一种掺硼肿瘤靶向药物及其制备方法和应用与流程
本发明属于生物医药技术领域,具体涉及一种掺硼药物及其制备方法和应用,尤其涉及一种掺硼肿瘤靶向药物及其制备方法和应用。
背景技术:
硼中子俘获治疗(bnct)是通过在肿瘤细胞内的原子核裂变反应来摧毁癌细胞的治疗方法。通过注射含硼化合物,使其聚集于肿瘤细胞,而其他组织内分布很少。当用一种超热中子射线进行照射时,癌细胞里的硼-10能发生中子捕获形成不稳定同位素,继而发生核裂变反应,释放出一种杀伤力极强的射线,从而杀死癌细胞。
含硼药物作为bnct疗法中重要的一员,在临床应用中表现尚不尽人意,因为bnct要求药物靶向肿瘤,并能在肿瘤细胞累积足量的含硼药物,具体来说是药物在肿瘤与正常组织或血液中的浓度比值应大于3.0,进入每克肿瘤的10b量平均要求为20-35μg,并对应于一个肿瘤细胞至少含有109个10b原子,但现有技术例如目前fda批准的两种bnct药物bsh(巯基多面体硼笼化物)和bpa(4-硼-l-苯丙氨酸)还达不到此水平,而且各种硼载荷的靶向药物例如低分子量硼化物、掺硼肽、抗体碎片与各种蛋白质、完整的抗体与基于抗体的结合物以及脂质体等等,也由于载药量低、穿透力差、系统毒性强等问题难以满足bnct的治疗要求。
cn106279231a公开了一种用于bnct的含硼化合物及其制备方法和用途,该发明提供的新的含硼化合物通过缩合、氨解及成环反应得到,易于制备、成本低,并具有良好的生物性能,在肿瘤细胞的摄取、积累和保留方面都优于bpa,在bnct方面具有良好的应用前景,还有望成为临床潜在的正电子发射断层扫描肿瘤显像剂,为恶性肿瘤的诊断、良恶性疾病的鉴别和全身转移病灶的探查等创造了有利条件,但该含硼化合物在肿瘤细胞靶向和积累方面依然不够理想。
cn1968705公开了一种低毒性的含硼化合物及其在肿瘤的治疗、观察和诊断中的使用方法,具体涉及到低毒性含碳硼烷5,10,15,20-四苯基卟啉化合物,特别是其在硼中子俘获治疗(bnct)和光动力疗法(pdt)用于治疗大脑、头部和颈部及其周围组织中的肿瘤时的应用方法。该发明所涉及的含硼化合物并不能运输足量的含硼药物进入肿瘤,实现良好的抗肿瘤效果。
cn100417421c公开了一种基于磁性纳米材料的有机金属碳硼烷靶向制剂的制备方法及其制成的靶向制剂,该发明将表面修饰和功能化的磁性纳米颗粒fe3o4与单/双金属中心碳硼烷通过自组装而制成有机金属碳硼烷靶向制剂。在外加磁场的作用下,通过纳米颗粒的磁性导航,靶向制剂能够“宏观”定向移动到肿瘤部位,实现了硼携带剂在靶向器官和细胞水平上的精确定位。但该制剂需要借助外界磁场的作用才能将其靶向至肿瘤部位,且可能存在载药量低和穿透力差等问题。
因此,开发一种满足bnct治疗要求,生物相容性好,且具有在肿瘤细胞快速高效的累积能力的掺硼靶向药物具有重要的意义。
技术实现要素:
针对现有技术的不足,本发明的目的在于提供一种掺硼药物及其制备方法和应用,尤其提供一种掺硼肿瘤靶向药物及其制备方法和应用。
为达到此发明目的,本发明采用以下技术方案:
一方面,本发明提供一种掺硼肿瘤靶向药物,所述掺硼肿瘤靶向药物包括多肽、位于多肽氮末端的疏水分子和位于多肽赖氨酸上的巯基十二硼烷二钠盐(bsh,巯基多面体硼笼化物);组成所述多肽的氨基酸包括磷酸化的l-酪氨酸、l-赖氨酸和l-苯丙氨酸。
本发明涉及的掺硼肿瘤靶向药物功能单元明确,其中磷酸化的l-酪氨酸作为磷酸酶响应单元,bsh作为硼提供单元,多肽氮末端的疏水分子和l-苯丙氨酸作为超分子组装单元。当所述含硼药物进入肿瘤细胞后,肿瘤细胞中过表达的磷酸酶能将磷酸化的l-酪氨酸去磷酸化,含硼药物通过氢键、π-π堆积作用在肿瘤细胞中原位自组装形成含硼组装体,从而实现含硼药物在肿瘤细胞内的特异性富集,每个癌细胞富集量可达37.3×109个b原子,满足bnct治疗的需要。正常细胞由于没有过表达的磷酸酶,因此不能导致l-酪氨酸去磷酸化,进而不能使含硼药物富集于正常细胞。因此实现含硼药物对肿瘤细胞的靶向。此外,该含硼药物的生物相容性好、低免疫原性、低毒性。
所述多肽中的l-酪氨酸、l-赖氨酸和l-苯丙氨酸可以根据实际需要任意排列其连接顺序,其数目也可以根据实际需要任意增减。
优选地,组成所述多肽的氨基酸还包括甘氨酸和/或l-精氨酸。
所述甘氨酸和l-精氨酸在多肽中的连接位置也根据实际需要任意选择,其数目也可以根据实际需要任意增减。
优选地,所述多肽的氨基酸残基数为3-6个,例如3个、4个、5个或6个。
优选地,所述疏水分子包括2-萘乙酸或芘乙酸。
另一方面,本发明提供一种如上所述的掺硼肿瘤靶向药物的制备方法,所述制备方法包括:通过固相合成法合成多肽,将疏水分子与制得的多肽进行连接,从载体树脂上脱除,并脱除侧链氨基保护基团,得到疏水分子-多肽化合物,再对疏水分子-多肽化合物中赖氨酸进行活化,并与巯基十二硼烷二钠盐连接,得到所述掺硼肿瘤靶向药物。
该制备方法采用经典的固相合成法,操作简单,所得掺硼肿瘤靶向药物具备高化学纯度以及高总产率等优点。
在本发明中,所述制备方法包括如下步骤:
(1)采用末端氨基和侧链氨基均得到保护的氨基酸为原料,根据所要合成多肽的氨基酸顺序,将第一个氨基酸与载体树脂进行反应并连接;
(2)脱去第一个氨基酸的保护基,加入第二个氨基酸与其反应并连接;
(3)脱去第二个氨基酸的保护基,加入下一个氨基酸与其反应并连接,重复此操作,直到完成所有氨基酸的缩合,得到多肽;
(4)将疏水分子与步骤(3)制得的多肽进行反应并连接;
(5)将步骤(4)得到的产物从载体树脂上脱除,并脱除侧链氨基保护基,得到疏水分子-多肽化合物;
(6)将疏水分子-多肽化合物中赖氨酸的氨基进行活化,并与巯基十二硼烷二钠盐进行反应,得到所述掺硼肿瘤靶向药物。
优选地,所述载体树脂在使用前进行溶胀。
优选地,所述载体树脂为2-氯-三苯甲基氯树脂。
优选地,所述溶胀的时间为20-40min,例如20min、25min、28min、30min、33min、35min或40min等,范围内的其他具体点值均可选择,在此便不再一一赘述。
优选地,步骤(1)所述将第一个氨基酸与载体树脂进行反应之前,先将其与n,n-二异丙基乙胺溶解于n,n-二甲基甲酰胺中。
优选地,步骤(1)所述反应在保护气氛下进行。
优选地,步骤(1)所述反应的温度为20-30℃,例如20℃、22℃、24℃、25℃、26℃、27℃、28℃或30℃等,时间为1.5-3h,例如1.5h、1.8h、2h、2.2h、2.4h、2.5h、2.6h、2.7h、2.8h或3h等,范围内的其他具体点值均可选择,在此便不再一一赘述。
优选地,步骤(1)所述反应结束后进行过滤,并用n,n-二甲基甲酰胺进行清洗,再将甲醇、n,n-二异丙基乙胺与二氯甲烷的混合液加入树脂中进行反应,过滤,并用n,n-二甲基甲酰胺进行清洗。优选地,所述甲醇与二氯甲烷的体积比为15:5:80。
优选地,步骤(2)所述脱去第一个氨基酸的保护基使用的是哌啶与n,n-二甲基甲酰胺的混合液,将其加入载体树脂中进行反应;优选地,所述哌啶与n,n-二甲基甲酰胺的体积比为1:4;优选地,所述反应的时间为20-40min(例如20min、22min、25min、30min、32min、35min、36min、38min或40min等);优选地,所述反应的温度为20-30℃(例如20℃、22℃、24℃、25℃、26℃、27℃、28℃或30℃等);优选地,反应结束后进行过滤,并用n,n-二甲基甲酰胺进行清洗。
优选地,步骤(2)所述加入第二个氨基酸之前,将其与苯并三氮唑-n,n,n',n'-四甲基脲六氟磷酸酯和n,n-二异丙基乙胺溶解于n,n-二甲基甲酰胺中。
优选地,步骤(2)所述反应在保护气氛下进行。
优选地,步骤(2)所述反应的温度为20-30℃,例如20℃、22℃、24℃、25℃、26℃、27℃、28℃或30℃等,时间为1.5-3h,例如1.5h、1.8h、2h、2.2h、2.4h、2.5h、2.6h、2.7h、2.8h或3h等,范围内的其他具体点值均可选择,在此便不再一一赘述。
优选地,步骤(3)所述脱保护条件与反应条件与步骤(2)一致。
优选地,步骤(4)所述将疏水分子与多肽进行反应之前,将其与苯并三氮唑-n,n,n',n'-四甲基脲六氟磷酸酯和n,n-二异丙基乙胺溶解于n,n-二甲基甲酰胺中。
优选地,步骤(4)所述反应在保护气氛下进行。
优选地,步骤(4)所述反应的温度为20-30℃,例如20℃、22℃、24℃、25℃、26℃、27℃、28℃或30℃等,时间为1.5-3h,例如1.5h、1.8h、2h、2.2h、2.4h、2.5h、2.6h、2.7h、2.8h或3h等,范围内的其他具体点值均可选择,在此便不再一一赘述。
优选地,步骤(4)所述反应结束后进行过滤,并用n,n-二甲基甲酰胺进行清洗。
优选地,步骤(5)的具体操作为:将三氟乙酸加入载体树脂中,进行反应,过滤,将滤液加入冰乙醚,过滤,得到白色固体即疏水分子-多肽化合物。
优选地,所述反应在保护气氛下进行。
优选地,所述保护气氛为氮气。
优选地,所述反应的时间为1.5-3h,例如1.5h、1.8h、2h、2.2h、2.4h、2.5h、2.6h、2.7h、2.8h或3h等,所述反应的温度为20-30℃,例如20℃、22℃、24℃、25℃、26℃、27℃、28℃或30℃等,范围内的其他具体点值均可选择,在此便不再一一赘述。
优选地,所述加入冰乙醚的方式为逐滴加入。
优选地,步骤(6)所述活化使用的活化剂包括3-马来酰亚胺基丙酸羟基琥珀酰亚胺酯(cas:55750-62-4)或5-马来酰亚胺戊酸羟基琥珀酰亚胺酯(cas:103750-03-4);操作可以为:将活化剂、疏水分子-多肽化合物分别溶于dmf中,混合均匀,加入dipea进行反应,半制备高效液相色谱进行提纯,得到白色固体即活化的疏水分子-多肽化合物。
优选地,步骤(6)所述活化的温度为20-30℃,例如20℃、22℃、24℃、25℃、26℃、27℃、28℃或30℃等,时间为12-24h,例如12h、14h、16h、18h、20h、22h或24h等,范围内的其他具体点值均可选择,在此便不再一一赘述。
优选地,步骤(6)所述反应的温度为20-30℃,例如20℃、22℃、24℃、25℃、26℃、27℃、28℃或30℃等,时间为12-24h,例如12h、14h、16h、18h、20h、22h或24h等,范围内的其他具体点值均可选择,在此便不再一一赘述。
步骤(6)所述反应的具体操作可以为:将活化产物与bsh溶于乙腈/水中,加入三乙胺进行反应,即得;优选地,乙腈/水的比例为1:1;优选地,所述反应在保护气氛下进行。
作为本发明的优选技术方案,所述掺硼肿瘤靶向药物的制备方法具体包括如下步骤:
(1)将载体树脂在二氯甲烷中进行溶胀20-40min;
(2)采用末端氨基均由fmoc保护、侧链氨基由boc保护的氨基酸为原料,根据所要合成的多肽的氨基酸顺序,将第一个氨基酸与n,n-二异丙基乙胺溶解于n,n-二甲基甲酰胺中,再加入载体树脂,通氮气,在20-30℃下与载体树脂进行1.5-3h反应并连接;过滤,并用n,n-二甲基甲酰胺进行清洗,再将甲醇、n,n-二异丙基乙胺与二氯甲烷以体积比15:5:80混合加入树脂中进行反应,过滤,并用n,n-二甲基甲酰胺进行清洗;
(3)将哌啶与n,n-二甲基甲酰胺以体积比1:4混合加入至载体树脂中,通氮气,在20-30℃下进行反应20-40min,过滤,并用n,n-二甲基甲酰胺进行清洗;
(4)将第二个氨基酸与苯并三氮唑-n,n,n',n'-四甲基脲六氟磷酸酯和n,n-二异丙基乙胺溶解于n,n-二甲基甲酰胺中,再加入载体树脂,通氮气,在20-30℃下与载体树脂进行1.5-3h反应并连接;过滤,并用n,n-二甲基甲酰胺进行清洗;
(5)将哌啶与n,n-二甲基甲酰胺以体积比1:4混合加入至载体树脂中,通氮气,在20-30℃下进行反应20-40min,过滤,并用n,n-二甲基甲酰胺进行清洗;
(6)再将下一个氨基酸加入载体树脂进行反应并连接,重复上述(4)和(5)操作,直到完成所有氨基酸的缩合,得到多肽;
(7)将疏水分子与苯并三氮唑-n,n,n',n'-四甲基脲六氟磷酸酯和n,n-二异丙基乙胺溶解于n,n-二甲基甲酰胺中,通氮气,在20-30℃下与制得的多肽进行1.5-3h反应并连接;过滤,并用n,n-二甲基甲酰胺进行清洗;
(8)将三氟乙酸加入载体树脂中,通氮气,在20-30℃下进行1.5-3h反应,过滤,将滤液逐滴加入冰乙醚,过滤,得到白色固体即疏水分子-多肽化合物;
(9)将活化剂、疏水分子-多肽化合物分别溶于dmf中,混合均匀,加入dipea,通氮气,在20-30℃下进行12-24h反应,得到白色固体即所述活化的疏水分子-多肽化合物;
(10)将步骤(9)中产物与bsh溶于1:1的乙腈/水中,加入三乙胺,通氮气,在20-30℃下进行12-24h反应,得到白色固体即所述掺硼肿瘤靶向药物。
再一方面,本发明提供一种如上所述的掺硼肿瘤靶向药物在制备抗肿瘤药物中的应用。
相对于现有技术,本发明具有以下有益效果:
(1)本发明所涉及的掺硼肿瘤靶向药物在肿瘤细胞内具有特异性、快速、高效的累积能力,每个细胞内的硼积累量在给药20μm时可达到2.17×109个,1000μm时可达到37.30×109个,是同等条件下传统药物bsh的6倍,能够更好地满足bnct的治疗需要。
(2)本发明所涉及的掺硼肿瘤靶向药物生物相容性好,无系统毒性。
(3)本发明所涉及的掺硼肿瘤靶向药物的制备采用经典的固相合成法,操作简单,所得掺硼肿瘤靶向药物具备高化学纯度以及高总产率等优点。
附图说明
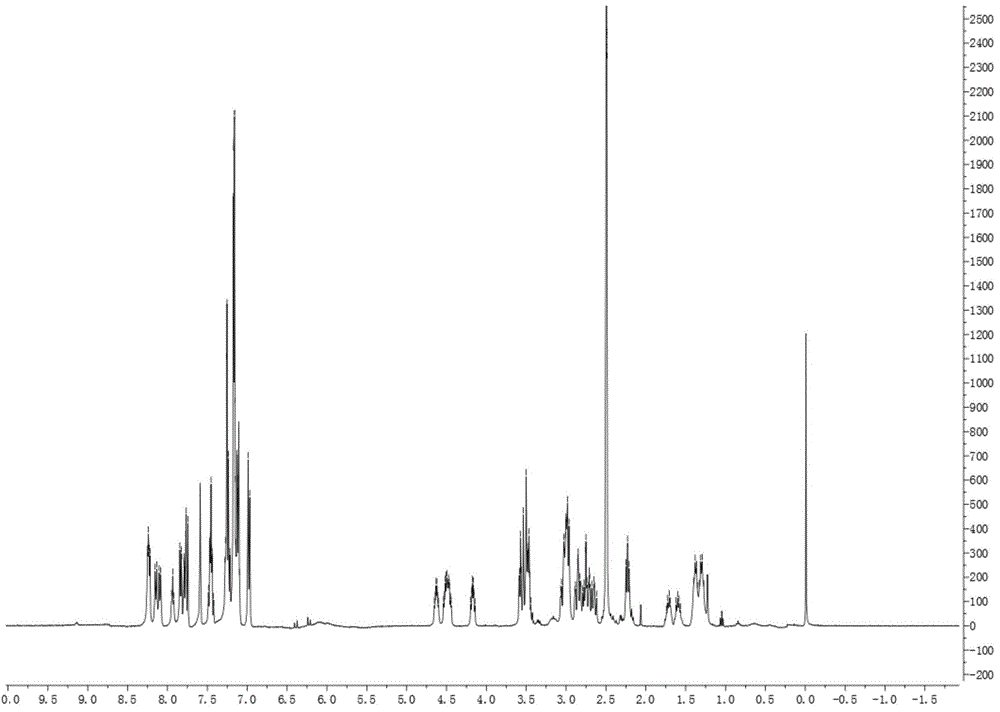
图1是实施例1所制得的掺硼肿瘤靶向药物的1h-nmr表征图;
图2是实施例1所制得的掺硼肿瘤靶向药物的电喷雾-质谱表征图;
图3是实施例1所制得的掺硼肿瘤靶向药物的电喷雾-质谱表征图;
图4是本发明涉及的掺硼肿瘤靶向药物和bsh分别在hela细胞内硼累积量与给药浓度的关系图;
图5是本发明涉及的掺硼肿瘤靶向药物和bsh分别对hela细胞毒性的统计结果图。
具体实施方式
下面通过具体实施方式来进一步说明本发明的技术方案。本领域技术人员应该明了,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。
实施例1
本实施例提供一种掺硼肿瘤靶向药物,包括多肽、位于多肽氮末端的疏水分子2-萘乙酸和连接于多肽赖氨酸上的bsh;所述多肽包括磷酸化的l-酪氨酸、l-赖氨酸和l-苯丙氨酸,其分子结构如下所示:
其制备方法包括如下步骤:
(1)在固相合成管中,将1.0g2-氯-三苯甲基氯树脂在10ml二氯甲烷中进行溶胀30min;
(2)采用末端氨基由fmoc保护的磷酸化的l-酪氨酸、l-苯丙氨酸、末端氨基和侧链氨基分别由fmoc和boc保护的l-赖氨酸为原料,根据上述多肽的氨基酸顺序,将1.0gfmoc和boc保护的l-赖氨酸与992μln,n-二异丙基乙胺溶解于5mln,n-二甲基甲酰胺中,再加入到载体树脂,通氮气,在25℃下与载体树脂进行2h反应并连接;过滤,并用n,n-二甲基甲酰胺进行清洗;将甲醇、n,n-二异丙基乙胺与二氯甲烷以体积比15:5:80混合加入树脂中进行反应30min,过滤,并用n,n-二甲基甲酰胺进行清洗;
(3)将哌啶与n,n-二甲基甲酰胺以体积比1:4混合(总体积为10ml)加入至载体树脂中,通氮气,在25℃下进行反应30min,过滤,并用n,n-二甲基甲酰胺进行清洗;
(4)再将下一个氨基酸即fmoc保护的l-苯丙氨酸与苯并三氮唑-n,n,n',n'-四甲基脲六氟磷酸酯、n,n-二异丙基乙胺溶解于n,n-二甲基甲酰胺中,加入载体树脂进行反应并连接,重复上述操作,直到完成所有氨基酸的缩合,得到多肽;
(5)将0.56g疏水分子2-萘乙酸与1.14g苯并三氮唑-n,n,n',n'-四甲基脲六氟磷酸酯和992μln,n-二异丙基乙胺溶解于5mln,n-二甲基甲酰胺中,再加入载体树脂,通氮气,在25℃下与制得的多肽进行2h反应并连接;过滤,并用n,n-二甲基甲酰胺进行清洗;
(6)将5ml三氟乙酸加入载体树脂中,通氮气,在25℃下进行2h反应,过滤,将滤液逐滴加入30ml冰乙醚,过滤,得到白色固体即疏水分子-多肽化合物;
(7)步骤(6)所得42.55mg疏水分子-多肽化合物与39.9mg3-马来酰亚胺基丙酸羟基琥珀酰亚胺酯分别溶于1mldmf中,混合均匀,加入11.35mgdipea,通氮气,在30℃下进行24h反应,半制备高效液相色谱进行提纯,得到白色固体即活化的疏水分子-多肽化合物;
(8)将步骤(7)中产物50.15mg与26.25mgbsh溶于1:1的乙腈/水中,加入10mg三乙胺,通氮气,在30℃下进行24h反应,半制备高效液相色谱进行提纯,得到35.28mg白色固体即所述掺硼肿瘤靶向药物,产率60.0%。
用1h-nmr和电喷雾-质谱(esi-ms)法分别对所制得的掺硼肿瘤靶向药物进行表征,结果如下:
1h-nmr表征结果为:1hnmr(dmso-d6,400mhz)δ(ppm):8.25-8.22(m,2h),8.15-8.08(q,2h),7.95-7.92(t,1h),7.84-7.74(m,3h),7.59(s,1h),7.49-7.42(m,2h),7.28-7.10(m,14h),6.99-6.97(d,2h),4.65-4.60(m,1h),4.54-5.44(m,2h),4.20-4.14(m,1h),3.59-3.44(m,5h),3.07-3.62(m,10h),2.25-2.23(t,2h),1.74-1.57(m,2h),1.40-1.27(m,4h)。核磁表征图如图1所示。
esi-ms表征结果为:c52h67b12n6o13ps,1178.53(m/z);[m-2h]-:588.26228。质谱图如图2和图3所示。
表明成功制得所述掺硼肿瘤靶向药物。
实施例2
将实施例1制得的掺硼肿瘤靶向药物和bsh进行在hela细胞内的累计性能测试,具体方法为:
选取hela细胞作为模型细胞,在接种有2×105个细胞的6孔板中,分别加入2ml含10-1000μm实施例1产品的dmem培养基或2ml含10-1000μmbsh的dmem培养基,并在37℃以及5%co2的条件下,孵育24h。将细胞用胰蛋白酶进行消化处理、计数、加入硝酸进行加热消解,最后用icp-ms测得累积硼的量,统计结果如图4所示。
由图4可以看出:本发明所涉及的掺硼肿瘤靶向药物在hela细胞中的累积硼量显著地大于bsh组在hela细胞中的累积硼量,且随着给药浓度的延长,这种优势愈发明显。
实施例3
将实施例1制得的掺硼肿瘤靶向药物和bsh进行细胞毒性测试,具体方法为:
选取hela细胞作为模型细胞,在接种有5000个细胞的96孔板中,分别加入0.1ml含10-1000μm实施例1产品或0.1ml含10-1000μmbsh的dmem培养基,并在37℃以及5%co2的条件下,孵育24h,然后用mtt法检测细胞毒性,统计结果如图5所示。
由图5可以看出:本发明所涉及的掺硼肿瘤靶向药物在10μm-500μm的范围内对hela细胞几乎没有细胞毒性,表明该含硼药物的生物相容性好,无系统毒性。
申请人声明,本发明通过上述实施例来说明本发明的一种掺硼肿瘤靶向药物及其制备方法和应用,但本发明并不局限于上述实施例,即不意味着本发明必须依赖上述实施例才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明产品各原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,这些简单变型均属于本发明的保护范围。
另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合,为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。
- 还没有人留言评论。精彩留言会获得点赞!