兰索拉唑肠溶微丸及其制备方法与流程
本发明涉及一种兰索拉唑肠溶微丸及其制备方法。
背景技术:
兰索拉唑是第二代质子泵抑制类抗溃疡药,于1994年由日本武田制药公司开发,1998年开始应用于我国临床治疗,主要用于治疗胃溃疡、十二指肠溃疡、反流性食管炎、佐-艾综合征(胃泌素瘤)等,可广泛用于治疗与胃酸分泌有关的各种消化功能紊乱性疾病。本品口服后选择性进入胃壁细胞,转化为次磺酰胺活性代谢产物,其活性代谢产物与h+/k+-atp酶上的巯基相互作用,形成共价结合的二硫键,对h+/k+-atp酶产生不可逆的抑制,进而达到抑制胃酸分泌的作用。兰索拉唑属于生物药剂学分类系统中的第二类药物,即低水溶性、高膜渗透性,并且兰索拉唑对酸、光、湿和热不稳定,极易降解,进一步影响了口服药物疗效的发挥,故现有的上市制剂为肠溶制剂。
现有的公开号为cn104188935a的中国发明专利申请中公开了一种兰索拉唑肠溶片及其制备方法,该方法采用将原辅料混合后湿法制粒、压片,再以肠溶树脂包衣制成,但缺点在于兰索拉唑肠溶片属于单单元制剂,存在下述局限性:①药物的作用和吸收容易受到消化物迁移的影响。②肠溶片剂会因为肠溶衣层的局部破损而造成药物整体失效,严重影响药效的发挥,这种行为极大地限制了兰索拉唑肠溶片的稳定性。
膜控微丸属于多单元缓控释制剂,与单单元剂型片剂相比,多单元膜控微丸更具临床应用优势:①因微丸“剂量分散”的制剂特点,每一个微丸都可以形成一个完整的释放单元,避免衣膜局部破损造成整体药物失效,因此释药规律具有重现性;②制成微丸可改变药物的某些性质,如成丸后流动性好、大小均匀,不易碎等,易于处理(如包衣、分剂量),并可作为制备片剂、胶囊剂等的基础。公开号为cn104013580a的中国发明专利申请中公开了一种兰索拉唑微丸、胶囊及其制备方法,该方法采用流化床包衣技术制备含药丸芯、隔离衣层及肠溶衣层,其缺点在于制备微丸时,需将兰索拉唑及辅料制成混悬乙醇水溶液,该溶液因辅料溶解度不同,极易造成流化床包衣机喷枪的堵塞,且需检测溶剂残留。公开号为cn104352494a的中国发明专利申请中公开了一种多潘立酮兰索拉唑微丸及其制备方法,该方法采用离心造粒技术制备含药丸芯、隔离衣层及肠溶衣层,其缺点在于制备肠溶衣层时,因离心造粒机的平板高速旋转易对微丸造成磨损。
技术实现要素:
本发明的目的之一是提供一种兰索拉唑肠溶微丸,用其制备的兰索拉唑肠溶微丸胶囊在释放度、耐酸力及稳定性方面明显提高;
本发明的目的之二是以提供一种上述兰索拉唑肠溶微丸的制备方法,该制备方法工艺简单,重现性好。
一种兰索拉唑肠溶微丸,其特别之处在于:所述微丸由内至外依次为空白丸芯、含药丸芯、隔离衣层和肠溶衣层,其中空白丸芯占微丸总重的15~30%,隔离衣层占微丸总重的5~20%,肠溶衣层占微丸总重的20~40%,余量为含药丸芯。
其中空白丸芯粒径为0.5~1mm。
其中按照重量份计:空白丸芯组成为蔗糖10-25份、淀粉2-8份、崩解剂0.5-3份;含药丸芯组成为兰索拉唑5-10份、稀释剂10-25份、稳定剂5-15份、崩解剂2-7份、增溶剂1-5份、粘合剂1-5份;隔离衣层组成为稀释剂3-10份、稳定剂1-5份、崩解剂0.2-2份、增溶剂0.1-0.5份、粘合剂0.1-0.5份、增塑剂0.1-0.5份、润滑剂0.1-0.5份;肠溶衣层组成为水溶性肠溶包衣材料10-30份、润滑剂2-6份、增塑剂2-10份。
其中空白丸芯中蔗糖与淀粉的重量比为4:1~6:1。
其中含药丸芯中所述粘合剂选用羟丙基纤维素、羟丙甲基纤维素、聚乙烯吡咯烷酮k30和聚乙烯吡咯烷酮k90中的至少一种;所述稀释剂选用蔗糖、玉米淀粉和微晶纤维素中的至少一种;所述崩解剂选用低取代羟丙纤维素、羧甲基淀粉钠、交联羧甲基纤维素钠和交联聚维酮中的至少一种;所述稳定剂选用碳酸氢钠、重质碳酸镁、碳酸钙和氧化镁中的至少一种;所述增溶剂选用十二烷基硫酸钠、葡甲胺和泊洛沙姆中的至少一种。
其中隔离衣层中所述粘合剂选用羟丙基纤维素、羟丙甲基纤维素、聚乙烯吡咯烷酮k30和聚乙烯吡咯烷酮k90中的至少一种;所述稀释剂选用蔗糖、玉米淀粉和微晶纤维素中的至少一种;所述崩解剂选用低取代羟丙纤维素、羧甲基淀粉钠、交联羧甲基纤维素钠和交联聚维酮中的至少一种;所述稳定剂选用碳酸氢钠、重质碳酸镁、碳酸钙和氧化镁中的至少一种;所述增溶剂选用十二烷基硫酸钠、葡甲胺和泊洛沙姆中的至少一种;所述增塑剂选用聚乙二醇、柠檬酸三乙酯和邻苯二甲酸二乙酯中的至少一种;所述润滑剂选用滑石粉、硬脂酸镁、聚乙二醇和单硬脂酸甘油酯中的至少一种。
其中肠溶衣层中所述水溶性肠溶包衣材料采用尤特奇l30-d55、尤特奇s100和尤特奇l100中的至少一种;所述增塑剂采用聚乙二醇、柠檬酸三乙酯和邻苯二甲酸二乙酯中的至少一种;所述润滑剂采用滑石粉、硬脂酸镁、二氧化钛和单硬脂酸甘油酯中的至少一种。
一种兰索拉唑肠溶微丸的制备方法,其特征在于,包括如下步骤:
(1)空白丸芯制备:
按照权利要求3中记载的配比,将蔗糖、淀粉及崩解剂混合均匀,放入离心式制丸机,以3~8%g/ml的粘合剂制备空白丸芯,工艺参数控制为0.1~0.3mpa的喷气压力、10~20r/min的挤出速度及15~20r/min的滚圆速度,制备完成后,将得到的空白丸芯于30~40℃烘箱干燥1~2h;
(2)含药丸芯制备:
将微粉化至300~500目的兰索拉唑与辅料,于混合机中混合5~20min后放入离心造粒机,以3~8%g/ml的粘合剂于步骤(1)得到的空白丸芯上层积兰索拉唑含药层,制备兰索拉唑含药丸芯,工艺参数控制为0.02~0.1mpa的喷气压力、30~40%的鼓风流量、3~8ml/min的喷浆速度及6~25r/min的喷粉速度,供粉完毕,将得到的微丸于30~40℃烘箱干燥2~3h,得到含药丸芯;
(3)隔离衣层制备:
将混合均匀的辅料及步骤(2)得到的含药丸芯放入离心造粒机,以3~8%g/ml的粘合剂制备微丸隔离衣层,工艺参数控制为0.02~0.1mpa的喷气压力、30~40%的鼓风流量、3~8ml/min的喷浆速度及6~25r/min的喷粉速度;
(4)肠溶衣层制备:
采用流化床包衣机,工艺参数控制为0.2~0.4mpa的喷气压力、13~40hz的鼓风频率、1~5ml/min的喷液速度、30~60℃的进风温度,30~60℃的进样器温度、30~60℃的物料温度,制备兰索拉唑微丸的肠溶衣层,得到兰索拉唑肠溶微丸。
步骤(4)之后将兰索拉唑肠溶微丸装入胶囊中制得胶囊剂,每一粒兰索拉唑肠溶微丸胶囊填充量为330~450mg。
本发明制备的兰索拉唑肠溶微丸制剂与目前现有的兰索拉唑肠溶片剂及肠溶微丸制剂相比,具有如下优点:1.与兰索拉唑肠溶片剂相比,每一个微丸都形成一个完整的释放单元,避免肠溶片剂衣膜局部破损造成整体药物失效,其释药规律具有重现性。2.与目前所用的流化床包衣技术相比,本方法采用离心造粒机,通过水溶性粘合剂将混合均匀的兰索拉唑及辅料喷到空白丸芯上形成含药微丸及隔离衣层,极大改善了流化床制粒时混悬物料对于喷枪的堵塞问题;采用流化床包衣机制备微丸肠溶衣层,解决了离心造粒机制备肠溶衣层时肠溶包衣液喷射不均匀、平板高速旋转对微丸的磨损及干燥时间长的问题。本发明制备的兰索拉唑肠溶微丸胶囊在释放度、耐酸力及稳定性方面明显提高,并且该制备工艺简单,重现性好。
附图说明
图1为兰索拉唑肠溶微丸结构示意图;
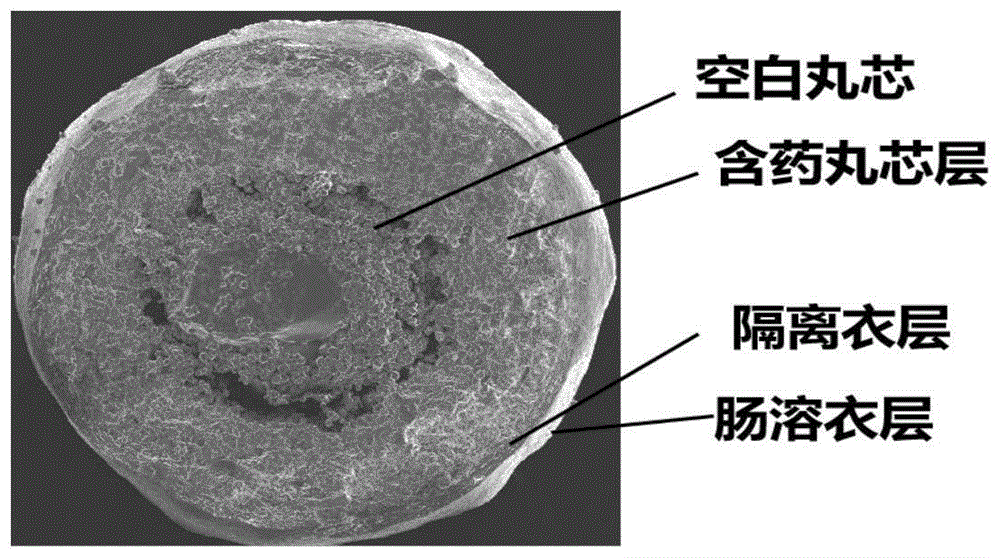
图2兰索拉唑肠溶微丸的表观及内部结构;
图3实施例1-3所制备的兰索拉唑肠溶微丸在ph6.8磷酸盐缓冲液中的释放度;
图4实施例2所制备的兰索拉唑肠溶微丸及参比制剂在(a)高温60℃,(b)高温40℃,(c)高湿(75%±5%),(d)光照(4500lx±500lx)影响因素条件下在ph6.8磷酸盐缓冲液中的释放度。
具体实施方式
本发明涉及一种质子泵抑制剂-兰索拉唑肠溶微丸及其制备方法。本发明应用粉末层积微丸制备方法制备兰索拉唑肠溶微丸。本发明通过结合离心造粒和流化床包衣制备技术,采用离心造粒技术在空白微丸上层积兰索拉唑、碱性稳定剂、崩解剂、稀释剂及增溶剂等辅料组成含药丸芯,再层积碱性稳定剂、崩解剂、稀释剂及增溶剂等组成的隔离衣层,最后采用流化床包衣技术制备微丸肠溶衣层。本发明所用制备技术改善了使用流化床制粒技术制备含药丸芯和隔离衣层时混悬物料对于流化床包衣机喷枪的堵塞问题,以及采用离心造粒技术制备肠溶衣层时,肠溶包衣液喷射不均及离心造粒机平板旋转对微丸的磨损问题。本发明制备的兰索拉唑肠溶微丸可以有效提高兰索拉唑在肠液中的释放度及稳定性,提高药物的口服生物利用度,并且该制备工艺简单,重现性好。
表1为实施例1-3所制备的兰索拉唑肠溶微丸与参比制剂的耐酸力及在ph6.8磷酸盐缓冲液中释放度。
表2为实施例2所制备的自制制剂及参比制剂在不同影响因素条件下的外观、吸湿增重及含量。
表3为实施例2所制备的自制制剂及参比制剂在不同影响因素条件下的有关物质。
表4实施例2所制备的自制制剂及参比制剂在不同影响因素条件下的耐酸力。
本发明采用粉末层积微丸制备方法,通过结合离心造粒技术及流化床包衣技术制备多单元口服膜控型微丸制剂。本发明所述兰索拉唑肠溶微丸由内至外,包含以下组成成分:
(1)空白丸芯:利用粘合剂将混合均匀的稀释剂及崩解剂制成空白丸芯。
(2)含药丸芯:利用粘合剂将混合均匀的兰索拉唑、稀释剂、崩解剂、稳定剂、增溶剂等药用辅料喷到空白丸芯上形成含药丸芯。
(3)隔离衣层:利用粘合剂将混合均匀的稀释剂、粘合剂、崩解剂、稳定剂、增溶剂、润滑剂、增塑剂等药用辅料喷到含药丸芯上形成隔离衣层。
(4)肠溶衣层:将水溶性肠溶包衣液与增塑剂及润滑剂制成混悬包衣液喷到隔离衣层上,形成肠溶衣层。
如权利要求1所述的兰索拉唑肠溶微丸,其特征在于:所制备的的空白丸芯粒径为0.5~1mm,优选为蔗糖与淀粉以4:1~6:1混合比例制得的微丸。
本发明所述的兰索拉唑肠溶微丸,含药丸芯中所述粘合剂选用羟丙基纤维素、羟丙甲基纤维素、聚乙烯吡咯烷酮k30、聚乙烯吡咯烷酮k90中的至少一种;所述稀释剂可选蔗糖、玉米淀粉、微晶纤维素中的至少一种;所述崩解剂选用低取代羟丙纤维素、羧甲基淀粉钠、交联羧甲基纤维素钠、交联聚维酮中的至少一种;所述稳定剂选用碳酸氢钠、重质碳酸镁、碳酸钙、氧化镁中的至少一种;所述增溶剂选用十二烷基硫酸钠,葡甲胺及泊洛沙姆中的至少一种。
本发明所述的兰索拉唑肠溶微丸,隔离衣层所述粘合剂选用羟丙基纤维素、羟丙甲基纤维素、聚乙烯吡咯烷酮k30、聚乙烯吡咯烷酮k90中的至少一种;所述稀释剂可选蔗糖、玉米淀粉、微晶纤维素中的至少一种;所述崩解剂选用低取代羟丙纤维素、羧甲基淀粉钠、交联羧甲基纤维素钠、交联聚维酮中的至少一种;所述稳定剂选用碳酸氢钠、重质碳酸镁、碳酸钙、氧化镁中的至少一种;所述增溶剂选用十二烷基硫酸钠、葡甲胺及泊洛沙姆中的至少一种;所述增塑剂选用聚乙二醇、柠檬酸三乙酯、邻苯二甲酸二乙酯中的至少一种;所述润滑剂选用滑石粉、硬脂酸镁、聚乙二醇、单硬脂酸甘油酯中的至少一种。
本发明所述的兰索拉唑肠溶微丸,肠溶衣层所述水溶性肠溶包衣材料为尤特奇l30-d55、尤特奇s100、尤特奇l100中的至少一种;所述增塑剂由聚乙二醇、柠檬酸三乙酯、邻苯二甲酸二乙酯中的至少一种;所述润滑剂为滑石粉、硬脂酸镁、二氧化钛、单硬脂酸甘油酯中的至少一种。
本发明所述的兰索拉唑肠溶微丸中,空白丸芯占微丸总重的15~30%,隔离衣层占微丸总重的5~20%,肠溶衣层占微丸总重的20~40%,余量为含药丸芯。
本发明所述的兰索拉唑肠溶微丸组成成分如下:
a.空白丸芯:
蔗糖10-25份
淀粉2-8份
崩解剂0.5-3份
b.含药丸芯:
c.隔离衣层:
d.肠溶衣层:
水溶性肠溶包衣材料10-30份
润滑剂2-6份
增塑剂2-10份
本发明所述的兰索拉唑肠溶微丸,其特征在于:兰索拉唑肠溶微丸的制备工艺如下:
(1)空白丸芯制备:
将处方量的蔗糖、淀粉及崩解剂混合均匀,放入wl350型离心式制丸机(温州市制药设备厂),以3~8%(g/ml)的粘合剂制备空白丸芯。工艺参数为0.1~0.3mpa的喷气压力、10~20r/min的挤出速度及15~20r/min的滚圆速度。制备完成后,将空白丸芯于30~40℃烘箱干燥1~2h。
(2)含药丸芯制备:
将微粉化至300~500目的兰索拉唑与辅料,于syh-5型混合机(常州彬达干燥制粒设备有限公司)中混合5~20min后放入bll-360型离心造粒机(北京长征天民高科技有限公司),以3~8%(g/ml)的粘合剂于步骤(1)得到的空白丸芯上层积兰索拉唑含药层,制备兰索拉唑含药丸芯。工艺参数为0.02~0.1mpa的喷气压力、30~40%的鼓风流量、3~8ml/min的喷浆速度及6~25r/min的喷粉速度,供粉完毕,将微丸于30~40℃烘箱干燥2~3h,得到含药丸芯。
(3)隔离衣层制备:
将混合均匀的辅料及含药丸芯放入离心造粒机,以3~8%(g/ml)的粘合剂制备微丸隔离衣层。工艺参数为0.02~0.1mpa的喷气压力、30~40%的鼓风流量、3~8ml/min的喷浆速度及6~25r/min的喷粉速度。
(4)肠溶衣层制备:
采用dpl-11型流化床包衣机(重庆精工机械有限公司),以0.2~0.4mpa的喷气压力、13~40hz的鼓风频率、1~5ml/min的喷液速度、30~60℃的进风温度,30~60℃的进样器温度、30~60℃的物料温度等工艺制备兰索拉唑微丸的肠溶衣层。
本发明所述的兰索拉唑肠溶微丸,兰索拉唑原料药需经过微粉化将药物粉碎至300~500目。
本发明所述的兰索拉唑肠溶微丸给药剂型为胶囊剂,每一粒兰索拉唑肠溶微丸胶囊中微丸填充量为330~450mg。
为了更好的阐述本发明的目的,技术方案和优点,下面结合附图和具体实施例,使本发明的优点和特征能更易于被本领域技术人员理解,以下实施例用于对本发明进行举例说明,并无意于限定本发明的范围。
实施例1:
(1)空白丸芯:
处方:
制备工艺:
将处方量的蔗糖、淀粉及低取代羟丙纤维素混合均匀,放入wl350型离心式制丸机(温州市制药设备厂),以5%(g/ml)的羟丙甲基纤维素制备空白丸芯。工艺参数为0.1~0.3mpa的喷气压力、10~20r/min的挤出速度及15~20r/min的滚圆速度。制备完成后,将空白丸芯于35℃烘箱干燥1h。
(2)含药丸芯:
处方:
制备工艺:
将粒径为30~40目的空白丸芯置于bll-360型离心造粒机(北京长征天民高科技有限公司)中,将处方量的兰索拉唑粉碎至500目,辅料过100目筛,于syh-5型三维混合机(常州彬达干燥制粒设备有限公司)中混合后,置于离心造粒机的供粉机内,以5%(g/ml)的羟丙甲基纤维素为粘合剂制备微丸。仪器操作参数:主机转速为90~120r/min、喷气压力为0.02~0.1mpa、鼓风流量为30%~40%、喷浆速度为3~8ml/min、喷粉速度为3~25r/min。供粉完毕,以2~5ml/min的喷浆速度继续喷粘合剂2~5min,抛光2~5min。取出微丸,于40℃烘箱内干燥2h,干燥后分别过30目及18目筛,弃去大于18目及小于30目的微丸,得到含药丸芯。
(3)隔离衣层:
处方:
制备工艺:
将粒径为18~30目含药丸芯置于bll-360型离心造粒机(北京长征天民高科技有限公司)中,将处方量的辅料分别过100目筛,混合5min,置于离心造粒机的供粉机内,以5%(g/ml)的羟丙甲基纤维素为粘合剂制备微丸隔离衣层。仪器操作参数:主机转速为90~120r/min、喷气压力为0.02~0.1mpa、鼓风流量为30%~40%、喷浆速度为3~8ml/min、喷粉速度为3~25r/min,保证药粉均匀层积在含药丸芯上。供粉完毕,以2~5ml/min的喷浆速度继续喷粘合剂2~5min,抛光2~5min,分别过16目及24目筛,弃去大于16目及小于24目的微丸,得包有隔离衣层的含药丸芯,隔离衣层增重为15%。
(4)肠溶衣层:
处方:
制备工艺:
将16~24目包过隔离衣层的微丸置于dpl-11型流化床包衣机(重庆精工机械有限公司)中,以20%(g/ml)的尤特奇s100及辅料制成的肠溶混悬液制备微丸的肠溶衣层。仪器操作参数:喷气压力0.2~0.4为mpa、鼓风频率为13~40hz、喷液速度为0.5~5ml/min、进风温度为30~60℃,进样器温度为30~60℃、物料温度为30~60℃。喷液完毕,微丸抛光3~5min,分别过16目及20目筛,弃去大于16目及小于20目的微丸,得包有肠溶衣层的微丸,肠溶衣层增重为20%。
将微丸封装于1号胶囊。
实施例2:
(1)空白丸芯:
处方:
制备工艺:空白丸芯制备工艺与实施例1空白丸芯制备工艺保持一致。
(2)含药丸芯:
处方:
含药丸芯制备工艺与实施例1含药丸芯制备工艺保持一致。
(3)隔离衣层:
处方:
制备工艺:隔离衣层制备工艺与实施例1隔离衣层制备工艺保持一致。
(4)肠溶衣层:
处方:
制备工艺:肠溶衣层的制备工艺与实施例1肠溶衣层的制备工艺保持一致。
实施例3:
(1)空白丸芯:
处方:
蔗糖43
淀粉7
聚乙烯吡咯烷酮k3010
羧甲基淀粉钠15
制备工艺:空白丸芯制备工艺与实施例1空白丸芯制备工艺保持一致。
(2)含药丸芯:
处方:
制备工艺:含药丸芯制备工艺与实施例1含药丸芯制备工艺保持一致。
(3)隔离衣层:
处方:
制备工艺:隔离衣层制备工艺与实施例1隔离衣层制备工艺保持一致
(4)肠溶衣层:
处方:
制备工艺:肠溶衣层的制备工艺与实施例1肠溶衣层的制备工艺保持一致。
以takepron为参比制剂,采用肉眼及扫描电镜观察自制制剂与参比制剂的外观及内部结构。微丸呈白色球形,表面光滑,无相互粘连现象。如图2所示,a行、b行为微丸表面形态,参比制剂外观呈球形,表面较为粗糙,有较多细小颗粒粘附,而自制制剂整体外观呈球形,表面凹凸不平,无颗粒粘附。c行、d行为微丸剖面形态,参比制剂与自制制剂均呈四层结构,由内之外,分别为空白丸芯层、含药丸芯层、隔离衣层及肠溶衣层。参比制剂的空白丸芯层与含药丸芯层中有空洞,而自制制剂无空洞呈现,说明自制制剂的空白丸芯层与含药丸芯层包裹较为紧密,但自制制剂的含药丸芯层颗粒粒径比参比制剂的大,呈不均匀分布,且自制制剂的隔离衣层增重比参比制剂少。
以takepron为参比制剂,将实施例1-3制备的不同兰索拉唑肠溶微丸胶囊进行体外释放度测定。在ph1.2盐酸介质中耐受2h,肠溶衣膜外观完整,如表1所示,所有微丸在酸中的释放度均小于标示量的7%。制剂在ph6.8磷酸盐缓冲液中的释放曲线如图3所示,实施例2所制备的微丸在缓冲液中累积释放度为82.7%,释放速率与参比制剂最为接近,说明该处方相较于其他处方较优。
以实施例2制备的兰索拉唑肠溶微丸胶囊进行影响因素试验,考察其外观、含量、有关物质及释放度变化,结果见表2、3、4及图4。如表2、3所示,参比制剂及自制制剂在高温60℃条件下放置5、10d,与0d的样品比较,微丸颜色均变为微红色,主药含量均呈增长趋势,且增长趋势相似,约增高了10%;参比制剂在该条件,杂质b含量呈下降趋势,杂质d含量呈增长趋势,总杂为0.25%,而自制制剂的杂质b、d的含量几乎未发生变化,总杂为0.16%。
参比制剂在高温40℃条件下放置5、10d,与0d的样品比较,微丸颜色无显著变化,主药含量呈增长趋势,而自制制剂外观及主药含量无明显变化。参比制剂在该条件,杂质b含量呈下降趋势,杂质d含量呈增长趋势,总杂为0.13%,而自制制剂的杂质b无明显变化,杂质d含量呈增长趋势,总杂为0.16%。
参比制剂及自制制剂在rh92.5%条件下放置5d后,均吸湿粘结成浆状物,故不做检查;参比制剂及自制制剂在rh75%条件下放置5、10d,与0d的样品比较,参比制剂的微丸颜色无变化,呈白色球形,但有明显吸潮现象,主药含量呈增长趋势,而自制制剂的微丸颜色变为微红色,有明显吸潮现象,5d时的主药含量无明显变化,10d的主药含量明显上升;参比制剂10d时,杂质b含量呈下降趋势,为0.05,杂质d含量呈增长趋势,为0.11,总杂为0.18%,而自制制剂的杂质b的含量未发生变化,为0.02,杂质d含量呈增长趋势,为0.09,总杂为0.21%。
参比制剂及自制制剂在光照(4500lx)条件下放置5、10d,与0d的样品比较,微丸颜色无明显变化,呈类白色,主药含量均呈增长趋势。参比制剂的杂质b含量呈下降趋势,杂质d含量呈增长趋势,总杂为0.25%,而自制制剂的杂质b的含量几乎无明显变化,杂质d含量呈增长趋势,总杂为0.19%。
耐酸力及释放度的考察:
将自制制剂及参比制剂按分别在高温(60、40℃)、高湿(rh75%)及光照(4500lx)条件下放置5、10d,测定其耐酸力及在ph6.8磷酸盐缓冲液中的释放度,并与0天样品进行对比,评价制剂的影响因素下耐酸力和释放度的稳定性。
如表4所示,在不同影响因素条件下,自制制剂的酸中释放度与0d样品相比,呈下降趋势,表明自制制剂的耐酸力增强,而参比制剂的的q2h与0d样品相比,无明显变化。如图4所示,在高温(60℃)、高湿(rh75%)及光照(4500lx)条件下,与0d样品相比,自制制剂在ph6.8的磷酸盐缓冲液中的释放速率无明显变化,而参比制剂呈上升趋势,约增加了5~10%。在高温40℃条件下,参比制剂及自制制剂在ph6.8的磷酸盐缓冲液中的释放速率均呈上升趋势。影响因素条件下的释放度考察结果表明,不同的影响因素条件可能改变了兰索拉唑肠溶微丸内部的粘附力,使微丸崩解速度发生变化。
在影响因素试验中,自制制剂的外观与参比制剂相同,均为白色或微红色微丸,含量变化趋势与参比制剂相同,均呈上升趋势,自制制剂在10d时的总杂小于参比制剂且释放度变化趋势与参比制剂相似,表明自制制剂相较于参比制剂,稳定性有明显提高。
- 还没有人留言评论。精彩留言会获得点赞!