载生物活性因子PLA/PLGA/CS复合膜及其制备方法与流程
本发明属于应用于脊髓损伤治疗的生物复合膜技术领域,涉及一种载生物活性因子pla/plga/cs复合膜及其制备方法。
背景技术:
脊髓损伤(spinalcordinjury,sci)是由于各种原因引起的椎管内脊髓或马尾神经不同程度的损伤。组织工程技术是目前治疗脊髓损伤最有前景的方法,通过选择具有良好的生物相容性、降解性、机械性能以及低神经毒性的生物材料,构建三维支架并负载促神经元生长或保护神经元的药物,为损伤脊髓的再生提供良好的微环境以达到脊髓修复的理想效果。中国专利申请cn102671238a公开了一种包括i型胶原蛋白、壳聚糖、明胶中的一种或多种原料制成的微孔支架,该支架具有内部孔径大小均匀、微管轴向平行排列高度仿真、邻近微管孔隙广泛沟通等结构优点。中国专利申请cn104399131a公开了一种以复合型红景天苷缓释微球与i型胶原蛋白为原料制备的多孔径柱状神经导管,该导管可以引导神经纤维定向生长,具有良好的降解性和生物相容性。文献1(yuzhuhe,yahuijin,xiumeiwang,etal.anantimicrobialpeptide-loadedgelatin/chitosannanofibrousmembranefabricatedbysequentiallayer-by-layerelectrospinningandelectrosprayingtechniques.nanomaterials.2018,8,3)利用壳聚糖明胶制备纤维膜和plga微球负载pac-525,制得促进骨成长的复合膜,该复合膜可成功引导骨再生药物释放时间约为30天。
上述支架均是将生物材料制成导管或块状引导神经生长,虽然具有较好的生物相容性,但却难以应用于临床。因为此类支架需对脊髓半侧切除后植入,由于临床病人的脊髓损伤范围不规则,多呈不完全性,难以应用,更不可能切除一段安置导管。而多数载药复合膜药物释放时间短,无法满足脊髓修复所需时间,其药物释放方向和速率也难以控制。因此选择合适的生物材料设计一种可方便应用于脊髓损伤临床治疗并能长时间稳定释放药物的支架材料具有重要的实践应用价值。
技术实现要素:
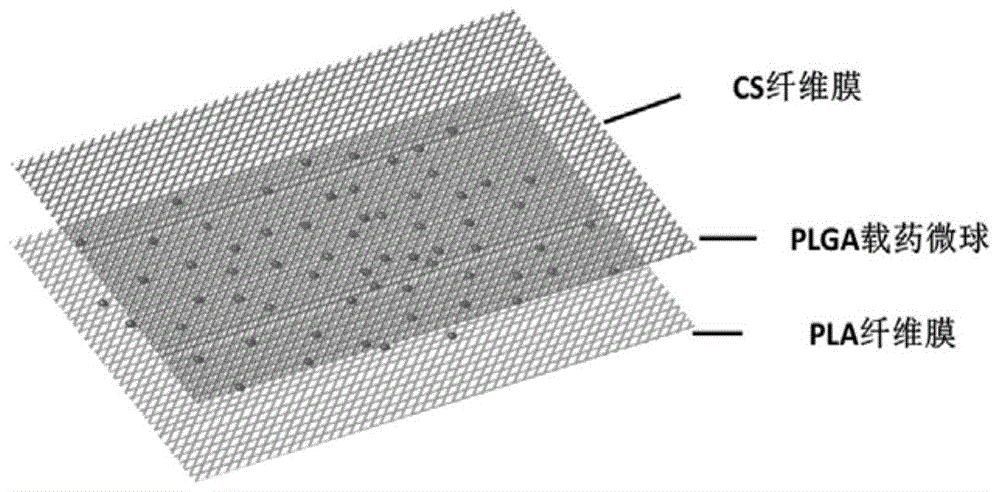
本发明的目的在于提供一种载生物活性因子聚乳酸/聚乳酸-羟基乙酸共聚物/壳聚糖(pla/plga/cs)复合膜及其制备方法。本发明利用静电纺丝和静电喷雾相结合的方式,制备一种膜/球/膜的三层结构。以pla膜作为密封层,利用pla膜的致密性防止药物扩散并提供一定的力学支撑;plga微球作为夹心层,负载药物;利用cs膜较大的孔隙率和较高的生物相容性作为调控缓释层,直接接触损伤区域,同时可以种植种子细胞以达到神经修复的目的。
实现本发明目的的技术方案如下:
载生物活性因子pla/plga/cs复合膜的制备方法,具体步骤如下:
(1)将聚乳酸(pla)溶于六氟异丙醇中配成7~8wt%的pla溶液;
(2)将聚乳酸-羟基乙酸共聚物(plga)溶于氯仿中得到6~7wt%的plga有机相溶液,将药物溶于水中制备水相溶液,然后,将水相溶液和plga有机相溶液混合,超声处理得到水油比为1:100~1:200的乳液;
(3)将壳聚糖(cs)溶于三氟乙酸/二氯甲烷(v/v=2:3)中配成6~7wt%的cs溶液;
(4)以pla溶液为纺丝溶液,采用静电纺丝技术,纺丝参数为:电压13~18kv,接收距离为12~15cm,流速为0.08~0.1mm/min,得到pla纤维膜;
(5)以pla纤维膜为接收板,步骤(2)的乳液为静电喷液,采用静电喷雾技术,喷雾参数为:8~12kv,接收距离为12~15cm,流速为0.06~0.08mm/min,得到pla纤维膜/plga载药微球;
(6)以pla纤维膜/plga载药微球为接收板,cs溶液为纺丝溶液,采用静电纺丝技术,纺丝参数为:电压为13~18kv,接收距离为12~15cm,流速为0.08~0.1mm/min,得到pla/plga/cs复合膜,冷冻干燥,制得载生物活性因子pla/plga/cs复合膜。
优选地,所述的pla溶液的浓度为8wt%,plga有机相溶液的浓度为6wt%,水油比为1:200,cs溶液的浓度为7wt%。
优选地,步骤(2)中,所述的超声处理次数为5次以上;每次超声处理时的功率为300w,超声处理时间为5s。
优选地,步骤(2)中,所述的药物选自单唾液酸神经节苷脂(gm1)、神经营养因子(nt-3)、胰岛素样生长因子(igf)和神经生长因子(ngf)中的一种或多种。
优选地,步骤(2)中,所述的乳液中,药物浓度为0.3~0.6μg/ml。
优选地,步骤(4)中,所述的静电纺丝时间为15~20h。
优选地,步骤(5)中,所述的静电喷雾时间为5~10h。
优选地,步骤(6)中,所述的静电纺丝时间为2~5h。
优选地,步骤(5)中,所述的plga载药微球的直径为6~11μm。
优选地,步骤(6)中,所述的载生物活性因子pla/plga/cs复合膜的厚度为190±20μm。
与现有技术相比,本发明具有以下优点:
(1)本发明的载生物活性因子pla/plga/cs复合膜结构中pla纤维膜结构致密,可防止药物扩散;plga载药微球球形度较好,药物分散均匀;cs纤维膜有较大的孔隙,可以控制药物缓释并延长药物释放时间。该复合膜结构稳定且具有一定的力学强度,拉伸强度为2.83±0.31mpa,断裂伸长率为53.25±0.18%;可以良好的负载药物并起到了很好的药物缓释作用,可以释放药物长达两个月以上,满足脊髓损伤修复所需要的时间。
(2)本发明的载生物活性因子pla/plga/cs复合膜无明显的细胞毒性,并成功诱导pc-12细胞在5天内分化为神经元,有助于神经修复。可以直接敷贴在损伤区域,同时占位效应小,对脊髓不产生明显压迫和二次损伤。
(3)本发明通过静电纺丝和静电喷雾逐层结合的方式制备,可直接将载药微球包裹于纤维膜中,过程简单,容易实现工业化生产。
附图说明
图1为实施例5中载生物活性因子pla/plga/cs复合膜示意图。
图2为实施例1中溶液浓度为(a)6wt%;(b)7wt%;(c)8wt%的pla纤维膜扫描电镜图。
图3为实施例2中溶液浓度分别为(a)5wt%;(c)6wt%;(e)7wt%的plga微球的扫描电镜图,溶液浓度分别为(b)5wt%;(d)6wt%;(f)7wt%plga微球的直径分布图。
图4为实施例3中水油比分别为(a)1:50;(c)1:100;(e)1:200的plga载药微球的扫描电镜图,水油比分别为(b)1:50;(d)1:100;(f)1:200的plga载药微球的直径分布图。
图5为实施例3中plga载药微球荧光图像。
图6为实施例4中溶液浓度为(a)6wt%,(b)7wt%cs纤维膜扫描电镜图。
图7为实施例5中(a)plga微球载于pla纤维上;(b)、(c)复合载药膜截面扫描电镜图。
图8为实施例5中复合载药膜的力学拉伸曲线。
图9为实施例5中复合载药膜60天释放效率曲线。
图10为实施例5中复合载药膜体外细胞毒性表征。
图11为实施例5中复合载药膜体外细胞分化实验。(a)空白对照组;(b)游离ngf组;(c)空白复合膜组;(d)复合载药膜组。
具体实施方式
下面结合实施例和附图对本发明作进一步详述。
实施例1:pla纤维膜的制备。
(1)将pla溶于六氟异丙醇中分别配成6wt%、7wt%和8wt%的pla溶液;
(2)分别将不同浓度的pla溶液装入容量为10ml的塑料注射器内,将注射器水平放置在注射泵上,在金属板上覆上一层铝箔作为接收器,注射器喷头选用20号针。电纺条件:电压为13-18kv,流速0.08-0.1mm/min,接收距离12-15cm;
(3)将(2)中所得纤维膜从铝箔上取下进行冷冻干燥,得到pla纤维膜。
实施例2:plga微球的制备。
(1)首先将plga溶于氯仿中配成5wt%、6wt%和7wt%的plga溶液,磁力搅拌至完全溶解;
(2)分别将不同浓度的plga溶液进行电喷雾,将电喷液装入容量为10ml的塑料注射器内,将注射器水平放置在注射泵上,在金属板上覆上一层铝箔作为接收器,注射器喷头选用22号针。电喷条件:电压为8-12kv,流速0.06-0.08mm/min,接收距离12-15cm。得到plga微球。
实施例3:制备plga氯仿溶液浓度为6wt%,水油比分别为1:50、1:100和1:200的载药微球。
(1)将plga溶于氯仿中,磁力搅拌至完全溶解,得到6wt%的plga有机相溶液;
(2)通过将ngf溶解在去离子水中来制备水相溶液。然后,将水相溶液和plga有机相溶液混合,超声处理(5个循环,10秒,300w)以形成水油比为1:50、1:100和1:200的乳液,药物含量为0.5μg/ml;
(3)分别将不同水油比的乳液进行电喷雾,将电喷液装入容量为10ml的塑料注射器内,将注射器水平放置在注射泵上,在金属板上覆上一层铝箔作为接收器,注射器喷头选用22号针。电喷条件:电压为8-12kv,流速0.06-0.08mm/min,接收距离12-15cm。得到plga载药微球。
实施例4:cs纤维膜的制备。
(1)将提纯后的cs溶于三氟乙酸/二氯甲烷(v/v=2:3)中分别配成6wt%、7wt%的cs溶液;
(2)分别将不同浓度的cs溶液装入容量为10ml的塑料注射器内,将注射器水平放置在注射泵上,在金属板上覆上一层铝箔作为接收器,注射器喷头选用20号针。电纺条件:电压为13-18kv,流速0.08-0.1mm/min,接收距离12-15cm;
(3)将(2)中所得纤维膜从铝箔上取下进行冷冻干燥,得到cs纤维膜。
实施例5:制备pla六氟异丙醇浓度8wt%,plga氯仿溶液浓度6wt%、水油比1:200,cs三氟乙酸/二氯甲烷(v/v=2:3)溶液浓度7wt%的载生物活性因子pla/plga/cs复合膜。
(1)将pla溶于六氟异丙醇中配成8wt%的pla溶液;
(2)将plga溶于氯仿中,磁力搅拌至完全溶解,得到6wt%的plga有机相溶液;将鼠神经生长因子溶解在去离子水中制备水相溶液。然后,将水相溶液和plga有机相溶液混合,超声处理(5个循环,10秒,300w)以形成体积分数(水油比)为1:200的乳液,药物含量为0.5μg/ml;
(3)将提纯后的cs溶于三氟乙酸/二氯甲烷(v/v=2:3)中配成7wt%的cs溶液;
(4)将铝箔覆于接收板表面作为收集器,通过20g针以0.08-0.1mm/min的流速静电纺丝pla溶液,电压设定为13-18kv,接收距离为12-15cm,电纺15小时,得到pla纤维膜;然后用(2)中乳液代替pla溶液,采用静电喷雾技术,在8-12kv的电压、0.06-0.08mm/min的流速及12-15cm的接收距离下从22g针电喷雾微粒5h,得到pla纤维膜/plga载药微球;随后将cs溶液以0.08-0.1mm/min的流速,经静电纺丝沉积在接收板上2小时,电压设定为15-18kv,接收距离为12-15cm;
(5)将(4)中所得复合膜进行冷冻干燥以保持构型,最终得到载生物活性因子pla/plga/cs复合膜。
载生物活性因子pla/plga/cs复合膜的相关表征和性能测试:
测试例1:sem:取实施例1得到的pla纤维膜,在金属离子溅射仪内喷金后,置于s-4800ⅱ场发射扫描电子显微镜下观察形貌,如图2所示。观察电镜图可以看出,静电纺丝法可以制备出光滑均匀的pla纤维,当纺丝液的浓度为6wt%和7wt%时,pla纤维呈现出明显的串珠状,这是因为纺丝液液喷射时溶液中固体含量较小,且电场中的静电力较大,溶液的表面张力较小,不足以形成连续的纤维。在纺丝液继续浓度提高时,串联现象明显减轻,纤维中的纺锤状结构变少。当浓度进一步提高到8wt%时,得到了粗细均匀、形貌最佳的纤维。随着溶液浓度的提高,pla纤维的直径也明显提高。
测试例2:sem:取适量实施例2中得到的plga载药微球在金属离子溅射仪内喷金后,置于s-4800ⅱ场发射扫描电子显微镜下观察形貌,如图3所示。三组样品均可制备出微球,直径尺寸在6~11μm范围内。可以看出随着电喷液浓度增大,相同面积内统计微球个数明显增多,成球率提高。5wt%的样品微球分散,甚至不足以覆盖铝箔表面,微球间由于表面张力而相互联接致使形状变得不规整;6wt%的样品微球密集规整,呈现出良好的球形;而7wt%的微球中出现了纤维结构,这是由于溶液中固体含量太高,这是因为固体含量较大时,分子链间的缠结较多对应着比较高的溶液黏度,此时电喷液喷射时的表面张力大于静电力,液滴难以被静电力彻底分散开,彼此相互串联。故溶液浓度为6wt%时最佳。
测试例3:sem:取适量实施例3中得到的plga载药微球在金属离子溅射仪内喷金后,置于s-4800ⅱ场发射扫描电子显微镜下观察形貌,如图4所示。三组样品均可制备出微球,无明显串珠状结构。可以看出水相体积比较高时微球成球率下降,球形度较低,这主要是因为水相比例较高时,微球表面含水量也相对较大,微球彼此之间倾向于相互融合。当水油比为1:200时样品微球密集规整,呈现出良好的球形,彼此之间粘度低。
测试例3:plga微球的荧光显微镜图:将实施例3中水油比为1:200的plga微球中的药物相替换成罗丹明b进行电喷雾,采用ix50/58f2型倒置式系统显微镜,在激光波长下观察荧光标记的plga微球。如图5所示,药物成功包裹于微球中。
测试例4:sem:实施例4中得到的cs纤维膜在金属离子溅射仪内喷金后,置于s-4800ⅱ场发射扫描电子显微镜下观察形貌,cs溶液在进行纺丝时表现出了不稳定,由于溶剂三氟乙酸挥发速度极快,纺丝液在离开针头后极短的时间内就形成了纤维。从图6中可以看出溶液浓度为6wt%时出现了明显的串珠状结构,纤维粗糙不流畅。7wt%的纤维更加均匀光滑,同时具有更大的直径。
测试例5:sem:实施例5中得到的载生物活性因子pla/plga/cs复合膜在金属离子溅射仪内喷金后,置于s-4800ⅱ场发射扫描电子显微镜下观察形貌,图7(a)为一层pla纤维上负载着一层plga微球,微球在纤维上仍然可以呈现良好的球形,并附着于纤维层表面。图7(b)的多层结构的截面图可以看到无规排列的纤维和埋在其中的plga微球,从图7(c)可以看出多层结构薄膜厚度约为190±20μm。
测试例5:单向拉伸测试:冻干的过程有利于材料整体结构的保持,为了判断冻干的过程是否对材料力学性能造成了影响,进行单向拉伸测试。取适量实施例5中得到的载生物活性因子pla/plga/cs复合膜,利用压力模具将冷冻干燥前后的支架制作成固定的形状,尺寸大小为20.0×5.0×0.19mm3,再使用拉伸强度试验机对其进行单向拉伸力学测试,拉伸测试以每分钟100mm的速度进行。图8的应力应变曲线显示,多层支架改性在冻干前后力学性能变化不大。冻干前后材料的拉伸强度分别为3.31±0.23mpa和2.83±0.31mpa,与之对应的断裂伸长率分别为44.75±0.43%及53.25±0.18%。从上述数据可以看出,冻干后材料强度略有下降,整体没有大的差异。
测试例6:支架材料载药量测试:取1×1cm2实施例5中的得到的载生物活性因子pla/plga/cs复合膜放入乙酸乙酯1ml中,加入去离子水10ml,充分振荡,静置后,用下清液萃取出ngf,重复操作3次,用elisa在450nm波长测定ngf的吸光度,带入标准曲线进行计算,得出ngf含量。计算公式:载药量=(测定的ngf含量/支架材料的质量)×100%。通过运算1×1cm2的支架载药量约为3.28×10-6%。
测试例7:支架材料体外释放测试曲线的测定:取1×1cm2实施例5中的得到的载生物活性因子pla/plga/cs复合膜,置于截留分子量为5万的透析袋中,加入2mlph=7.4的模拟体液(pbs),透析袋置于装有8mlpbs溶液的离心管中,在170rpm,37℃的水浴摇床中释放。每隔一段时间(0.5、1、2、4、8、16、24h、2、4、7、10、14、21、28d、5、6、7、8w)取出2ml样品,再补充相同量的pbs溶液。取出的样品用elisa在450nm波长测定ngf的吸光度,所测得的吸光度根据标准曲线计算其中ngf的浓度。以时间为横坐标,以ngf的累计释放百分率为纵坐标作图,即可得到ngf的体外控制释放曲线。从图9中可以看出多层支架的释放曲线图在24小时呈现出爆发性的药物释放,因为cs膜的孔隙率较大,靠近cs膜的一层plga微球首先释放出药物,且水相的药物倾向于分布在微球表面,因此微球外层的药物也能快速释放出来,24小时的释放率达到29.66%。随后的2个月里,支架内部微球中的药物仍然在持续稳定的释放,释放率为50.68%。
测试例8:体外细胞毒性实验:将pc12细胞接种在24孔板的孔底部,温育24小时后,将实施例5中得到的载生物活性因子pla/plga/cs复合膜剪成直径为0.65cm的圆盘,置于细胞嵌入皿中,将嵌入皿置于24孔板的孔中,设置空白对照组。将培养板放入培养箱中,在37℃,5%co2饱和湿度条件下培养。于培养第1、3、5、7天进行检测,每孔加入cck8溶液,37℃继续孵育1h,终止培养。选择450nm检测波长,在酶标仪上测定各孔吸光度值。如图10所示,复合膜在一周内无明显细胞毒性。
测试例9:细胞分化实验:将pc12细胞接种在24孔板的孔底部,温育24小时后,换分化培养基。将实施例5得到的载生物活性因子pla/plga/cs复合膜剪成直径为0.65cm的圆盘,置于细胞嵌入皿中,将嵌入皿置于24孔板的孔中。然后在保持在37℃,5%co2的细胞培养室中培养。对于对照组,设置空白对照组、空白合膜组、及含有游离ngf组。
在孵育的第5天,采用ix50/58f2型倒置式系统显微镜观察复合载药膜对pc-12细胞分化的促进情况。从图11中可以看出,空白对照组及空白复合膜组细胞无明显变化,而复合载药膜组及游离ngf组pc-12细胞均出现了明显的神经元显性,证明载生物活性因子pla/plga/cs复合膜能良好的释放具有活性的药物以促进细胞分化。
- 还没有人留言评论。精彩留言会获得点赞!