脱氧核糖核酸编码化方法及其编码化方法的医药生命工程用途与流程
本发明涉及脱氧核糖核酸(DNA)的密码标准化方法及其方法的最优化的医药生命工程用途。
背景技术
在生物体中以遗传物质存在的脱氧核糖核酸(DNA,DeoxyriboNucleic Acid)由通过蛋白质表达的基因部位和非基因部位构成。在脱氧核糖核酸的化学结构中,在作为脱氧核糖(Deoxyribose)的戊糖的5′碳连接磷酸基且在1′碳连接碱基(base)来形成称为核甘酸(Nucloeotide)的单体,此时,根据与核甘酸连接的碱基的种类确定脱氧核糖核酸的序列。
碱基的种类分为2种类型,具有环状结构为2个的嘌呤碱基和环状结构为1个的嘧啶类。嘌呤类具有鸟嘌呤(G)和腺嘌呤(A),嘧啶类具有胞嘧啶(C)和胸腺嘧啶(T)等,在核糖核酸(RNA)的情况下,具有如下差异,即,戊糖的2′碳与-OH基连接,碱基的组成被尿嘧啶(U)取代,而不是被胸腺嘧啶取代。嘌呤类的G通过氢键与作为嘧啶的C形成互补对,A与T形成对。此时,G与C的互补结合通过3个氢键连接,因此,形成与形成2个氢键的A与T的结合相比强的键合。
在脱氧核糖核酸的核甘酸单体中,与5′碳连接的磷酸基通过磷酸双酯键(Phosphodiester bond)与另一单体的3′碳-OH基连接来形成单链。通过磷酸双酯键连接的2个互补单链通过互补碱基的氢键形成双螺旋结构。这种双螺旋结构在1953年由Watson和Crick介绍。[Watson,J.D.,&Crick,F.H.(1953).Molecular structure of nucleic acids.Nature,171(4356),737-738.]
在脱氧核糖核酸中的基因部位的碱基序列中,3个碱基密码被翻译为构成蛋白质的一个氨基酸(Amino acid)并连接,由此在合成蛋白质时起到重要作用。脱氧核糖核酸转录为信使核糖核酸(mRNA)后,根据碱基序列的顺序翻译成20种氨基酸,被翻译的氨基酸通过转运核糖核酸(tRNA)连接并形成蛋白质,由此作为细胞内的组合物质存在,并起到介导体内各种反应的酶的作用。
在人的脱氧核糖核酸的情况下,具有30亿个碱基对(bp),每人具有GB单位的数据容量。若将该容量换算成人口数量,则PB单位也不够。因此,与分析人的所有脱氧核糖核酸序列(DNA sequence)相比,分析疾病特异性单核苷酸多态性(Single Nucleotide polymorphism,碱基多态性)位点等,来利用短的脱氧核糖核酸片段的序列(sequence)预测分析疾病,即使这样也无法分析所有基因的单核苷酸多态性位点,由此需要用于分析其的各种程序。
现有专利文献
韩国公开专利:10-2016-0001455
技术实现要素:
技术问题
本发明使为了解决上述问题且根据上述需要提出的,本发明的目的在于,提供如下的最优化的方法:将脱氧核糖核酸碱基标准化为考虑各碱基的分子量的二进制密码(每1个碱基为2bit),由此掌握存在于碱基序列内的特定模式。
本发明地再一目的在于,提供利用碱基序列的密码之和来掌握是否互补结合及模式的方法,提供容易预测脱氧核糖核酸片段或脱氧核糖核酸适配体的模式及功能的方法。
本发明的另一目的在于,提供仅利用碱基序列的密码也可容易掌握序列之间的分子量比例和各碱基的比例等的方法。
本发明的还有一目的在于,提供容易掌握碱基序列内的变异的方法,提供利用单核苷酸多态性等的疾病特异性序列变异来容易预测疾病的方法。
技术方案
为了实现如上所述的目的,本发明提供脱氧核糖核酸的密码标准化方法,其包括:步骤(a),将C、T、A、G的4种碱基分别命名为00、01、10、11;步骤(b),当各碱基形成G和C以及A和T的碱基对时,在从5′到3′的方向,分别在G和C的情况下命名为1100,在C和G的情况下命名为0011,在A和T的情况下命名为1001,在T和A的情况下命名为0110。
并且,本发明提供利用脱氧核糖核酸的密码标准化来提供用于确认特定脱氧核糖核酸片段或适配体的特定模式或二级结构的最优化的信息的方法,其包括:步骤(a),将特定脱氧核糖核酸片段碱基序列的C、T、A及G分别命名为00、01、10、11;以及步骤(b),比较以上述数值命名的密码的排列与各密码和的排列。
在本发明的一实例中,优选地,本发明的特征在于,利用脱氧核糖核酸的密码标准化来提供用于确认特定脱氧核糖核酸片段或适配体的特定模式或二级结构的最优化的信息的方法中,在上述比较密码的排列与各密码和的排列的步骤中,将上述步骤(a)的00、01、10及11的二进制数的数组转换为十进制数后,当在两端排列两对以上的各序列之和等于3的密码排列时,判断可形成茎状结构,当在中心连接3个以上的相向的序列的密码之和大于或小于3而无法形成互补结合的序列时,判断为形成环状结构,但并不限定于此。
并且,本发明提供利用脱氧核糖核酸的密码标准化来提供与在特定脱氧核糖核酸片段中是否存在碱基序列变异有关的信息的方法,包括:步骤(a),将特定脱氧核糖核酸片段碱基序列的C、T、A及G分别命名为00、01、10、11;以及步骤(b),比较由上述数值命名的密码之和。
在本发明的一实例中,优选地,本发明的特征在于,在上述比较密码之和的步骤中,将上述步骤(a)的00、01、10及11的二进制数的数组转换为十进制数后,求得其和,并与正常序列进行比较,,当存在1至3的差异时,判断为存在变异,但并不限定于此。
在本发明的另一实例中,优选地,上述方法可通过对将特定脱氧核糖核酸片段的碱基序列的C、T、A及G分别命名为00、01、10、11来获取的密码各自的数值进行比较来确认变异序列的位置,但并不限定于此。
并且,本发明提供存储在计算机可读介质的计算机程序,使计算机执行下述步骤,以提供用于确认特定脱氧核糖核酸片段或适配体的特定模式或二级结构的最优化的信息,上述步骤包括:步骤(a),将特定脱氧核糖核酸片段的碱基序列的C、T、A及G分别命名为00、01、10、11;以及步骤(b),将上述步骤(a)的00、01、10及11的二进制数的数组转换为十进制数后,当在两端排列两对以上的各序列之和等于3的密码排列时,判断为可形成茎状结构,当在中心连接3个以上的相向的序列之和大于或小于3而无法形成互补结合的序列时,判断为形成环状结构。
并且,本发明提供存储在计算机可读介质的计算机程序,使计算机执行下述步骤,以提供与在特定脱氧核糖核酸片段中是否存在碱基序列的变异有关的信息,上述步骤包括:步骤(a),将特定脱氧核糖核酸片段的碱基序列的C、T、A及G分别命名为00、01、10、11;以及步骤(b),将上述步骤(a)的二进制数的数组转换为十进制数后,求得其和,并与正常序列进行比较,当存在1至3的差异时,判断为存在变异。
并且,本发明提供存储在计算机可读介质的计算机程序,使计算机执行下述步骤,以提供与特定脱氧核糖核酸片段的碱基序列变异序列的位置有关的信息,上述步骤包括:步骤(a),将特定脱氧核糖核酸片段的碱基序列的C、T、A及G分别命名为00、01、10、11;以及步骤(b),通过对上述步骤(a)的将特定脱氧核糖核酸片段的碱基序列的C、T、A及G分别命名为00、01、10、11来获取的密码各自的数值进行比较来确认变异序列的位置。
以下,说明本发明。
本发明提供如下的方法:以脱氧核糖核酸的各个分子量小的顺序将C、T、A、G的4种碱基分别命名为00、01、10、11的密码,当各碱基形成G和C以及A和T的碱基对时,以使各个分子量之和与密码和的比例一致的方式命名密码。
并且,本发明构建如下的系统:通过密码使与利用指数富集配体进化技术(SELEX)确认的各化合物特异性的适配体标准化,由此掌握与在各化合物中存在的反应基团结合的特定模式,可利用大数据进行预测。
并且,本发明提供如下的方法:通过密码使脱氧核糖核酸的序列标准化后,将各序列的值转换为十进制数并导出其总和,由此确认各序列是否变异,从而可快速掌握是否存在特定疾病的单核苷酸多态性。
本发明提供如下的方法:通过密码使脱氧核糖核酸标准化,由此容易掌握在碱基序列内存在的特定模式。
本发明提供如下的信息:掌握与特定靶及化学结构结合的脱氧核糖核酸序列模式,将其用作大数据,由此预测与相应化学结构单位结合的适配体(Aptamer),并提供指数富集配体进化技术(SELEX,Systematic evolution of ligands by exponential enrichment)模拟程序化所需的信息。
并且,本发明提供如下的最优化的方法:通过反映碱基分子量的密码使脱氧核糖核酸标准化,仅通过碱基序列的密码掌握序列之间的分子量比例和各碱基的比例等。
并且,本发明提供如下的方法:通过反映碱基分子量的密码使脱氧核糖核酸标准化,由此容易掌握碱基序列内的变异,并且,提供被最优化以对密码之和与排列顺序进行比较的方法,从而提供可掌握单核苷酸多态性等的疾病特异性变异且容易预测疾病的方法。
本发明的效果
通过本发明可知,本发明的脱氧核糖核酸密码标准化方法提供容易掌握碱基序列内的变异的方法,提供利用单核苷酸多态性等的疾病特异性序列变异来容易预测疾病等的容易掌握在碱基序列内存在的特定模式的方法。
附图说明
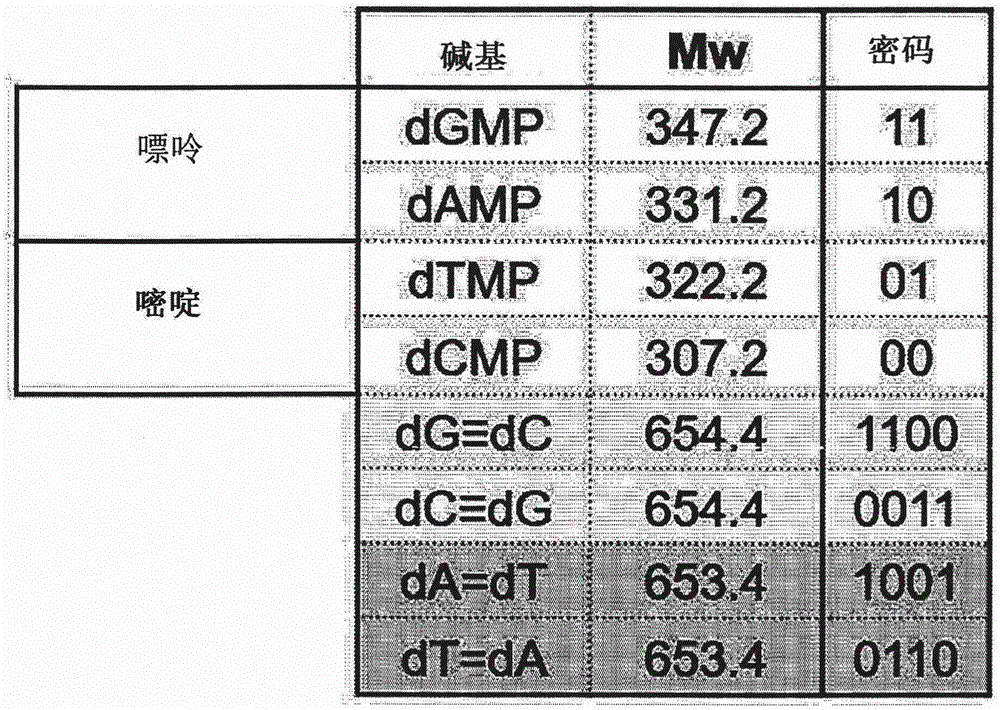
图1为示出通过反映脱氧核糖核酸的分子结构及结合质量比的原理来指定的密码值以从分子量小的碱基至大的顺序将C、T、A、G指定为00、01、10、11值的二进制数的图。
图2为示出当指定的二进制数的密码分别形成G和C、A和T的碱基对时,以各密码和的比例具有1∶1的与实际质量比相同的比例的方式设计的图。
图3示出6种序列的密码转换值,为比较示出各序列的密码和与各序列的分子量的图。
图4利用脱氧核糖核酸序列的密码来确认例示序列的模式,根据各序列的密码和确认是否可以互补结合,根据与其结合的数连接的碱基的数量确认茎环结构形成和模式的图。
图5向在乳腺癌患者中确认的单核苷酸多态性序列适用密码来确认本发明的密码标准化效率,将距外显子(Exon)2位于第14位的A碱基变异为G的单核苷酸多态性序列转换为密码,以二进制数的数组配置后,求得密码和来比较正常序列与变异序列的密码和的图。
具体实施方式
以下,通过非限制性实施例详细说明本发明。但是,下述实施例为了例示本发明而记载,不应解释为本发明的范围局限于下述实施例。
实施例1:根据各碱基的分子量的密码标准化
为了通过作为计算机语言的二进制两位数示出确定脱氧核糖核酸的序列的各4种碱基来通过密码使其标准化,分析各碱基的分子量并在图1示出。将各个碱基G、A、T、C与1个磷酸基连接的脱氧核苷酸(deoxyribonucleotide)分别表示为dGMP、dAMP、dTMP、dCMP。
各碱基以G、A、T、C的顺序具有大值,将通过氢键与G成对的C以及与A互补结合的T的分子量分别相加并比较的结果为654.4(=347.2+307.2)和653.4(=331.2+322.2),确认了在具有大致1∶1的等同的分子质量的状态下成对。A与T的分子量之和与G与C的分子量之和相比小1的原因在于,G≡为氮(N),A=T为碳(C)、氢(H)与其他结合对相比多一个,具有与N的分子量与C+H的分子量之和的差异(14>12+1)相同的各对的分子量之和的差异(=1)。因此具有如下的特性:A和T由可形成氢键的O或N的部件形成2个氢键,由此形成与形成3个氢键的G≡结合弱的结合。
因此,通过反映上述脱氧核糖核酸的分子结构及结合质量比的原理来指定各碱基的密码。对于所赋予的各碱基的密码,以分子量小的碱基至大的顺序将C、T、A、G指定为00、01、10、11值的二进制数(图1)。
以如下的方式设计指定的密码的值:当G和C、A和T的碱基分别成对时,各自的密码和比例为1∶1,具有与实际质量比相同的比例(图2)。
密码和示出将各碱基的密码转换为十进制数后的各密码值的和,G和C、A和T各自的密码和为相同的“3”。
实施例2:脱氧核糖核酸片段及适配体(Aptamer)的分子量比例反映最优化
根据脱氧核糖核酸的各碱基分子量,以质量低到高的顺序指定密码,因此,脱氧核糖核酸片段的总密码和通过反映各序列的分子量比例来计算(图3)。通过确认密码的分子量反映比例来利用6个例示序列比较密码和与分子量。
上述例示序列为为了确认密码的分子量反映比例而例示的序列,不应解释为范围局限于SEQ ID NO:1至SEQ ID NO:6的序列。
上述SEQ ID NO:1至SEQ ID NO:6的序列如下。
5′AGAGCTCGCGCCGGAGTTCTCAATGCAAGAGC 3′(SEQ ID NO:1)
5′GCGGCGGTGGCCTGAAGTCTGGCGGTGGCCCC 3′(SEQ ID NO:2)
5′GCGGCGGTGGCCAGAAGTCTCGCGGTGGCGGC 3′(SEQ ID NO:3)
5′GTGGAGGCGGTGGCCAGTCTCGCGGTGGCGGC 3′(SEQ ID NO:4)
5′GTGGCGGTGGCCAGCATAGTGGCGGTGGCCAG 3′(SEQ ID NO:5)
5′GTGGAGGCGGTGGCCGTGGAGGCGGAGGCCGC 3′(SEQ ID NO:6)
上述6个例示序列为32mer的碱基序列,碱基的长度相同,但由不同的碱基种类和顺序组成,在图3示出各碱基的密码转换值。密码和为将各碱基的密码转换为十进制数后求得的总和,根据各序列的碱基组成反映密码和或各序列的分子量来计算。
当与各序列的分子量(Mw)进行比较时,确认了若分子量越小,则密码和的值越小,在分子量大的序列的情况下,所计算的密码和为大的值(图3)。
如上所述,通过反映分子量的比例来指定密码并转换的结果,利用密码和来最优化,由此比较各序列的分子量之比。
实施例3:脱氧核糖核酸片段及适配体的模式确认的最优化
进行下述方式最优化:将脱氧核糖核酸片段及适配体的序列转换为二进制数碱基密码,通过比较各序列来掌握包含在序列内的特定模式及二级结构(secondary structure)等。为了掌握其,将由9个碱基序列组成的脱氧核糖核酸序列用作例示序列(图4)。
上述例示序列为了例示密码的模式而记载,不应解释为其范围局限于SEQ ID NO:7的例示序列。
上述SEQ ID NO:7的例示序列如下。
5′GCGGTGGCG 3′(SEQ ID NO:7)
将上述例示序列转换为碱基密码来罗列的数如下。
11 00 11 11 01 11 11 00 11(例示序列密码1)
各碱基以使可形成氢键的与互补碱基的密码和成为“3”的方式设计密码,这种序列的排列可在脱氧核糖核酸适配体序列中形成茎状结构(图4;茎状(Stem))。
脱氧核糖核酸的茎环(Stem-loop)结构的模式大部分在量末端连接两个以上的可形成茎状结构的碱基,当相向的序列的密码和大于或小于3而在中心连接3个以上的无法形成互补结合的序列时,具有可形成环状结构的特性。
上述例示序列可形成两种茎环结构,这可通过碱基密码排列简单确认。可与第一个11碱基密码互补结合的序列为除相邻的00密码之外的第8个00密码的碱基(图4,①红色箭头),可与第二个00密码互补结合的碱基有第6个11(图4,③绿色箭头)和第7个11、第9个11密码。与其相同地,第3个11密码的碱基可与第8个00(图4,②蓝色箭头)密码互补结合。此时,茎环结构的茎状部位需使两个以上的碱基连接才能形成结构,因此,在图3中,通过红色箭头连接的碱基的互补结合或通过蓝色箭头连接的碱基的互补结合可形成茎状结构(图4,虚线的圆形),绿色箭头的互补结合无法通过单一的互补结合形成茎状结构。在可形成茎状结构的两种情况下,在中间均存在可形成环状结构的4个碱基,因此预测可形成茎环结构。
如上所述,确认了,通过密码将各碱基标准化,由此可根据碱基密码和预测是否可与各碱基互补结合,可根据各序列的互补结合的数和与其连接的碱基的数容易预测脱氧核糖核酸序列的二级结构及模式等。
实施例4:通过密码标准化掌握单核苷酸多态性的最优化
进行最优化,以将脱氧核糖核酸序列转换为密码,通过比较各序列的密码和来掌握在特定脱氧核糖核酸片段中是否发生碱基序列的变异。单核苷酸多态性序列为一个碱基变异的脱氧核糖核酸片段序列,因此,将密码适用于单核苷酸多态性序列并与正常序列进行比较,由此确认了容易掌握是否存在变异和位置。适用于作为各种单核苷酸多态性序列中的一种且在84%的乳腺癌患者中确认的CD44基因的单核苷酸多态性序列,由此确认了密码标准化的效率。[Zhou,J.,Nagarkatti,P.S.,Zhong,Y.,Creek,K.,Zhang,J.,&Nagarkatti,M.(2010).Unique SNP in CD44 intron 1 and its role in breast cancer development.Anticancer research,30(4),1263-1272.]
上述乳腺癌患者的单核苷酸多态性序列的存在于基因的第一个内含子(intron 1)的位置的序列中的从外显子(Exon 2)位于第14位的A碱基变异为G,将该序列转换为密码来以二进制数的排列配置后,求得密码和,并比较正常序列与变异序列的密码和(图5)。
当将正常序列和变异序列的密码分别转换为十进制数后求得和时,正常序列为39,变异序列为40,确认了变异序列比正常序列大1。如上所述,仅可通过密码和确认在脱氧核糖核酸片段内是否存在变异,根据此时变异的碱基的种类,密码和可具有1~3左右的差异。并且,可通过比较变异的密码的各个数值来确认序列的位置。
如上所述,将在正常对照组种确认的脱氧核糖核酸片段序列和在疾病实验组中确认的特定变异序列转换为密码,通过比较密码和来快速确认序列之间的差异,并可寻找是否存在单核苷酸多态性,可通过向确认的单核苷酸多态性序列适用密码和来用于诊断疾病。
- 还没有人留言评论。精彩留言会获得点赞!