一种核-壳结构树状大分子CT/MR成像造影剂的制备方法与流程
本发明属于分子影像学领域,特别涉及一种核-壳结构树状大分子ct/mr成像造影剂的制备方法。
背景技术:
近年来,随着纳米技术与分子影像学的快速发展,出现了多种应用于肿瘤诊断领域的纳米造影剂,尤其是纳米载体的应用使构建多功能多模态纳米探针(同一纳米平台整合多种造影剂)用于肿瘤精准诊断成为现实。ct成像技术具有较高的空间分辨率,较短的图像采集时间,但是其存在对软组织分辨率差、较高的放射性对人体存有危害的缺点。而磁共振成像(mr)对软组织有极好的分辨率且对人体没有辐射损伤,但是其费用昂贵,图像采集时间长。鉴于每种成像技术都有其自身的不足之处,单一的成像技术已经不能满足对疾病的精准诊断的需要。因此,结合两种或两种以上的成像技术可以克服单一成像的弊端,增强诊断信息的准确性和可靠性。
发展一种多功能的ct/mr成像造影剂,结合ct和mr两种成像技术各自的优点从而大大提高诊断的灵敏度和准确度,同时可以克服传统医用成像造影剂的缺陷,在临床应用中将具有极大的价值,它一方面可以减少造影剂对患者的毒副作用,另一方面可以提供更加全面的诊断信息。
为了在同一平台同时整合两种造影剂并赋予其多种功能,通常需要载体。在众多载体材料中,聚酰胺-胺树状大分子(pamam)因其表面有大量可以被修饰的官能团,内部有较大的疏水空腔,被广泛用作载体来负载造影剂或药物用于肿瘤的诊断和治疗。相比于低代树状大分子,高代树状大分子具有更好的三维尺寸,更稳定的分子结构。但由于现阶段合成方法的限制,纯化方法的复杂,高代的树状大分子的合成面临巨大的挑战。因此,有人提出以较低代的树状大分子作为反应的单体,然后将这些单体通过一定的方法连接起来,以达到快速合成高代树状大分子的目的。j.li等人以表面氨基的高代树状大分子为核,表面羧基的较低代的树状大分子为壳,在1-(3-二甲氨基丙基)-3-乙基碳二亚胺(edc)的催化下,高代树状大分子表面的氨基与低代树状大分子表面羧基之间发生键和,合成出了核-壳结构树状大分子(core-shelltectodendrimers)(j.li,d.r.swanson,etal.langmuir,15(21)(1999)7347-7350)。chen等人通过环糊精与金刚烷的超分子主客体自组装作用构建了一种表面氨基的核-壳结构树状大分子(fengchen,etal.j.mater.chem.b.,2017,5,8459)。这种核-壳结构的树状大分子的表面有更多的氨基可供功能化修饰,内部有更多的疏水性空腔可供负载目标分子,因此十分适合用于多功能化纳米造影剂的载体材料。
然而,面对人体复杂的免疫系统,载体材料还需要具备阻抗蛋白质非特异性吸附,从而逃离网状内皮系统(res)的识别,延长体内血液循环时间,增大纳米材料到达病灶的几率(fangc,bhattarain,sunc,etal.small,2010,5(14):1637-1641)。近期研究两性离子修饰的树状大分子可以有效延长其在体内的血液循环时间(liujy,xiongzj,etal.acsappl.mater.interfaces2019,11,1521-215221)。这种新的表面修饰使得材料具有极低的非特异性蛋白吸附,为材料在体内的应用拓宽了道路。
检索国内外有关ct/mr双模态造影剂方面的文献和专利结果表明:目前尚未发现在核-壳结构树状大分子表面修饰靶向分子rgd和两性离子1,3-ps、螯合钆离子,内部包裹金纳米颗粒的核-壳结构树状大分子的制备及其在ct/mr成像方面的报道。
技术实现要素:
本发明所要解决的技术问题是提供一种核-壳结构树状大分子ct/mr成像造影剂的制备方法,制备的造影剂具备良好的分散性和生物相容性以及抵抗非特异性蛋白吸附功能和良好的靶向ct/mr成像效果。
本发明提供了一种核-壳结构树状大分子ct/mr成像造影剂的制备方法,包括:
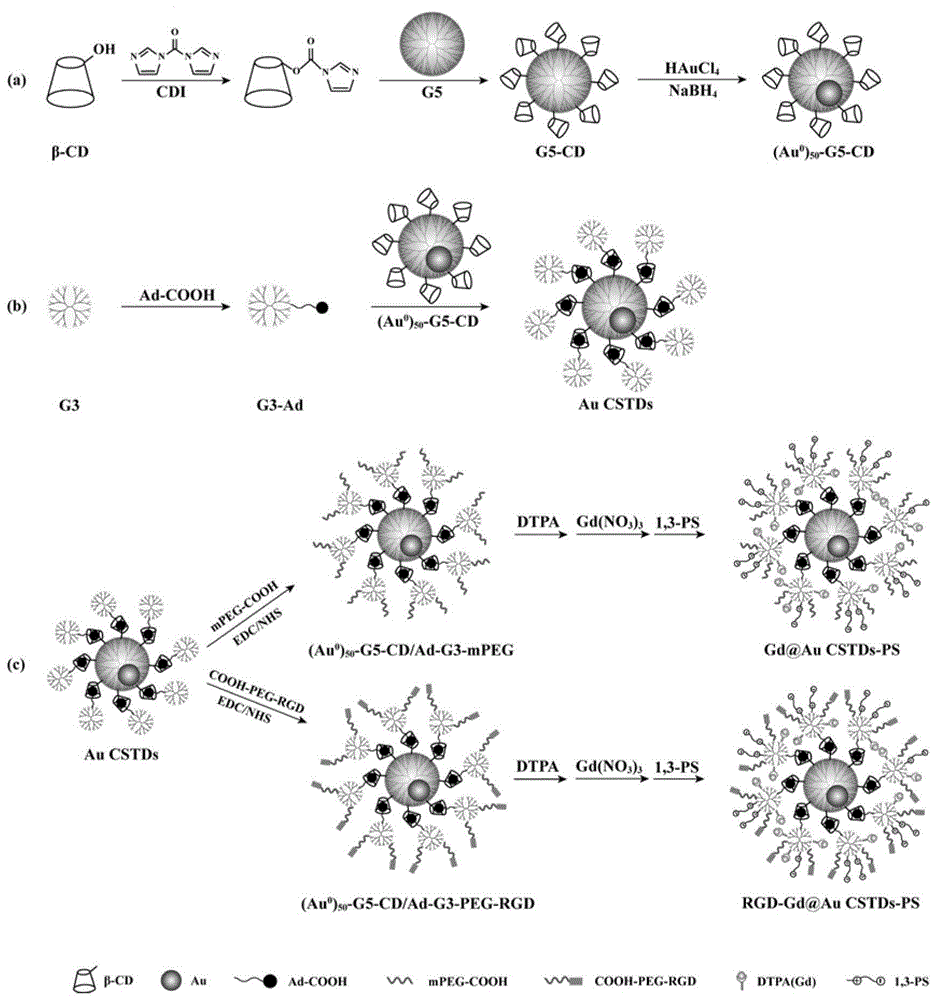
(1)将β-环糊精β-cd和n,n’-羰基二咪唑cdi以及第五代聚酰胺-胺树状大分子g5分别溶解在二甲基亚砜dmso溶液中,将β-cd和cdi溶液混合搅拌反应6-8h,得到反应液;再将反应液滴加到g5溶液中,室温搅拌反应70-80h,透析,冷冻干燥,得到g5-cd;在g5-cd的水溶液中逐滴加入氯金酸水溶液,搅拌反应15-30min,然后加入预冷的硼氢化钠水溶液,冰浴搅拌反应3-4h,透析,冷冻干燥,得到主体分子(au0)50-g5-cd;
(2)将1-金刚烷乙酸ad-cooh和edc以及nhs分别溶解在dmso溶液中,将edc加入到ad-cooh溶液中室温搅拌反应30-40min,再加入nhs室温搅拌反应3-4h,得到活化的ad-cooh溶液;然后逐滴滴加到溶有第三代聚酰胺-胺树状大分子g3的dmso溶液中,室温搅拌反应3-4天,透析,冷冻干燥,得到客体分子g3-ad;
(3)将主体分子(au0)50-g5-cd和客体分子g3-ad分别溶解在超纯水中,混合室温搅拌反应20-30h,通过超分子主客体作用得到核-壳结构树状大分子(au0)50-g5-cd/ad-g3溶液,透析,冷冻干燥,得到(au0)50-g5-cd/ad-g3,记为aucstds;
(4)将cooh-peg-rgd溶于dmso中,加入edc和nhs活化,得到活化的cooh-peg-rgd溶液;然后逐滴滴加到aucstds的dmso溶液中,于室温条件下搅拌反应3-4天;最后透析,冷冻干燥,得到(au0)50-g5-cd/ad-g3-peg-rgd;
(5)将(au0)50-g5-cd/ad-g3-peg-rgd和螯合剂二乙烯五乙酸盐dtpa分别溶解在超纯水中,混合室温搅拌反应24-30h,透析,冷冻干燥,得到(au0)50-g5-cd/ad-g3-peg-rgd-dtpa;
(6)将(au0)50-g5-cd/ad-g3-peg-rgd-dtpa和硝酸钆分别溶解在超纯水中,混合室温搅拌反应24-30h,然后滴加1,3-ps溶液搅拌反应24-30h,透析,冷冻干燥,得到(au0)50-g5-cd/ad-g3-peg-rgd-dtpa(gd)-ps,记为rgd-gd@aucstds-ps,即核-壳结构树状大分子ct/mr成像造影剂。
所述步骤(1)中的cdi与β-cd的摩尔比为10-15:1;β-cd与g5的摩尔比为18-20:1。
所述步骤(1)中的g5-cd和氯金酸的摩尔比为1:50-52;硼氢化钠和氯金酸的摩尔比为3-4:1。
所述步骤(2)中的edc、nhs和ad-cooh的摩尔比为10-15:10-15:1;g3和ad-cooh的摩尔比为1:1.3-1.5。
所述步骤(3)中的(au0)50-g5-cd和g3-ad的摩尔比为1:10。
所述步骤(4)中的cooh-peg-rgd和aucstds的摩尔比为10-12:1;edc、nhs和cooh-peg-rgd的摩尔比为10-15:10-15:1。
所述cooh-peg-rgd的制备过程如下:
将一端为羧基另一端为马来酰亚胺基团的聚乙二醇分子mal-peg-cooh和多肽rgd分别溶于dmso中,混合均匀后,室温搅拌反应24-30h,透析,冷冻干燥后得到多肽rgd修饰的聚乙二醇分子cooh-peg-rgd;其中,mal-peg-cooh和rgd的摩尔比为1:1.2-1.5。
所述步骤(5)中的(au0)50-g5-cd/ad-g3-peg-rgd和dtpa的摩尔比为1:28-30。
所述步骤(6)中的(au0)50-g5-cd/ad-g3-peg-rgd-dtpa、硝酸钆和1,3-ps的摩尔比为1:28-30:90-100。
本发明分别合成表面修饰β-环糊精、内部包裹金纳米颗粒的第五代树状大分子g5和表面修饰金刚烷第三代树状大分子g3,并利用主客体识别作用构建核-壳结构的树状大分子纳米平台,然后在该核-壳结构树状大分子表面修饰靶向分子rgd和两性离子1,3-ps、表面螯合钆离子,得到具有特异性靶向功能和抵抗非特异性蛋白吸附功能的ct/mr造影剂。
本发明通过核磁共振(1hnmr)对修饰在树状大分子上的β-cd(主体分子)和ad(客体分子)的数量进行了表征;二维核磁(2droesy)对通过主客体相互作用构建的核-壳结构树状大分子进行表征;透射电子显微镜(tem)、原子力显微镜(afm)对主客体单元以及构建的核-壳结构树状大分子的表面形貌和尺寸大小进行了表征;使用紫外可见吸收光谱(uv-vis)、电感耦合等离子体原子发射光谱法(icp-oes)、zeta电势、水合粒径测试和透射电镜测试等方法表征材料的物理化学性质;然后通过cck8法来评价该造影剂的细胞毒性,体外蛋白吸附试验评价抗非特异性蛋白吸附性能;最后利用体内外ct/mr成像来表征制备的具有双模态造影功能的核-壳结构树状大分子对肿瘤细胞和肿瘤组织的诊断效果。
有益效果
(1)本发明合成工艺简单,反应条件温和可控,易于操作,成本较低,所用均为环境友好材料,制备的ct/mr造影剂具有良好的生物相容性;
(2)本发明制备的ct/mr造影剂通过超分子主客体的方法以g5为核g3为壳,表面修饰靶向分子和两性离子,其具有良好的稳定性,尺寸较一般的纳米颗粒大,可以有效的在肿瘤部位富集,使其具有良好的ct/mr成像效果;
(3)本发明制备的核-壳结构树状大分子ct/mr造影剂在分子影像诊断领域有着潜在的应用。
附图说明
图1为本发明工艺流程示意图;
图2为本发明制备的g5-cd(a)、g3-ad(b)的1hnmr图谱;
图3为本发明制备的aucstds的2dnoesy图谱;
图4为本发明制备的cooh-peg-rgd(a)、(au0)50-g5-cd/ad-g3-peg-rgd(b)和(au0)50-g5-cd/ad-g3-peg-rgd-dtpa(c)的1hnmr图谱;
图5为本发明制备的(au0)50-g5-cd/ad-g3-mpeg(a)和(au0)50-g5-cd/ad-g3-mpeg-dtpa(b)的1hnmr图谱;
图6为本发明制备的(au0)50-g5-cd、aucstds(a)、gd@aucstds-ps、rgd-gd@aucstds-ps(b)的uv-vis谱图;
图7为本发明制备的gd@aucstds-ps(a)和rgd-gd@aucstds-ps(b)的高分辨tem图和粒径分布直方图;
图8为本发明制备的g3-ad(a)、(au0)50-g5-cd(b)、aucstds(c)、gd@aucstds-ps(d)和rgd-gd@aucstds-ps(e)的原子力显微镜(afm)图;
图9为本发明制备的gd@aucstds-ps和rgd-gd@aucstds-ps尺寸稳定性图;
图10为bsa蛋白的uv-vis光谱图(a)和本发明制备的不同浓度的rgd-gd@aucstds和rgd-gd@aucstds-ps与bsa(1mg/ml)孵育2h后,离心去沉淀,测量其在278nm处的吸光值,吸收差值为离心前后紫外的吸收差值(b);
图11为本发明制备的gd@aucstds-ps和rgd-gd@aucstds-ps在钆浓度为0.08-0.64mm范围内的和mr成像图(a)和1/t1与gd浓度的线性关系图(b);
图12为本发明制备的gd@aucstds-ps和rgd-gd@aucstds-ps纳米颗粒在金浓度为0.005-0.08m范围内的ct成像图(a)和x射线衰减与金或碘的浓度线性关系图(b);
图13为本发明制备的gd@aucstds-ps和rgd-gd@aucstds-ps经cck8法测得4t1细胞在不同金浓度(0-200μm)下处理24小时后的细胞活力(a);4t1细胞经过gd@aucstds-ps和rgd-gd@aucstds-ps在不同金浓度(0-200μm)下处理4小时后的细胞吞噬材料的au含量(***,p<0.001)(b);
图14为尾静脉注射gd@aucstds-ps和rgd-gd@aucstds-ps(au:0.1m,150μl)后不同时间点小鼠肿瘤部位的mr成像图(a)、ct成像图(b)、mr值(c)和ct值(d);图15为尾静脉注射gd@aucstds-ps(a)和rgd-gd@aucstds-ps(b)在小鼠心、肝、脾、肺、肾以及肿瘤中的分布。
具体实施方式
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。此外应理解,在阅读了本发明讲授的内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
实施例1
称取16.36mgβ-cd和23.38mgcdi分别溶于5mldmso中,混合搅拌反应6-8h。称取g515mg溶于5mldmso中,然后将β-cd和cdi的混合液缓慢滴加到g5的dmso溶液中,室温下搅拌反应70-80h。待反应结束后,将反应液转移至截留分子量为10000da的透析袋中,在超纯水中透析3天。最后经冷冻干燥得到固体产物g5-cd,储存于-20℃。
称取15mgg5-cd溶解在15ml超纯水中,在冰浴条件下逐滴加入396μl氯金酸水溶液(30mg/ml),搅拌反应15-30min。称取5.45mg的硼氢化钠,将预冷的1ml超纯水倒入其中,充分溶解后,快速加入到上述混合溶液中,冰浴条件下搅拌反应3-4h。待反应结束后,将反应液转移至截留分子量为1000da的透析袋中,在超纯水中透析3天。最后经冷冻干燥得到固体产物(au0)50-g5-cd,储存于-20℃。
称取1-金刚烷乙酸(ad-cooh)2.04mg,溶于2mldmso。分别称取edc和nhs20.14mg、12.09mg,溶于3mldmso溶液。将edc加入到ad-cooh溶液中室温搅拌反应30-40min,再加入nhs溶液室温搅拌反应3-4h,得到活化的ad-cooh溶液。称取g350mg,溶于5mldmso中。然后将edc/nhs活化的ada-cooh溶液逐滴加入到g3溶液中,磁力搅拌下反应3天。待反应结束后,使用截留分子量为1000da的透析袋在超纯水中透析,最后经冷冻干燥得到固体产物g3-ad,储存于-20℃。
按照(au0)50-g5-cd和g3-ad的摩尔比为1:10,称取干燥的g3-ad25.4mg,(au0)50-g5-cd15.23mg,分别在5ml超纯水中溶解,然后将两者混合,磁力搅拌下室温反应20-30h,待反应结束后,使用截留分子量为10000da的透析袋在超纯水中透析,最后经冷冻干燥得到固体产物aucstds,储存于-20℃。
称取13.34mgmal-peg-cooh和4.61mgrgd分别溶解于3mldmso中,完全溶解后,于室温下混合搅拌反应24-30h;待反应结束后,用截留分子量为1000da的透析袋透析3天,最后经冷冻干燥得到固体产物cooh-peg-rgd,储存于-20℃。
称取12mgcooh-peg-rgd、12.79mgedc和7.68mgnhs分别溶解于3mldmso中,在匀速搅拌的情况下,先加入edc活化30-40min,再加入nhs活化3-4h,得到活化的cooh-peg-rgd溶液;称取20mg(au0)50-g5-cd/ad-g3溶解在5mldmso中,然后将活化的cooh-peg-rgd溶液缓慢加入到其中,于室温条件下搅拌反应3-4天。待反应结束后,用截留分子量为3000da的透析袋透析3天,最后经冷冻干燥得到固体产物(au0)50-g5-cd/ad-g3-peg-rgd,储存于-20℃。
称取22mg(au0)50-g5-cd/ad-g3-peg-rgd和7.11mgdtpa分别溶解在4ml超纯水中,混合均匀后,室温搅拌反应24-30h。待反应结束后,用截留分子量为1000da的透析袋透析3天,最后经冷冻干燥得到固体产物(au0)50-g5-cd/ad-g3-peg-rgd-dtpa,储存于-20℃。
称取30mg(au0)50-g5-cd/ad-g3-peg-rgd-dtpa和5.36mg硝酸钆分别溶解在4ml超纯水中,在室温条件下搅拌反应24-30h,然后滴加450μl1,3-ps溶液(13.7mg/ml)搅拌反应24-30h。待反应结束后,用截留分子量为1000da的透析袋透析3天,最后经冷冻干燥得到固体产物rgd-gd@aucstds-ps,储存于-20℃。
对比例1
称取10mgmpeg-cooh、9.59mgedc和5.75mgnhs分别溶解于3ml超纯水中,在匀速搅拌的情况下,先加入edc活化30-40min,再加入nhs活化3-4h,得到活化的mpeg-cooh溶液;称取30mg(au0)50-g5-cd/ad-g3溶解在5ml超纯水中,然后将活化的mpeg-cooh溶液缓慢加入到其中,于室温条件下搅拌反应3-4天。待反应结束后,用截留分子量为3000da的透析袋透析3天,最后经冷冻干燥得到固体产物(au0)50-g5-cd/ad-g3-mpeg,储存于-20℃。
称取25mg(au0)50-g5-cd/ad-g3-mpeg和8.25mgdtpa分别溶解在3ml超纯水中,混合均匀后,室温搅拌反应24-30h。待反应结束后,用截留分子量为1000da的透析袋透析3天,最后经冷冻干燥得到固体产物(au0)50-g5-cd/ad-g3-mpeg-dtpa,储存于-20℃。
称取30mg(au0)50-g5-cd/ad-g3-mpeg-dtpa和5.36mg硝酸钆分别溶解在4ml超纯水中,在室温条件下搅拌反应24-30h,然后滴加450μl1,3-ps溶液搅拌反应24-30h。待反应结束后,用截留分子量为1000da的透析袋透析3天,最后经冷冻干燥得到固体产物gd@aucstds-ps,储存于-20℃。
实施例2
分别称取实施例1中g5-cd、g3-ad、aucstds、cooh-peg-rgd、(au0)50-g5-cd/ad-g3-peg-rgd和(au0)50-g5-cd/ad-g3-peg-rgd-dtpa各5mg,分别溶于500μld2o中,进行氢谱分析。g5-cd的1hnmr表征结果如图2(a)所示,在化学位移2.2-3.4ppm处的质子峰代表g5的亚甲基质子峰,在化学位移3.5-4.1ppm以及5.0ppm处的质子峰代表β-cd分子结构中的质子峰,表明β-cd已成功修饰在g5表面。通过对g5和β-cd的质子峰区域进行积分,计算出每个g5表面修饰了10.9个β-cd分子。g3-ad的1hnmr表征结果如图2(b)所示,在化学位移1.6-1.9ppm处的质子峰代表金刚烷基团分子结构中的质子峰,化学位移2.3-3.2ppm处的质子峰代表g3中酰胺键上的质子峰,表明ad-cooh已成功修饰在g3表面。通过对g3和ad-cooh的质子峰区域进行积分,计算出每个g3表面修饰了1.2个ad-cooh分子。
aucstds的2dnoesy表征结果如图3所示,在化学位移1.6-1.9ppm处的金刚烷基团与化学位移3.5-4.1ppm处β-cd基团出现了明显的相关交叉信号,由此可以说明金刚烷基团与β-cd发生了相互作用,紧密结合。同时证明了主体分子(au0)50-g5-cd和客体分子g3-ad通过金刚烷与环糊精的主客体作用成功构建了核-壳结构树状大分子aucstds。cooh-peg-rgd的1hnmr表征结果如图4(a)所示,出现在3.4-3.6ppm和6.75-7.05ppm处的化学位移峰分别对应为聚乙二醇和rgd的特征峰。经积分可得每个聚乙二醇上修饰了0.76个rgd分子。(au0)50-g5-cd/ad-g3-peg-rgd的1hnmr表征结果如图4(b)所示,出现在3.4-3.6ppm和2.2-3.3ppm处的化学位移峰分别对应为聚乙二醇和树状大分子的特征峰,6.75-7.05ppm的化学位移峰对应为rgd的特征峰。经积分可得每个核-壳结构树状大分子上修饰了8.28个peg和6.3个rgd分子。(au0)50-g5-cd/ad-g3-peg-rgd-dtpa的1hnmr表征结果如图4(c)所示,出现在3.4-3.6ppm和2.2-3.3ppm以及6.75-7.05ppm处的化学位移峰分别对应为聚乙二醇、树状大分子和rgd的特征峰,3.78ppm处的化学位移峰对应为dtpa的特征峰。经积分可得每个核-壳结构树状大分子上修饰了26.7个dtpa分子。
实施例3
分别称取对比例1中(au0)50-g5-cd/ad-g3-mpeg和(au0)50-g5-cd/ad-g3-mpeg-dtpa各5mg,分别溶于500μld2o中,进行氢谱分析。(au0)50-g5-cd/ad-g3-mpeg的1hnmr表征结果如图5(a)所示,出现在3.5-3.65ppm和2.2-3.3ppm处的化学位移峰分别对应为聚乙二醇和树状大分子的特征峰,经积分可得每个核-壳结构树状大分子上修饰了10.4个mpeg。(au0)50-g5-cd/ad-g3-mpeg-dtpa的1hnmr表征结果如图5(b)所示,出现在3.5-3.65ppm和2.2-3.3ppm处的化学位移峰分别对应为聚乙二醇和树状大分子的特征峰,3.78ppm处的化学位移峰对应为dtpa的特征峰。经积分可得每个核-壳结构树状大分子上修饰了21.7个dtpa分子。
实施例4
分别称取实施例1中g5-cd、g3-ad、(au0)50-g5-cd、aucstds、rgd-gd@aucstds-ps和对比例1中的gd@aucstds-ps各0.5mg,分别溶于的1ml超纯水中进行水动力学尺寸及表面zeta电势测试(如表1所示)。其水合粒径分别为119.3nm、110.2nm、94.0nm、105.7nm、135.3nm和115.8nm。电势分别为29.8mv、23.4mv、26.5mv、37.7mv、8.3mv和5.2mv。根据数据可以看出,材料的表面电势从37.7mv降低到8.3mv,这为材料的低细胞毒性提供了保障。从纳米颗粒的水动力学粒径可以看出,树状大分子组装平台尺寸较小并且具有良好的分散性,并且表面修饰没有引起材料水合粒径的显著增大。
表1样品的表面电势和水合粒径
实施例5
分别称取实施例1中(au0)50-g5-cd、aucstds、rgd-gd@aucstds-ps和对比例1中的gd@aucstds-ps各0.1mg,分别溶于的1ml超纯水中进行紫外可见吸收光谱测试。如图6所示,(au0)50-g5-cd、aucstds(a)和gd@aucstds-ps、rgd-gd@aucstds-ps(b)四种纳米颗粒的水溶液在500-550nm波长范围内均显示出明显的特征吸收峰,特别是在520nm左右处显示了金纳米颗粒的表面等离子体共振吸收峰,表明所合成的两种载体材料均成功包裹了金纳米颗粒。
实施例6
分别称取实施例1中rgd-gd@aucstds-ps和对比例1中的gd@aucstds-ps各0.5mg,分别溶于的1ml超纯水中进行tem测试。如图7所示,gd@aucstds-ps(图7a)和rgd-gd@aucstds-ps(图7b)的金核粒径分别为2.2nm、2.4nm。分析结果可知,两种材料金核尺寸并无较大差异,且分布较窄,具有良好的单分散性。
实施例7
分别称取实施例1中g3-ad、(au0)50-g5-cd、aucstds、rgd-gd@aucstds-ps和对比例1中的gd@aucstds-ps各0.5mg,分别溶于的1ml超纯水中,滴加在硅片上进行afm测试。如图8所示,制备的g3-ad(a)、(au0)50-g5-cd(b)、aucstds(c)、gd@aucstds-ps(d)、rgd-gd@aucstds-ps(e)形状接近球形,粒径分别为4.20nm、5.78nm、10.75nm、11.13nm和11.61nm,由此可以进一步证明g3-ad和(au0)50-g5-cd通过主客体的自组装作用形成了粒径更大的核-壳结构树状大分子aucstds。
实施例8
分别称取实施例1中rgd-gd@aucstds-ps和对比例1中的gd@aucstds-ps各0.5mg,分别溶于的1ml超纯水中进行水动力学尺寸测试。如图9所示,两种材料在水溶液中放置10天内的水合粒径没有发生明显的变化,说明材料具有良好的稳定性能。
实施例9
通过将实施例1中制备的rgd-gd@aucstds和rgd-gd@aucstds-ps进行蛋白吸附实验测试,能够比较两种材料抗非特异性蛋白吸附的效果。称取牛血清白蛋白(bsa)1mg溶解于pbs中,然后用紫外吸收光谱仪对其进行扫描,得到的结果如图10(a)所示,bsa在278nm处有吸收峰。然后称取实施1中制备的rgd-gd@aucstds和rgd-gd@aucstds-ps各2mg,配置成浓度为2mg/ml的pbs溶液,然后分别稀释成1mg/ml、0.5mg/ml和0.25mg/ml。称取bsa2mg,配置成1mg/ml的pbs溶液。吸取0.5ml的bsa溶液分别加入刚刚配置好的rgd-gd@aucstds和rgd-gd@aucstds-ps的溶液中,充分混匀后,分别测试其在278nm处的紫外吸收值。将混合溶液放置于37℃培养箱中2h,然后8000rpm离心3min,然后去除沉淀,测量上清液在278nm处的紫外吸收值。将rgd-gd@aucstds和rgd-gd@aucstds-ps所对应的孵育离心前后紫外吸收值相减即得紫外吸收差值。从图10(b)可以看出,随着材料浓度的增加,材料的紫外吸收差值也在增加,在达到最高材料浓度时更明显,从而可以说明1,3-ps的修饰能够很好地降低材料的非特异性蛋白吸附。
实施例10
取实施例1中制得的靶向rgd-gd@aucstds-ps和对比例1中制得的非靶向gd@aucstds-ps材料,以超纯水为溶剂,配制gd浓度为0.64mm、0.32mm、0.16mm、0.08mm和0.04mm的纳米颗粒水溶液,用t1磁共振成像仪测定两种材料的t1弛豫时间并且扫描mr成像图片。测试结果如图11所示,制备的rgd-gd@aucstds-ps颗粒的弛豫时间倒数随gd浓度的增加而增加,呈现良好的线性关系,通过计算知rgd-gd@aucstds-ps颗粒的弛豫率为9.414mm-1s-1,对比例1中所得的gd@aucstds-ps颗粒的弛豫率为8.795mm-1s-1。实验结果表明,制备的两种纳米颗粒均具备良好的mr成像造影功能。
实施例11
用医用ct扫描仪对实施例1制备的材料rgd-gd@aucstds-ps和对比例1中制得的gd@aucstds-ps以及医用造影剂omnipaque的体外ct成像效果进行观察和比较。称取实施例1样品rgd-gd@aucstds-ps和对比例1中制得的gd@aucstds-ps在超纯水中配制au浓度为0.08m的溶液,再分别稀释至为0.04m、0.02m、0.01m和0.005m溶液各100μl。以同样的方法制备相同碘浓度梯度的omnipaque。用ct测试仪测试各个样品的ct值并得出x-射线衰减与金浓度的线性关系。如图12所示,相比于omnipaque拟合曲线的斜率,rgd-gd@aucstds-ps和gd@aucstds-ps具有更大的斜率,进一步说明材料具有比临床所用的omnipaque更强的ct造影效果。
实施例12
通过cck8比色法,以4t1细胞为模型细胞评价实施例1制备的材料rgd-gd@aucstds-ps和对比例1中gd@aucstds-ps对细胞存活的影响。取rgd-gd@aucstds-ps分散在无菌pbs缓冲液中配制成金浓度为2000μm的母液,并用紫外照射过夜杀菌。另以同样的方法准备对比例1中制得的非靶向gd@aucstds-ps的样品溶液。取培养好的4t1细胞种于96孔板中,按照1万细胞/孔的密度接种,每孔体积100μl。培养过夜后,加入各稀释梯度的样品,每孔au浓度最终分别为10、25、50、100、200μm,与细胞共培养24h。每个梯度做5个平行孔,以pbs缓冲液作为空白对照。随后用cck-8法检测细胞活力,每孔加cck-8溶液,在37℃下培养4小时。之后用酶标仪检测450nm处吸光度。cck-8测试结果如图13(a)所示,gd@aucstds-ps和rgd-gd@aucstds-ps在浓度0到200μm范围内,细胞存活率都在85%以上,在达到最高浓度200μm时,4t1细胞对应两种材料的存活率分别为85.6%和97.5%,说明所制备的材料具有良好的细胞相容性,可以安全应用于生物体ct/mr成像。
实施例13
取实施例1制得的靶向rgd-gd@aucstds-ps用无菌pbs缓冲液配制成au浓度为2000μm的母液,并用紫外照射过夜杀菌。之后吸取培养好的4t1细胞种于12孔板中,按照20万细胞/孔的密度接种,每孔体积为1ml。用1640培养基分别将母液稀释为au浓度10、25、50、100、200μm的溶液。加入上述稀释后的样品与4t1细胞在37℃下共培养4h。共培养后细胞用pbs洗三次,再用胰酶消化并离心,弃上清液,得到的沉淀用1ml王水消化24h,消化完全后加入1ml超纯水,用icp-oes检测样品中的au浓度。以同样的方法测定对比例1中非靶向gd@aucstds-ps与4t1细胞共培养4h后的吞噬情况。还需要做靶向阻断实验,称取rgd-gd@aucstds-ps分散在无菌pbs缓冲液中配制成材料浓度为2000μm的pbs缓冲液,并用紫外照射过夜杀菌。然后在超净工作台中用无菌pbs缓冲配制浓度为1000、500、250、100μm的rgd-gd@aucstds-ps溶液。4t1细胞与rgd溶液(5μm)共同种植于12孔板过夜,弃去培养基,每孔加入900μl新鲜培养基与100μl的不同浓度的rgd-gd@aucstds-ps溶液,使得材料浓度最终为10、25、50、100、200μm,在37℃下共培养4小时。共培养后细胞用pbs洗三次,再用胰酶消化并离心,弃上清液,得到的沉淀用1ml王水消化24h,消化完全后加入1ml超纯水,用icp-oes检测样品中的au浓度。细胞靶向性测试结果如图13(b)所示,在不同浓度下,4t1细胞对材料rgd-gd@aucstds-ps的吞噬量总是多于对gd@aucstds-ps和经rgd阻断之后的rgd-gd@aucstds-ps的吞噬量,在最高材料浓度时,gd@aucstds-ps、经rgd阻断之后的rgd-gd@aucstds-ps和rgd-gd@aucstds-ps的吞噬量分别为:15.8pg/细胞、21.1pg/细胞和47.9pg/细胞。本实验充分证明了rgd的靶向性能。
实施例14
将实施例1制备的材料rgd-gd@aucstds-ps和对比例1中gd@aucstds-ps配置成钆浓度为0.01m的0.1ml生理盐水溶液。在对小鼠腹腔注射麻药麻醉后,分别通过尾静脉注射每只小鼠100μl的两种不同的生理盐水溶液,并在注射前后不同时间进行mr扫描,用以来评价材料的mr成像效果。如图14(a)和(c)所示,在注射30min后,小鼠肿瘤部位信号增强,在1h达到峰值,gd@aucstds-ps和rgd-gd@aucstds-ps对应的肿瘤部位的信噪比分别为:65和52。在同一时间点,rgd-gd@aucstds-ps的信号增强效果显著高于gd@aucstds-ps,充分证明了经rgd修饰之后,材料可以特异性靶向到肿瘤组织,增强成像效果。
实施例15
将实施例1制备的材料rgd-gd@aucstds-ps和对比例1中gd@aucstds-ps配置成金浓度为0.1m的0.1ml生理盐水溶液。在对小鼠腹腔注射麻药麻醉后,分别通过尾静脉注射每只小鼠100μl的两种不同的生理盐水溶液,并在注射前后不同时间进行ct扫描,用以来评价材料的ct成像效果。如图14(b)和(d)所示,在注射30min后,小鼠肿瘤部位信号增强,在1h达到峰值,gd@aucstds-ps和rgd-gd@aucstds-ps对应的肿瘤部位的ct值分别为:60.8hu和51.1hu。在同一时间点,rgd-gd@aucstds-ps的信号增强效果要高于gd@aucstds-ps,说明材料具有增强成像效果。
实施例16
将实施例1制备的材料rgd-gd@aucstds-ps和对比例1中gd@aucstds-ps配置成金浓度为0.1m的0.1ml生理盐水溶液。在对小鼠腹腔注射麻药麻醉后,分别通过尾静脉注射每只小鼠100μl金浓度为0.1m的两种不同的生理盐水溶液,分别在注射后不同的时间点(1、6、12、24h)将小鼠处死,取出主要器官(心、肝、脾、肺、肾和肿瘤)称重,并经过王水消化一周,然后经icp-oes测定各个器官中au的含量。取未注射的小鼠作为对照组,作为0h,每个时间点平行做三只老鼠。结果如图15(a)和(b)所示,在尾静脉注射完各组材料24小时之后,各器官(心、肝、脾、肺、肾)的金元素含量先增加后降低,这些结果证明了制备的两组材料(gd@aucstds-ps和rgd-gd@aucstds-ps)能够在小鼠体内正常代谢清除。从图中可以看出,随着时间的延长,肿瘤部位材料的浓度在缓慢的下降,并且在同一时间点靶向材料rgd-gd@aucstds-ps比非靶向材料gd@aucstds-ps更多的在肿瘤部位富集,这也在动物水平上证实了材料的靶向性能。
- 还没有人留言评论。精彩留言会获得点赞!