因被下调(表s1)[9]。ep400耗竭也导致mdm2e3连接酶和ck1α水平降低(图2b,s2a-c)。mdm2和mdm4包含n末端p53结合结构域,该结构域直接与p53的反式激活结构域结合,从而阻断p53激活[11]。mdm4可以调节其自身对p53的活性。mdm4的n末端p53结合结构域与其中心w基序区形成分子内相互作用,从而减少与p53的结合[12]。ck1α(csnk1a)是一种丝氨酸/苏氨酸激酶,可结合并磷酸化mdm4,从而阻止这种自抑制相互作用并激活mdm4[13]。rt-qpcr和蛋白质印迹证实,在mkl-1细胞中ep400敲低后,p21水平升高,而mdm2和ck1α水平降低(图2b,s1c)。
使用针对st、max(与mycl二聚)和ep400的抗体,chip-seq揭示了mdm2和ck1α启动子的富集(s2e)[15]。诸位发明人对max、st和ep400进行了chip-qpcr,并观察到了这些启动子的特异性富集(图2c)。诸位发明人在mkl-1细胞中使用特异性shrna耗竭了st或st和lt,并且发现mdm2和ck1α的水平降低了shrna的表达(图2d)[16]。该结果与rt-qpcr和chip数据一起表明mdm2和ck1α是st-mycl-ep400复合物的直接转录靶标。值得注意的是,st耗竭后mdm4水平降低,尽管诸位发明人没有发现st直接激活mdm4的证据。st增加了可激活mdm4对p53的活性的ck1α的水平。
由于mdm2是p53靶基因,因此mcvt抗原可能通过激活p53间接增加mdm2水平[9]。为了排除这种可能性,诸位发明人向imr90细胞中引入了p53的显性负性型(p53dd),其结合内源性p53并使内源性p53失活[17]。用mycl和带有st的mcvlt-l21进一步转导imr90-p53dd细胞[17]。诸位发明人通过chip-qpcr检测到st与mdm2和ck1α启动子的结合,并观察到在mcvt抗原存在下,ep400对mdm2启动子的富集增加(图2e,s2f)。用努林-3进行的mdm2抑制剂治疗并未增加p53、p-p53和puma水平,指示p53激活(图2f,s1d)。为了进一步确定st-mycl-ep400复合物独立于p53而与这些启动子结合,诸位发明人用shrna使mkl-1中的p53耗竭,并进行了ep400和st的chip-qpcr(s2g)。诸位发明人发现p53不影响ep400和st对mdm2和ck1α启动子的富集。这些结果表明,mcvst独立于p53反式激活mcc和imr90细胞中的mdm2和ck1α。
mdm4在病毒阳性mcc中过表达。
在某些具有野生型p53的癌症中可以发现mdm4的过表达[18]。诸位发明人使用三组mdm4引物来评估mcc细胞系中的总mdm4(所有剪接变体)、mdm4-fl(全长)和mdm4-s(缺失ring结构域的短变体)水平[18]。病毒阳性(mkl-1、mkl-2、ms-1、waga、peta和broli)mcc细胞系相对于病毒阴性(uiso、mcc13和mcc26)mcc细胞系具有显著更高的总mdm4和mdm4-fl水平)(图3a)。此外,在病毒阳性细胞系(s3a)中,mdm4-fl与mdm4-s的比率更高。诸位发明人评估了这些mcc细胞系中的mdm4-fl蛋白水平。与病毒阴性mcc细胞系相比,病毒阳性mcc细胞系表达高水平的mdm4-fl(图3b)。值得注意的是,病毒阴性uiso细胞系具有高mdm4-fl与mdm4-s比率,但不表达丰度水平的mdm4蛋白(图3a,s3a)。在病毒阳性mcc中p53的lt激活可能会导致对mdm2和mdm4表达的依赖性。101个mcc肿瘤的靶向下一代测序(oncopanel)显示,无论是否存在mcv,mdm4的拷贝增益(3-8拷贝)频率低(s3b)[19]。
抑制mdm2和mdm4可激活mcc中的p53。
诸位发明人观察到努林-3mdm2抑制剂治疗激活了p53,如含有野生型p53的mkl-1、waga、peta和brolimcc细胞系中总p53、p-p53、ac-p53、p21和puma的水平升高,但在具有失活的p53突变的ms-1细胞中却没有所表明的(图2f,图4a,s2b)[20]。最近的报道表明,在来那度胺存在下,crbn(cereblon)e3连接酶可以特异性靶向ck1α进行泛素化[21,22]。诸位发明人评估来那度胺是否可以降低mcc细胞中的ck1α水平并激活p53。加或不加环己酰亚胺,将mkl-1细胞用来那度胺处理,以阻断蛋白质合成。尽管环己酰亚胺处理6小时后ck1α水平没有明显变化,但加入来那度胺(s4a)后,水平迅速下降。
单独来那度胺处理对p53水平有中等程度的积极效果。然而,当将努林-3和来那度胺组合时,在具有野生型p53的细胞系中观察到p53和p53靶基因的较大增加(图4a,s4b)。诸位发明人通过定量蛋白质印迹法(s4d)评估了在环己酰亚胺存在下用努林-3、来那度胺或两者处理的mkl-1细胞中的p53稳定性。在努林-3存在下,来那度胺显著增加了p53的稳定性[23]。来那度胺耗竭ck1α可能会降低mdm4对p53的活性。诸位发明人测试了来那度胺是否增强了表达很少mdm2和mdm4的病毒阴性和p53野生型uiso细胞中的p53活化(图3a-3b)。诸位发明人观察到来那度胺降低了uiso细胞中ck1α水平,但是未能增强努林-3对p53的激活(图4b)。
crbn的结构分析显示来那度胺可以强烈促进与ck1α的相互作用,而相关化合物沙利度胺和泊马度胺募集ck1α到crbn的能力要低得多[22]。与来那度胺相比,泊马度胺和沙利度胺不会降低mkl-1细胞中的ck1α蛋白水平(s4c)。此外,泊马度胺或沙利度胺未增强mkl-1细胞中p53对努林-3的反应(图4c)。为了测试ck1α对mdm4与p53结合的贡献,诸位发明人用从用努林-3和来那度胺处理40小时的mkl-1细胞制备的裂解物对mdm4、p53和ck1α进行了免疫沉淀(ip)(图4d和s4e)。与仅努林-3相比,来那度胺和努林-3降低了ck1α水平并增加了p53水平。尽管p53的水平增加,但与未处理或努林-3处理的对照相比,来那度胺与努林-3共处理并未增加mdm4、ck1α或激活的mdm2对p53的共沉淀。这表明ck1α的水平降低减少mdm4与p53的结合。
如果ck1α能够使mdm4与p53结合,则诸位发明人预期ck1α的缺失将减少mdm4与p53的结合并增强p53激活[13]。为了测试这一点,诸位发明人通过两个独立的crisprsgrna耗竭了mkl-1中的ck1α,并评估了努林-3处理后的p53激活(图4e)[24]。crispr敲除ck1α导致p53活性增强(通过激活的mdm2(磷酸-丝氨酸166,p-mdm2)、p-p53、p53和p21的增加的水平测定),表明来那度胺或sgrna降低ck1α增强了p53激活[25]。值得注意的是,来那度胺的添加降低了crispr并未完全耗尽的ck1α蛋白,并进一步增加了p53激活的标记。诸位发明人测试了新开发的mdm4抑制剂(sc-24-ur99,ur99)是否可以增强mdm2抑制剂努林-3或hdm201的活性(图4f)[26]。ur99特异性阻断mdm4与p53的结合。诸位发明人发现ur99对mdm4的抑制增加了mdm2抑制剂对p53的激活。
来那度胺与mdm2抑制剂协同作用,以诱导mcc细胞系的细胞凋亡。
为了探查mdm2抑制对mcc细胞活力的影响,诸位发明人用mdm2抑制剂努林-3、rg7388或amg232将mkl-1(p53野生型)和ms-1(p53突变体)细胞处理了96小时,并进行了xtt活力测定[27]。mkl-1而非ms-1细胞对所有三种mdm2抑制剂均敏感(图5a)。诸位发明人测试了来那度胺与努林-3、rg7388和amg232的组合的效果,并且观察到当与来那度胺(s5a-d)一起使用时,所有三种mdm2抑制剂的细胞毒性均显著改善。使用compusyn(s6a)和bliss(图5b)方法进行的协同测试揭示,努林-3或rg7388与来那度胺或ur99(s6b-f)的组合具有协同活性[28-31]。相反,沙利度胺与努林-3组合使用时没有协同作用的证据,这与它对ck1α水平的相对减少的影响一致(图5b,s5e)。
为了确定在mcc细胞中来那度胺是否会影响努林-3通过细胞凋亡引起的细胞死亡的能力,诸位发明人进行了bh3谱分析,测量了其对游离bh3肽的敏感性[32]。将来那度胺添加到努林-3增强对细胞凋亡的启动作用(图5c)。为了确定mdm2和mdm4的体内双重抑制,加或不加来那度胺,诸位发明人用hdm201(适用于体内功效研究)处理mkl-1mcc异种移植物(图5d,表s3)。诸位发明人发现添加来那度胺大大增强了hdm201的功效,为联合治疗表达mdm2和mdm4的p53野生型肿瘤提供了临床应用的潜力。诸位发明人提出了一种模型,其中mcvt抗原增加了对mdm2、mdm4和ck1α的依赖性,以抑制p53活性,在病毒阳性mcc中具有治疗潜力(图5e)。
来那度胺已被用于治疗恶性肿瘤、骨髓增生异常综合征(mds)和多发性骨髓瘤(mm)[21]。据报道,突变体csnk1a1预测来那度胺治疗的mds的预后不良[33]。此外,与野生型p53的mds相比,具有突变体p53的mds对来那度胺的响应不太好,并且更可能进展为急性髓细胞性白血病,这表明来那度胺的作用可能部分取决于mdm4的失活[34]。诸位发明人的工作提供了来那度胺与mdm2抑制剂的组合在mcc和其他实体瘤和含有野生型p53的血液恶性肿瘤中的原理。
为了供人类使用,根据revlimid的处方信息,来那度胺可以例如以胶囊的形式口服施用,例如对于mm组合疗法:在28天的重复周期中的第1-21天,每天口服一次25mg;对于mm维持治疗,在自体hsct之后:在28天的重复周期的第1-28天,连续地每天一次10mg;对于mds:每天一次10mg;对于mcl:在28天的重复周期的第1-21天,每天口服一次25mg。基于临床和实验室检查结果继续或调整给药,在肾损害的情况下根据肌酐清除率值调整起始剂量。
为了供人类使用,hdm201可以例如以胶囊的形式口服施用。特别地,口服施用可以使用高剂量间歇方案[例如,方案a(在3周周期的第1天施用50mg-400mghdm201)或方案b(在4周周期的第1天和第8天施用50mg-150mghdm201)或方案c(在4周周期的第1天施用50mg-500mghdm201]或低剂量延长方案[例如,方案d(在4周周期的前2周中每天一次10mg-30mghdm201)或方案e(在4周周期的第一周中每天一次15mg-50mghdm201)]。
为了供人类使用,伊达努林可以口服施用,例如在每个28天的治疗周期的第1-5天,每天一次或每天两次,以胶囊的形式口服施用。日剂量可以是从100mg至1000mg。
为了供人类使用,sc-24-ur99可以例如以溶液或胶囊的形式静脉内注射或口服施用。施用可以使用高剂量间歇方案[例如,方案a(在3周周期的第1天施用10mg-1000mgsc-24-ur99)或方案b(在4周周期的第1天和第8天施用5mg-500mgsc-24-ur99)或方案c(在4周周期的第1天施用100mg-1000mgsc-24-ur99]或低剂量延长方案[例如,方案d(在4周周期的前2周中每天一次1mg-100mgsc-24-ur99)或方案e(在4周周期的第一周中每天一次1mg-200mgsc-24-ur99)]。
化学式
本文提及的化合物也可以由以下化学式表示。
来那度胺
hdm201
伊达努林
sc-24-ur99
表
表s1.p53靶向ep400shrna存在下显著差异表达的基因
表s2.rt-和chip-qpcr引物
表s3.
肿瘤体积变化值,%ch=(t-t0)/t0,绝对ch=t-t0,t-当前值,t0-初始值
定义
如本文所用,冠词“一个”和“一种”是指一个或多于一个(例如,至少一个)该冠词的语法宾语。
除非上下文另有明确说明,否则术语“或”在本文中用于表示术语“和/或”并且可与术语“和/或”互换使用。
“约”和“大约”通常表示在给定测量的性质或精度的情况下测量的量的可接受的误差度。示例性误差度在给定值或值范围的20%内,典型地在10%内,并且更典型地,在5%内。
如本文所用,冠词“一个”和“一种”是指一个或多于一个(例如,至少一个)该冠词的语法宾语。
除非上下文另有明确说明,否则术语“或”在本文中用于表示术语“和/或”并且可与术语“和/或”互换使用。
“约”和“大约”通常表示在给定测量的性质或精度的情况下测量的量的可接受的误差度。示例性误差度在给定值或值范围的20%内,典型地在10%内,并且更典型地,在5%内。
所谓“组合”或“与……组合”并不旨在暗示疗法或治疗剂必需同时施用和/或将这些疗法或治疗剂配制用于一起递送,尽管这些递送方法也在本文所述的范围内。组合中的治疗剂可以与一种或多种其他另外的疗法或治疗剂同时、在其之前或之后施用。这些治疗剂或治疗方案可以以任何顺序施用。通常,每种药剂将以针对该药剂确定的剂量和/或日程表施用。还应理解,该组合中使用的另外的治疗剂可以按单一组合物一起施用或按不同组合物单独施用。通常,预期组合中使用的其他治疗剂以不超过它们单独使用时的水平使用。在一些实施例中,组合中使用的水平将低于单独使用的水平。
在实施例中,另外的治疗剂以治疗剂量或低于治疗剂量给予。在某些实施例中,当将第二治疗剂与第一治疗剂组合施用时,实现抑制例如生长抑制所需的第二治疗剂的浓度降低。在某些实施例中,当将第二治疗剂与第一治疗剂组合施用时,实现抑制所需的第一治疗剂的浓度比单独施用第一治疗剂时低。在某些实施例中,在组合疗法中,实现抑制所需的第二治疗剂的浓度比作为单一疗法的第二治疗剂的治疗剂量低,例如低10%-20%、20%-30%、30%-40%、40%-50%、50%-60%、60%-70%、70%-80%、或80%-90%。在某些实施例中,在组合疗法中,实现抑制所需的第一治疗剂的浓度比作为单一疗法的第一治疗剂的治疗剂量低,例如低10%-20%、20%-30%、30%-40%、40%-50%、50%-60%、60%-70%、70%-80%、或80%-90%。
术语“抑制”、“抑制剂”或“拮抗剂”包括给定分子(例如免疫检查点抑制剂)的某些参数(例如活性)的减小。例如,此术语包括活性,例如给定分子,例如抑制性分子的活性的至少5%、10%、20%、30%、40%或更多的抑制。因此,抑制不必是100%。
术语“抗癌作用”是指可以通过各种手段显现的生物学作用,包括但不限于例如肿瘤体积减少、癌细胞数量减少、转移数量减少、预期寿命延长、癌细胞增殖减少、癌细胞存活率降低、或改善与癌症相关的各种生理症状。“抗癌作用”还可以通过肽、多核苷酸、细胞和抗体首先预防癌症发生的能力来显现。
术语“抗肿瘤作用”是指可以通过各种手段显现的生物学作用,包括但不限于例如肿瘤体积减少、肿瘤细胞数量减少、肿瘤细胞增殖减少、或肿瘤细胞存活率降低。
术语“癌症”是指以异常细胞的快速和不受控制的生长为特征的疾病。癌细胞可以局部或通过血流和淋巴系统扩散到身体的其他部位。本文描述了各种癌症的实例,并且包括但不限于乳腺癌、前列腺癌、卵巢癌、宫颈癌、皮肤癌、胰腺癌、结肠直肠癌、肾癌、肝癌、脑癌、淋巴瘤、白血病、肺癌等。术语“肿瘤”和“癌症”在本文中可互换使用,例如,这两个术语包括实体和液体瘤,例如,弥散或循环肿瘤。如本文所用,所述术语“癌症”或“肿瘤”包括恶化前以及恶性癌症和肿瘤。如本文所用的术语“癌症”包括原发性恶性细胞或肿瘤(例如其细胞未迁移到受试者体内除原始恶性肿瘤或肿瘤部位以外的部位的那些)和继发性恶性细胞或肿瘤(例如由转移(恶性细胞或肿瘤细胞迁移到与原始肿瘤部位不同的第二部位)引起的那些)。
如本文所用,术语“治疗(treat、treatment和treating)”是指由施用一种或多种疗法导致的障碍(例如增殖性障碍)的进展、严重程度和/或持续时间的减少或缓解,或者障碍的一种或多种症状(优选地,一种或多种可辨别的症状)的缓解。在具体实施例中,术语“治疗(treat、treatment和treating)”是指增殖性障碍的至少一种可测量的物理参数(诸如肿瘤的生长)的改善,但该物理参数不一定是患者可辨别的。在其他实施例中,所述术语“治疗(treat、treatment和treating)”是指通过例如稳定可辨别的症状来物理地,或通过例如稳定物理参数来生理地,或通过两者,抑制增殖性障碍的进展。在其他实施例中,所述术语“治疗(treat、treatment和treating)”是指减少或稳定肿瘤大小或癌细胞计数。
术语“受试者”在本文中是指人类。人类可以是成人、青少年、儿童或婴儿。
本文中的术语mdm4抑制剂是指优先结合mdm-4、破坏p53-mdm4结合、从而释放螯合的p53蛋白的抑制剂。
参考文献
1.beckerjccetal.(2017)merkelcellcarcinoma.natrevdisprimers3:17077.
2.paulsonkgetal.(2017)merkelcellcarcinoma:currentusincidenceandprojectedincreasesbasedonchangingdemographics.j.am.acad.dermatol.
3.fengh,shudam,changy,mooreps(2008)clonalintegrationofapolyomavirusinhumanmerkelcellcarcinoma.science(newyork,n.y.)319(5866):1096-1100.10.1126/science.1152586.
4.rodigsjetal.(2012)improveddetectionsuggestsallmerkelcellcarcinomasharbormerkelpolyomavirus.thejournalofclinicalinvestigation122(12):4645-4653.10.1172/jci64116.
5.shudam,kwunhj,fengh,changy,mooreps(2011)humanmerkelcellpolyomavirussmalltantigenisanoncoproteintargetingthe4e-bp1translationregulator.thejournalofclinicalinvestigation121(9):3623-3634.10.1172/jci46323.
6.chengj,rozenblatt-roseno,paulsonkg,nghiemp,decaprioja(2013)merkelcellpolyomaviruslargetantigenhasgrowth-promotingandinhibitoryactivities.journalofvirology87(11):6118-6126.10.1128/jvi.00385-13.
7.starrettgjetal.(2017)merkelcellpolyomavirusexhibitsdominantcontrolofthetumorgenomeandtranscriptomeinvirus-associatedmerkelcellcarcinoma.mbio8(1).10.1128/mbio.02079-16.
8.harmspwetal.(2015)thedistinctivemutationalspectraofpolyomavirus-negativemerkelcellcarcinoma.cancerresearch75(18):3720-3727.10.1158/0008-5472.can-15-0702.
9.allenmaetal.(2014)globalanalysisofp53-regulatedtranscriptionidentifiesitsdirecttargetsandunexpectedregulatorymechanisms.elife3.10.7554/elife.02200.
10.batessetal.(1998)p14arflinksthetumoursuppressorsrbandp53.nature395(6698):124-5.
11.nomuraketal.(2017)structuralanalysisofmdm2ringseparatesdegradationfromregulationofp53transcriptionactivity.nat.struct.mol.biol.24(7):578-587.
12.bistam,petrovichm,fershtar(2013)mdmxcontainsanautoinhibitorysequenceelement.proc.natl.acad.sci.u.s.a.110(44):17814-9.
13.chenletal.(2015)autoinhibitionofmdmxbyintramolecularp53mimicry.proc.natl.acad.sci.u.s.a.112(15):4624-9.
14.beliveaua,yaswenp(2007)soothingthewatchman:telomerasereducesthep53-dependentcellularstressresponse.cellcycle(georgetown,tex.)6(11):1284-1287.10.4161/cc.6.11.4298.
15.chengjetal.(2017)merkelcellpolyomavirusrecruitsmycltotheep400complextopromoteoncogenesis.plospathogens13(10):1-31.
16.aroraretal.(2012)survivinisatherapeutictargetinmerkelcellcarcinoma.scitranslmed4(133):133ra56.
17.berrioscetal.(2016)merkelcellpolyomavirussmalltantigenpromotespro-glycolyticmetabolicperturbationsrequiredfortransformation.plospathogens12(11).10.1371/journal.ppat.1006020.
18.lenosketal.(2012)alternatesplicingofthep53inhibitorhdmxoffersasuperiorprognosticbiomarkerthanp53mutationinhumancancer.cancerresearch72(16):4074-4084.10.1158/0008-5472.can-12-0215.
19.sholllmetal.(2016)institutionalimplementationofclinicaltumorprofilingonanunselectedcancerpopulation.jciinsight1(19):e87062.
20.houbenretal.(2013)mechanismsofp53restrictioninmerkelcellcarcinomacellsareindependentofthemerkelcellpolyomavirustantigens.thejournalofinvestigativedermatology133(10):2453-2460.10.1038/jid.2013.169.
21.
22.petzoldg,fischeres,
23.spiottomtetal.(2010)imagingtheunfoldedproteinresponseinprimarytumorsrevealsmicroenvironmentswithmetabolicvariationsthatpredicttumorgrowth.cancerres.70(1):78-88.
24.doenchjgetal.(2016)optimizedsgrnadesigntomaximizeactivityandminimizeoff-targeteffectsofcrispr-cas9.nat.biotechnol.34(2):184-191.
25.zhoubetal.(2001)her-2/neuinducesp53ubiquitinationviaakt-mediatedmdm2phosphorylation.nat.cellbiol.3(11):973-82.
26.furetpetal.(2016)discoveryofanovelclassofhighlypotentinhibitorsofthep53-mdm2interactionbystructure-baseddesignstartingfromaconformationalargument.bioorg.med.chem.lett.26(19):4837-4841.
27.scudierodetal.(1988)evaluationofasolubletetrazolium/formazanassayforcellgrowthanddrugsensitivityincultureusinghumanandothertumorcelllines.cancerres.48(17):4827-33.
28.chout,motzerr,tongy,boslg(1994)computerizedquantitationofsynergismandantagonismoftaxol,topotecan,andcisplatinagainsthumanteratocarcinomacellgrowth:arationalapproachtoclinicalprotocoldesign.j.natl.cancerinst.86(20):1517-24.
29.ianevskia,hel,aittokalliot,tangj(2017)synergyfinder:awebapplicationforanalyzingdrugcombinationdose-responsematrixdata.bioinformatics33(15):2413-2415.
30.dingqetal.(2013)discoveryofrg7388,apotentandselectivep53-mdm2inhibitorinclinicaldevelopment.journalofmedicinalchemistry56(14):5979-5983.10.1021/jm400487c.
31.sundetal.(2014)discoveryofamg232,apotent,selective,andorallybioavailablemdm2-p53inhibitorinclinicaldevelopment.journalofmedicinalchemistry57(4):1454-1472.10.1021/jm401753e.
32.monterojetal.(2015)drug-induceddeathsignalingstrategyrapidlypredictscancerresponsetochemotherapy.cell160(5):977-89.
33.smithaeetal.(2015)csnk1a1mutationsandisolateddel(5q)abnormalityinmyelodysplasticsyndrome:aretrospectivemutationalanalysis.lancethaematol2(5):e212-21.
34.saftletal.(2014)p53proteinexpressionindependentlypredictsoutcomeinpatientswithlower-riskmyelodysplasticsyndromeswithdel(5q).haematologica99(6):1041-9.
35.croninj,zhangxyy,reiserj(2005)alteringthetropismoflentiviralvectorsthroughpseudotyping.currentgenetherapy5(4):387-398.10.2174/1566523054546224.
36.pallismetal.(2016)complementarydynamicbh3profilespredictco-operativitybetweenthemulti-kinaseinhibitortg02andthebh3mimeticabt-199inacutemyeloidleukaemiacells.oncotarget.10.18632/oncotarget.8742.
37.schmidtdetal.(2009)chip-seq:usinghigh-throughputsequencingtodiscoverprotein-dnainteractions.methods48(3):240-248.10.1016/j.ymeth.2009.03.001.
实例
材料和方法。
质粒
gfp和t抗原cdna被gateway(英杰公司(invitrogen))克隆到分别从davidroot(addgene质粒]41394)和ericcampeau(addgene质粒17486)获得的plix-402诱导型空载体或plenticmvblast空载体(w263-1)中。ck1α的sgrna克隆(brdn0001149315、brdn0001145680)获得自johndoench和davidroot(addgene质粒76188、76189)。plko-p53-shrna-941获得自toddwaldman(addgene质粒25637)。
细胞和细胞培养
为了生成稳定表达这些构建体的细胞系,使用三载体慢病毒转导系统转导了imr90人肺成纤维细胞、hct116结肠癌或mkl-1mcc细胞(15)。mcc细胞系获得自masashuda(宾夕法尼亚州匹兹堡大学(universityofpittsburgh,pa))、jürgenbecker(奥地利格拉茨医学大学(medicaluniversitygraz,austria))和rolandhouben(德国维尔茨堡大学(universityofwuerzburg,germany))。293t、hct116和imr90细胞获得自atcc。诱导表达shrna的mkl-1mcc细胞系的生成和使用p53dd、mycl和htert构建体的imr90转化先前已有描述(17)。在补充有15%fbs、抗生素和非必需氨基酸的dmem中培养imr90和人原发包皮成纤维细胞(hff),并在补充有10%fbs和抗生素的rpmi中使mcc细胞系生长。
细胞活力测定
努林-3(开曼化学公司(caymanchemical))、来那度胺(开曼化学公司)、沙利度胺(圣克鲁斯生物技术公司(santacruzbiotechnology))、泊马度胺(赛力克化学公司(selleckchemicals))、amg232(medchemexpress公司)、rg7388(来自艾勒朗制药公司(ailerontherapeutics))、hdm201(诺华制药公司(novartispharmaceuticals))、sc-24-ur99(诺华制药公司)、环己酰亚胺(西格玛奥德里奇公司(sigma-aldrich))和mg132(bostonbiochem公司)在dmso中重构、并直接添加到培养基中。根据制造商的说明,使用细胞增殖分析试剂盒(cellproliferationkitii)(xtt)(罗氏公司(roche))进行了细胞活力测定,并且bh3谱如pallis等人描述(36)。使用compusyn和synergyfinder(28,29)进行协同测试。
免疫沉淀、免疫印迹和抗体。
用冰冷的磷酸盐缓冲盐水(pbs)洗涤融合的细胞培养物,并重悬于ebc裂解缓冲液(50mmtris,ph8.0,150mmnacl,0.5%np-40,1:10,000-巯基乙醇,0.5mmedta)在冰上放置10min,然后离心。将澄清的裂解物与抗体和蛋白a/g磁珠(pureproteome磁珠,emdmillipore公司)孵育过夜。将磁珠用高盐ebc缓冲液(50mmtris,ph8.0,300mmnacl,0.5%np-40,0.5mmedta)洗涤5次,并在laemmli样品缓冲液中煮沸,然后再进行sds聚丙烯酰胺凝胶电泳。使用针对mdm4(17914-1-ap;proteintechgroup公司)、mdm2(smp14,圣克鲁斯生物技术公司)、ck1α(17125-1-ap;proteintechgroup公司)、磷酸-ser166mdm2(]3521,细胞信号传导技术公司(cellsignalingtechnology))、磷酸-ser15p53(]9284;细胞信号传导技术公司)、乙酰基-lys382p53(]2525;细胞信号传导技术公司)、p21(]2946;细胞信号传导技术公司)、puma(]12450;细胞信号传导技术公司)、vps39(ab107570;艾博抗公司(abcam))、parp(]9542;细胞信号传导技术公司)、gfp(]2555;细胞信号传导技术公司)、mycl(14584-1-ap;proteintechgroup公司)、p53(do-1;赛默飞世尔公司(thermoscientific)/lab视觉公司)和rb1(g3-245;bdbiosciences公司)的抗体进行免疫沉淀和蛋白质印迹。针对mcv大t抗原残基1至260产生了针对mcvt抗原的小鼠单克隆抗体ab3和ab5,并在细菌中以谷胱甘肽s-转移酶(gst)融合蛋白的形式产生(17)。使用odyssey近红外系统(li-corbiotechnology公司)进行定量蛋白质印迹分析。
chip-和rt-qpcr
chip方法改良自schmidt等人描述的方案(37)。使用双琥珀酰亚胺戊二酸酯(dsg)和甲醛进行双重交联,将mkl-1细胞或imr90细胞交联。交联后,使用simplechip缓冲液a和b(细胞信号传导公司)裂解细胞,并在37℃用微球菌核酸酶(纽英伦生物技术公司(newenglandbiolabsinc.))处理dna30分钟,然后在4℃以20秒脉冲超声处理5次。为了
rt-qpcr,使用rneasyplusmini试剂盒(凯杰公司(qiagen))纯化总rna,并使用高容量cdna逆转录试剂盒(high-capacitycdnareversetranscriptionkit)(应用生物系统公司(appliedbiosystems))合成cdna。使用brilliantiiiultra-fastsybrgreenqpcrmastermix(安捷伦科技公司(agilenttechnologies))进行定量pcr。引物信息可在附表2中找到。
异种移植功效研究
给7至9周龄的nsg小鼠(杰克逊实验室(jacksonlaboratory))注射1.00e+007mkl-1mcc细胞。当肿瘤大小达到200mm3时,每天用媒介物、hdm201(来自诺华制药公司,40mg/kg,0.5%甲基纤维素(400cp)于50mm磷酸盐缓冲液中,ph6.8,口服)、来那度胺(medchemexpress公司,50mg/kg,0.5%cmc+0.25%tween80,口服)或hdm201和来那度胺两者处理每组8只小鼠。当肿瘤体积达到2000mm3的最大允许大小时,研究终止。
本文进行的一些实验和获得的结果在本发明的具体实施方式部分以及附图及其描述中披露。
实例1:
下表提供了药物/化合物各种组合在降低mkl-1mcc细胞活力方面的协同测试数据。将mkl-1(具有野生型p53的病毒阳性默克尔细胞癌细胞系)处理96小时。进行xtt测定以确定相对于未处理细胞归一化的相对活力(%反应)。单独的表格包含组合的数据:1.努林-来那度胺2.努林-ur993.hdm201-ur994.努林-3+/-来那度胺;rg7388+/-来那度胺;仅来那度胺5.hdm201+来那度胺。
通过引用并入
本文提及的所有出版物、专利和登录号均通过援引以其全文特此并入,如同每个单独的出版物或专利被明确且单独地表明通过援引而并入。
等同物
虽然已经讨论了本发明的特定实施例,但上述说明是说明性而非限制性的。在综述本说明书和以下权利要求书之后,本发明的许多修改对于本领域的技术人员将是显而易见的。应当通过参考权利要求书及其等同形式的全范围以及说明书连同此类变化来确定本发明的全范围。
- 还没有人留言评论。精彩留言会获得点赞!