一种靶点预测方法与流程
1.本发明涉及计算生物学,特别涉及一种药物-靶点关系预测方法。
背景技术:
2.一种新的药物从筛选到上市成功,据统计,花费高达2.0-3.5亿美元,且平均需要历经10到14年的时间。目前,临床阶段候选药物的淘汰率高达90%。其中,发现和确认药物靶点是一个特别复杂的过程,确认一个简单的药物靶点都需要花费巨大的金钱和时间成本。因此,如何缩小发现药物靶点到药物成功上市(r&d)这一过程的成本,提升药物靶点发现到临床研究的速度就成为了各大制药公司和学术机构的研究重点。
3.药物研究过程中一个重要的环节就是新药的筛选,而建立一个新药的筛选系统的关键就是寻找药物作用靶点来提供干预治疗。药物靶点是药物与机体生物大分子作用而产生药理学作用并达到防治疾病目的的由生物分子形成的特殊位点,是药物发挥作用的基础,在新药筛选中具有十分重要的意义。预测药物靶点不但对药物分子初期成药性的评价有着不可替代的作用,而且对药物成熟后老药新用等领域都有着重大的意义,但因为药物的通量、准确度、成本等限制,实验方法的应用难以广泛地展开。而作为一类低成本且快速的方法,利用计算机技术预测药物靶点的方法正受到越来越多的重视。
4.药物靶点的预测主要是为了新药物的发现,而新药物的发现是化学信息学最主要的应用。化学信息学是建立在多学科基础上的一个新分支。从化学信息学的角度出发药物靶点预测可分为3类:基于配体结构特征的预测、基于受体结构特征的预测、基于配体受体相互作用的预测。基于配体的结构特征的预测主要包括化学相似性搜索、反向药效团搜索以及采用数据挖掘的方法。化学相似性搜索的原理是结构或物理化学性质相似的小分子化合物可能与性质相同或相近的靶点结合,通过比较查询分子与己知靶点活性分子的结构或物理化学性质的相似程度来预测查询分子的潜在作用靶点。基于受体结构特征的药物作用靶点预测一般采用的是反向分子对接方法。该方法是将查询分子与靶点数据库中的若干个靶点进行分子对接,根据打分函数等指标对结合的作用靶点进行排序来确定查询分子的潜在靶点。基于数据挖掘的方法发现药物的作用靶点:第一步是将收集的数据集进行预处理后再进一步划分训练集与测试集。第二步是选择合适的机器学习算法如随机森林、决策树、人工神经网络等,训练集用于构建模型,测试集验证模型的预测性能。
5.目前基于数据挖掘的药物靶点预测算法效果最好。基于数据挖掘的药物靶点预测算法主要分为两类:一类是基于传统二部图模型的靶点预测算法;另一类是基于传统逻辑矩阵分解的靶点预测算法。而且,在进行靶点预测时,这些算法往往仅限于单个药物分子对那些靶点有效,而没有预测药物分子与靶点之间的联系以及联系的强弱。基于传统二部图模型的靶点预测算法,主要将药物和靶点视为图的两个顶点,而药物-靶点的相互作用视为图的边(其中药物和药物的关系、靶点和靶点的关系不体现在图的边上),通过对边关系的学习进修药物-靶点关系的预测。基于二部图模型的药物-靶点相互作用关系预测普遍存在一个问题,即不能预测新加入药物或者新加入靶点的问题,并且用于解决该问题的方法大
部分是通过引入药物相似性矩阵和靶点相似性矩阵,而相似性度量的选择对未知药物-靶点的关系预测准确度影响非常大,算法泛化能力。基于传统逻辑矩阵分解的靶点预测算法,主要通过将药物-靶点关系矩阵作为研究对象,药物-靶点相关矩阵是稀疏并且低秩的,因此利用药物-靶点关系矩阵的稀疏低秩属性,来恢复丢失的药物-靶点相互作用关系。基于传统逻辑矩阵分解的靶点预测算法在进行靶点预测时,需要进行参数寻优,而且当药物-靶点矩阵非常大时算法对药物-靶点关系预测所需时间较长。本发明在基于矩阵分解的药物-靶点关系预测基础上进行改进。
技术实现要素:
6.本发明实施例的目的在于提供一种药物-靶点关系预测方法,旨在解决基于传统二部图模型的靶点预测算法对未知药物-靶点的关系预测较差,而基于传统逻辑矩阵分解的靶点预测算法预测所需时间较长的问题。
7.为达到上述目的,本发明实施例采用了如下技术方案:
8.第一方面,公开了一种新的基于逻辑矩阵分解的药物-靶点关系预测方法,包括:利用多种药物分子系数加权求闭包计算药物和靶点相似性,通过高斯映射将药物相似性矩阵和靶点相似性矩阵映射到非线性空间以获取新的药物、靶点相似性矩,并利用上述的相似性矩阵进行逻辑矩阵分解,然后通过贝叶斯优化进行参数选优,最后利用药物、靶点的相似性矩阵对预测的药物-靶点关系矩阵进行优化获得最终的药物-靶点关系。
9.第二方面,结合第一方面公开了一种药物和靶点相似性计算方法,包括:利用药物分子二维结构的杰卡德相似系数、偏好连接系数、皮尔逊相关系数、卡兹系数进行均值加权得到药物分子的关系图,再对药物分子关系图求闭包获得最终的药物分子相似性矩阵,同理获得靶点相似性矩阵。
10.第二方面所述的药物分子二维结构为一个向量,在对药物分子二维结构的四种系数进行均值加权得到药物分子的关系图,区别于只采用其中一种系数获得的关系图,具有更全面和更好的药物分子相似性衡量,而关系图求闭包可以很好的消除孤立关系。
11.第三方面,结合第一方面公开了一种药物-靶点预测算法,包括:将药物相似性矩阵和靶点相似性矩阵与高斯核函数进行卷积得到新的药物相似性矩阵和靶点相似性矩阵,利用新的药物相似性矩阵和靶点相似性矩阵做逻辑矩阵分解,从而获得药物-靶点关系矩阵。
12.第三方面所述的将药物相似性矩阵和靶点相似性矩阵与高斯核函数进行卷积得到新的药物相似性矩阵和靶点相似性矩阵,是指对药物相似性矩阵和靶点相似性矩阵进行非线性映射以提高药物-靶点关系预测的精度。
13.在第三方面所述的逻辑矩阵分解中,逻辑矩阵分解是常用的靶点预测方法之一,与传统算法不同的,本发明在进行逻辑矩阵分解时,将药物相似性矩阵和靶点相似性矩阵也作为参考条件进行逻辑矩阵的概率计算,从而获得更为精确的药物-靶点关系预测值。
14.在第三方面所述的逻辑矩阵分解中,在进行药物-靶点关系预测时需要对大量的参数进行调参,为了优化矩阵分解中所需要的参数,从而节省靶点预测的时间,本发明采用贝叶斯优化方法进行参数优化获得逻辑矩阵分解所需最优参数。
15.第四方面,结合第一方面和第三方面公开了一种药物-靶点关系预测精度优化方
法,包括:利用药物和靶点的相似矩阵的局部信息对药物-靶点关系矩阵进行改进。所述的相似性矩阵的局部信息是指药物、靶点相似性矩阵中每个元素的相似元素。对于药物和靶点的相似矩阵中的元素,首先寻找其所对应的药物分子与药物分子的相似性矩阵中最为相似的n个药物分子,然后在药物-靶点的关系矩阵中查询这n个的药物分子看是否有靶点与之相匹配,如果有,则设共有m个靶点与之匹配,其对应的药物-靶点关系值v
i
,计算所有v
i
的均值avg,则将此药物和靶点的关系值设置为原始关系值和avg的均值;若无靶点与之相匹配,则不改变药物和靶点的相似矩阵中此元素相应关系值,遍历完整个药物-靶点关系矩阵,最终得到改善以后的药物-靶点关系矩阵。同理,可以利用靶点相似性矩阵进行药物-靶点关系矩阵改善。
16.本发明实施例通过新的药物和靶点相似性矩阵计算方法,以贝叶斯优化的逻辑矩阵分解进行药物-靶点的关系预测,最后通过利用药物和靶点的相似矩阵的局部信息对药物-靶点关系矩阵进行改进,最终预测的药物-靶点关系精度更高,预测时间更短。
附图说明
17.为了更清楚地说明本发明实施例中的技术方案,下面将对实施例描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
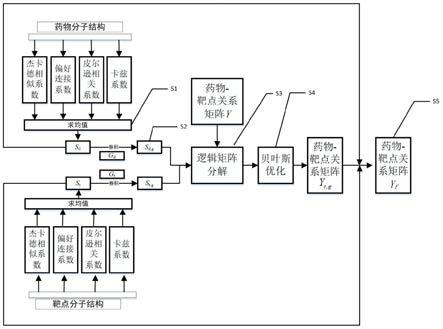
18.图1是本发明实施例提供的一种靶点预测方法的流程图;
具体实施方式
19.为使本发明的目的、技术方案和优点更加清楚,下面将结合附图对本发明实施方式作进一步地详细描述。显然,所描述的实施例仅仅是本发明的一部分实施例而不是全部的实施例。基于本发明中的实施例本领域普通技术人员在没有付出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
20.如图1所示,本发明实施例提供了一种靶点预测方法,其具体实现过程为:
21.步骤s1:新的药物相似性矩阵和靶点相似性矩阵计算方法,所述计算方法包括对每个药物分子(药物分子二维结构为一个向量)利用其二维结构计算它们的杰卡德相似系数、偏好连接系数、皮尔逊相关系数、卡兹系数进行均值加权得到各个药物分子的关系矩阵图,即
22.杰卡德相似系数:
23.偏好连接系数:
24.皮尔逊相关系数:
25.卡兹系数:k(x
i
,x
j
)=(i-ηa)-1-i
ꢀꢀꢀ
(4)
26.各个药物分子的关系矩阵图:其中,x
i
,x
j
是任意两个药物分子向量,分别表示x
i
,x
j
所作用的靶点的个数,cov(x
i
,x
j
)为x
i
,x
j
的协方差矩阵,为x
i
,x
j
的标准差,η=0.5,a为x
i
,x
j
的邻接矩阵。
27.然后对药物分子关系矩阵图求闭包获得最终的药物分子相似性矩阵s
d
,同理获得靶点相似性矩阵s
t
,与只采用其中一种系数获得的关系图相比,新的相似性矩阵计算方法使获得的药物和药物之间的相似性矩阵与靶点和靶点之间获得的相似性矩阵具有更好的相似性度量,有利于提高后期药物与靶点之间的关系预测的准确率。
28.步骤s2:新的药物相似性矩阵和靶点相似性矩阵计算方法,还包括对药物分子相似性矩阵s
d
和靶点相似性矩阵s
t
进行高斯卷积,卷积过程相当于对药物相似性矩阵和靶点相似性矩阵进行非线性映射,通过高斯非线性映射后得到的新的药物相似性矩阵和靶点相似性矩阵具有更好的泛化能力,有助于药物-靶点预测算法可以广泛的应用于不同的数据库中,且取得较为理想的预测效果。最终得到药物分子相似性矩阵s
d,g
和靶点相似性矩阵s
t,g
。
29.步骤s3:对药物分子相似性矩阵s
d,g
和靶点相似性矩阵s
t,g
和经过验证的药物-靶点关系矩阵y相结合进行逻辑矩阵分解,从而得到全部药物-靶点关系矩阵y
t,g
。
30.设药物和靶点之间的关系的概率分数可以用下式进行计算:
[0031][0032]
其中,u,v为药物-靶点之间的隐式关系矩阵。则上述公式最终可以通过贝叶斯公式转换为求解下式
[0033][0034]
最终通过求解(6)得到药物-靶点关系矩阵y
t,g
=uv
t
。
[0035]
步骤s4:对步骤s3中的逻辑矩阵分解进行贝叶斯优化。贝叶斯优化通过基于目标函数的过去评估结果建立替代函数(概率模型),来找到最小化目标函数的值。贝叶斯方法与随机或网格搜索的不同之处在于,它在尝试下一组超参数时,会参考之前的评估结果,因此可以省去很多无用功,缩短靶点预测时间。
[0036]
步骤s5:对于药物和靶点的关系矩阵中的元素,首先寻找其所对应的药物分子与药物分子的相似性矩阵s
d
中最为相似的n个药物分子,然后在药物-靶点的关系矩阵y
t,g
中查询这n个的药物分子看是否有靶点与之相匹配,如果有,则设共有m个靶点与之匹配,按照药物-靶点关系强度从大到小地记录这些匹配的靶点和对应的药物-靶点关系值v
i
,计算所有v
i
的均值avg,则将此药物和靶点的关系值设置为原始关系值和avg的均值;若无靶点与之相匹配,则不改变药物和靶点的关系矩阵中此元素相应关系值,遍历完整个药物-靶点关系矩阵y
t,g
,最终得到改善以后的药物-靶点关系矩阵y
f1
。同理,可以利用靶点相似性矩阵s
t
进行药物-靶点关系矩阵改善,即,首先寻找其所对应的靶点与靶点相似性矩阵s
t
中最为相
似的n个药物分子,然后在药物-靶点的关系矩阵中查询这n个的靶点看是否有药物分子与之相匹配,如果有,则设共有m个药物分子与之匹配,按照药物-靶点关系强度从大到小地记录这些匹配的药物分子和对应的药物-靶点关系值v
i
,计算所有v
i
的均值avg,则将此药物和靶点的关系值设置为原始关系值和avg的均值;若无药物分子与之相匹配,则不改变药物和靶点的关系矩阵中此元素相应关系值,遍历完整个药物-靶点关系矩阵y
f1
,最终得到改善以后的药物-靶点关系矩阵y
f
。
[0037]
综上所述,本发明实施例所提供的靶点预测方法,利用四种相似性系数加权平均求闭包的策略计算药物和药物、靶点与靶点的相似性矩阵,然后利用高斯核对药物和药物、靶点与靶点的相似性矩阵进行改善,并使用基于贝叶斯优化的逻辑矩阵分解求得药物和靶点的关系,利用药物和靶点的相似矩阵的局部信息对药物-靶点关系矩阵进行改进,提升药物-靶点关系预测精度。
[0038]
利用日本京都大学的公开测试集,通过10折交叉验证方式进行模型训练和数据测试,测试数据中enzymes的auc=0.95,ic的auc=0.96,gpcr的auc=0.94,nr的auc=0.90。而测试时间使用贝叶斯优化比不使用快了8倍。这充分的说明了算法的可行性和有效性。
[0039]
以上仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1