定量构效关系辅助匹配分子对分析方法与流程
本发明涉及计算机辅助药物设计的技术领域,特别涉及一种定量构效关系辅助匹配分子对分析方法。
背景技术:
先导化合物的优化是一项复杂、耗时的任务,优化的目的是为了找到更理想的理化性质,良好的药代动力学性质,提高化合物的生物利用度,降低毒性等等,使其值得让一个分子进入到药物发现的候选阶段。如果药物化学家能够设想一个合适的取代基来修饰核心骨架来解决以上的所有问题,那么一种新的潜在的价值数十亿美元的产品就可能被发明并获得专利。而药物化学家们的灵感主要取决于已知的药物、文献知识、过去的合成经验或者是其他项目的同事。在这个过程中,总结经验知识,掌握这些化学结构的变化规律对制药企业和药物设计者合理的改造先导化合物,提高药物优化是至关重要的。
匹配分子对对分析(matchedmolecularpairanalysis,mmpa)作为一个从大量实验数据中,提出药物化学知识并指导先导化合物的改造的工具脱颖而出。一个匹配分子对(matchedmolecularpair,mmp)定义为一对化合物,它们只有一个位点发生结构改变。形成匹配分子对的化合物通过两个子结构的交换而相互转化,这称为化学转换。对于由同一转换连接的所有对,我们通过匹配分子对分析计算属性差异并汇总统计信息,从而获得使用化学语言描述的规则指导先导化合物的优化。但是,进行匹配分析对分析的数据缺少的问题,研究者们并没有关注。目前大多数关于匹配分子对分析的出版物都是来自于拥有庞大实验室数据的制药公司。虽然有来自学术团队的匹配分析对分析的出版物存在,但是相对较少,最可能的原因就是在这个以专利为基础的竞争环境下,药物公司只公开内部产生的所有信息中的部分数据。对于小数据集,它们缺少分子对,缺乏特定的化学转化的知识,从而不足以执行匹配分子对分析。导致有许多有价值的化学规则还未被挖掘或者开发。因此,基于以上分析,数据的缺乏成为了限制mmpa应用的一个关键问题。
技术实现要素:
本发明提供了一种定量构效关系辅助匹配分子对分析方法,其目的是为了解决匹配分子对分析在小数据上应用得到限制的问题。
为了达到上述目的,本发明的实施例提供了一种定量构效关系辅助匹配分子对分析方法,包括:
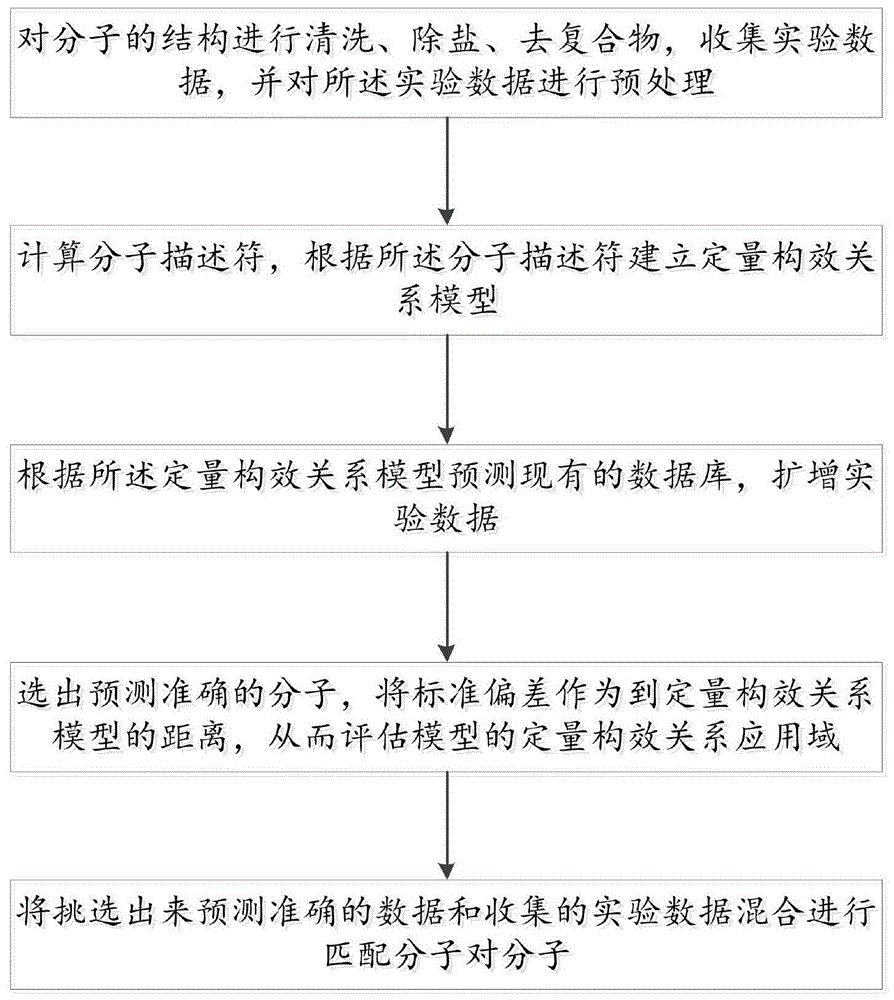
步骤1,对分子的结构进行清洗、除盐、去复合物,收集实验数据,并对所述实验数据进行预处理;
步骤2,计算分子2d描述符,根据所述分子2d描述符建立定量构效关系模型;
步骤3,根据所述定量构效关系模型预测现有的数据库,扩增实验数据;
步骤4,选出预测准确的分子,将标准偏差作为到定量构效关系模型的距离,从而评估模型的应用域;
步骤5,将挑选出来预测准确的数据和收集的实验数据混合进行匹配分子对分子。
其中,所述步骤1还包括:
对分子结构进行标准化处理,如一个分子对应多个目标值,则取中位数。
其中,所述步骤2具体包括:
使用极度梯度提升树、支持向量机、随机森林、cubist、相关向量机、梯度提升树、深度学习、高斯建立分类或回归预测模型;
对上述方法进行排列组合,通过求算数平均值建立共识模型;
选择预测能力最好的共识模型作为最后的预测模型。
其中,所述步骤3具体包括:
预测小分子化合物数据库,将收集的实验数据集按照80%和20%的比例进行随机批分,使用80%训练上述得到的最后的预测模型来预测数据库的数据,重复多次后,计算预测多次的均值和标准差。
其中,所述步骤4具体包括:
将收集的数据集按照80%和20%进行随机批分,使用80%训练上述得到的最好的模型,预测剩下的20%的数据,重复多次后,计算数据集中预测结果的标准差std:
其中:xi为预测值,μ为预测的均值,n为循环的次数;
若标准差小于预设值,则表明预测的结果准确;
若标准差大于预设值,则表明预测结果与实际偏差大。
其中,所述步骤5具体包括:
将分子所有的非环单键打断,一部分碎片作为key,另一部分作为value;若一个分子被打断三次以下,key为固定不变的部分,value为分子中可以替换的部分;
搜索具有相同key的两个分子作为一个匹配分子对,两个分子之间的局部不同则为一个转换,再匹配上相应的两个分子的活性或者物理化学性质,得到该转换导致性质变化;
统计每一个转换的分子对数量,若数量大于10,则进行秩和检验,检验所述转换在统计上是否改变了分子的活性或其他性质;
统计检验为有意义的为新的化学规则,用于改造先导化合物往预期的方向发展。
本发明的上述方案有如下的有益效果:
本发明的上述实施例所述的定量构效关系辅助匹配分子对分析方法基于定量构效关系预测后的数据进行匹配分子对分析,扩大了现有的化学规则,使得部分无统计意义的转换由于分子对数增加变得有统计意义成为化学规则,同时挖掘新的在原数据集中未出现的转换,有更多的知识指导先导化合物的优化,加速了药物设计周期,为数据集的匹配分子对分析提供了新的途径,解决了小数据上进行匹配分子对的限制,打破了匹配分子对分析只能基于实验数据的局限性。
附图说明
图1为本发明的定量构效关系辅助匹配分子对分析方法的流程图;
图2a为本发明的根据logd7.4数据集的预期误差与std分布图;
图2b为本发明的logd7.4数据集,chembl和specs数据库std的分布图;
图3a为本发明的变换对的数目的分布图;
图3b为本发明的specs数据的预测转换示意图;
图3c为本发明的chembl数据的预测转换示意图;
图3d为本发明的混合数据的转换分布示意图;
图4a为本发明的在logd7.4数据集和sepcs预测数据集中均有统计意义的25个规则的平均效应大小示意图;
图4b为本发明的59个在logd7.4数据集和chembl预测数据规则的logd7.4平均效应大小示意图;
图5a为本发明的实验数据得出的规则与来自混合数据集规则的大小相关图;
图5b为本发明的实验数据得出的规则与来自混合数据集规则的标准偏差的相关图。
具体实施方式
为使本发明要解决的技术问题、技术方案和优点更加清楚,下面将结合附图及具体实施例进行详细描述。
本发明针对现有的匹配分子对分析在小数据上应用得到限制的问题,提供了一种定量构效关系辅助匹配分子对分析方法。
如图1所示,本发明的实施例提供了一种定量构效关系辅助匹配分子对分析方法,包括:
步骤1,对分子的结构进行清洗、除盐、去复合物,收集实验数据,并对所述实验数据进行预处理;
步骤2,计算分子2d描述符,根据所述分子2d描述符建立定量构效关系模型;
步骤3,根据所述定量构效关系模型预测现有的数据库,扩增实验数据;
步骤4,选出预测准确的分子,将标准偏差作为到模型的距离,从而评估模型的定量构效关系应用域;
步骤5,将挑选出来预测准确的数据和收集的实验数据混合进行匹配分子对分子。
本发明的描述符包括计算分子的moe类型的二维(2d)描述符,cdk描述符,rdkit描述符,pybel描述符,e-state描述符;所述定量构效关系英文缩写为qsar;且所述评估模型的应用域也不限于只以标准偏差作为评估应用域的方法。其中,所述步骤1还包括:
对分子结构进行标准化处理,如一个分子对应多个目标值,则取中位数。
其中,所述步骤2具体包括:
使用极度梯度提升树、支持向量机、随机森林、cubist、相关向量机、梯度提升树、深度学习、高斯建立分类或回归预测模型;
对上述方法进行排列组合,通过求算数平均值建立共识模型;
选择预测能力最好的共识模型作为最后的预测模型。
其中,所述步骤3具体包括:
预测小分子化合物数据库,包括:chembl,pubchem,ochem,zinc,chemspider和specs数据库;将收集的实验数据集按照80%和20%的比例进行随机批分,使用80%训练上述得到的最后的预测模型来预测数据库的数据,重复多次后,计算预测多次的均值和标准差。
其中,所述步骤4具体包括:
将收集的数据集按照80%和20%进行随机批分,使用80%训练上述得到的最好的模型,预测剩下的20%的数据,重复多次后,计算数据集中预测结果的标准差std:
其中:xi为预测值,μ为预测的均值,n为循环的次数;
若标准差小于预设值,则表明预测的结果准确;
若标准差大于预设值,则表明预测结果与实际偏差大。
将样本的由std从小到大排序,分成相同等分的几个小组,拟合各个小组的预测误差从而得出均值和方差,该方差则为预测的误差。通过设置预测误差小于0.3,找到相对于的std,选出在预测数据std在该范围内的分子。这些分子的预测误差我们就认为在0.3之内。0.3为实验误差的控制范围。
其中,所述步骤5具体包括:
将分子所有的非环单键打断,一部分碎片作为key,另一部分作为value;若一个分子被打断三次以下,key为固定不变的部分,value为分子中可以替换的部分;
搜索具有相同key的两个分子作为一个匹配分子对,两个分子之间的局部不同则为一个转换,再匹配上相应的两个分子的活性或者物理化学性质,得到该转换导致性质变化;
统计每一个转换的分子对数量,若数量大于10,则进行秩和检验,检验所述转换在统计上是否改变了分子的活性或其他性质;
统计检验为有意义的为新的化学规则,用于改造先导化合物往预期的方向发展。
实施例1:
以分子的logd7.4来说明该方法:logd7.4是药物分子一个重要的性质,影响药物的细胞渗透率;
第一步:收集数据,数据预处理。我们从chembl数据库,ochem数据库还有现有的文献中收集了分子的logd数据。选取ph为7.4条件下的数据。提取这些分子的smile进行清洗,除盐,除复合物;对smile进行标准化。通过标准化后的smile计算分子的inchikey。inchikey相同则认为是同一个分子。将大于10,小于-10的数据移除。移除含有大于号,小于号这些不确定的限定符的数据。如果一个分子具有多个logd7.4值则由以下步骤进行处理:(1);最大值和最小值之间超过0.3,则该分子被移除;(2)最大值和最小值在0.3以内,则以中位数作为该分子的目标值。通过数据的预处理后,获得了10367个分子。
第二步:计算分子描述符,进行特征选择。使用moe软件计算了10367个分子的2d描述符。将方差为0,描述符的相关性大于0.95的描述符移除之后,得到了161个描述符。对这些描述符进行特征选择,选用基于随机森林的递归式特征消除法(rf-rfe)。最后得到44个描述符去建立定量构效关系模型。
第三步:模型的评估。该实例建立的是logd7.4回归模型,使用五折交互检验模型评估模型的稳健性。将数据集随机批分出80%建立预测模型,预测剩下的20%,重复1000次。求出logd7.4数据集中每个分子预测值的均值,std。模型评估使用以下的评估参数:交互检验的决定系数(q2),均方根误差(rmsecv),平均绝对误差(maecv),测试集的决定系数(rt2),均方根误差(rmset),平均绝对误差(maet)。各个评估参数公式如下所示:
其中,yi为实验logd7.4值,
第三步:基于10376个分子logd7.4数据和计算的44个2d描述符来建立logd7.4预测模型。由极度梯度提升树(xgboost),支持向量机(svm),随机森林(rf),cubist,梯度提升树(gb),深度学习(dl),高斯(gp)这7种机器学习方法建立logd7.4的预测模型。在建立模型的过程中使用五折交互检验和网格搜索的方法搜索这些模型的最佳参数。得到的最佳参数结构如表1所示。
表1.用来建立logd7.4预测模型的最优参数
为了进一步提高的模型预测性能,将这7种机器学习方法进行排列组合,求算数平均值建立121个共识模型。选出预测性能最高的模型作为最终的logd7.4的预测模型。另外,还使用非支配排序遗传算法ii(nsga-ii)通过搜索最优的r2,计算出7个模型最佳权重。这些模型的统计结果如表2所示。
表2.不同的模型的评估结果
从表2中可以得出组合了不同原理的机器学习算法时,共识模型的结果明显优于单个模型的结果。基于遗传算法得到的共识模型con_nsga与sxbd共识模型的预测性能不相上下。sxbd共识模型是通过计算svm,xgboost,gb,dl这四种单个模型的算数平均值得到的。最后,我们决定以sxbd为最终的logd7.4预测模型。
第四步:预测两个大型的药物化学数据库。在这里我们选择chembl和specs数据库。将chembl数据库中与logd7.4数据集中重复的分子去掉之后得到904588个分子。将specs数据库去除重复分子之后得到212404个分子。两个数据库采取与第一步同样的预处理步骤。随机选取logd7.4数据集中80%的数据建立sxbd训练模型,分别预测两个数据库的数据,重复1000次。计算每一个分子预测值的均值和std。以预测的均值作为我们的预测值。
第五步:选出预测准确的分子。要选出预测准确的分子,首先要确定模型的应用域,在什么std范围内,我们认为分子是预测准确的。将logd7.4数据集内的分子的照std从小到大的顺序排列,并计算预测误差。基于混合高斯分布(mgd)的方法,评估在一定的std范围内的预测误差。将数据集中的分子分成十组,拟合每个组的预测误差的高斯分布。拟合高斯分布的公式为:
其中,e为分子的预测误差。拟合出来的方差为σ该组总体的预测误差。从而得出σ会随着std的增大而增大。以σ<0.3为阈值,此时sxgd模型的std<0.104。specs数据库中有1601个分子的std<0.104,chembl数据库中9454个分子的std<0.104。这些分子可以被sxbd模型准确的预测,其预测误差在0.3以内。根据logd7.4数据集的预期误差与std分布如图2a所示。logd7.4数据集,chembl和specs数据库std的分布如图2b所示。
第六步:将挑选出来预测准确的数据和收集的数据混合进行匹配分子对分子。匹配分子对分析是基于husssain和rea开发的碎片和索引的方法。匹配分子对分子的过程:(1)将这些分子所有的环外单键可以被打断,生成从而一部分碎片作为key,另一部分作为value,一个分子被打断三次以下,key为固定不变的部分,value为分子中可以替换的部分;(2)搜索具有相同key的两个分子作为一个匹配分子对,两个分子之间的局部不同则为一个转换,再匹配上相应的两个分子的logd7.4的值,我们可以得到该转换导致的分子logd7.4变化情况;(3)统计每一个转换的分子对数量,数量大于10则进行秩和检验,检验该转换在统计上是不是真的改变了分子的logd7.4值;(4)统计检验有意义的则为化学规则,用于改造先导化合物往预期的logd7.4方向发展。
对定量构效关系辅助匹配分子对分析方法的评估:
1.1对数据集进行匹配分子对分析的结果。
将收集到的实验的logd7.4数据执行匹配分子对分析,其中只有5364种转换拥有的分子对的个数大于10,这5364种转换中只有290中转换通过了wilcoxon符号秩和检验成为了化学规则。图3a中示出了变换对的数目的分布。90%的转化仅被观察到一次,只有4%的转化被观察到十次或更多次。显然,由于转换频率较低,无法进行更严格的统计分析。图3b和3c显示了specs和chembl数据的预测转换。其中,只有49个转换符合我们的严格统计。对于9454个预测准确的chembl数据,141个转换有显著统计意义。表3总结了针对不同打断类型的转换数。在cut1类型中,logd7.4数据集,chembl和specs预测数据的转换数分别为175、99和31。在cut3类型中,specs和chembl数据中没有转换通过的wilcoxon符号秩和检验。
表3.不同数据集的具有统计意义的化学规则统计结果。(括号中表示方向相同的规则的数量)
1.2评估预测规则
为了测试定量构效关系辅助mmpa方法的鲁棒性,我们将logd7.4数据集与specs和chembl预测数据中相同规则的平均logd7.4差异进行了比较。
为了研究预测转换趋势的正确性(即增加或减小),比较wilcoxon符号秩和检验的结果。如表3所示,两个预测数据集和logd7.4数据集中存在的相同规则在变化趋势上非常一致。sepcs预测数据有25个与实验数据相同的规则,且在方向上一致。chembl预测数据有59个与实验数据相同的规则,且在方向上一致。这清楚地表明,可以基于定量构效关系模型准确地预测转换的符号。
为了验证预测规则的数值大小的准确性,计算预测的规则和实验规则之间的相关性。在logd7.4数据集和sepcs预测数据集中均有统计意义的25个规则的平均效应大小显示在图4a中。在预测规则与实验规则的相关性达到0.94。图4b显示59个在logd7.4数据集和chembl预测数据规则的logd7.4平均效应大小。它们之间的相关性为0.92,这表明基于qspr模型预测变换效果的大小是准确的。误差线表示平均logd7.4差异的sem。另外,我们发现这些规则中,有一对规则(羟基转化成甲基)在实验数据中有较大的sd且预测偏差较大,在图2b突出显示。显然,这个规则预测的sd较小。这很可能由于该规则容易受到化学环境的影响。
总的来说,通过比对来自于实验数据和预测数据的相同规则的方向和大小,得出预测的规则与实验规则在方向上一致,变化的大小的相关大于0.9。这表明通过qsar辅助匹配分子对分析的方法得出的预测规则是非常可靠的。
1.3评估新增的规则
为了评估将来自chembl和specs数据的预测分子添加到实验的logd7.4数据集中是否会增加logd7.4的规则,还是通过添加噪音来破坏logd7.4数据集的实验规则的统计信息。为了对此进行检验,我们生成了一个混合数据,该数据将chembl,specs预测数据添加到logd7.4实验数据集中,并重新进行匹配分子对分析。混合数据的转换分布如图3d所示。灰色条表示新增加的转换类型。在添加预测数据后logd7.4的转换类型的增益为125%。这表明预测数据创建了较大的匹配对集并增加了化学多样性。尤其是,拥有十对或十对以上的转换数量增加了两倍。经过统计检验,我们发现698个转换在混合数据中具有统计意义,与logd7.4测量数据相比,增加了140%。其中有341种新的规则,只有在考虑预测数据后才能识别出来的。图5a中显示了357个常见规则,用于比较每个规则的混合均值与测量均值。除了在实验数据集中识别出来的290个规则中,另有67个在混合数据才变得具有统计意义的规则。logd7.4实验数据集中原有的转换以灰色圆点显示(n=290);在混合数据中变得有统计意义的转换用黑色圆点显示(n=67)。
在logd7.4实验数据集中,有291个规则对logd7.4具有重大影响。对于混合数据,获得了新颖的341个转换类型。这清楚地表明,将预测数据添加到实验数据可以增加数据集的规则。表4列出了用于混合数据集中新增规则的示例,并将它们与之前已经发表的文献中相同的规则进行了比较。我们发现这些在混合数据中新增的规则与其之前文献上发表的结果非常一致。
表4.以往的研究得出的规则与混合数据集中的新增规则比较
第一对,在先前的研究中发现苯基取代为间氯苯基倾向于使logd7.4增加0.58个log单位。由于氯是亲脂基团,因此增加了logd7.4。与我们在混合数据集中发现的趋势相符:在混合物数据集中,此规则平均使logd7.4增加0.57个log单位。
第二对,在先前的研究中发现将苯转化为间吡啶时,logd7.4平均降低了0.92个log单位。logd7.4的降低应归因于强疏水基团(苯基)被h键受体芳香环(间吡啶)取代。在我们的混合数据集中,此规则平均使logd7.4减少1.05个log单位。
第三对,在先前的报道的同类规则中,我们发现氟原子取代甲基上的氢原子会降低logd7.4。在我们的混合数据集中,乙基与氟原子的交换平均使logd7.4降低0.59个log单位。
第四对,在先前的研究中发现氟化芳香族的乙基使得logd7.4增加0.21个log单位。我们在混合数据集中发现了同类转换。从乙基到三氟乙基的取代使logd7.4增加0.27个log单位。
通过分析上述四个混合数据集中新增的规则与先前的文献规则,说明混合数据集新增规则是非常可靠的。从而证明了混合实验数据集和预测数据集不会为随后的匹配分子对分析引入噪音,相反可以扩大原数据集的规则类型。本发明的上述实施例所述的定量构效关系辅助匹配分子对分析方法基于定量构效关系预测后的数据进行匹配分子对分析,扩大了现有的化学规则,部分无统计意义的转换由于分子对数增加变得有统计意义成为化学规则,同时挖掘新的在原数据集中未出现的转换,有更多的知识指导先导化合物的优化,加速了药物设计周期,为数据集的匹配分子对分析提供了新的途径,解决了小数据上进行匹配分子对的限制,打破了匹配分子对分析只能基于实验数据的局限性。
以上所述是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明所述原理的前提下,还可以作出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!