一种检测拷贝数变异的方法、装置和存储介质与流程
本申请涉及拷贝数变异检测领域,特别是涉及一种检测拷贝数变异的方法、装置和存储介质。
背景技术:
拷贝数变异(copynumbervariation,cnv)是由基因组发生重排而导致的,一般指长度为1kb以上的基因组大片段的拷贝数增加或者减少,主要表现为亚显微水平的缺失和重复。cnv是基因组结构变异(structuralvariation,sv)的重要组成部分。cnv位点的突变率远高于snp(singlenucleotidepolymorphism),是人类疾病的重要致病因素之一。
随着二代测序技术的快速发展,其逐渐应用于癌症方向及遗传病的科学研究和临床检测,使我们对癌症及遗传病的发生发展、临床表现及发病机理有了全新的认识。大量研究表明,遗传病的产生是与拷贝数变异(cnv)密切相关,并且这种拷贝数变异呈现出区域大小的不稳定性。有的拷贝数变异可能只涉及单个外显子,而有的却涉及多个基因或更有甚者涉及整个染色体的长臂/短臂区域。拷贝数变异的研究为疾病病程发展及治疗提供了新的方向和重要的参考依据。
目前主流的检测拷贝数变异的算法主要是基于染色体微阵列分析(cma)来实现。这种方法存在以下不可避免的内在局限性:1、无法明确找到断点信息;2、通量低,一次只能做较少区域的检测,无法进行高通量的实验;3、只能针对目标区域设计探针进行检测,无法达到全基因组筛查的目的。
随着二代测序技术的快速发展,高深度的二代测序技术也可以检测拷贝数变异。但是,基于二代测序的拷贝数变异检测方法同样存在不可避免的局限性:1、由于基因组中同源序列的存在,导致比对错误;2、由于实验的pcr过程会对不同dna模板有不同的扩增效率,导致凭借深度判断拷贝数变异会有极大的假阳性;3、基于二代测序的染色体拷贝数变异检测(缩写cnv-seq)依赖于待检测样本的父/母正常对照样本,在单个样本情况下很难进行检测。
因此,亟需研发一种能够更准确区分真假拷贝数变异,且能够实现单个样本检测而无需亲缘对照样本的检测技术,以满足拷贝数变异研究和临床检测使用的需求。
技术实现要素:
本申请的目的是提供一种新的检测拷贝数变异的方法、装置和存储介质。
本申请具体采用了以下技术方案:
本申请的第一方面公开了一种检测拷贝数变异的方法,包括以下步骤:
数据获取步骤,包括获取待测样本dna测序数据及其在人类参考基因组上各个位置的比对数据;
深度统计步骤,包括根据数据获取步骤获取的待测样本dna测序数据,统计待测样本dna在固定窗口区域内的深度;
异常数据提取步骤,包括根据数据获取步骤中获取的待测样本dna在人类参考基因组上各个位置的比对数据,提取比对异常的数据序列,作为支持拷贝数变异的第一维度结果;
窗口深度对比步骤,包括根据深度统计步骤的结果,将待测样本dna的窗口深度与统计的健康人群dna窗口深度的分布统计模型进行比较,获得与分布统计模型显示出差异的窗口结果;可以理解,该步骤中具体与待测样本dna的窗口深度进行比较的是分布统计模型中相应的窗口的深度分布模型;
拷贝数变异判定步骤,包括根据窗口深度对比步骤的结果,分析与分布统计模型显示出差异的窗口,判断其差异是否显著;如果差异显著,且能够得到异常数据提取步骤中第一维度结果的支持,则判断为阳性拷贝数变异;
检测结果输出步骤,包括根据拷贝数变异判定步骤的判断结果,输出待测样本dna的所有阳性拷贝数变异。
需要说明的是,本申请的拷贝数变异检测方法中,健康人群dna窗口深度的分布统计模型,是根据健康人群的dna测序数据统计的,健康人群的dna在固定窗口区域的深度分布情况,以此作为拷贝数变异的一个区分维度,能够在不需要亲缘对照样本的情况下,对单个样本进行拷贝数变异检测。
本申请的一种实现方式中,健康人群dna窗口深度的分布统计模型的构建方法包括,健康人群dna数据获取步骤,包括获取健康人群dna测序数据及健康人群中各单个样本dna在人类参考基因组上各个位置的比对数据;健康人群dna窗口深度统计步骤,包括根据健康人群dna数据获取步骤获取的健康人群dna测序数据,统计各单个样本dna在固定窗口区域内的深度,得到每个窗口的深度在健康人群的分布情况,统计所有窗口的深度分布情况,即获得本申请的分布统计模型。
在首次采用本申请的检测拷贝数变异的方法时,需要构建健康人群dna窗口深度的分布统计模型;其中,健康人群dna数据获取步骤可以与本申请拷贝数变异检测方法中的数据获取步骤整合在一起,同时对测样本dna测序数据和健康人群dna测序数据进行提取;同样的,健康人群dna窗口深度统计步骤可以与本申请拷贝数变异检测方法中的深度统计步骤整合在一起,即统计待测样本dna在固定窗口区域内的深度,同时统计健康人群中各单个样本dna在相应的固定窗口区域内的深度,得到每个窗口的深度在健康人群的分布统计模型。可以理解,构建好健康人群dna窗口深度的分布统计模型后,该分布统计模型可以重复使用,后续进行拷贝数变异检测时,则不需要重复的进行分布统计模型构建。因此,本申请的拷贝数变异检测方法中并没有将健康人群dna数据获取步骤以及健康人群dna窗口深度统计步骤整合到本申请的检测方法中。
其中,健康人群dna测序数据的作用是用于构建健康人群基因组中每个固定窗口区域内的深度分布统计模型,原则上,所采用的健康人群dna测序数据越多,即采集的健康人群的数量越多,相应的深度分布统计模型也越准确,但是数据处理量和成本也会相应增加,因此,具体健康人群dna测序数据的量可以根据检测条件和所需达到的准确性程度而调整,在此不作具体限定。
还需要说明的是,本申请的拷贝数变异检测方法,利用健康人群dna测序数据,确定健康人群的基因组中每个固定窗口区域内的深度变化范围,从而准确地将拷贝数变异的测序片段筛选出来,实现假阳性拷贝数变化与真实的拷贝数变异区分;并且,结合第一维度结果支撑,从不同的角度确保拷贝数变异检测结果的准确性,降低了拷贝数变异检测的假阳性或假阴性。
可以理解,本申请检测方法的数据获取步骤中,待测样本dna测序数据可以是正常进行测序获得的数据,也可以是事先存储的测序数据;同样的,健康人群dna测序数据,也可以是采用待测样本dna相同的方法测序获得的数据,或者是事先构建好的健康人群的dna测序数据,如果是事先已经准备好的健康人群的dna测序数据,优选采用与待测样本dna相同的测序方法获得的数据;即保障健康人群dna测序数据与待测样本dna测序数据为平行测序数据,以避免其它因素对本申请检测方法的准确性造成影响。
优选的,数据获取步骤包括以下子步骤,
获取子步骤,包括获取待测样本dna测序数据;
过滤子步骤,包括对获取子步骤获取的待测样本dna测序数据进行质检,过滤去除低质量的测序数据;
比对子步骤,包括将经过过滤子步骤的测序数据与人类参考基因组比对,获取待测样本dna的测序片段在人类参考基因组中的位置。
优选的,数据获取步骤还包括以下子步骤,
去重子步骤,包括根据比对子步骤的结果,去除比对到人类参考基因组相同位置的重复的测序片段;
校正子步骤,包括根据去重子步骤的结果,重新校正测序数据与人类参考基因组的比对结果。
优选的,深度统计步骤,还包括统计待测样本dna在固定窗口区域内深度的负二项分布情况。
优选的,异常数据提取步骤中,比对异常的数据序列包括插入片段大小异常的序列数据、比对方向异常的序列数据、序列数据比对到不同位置的序列数据。
需要说明的是,本申请的检测方法中,比对异常的数据序列主要是指插入片段大小异常、比对方向异常、同时比对到人类参考基因组上不同位置,这三种情况;这三种情况是拷贝数变异所引起的比较常见的三种情况,因此,将出现这三种情况的测序数据作为支持拷贝数变异的第一维度结果。
优选的,拷贝数变异判定步骤中,判断差异是否显著具体包括,提出零假设和备择假设,并以贝叶斯因子为判别条件,当贝叶斯因子小于设定值时,认为备择假设成立而零假设不成立,即判定为差异显著。
需要说明的是,判断差异是否显著,主要是指待测样本dna测序数据在某个固定窗口的深度与统计的健康人群dna测序数据在相应的固定窗口的深度范围之间是否存在显著差异,如果存在显著差异,则有可能是拷贝数变异引起。因此,本申请优选采用贝叶斯因子作为判断条件,不排除还可以采用其它显著差异的判断方法。
本申请的拷贝数变异检测方法中,健康人群dna窗口深度的分布统计模型可以仅在首次采用本申请检测方法的时候构建,后续则直接对待测样本dna测序数据进行分析后与构建好的分布统计模型进行差异显著性分析即可。
因此,本申请的第二方面提供了一种健康人群dna窗口深度分布统计模型的构建方法,包括以下步骤,
健康人群dna数据获取步骤,包括获取健康人群dna测序数据及健康人群中各单个样本dna在人类参考基因组上各个位置的比对数据;
健康人群dna窗口深度统计步骤,包括根据健康人群dna数据获取步骤获取的健康人群dna测序数据,统计各单个样本dna在固定窗口区域内的深度,得到每个窗口的深度在健康人群的分布情况,统计完成所有窗口,即获得所述分布统计模型。
采用本申请的构建方法可以获得用于拷贝数变异检测的健康人群dna窗口深度分布统计模型,该分布统计模型可以用于本申请的拷贝数变异检测方法。
优选的,本申请健康人群dna窗口深度分布统计模型的构建方法中,健康人群dna窗口深度统计步骤,还包括统计健康人群中各单个样本dna在固定窗口区域内深度的负二项分布情况,以此得到健康人群的各窗口的深度分布统计模型。
优选的,本申请健康人群dna窗口深度分布统计模型的构建方法还包括分布统计模型校正步骤,包括根据健康人群dna窗口深度统计步骤获得的分布统计模型,对每个固定窗口的健康人群中各单个样本dna的深度进行评估,舍去明显偏离的位点,并根据没有舍去的位点的窗口深度分布情况,得到校正的分布统计模型。
优选的,本申请健康人群dna窗口深度分布统计模型的构建方法中,分布统计模型校正步骤,还包括统计没有舍去的位点的窗口深度的负二项分布情况,以此得到校正的分布统计模型。
可以理解,本申请的拷贝数变异检测的方法或者健康人群dna窗口深度分布统计模型的构建方法,其全部或部分功能可以通过硬件的方式实现,也可以通过计算机程序的方式实现。当通过计算机程序的方式实现时,该程序可以存储于一计算机可读存储介质中,存储介质可以包括:只读存储器、随机存储器、磁盘、光盘、硬盘等,通过计算机执行该程序以实现本申请的方法。例如,将程序存储在设备的存储器中,当通过处理器执行存储器中程序,即可实现本申请的方法。当本申请的方法中全部或部分功能通过计算机程序的方式实现时,该程序也可以存储在服务器、另一计算机、磁盘、光盘、闪存盘或移动硬盘等存储介质中,通过下载或复制保存到本地设备的存储器中,或对本地设备的系统进行版本更新,当通过处理器执行存储器中的程序时,即可实现本申请拷贝数变异检测方法或者健康人群dna窗口深度分布统计模型的构建方法的全部或部分功能。
因此,本申请的第三方面公开了一种拷贝数变异检测的装置,包括数据获取模块、深度统计模块、异常数据提取模块、窗口深度对比模块、拷贝数变异判定模块和检测结果输出模块;
数据获取模块,包括用于获取待测样本dna测序数据及其在人类参考基因组上各个位置的比对数据;
深度统计模块,包括根据数据获取模块获取的待测样本dna测序数据,统计待测样本dna在固定窗口区域内的深度;
异常数据提取模块,包括用于根据数据获取模块中获取的待测样本dna在人类参考基因组上各个位置的比对数据,提取比对异常的数据序列,作为支持拷贝数变异的第一维度结果;
窗口深度对比模块,包括用于根据深度统计模块的结果,将待测样本dna的窗口深度与统计的健康人群dna窗口深度的分布统计模型进行比较,获得与分布统计模型显示出差异的窗口结果;
拷贝数变异判定模块,包括用于根据窗口深度对比模块的结果,分析与分布统计模型显示出差异的窗口,判断其差异是否显著;如果差异显著,且能够得到第一维度结果的支持,则判断为阳性拷贝数变异;
检测结果输出模块,包括用于根据拷贝数变异判定模块的判断结果,输出待测样本dna的所有阳性拷贝数变异。
需要说明的是,本申请的拷贝数变异检测的装置,实际上就是通过各模块实现本申请的拷贝数变异检测的方法的各步骤,从而实现拷贝数变异的自动化分析和检测。
优选的,数据获取模块包括以下子模块,
获取子模块,包括用于获取待测样本dna测序数据;
过滤子模块,包括用于对获取子模块获取的待测样本dna测序数据进行质检,过滤去除低质量的测序数据;
比对子模块,包括用于将经过过滤子模块的测序数据与人类参考基因组比对,获取待测样本dna的测序片段在人类参考基因组中的位置。
优选的,数据获取模块还包括以下子模块,
去重子模块,包括用于根据比对子模块的结果,去除比对到人类参考基因组相同位置的重复的测序片段;
校正子模块,包括用于根据去重子模块的结果,重新校正测序数据与人类参考基因组的比对结果。
需要说明的是,本申请的一种实现方式中,为了确保拷贝数变异检测的准确性,本申请的数据获取模块主要由获取子模块、过滤子模块、去重子模块和校正子模块组成,通过对低质量测序数据和重复测序数据的过滤和去除,一方面,降低了后续步骤或处理模块的数据处理量;另一方面,也提高了检测结果的准确性和可靠性。
优选的,本申请的深度统计模块,还包括用于统计待测样本dna在固定窗口区域内深度的负二项分布情况。
优选的,本申请的异常数据提取模块中,比对异常的数据序列包括插入片段大小异常的序列数据、比对方向异常的序列数据、序列数据比对到不同位置的序列数据。
需要说明的是,在本申请的一种实现方式中,本申请的异常数据提取模块在对插入片段大小异常、比对方向异常、同时比对到人类参考基因组上不同位置,这三种情况的异常数据序列进行提取,分别采用的不同的子模块;例如,通过异常插入片段提取子模块,提取插入片段大小异常的序列数据;通过异常比对方向提取子模块,提取比对方向异常的序列数据;通过异常分割片段提取子模块,提取序列数据比对到不同位置的序列数据。
优选的,本申请的拷贝数变异判定模块中,判断差异是否显著具体包括,提出零假设和备择假设,并以贝叶斯因子为判别条件,当贝叶斯因子小于设定值时,认为备择假设成立而零假设不成立,即判定为差异显著。
优选的,本申请检测拷贝数变异的装置还包括分布统计模型构建模块,分布统计模型构建模块包括健康人群dna数据获取子模块和健康人群dna窗口深度统计子模块;健康人群dna数据获取子模块,包括用于获取健康人群dna测序数据及健康人群中各单个样本dna在人类参考基因组上各个位置的比对数据;健康人群dna窗口深度统计子模块,包括用于根据健康人群dna数据获取子模块获取的健康人群dna测序数据,统计各单个样本dna在固定窗口区域内的深度,得到每个窗口的深度在健康人群的分布情况,统计完成所有窗口,即获得分布统计模型。
需要说明的是,分布统计模型构建模块由于只在首次采用本申请的拷贝数变异检测方法时构建健康人群dna窗口深度的分布统计模型使用,因此,对于本申请的拷贝数变异检测装置,分布统计模型构建模块并非必须的模块。在已经构建好健康人群dna窗口深度的分布统计模型的情况下,可以直接将构建好的分布统计模型导入本申请的拷贝数变异检测装置中,则不需要分布统计模型构建模块。其中,将构建好的分布统计模型导入本申请的拷贝数变异检测装置中的方式可以是利用存储介质将分布统计模型拷贝到本申请的检测装置中,或者通过网络下载获得,在此不作具体限定。
可以理解,参考本申请拷贝数变异检测方法中,健康人群dna数据获取步骤整合到数据获取步骤,健康人群dna窗口深度统计步骤整合到深度统计步骤的方案;本申请的分布统计模型构建模块中,各子模块同样可以整合到本申请拷贝数变异检测装置的其它模块中。例如,将健康人群dna数据获取子模块整合到本申请检测装置的数据获取模块中,在提取待测样本dna测序数据的同时提取健康人群dna测序数据;将健康人群dna窗口深度统计子模块整合到本申请检测装置的深度统计模块,即统计待测样本dna在固定窗口区域内的深度,同时统计健康人群中各单个样本dna在相应的固定窗口区域内的深度,得到每个窗口的深度在健康人群的分布统计模型。整合后的数据获取模块和深度统计模块,只是在功能上增加了健康人群dna测序数据提取或深度统计的功能,在已经构建好分布统计模型的情况下,这些新增加的功能也可以不使用。
优选的,分布统计模型构建模块中,健康人群dna窗口深度统计子模块,还包括用于统计健康人群中各单个样本dna在固定窗口区域内深度的负二项分布情况,以此得到健康人群的各窗口的深度分布统计模型;
优选的,分布统计模型构建模块还包括分布统计模型校正子模块,分布统计模型校正子模块包括用于根据健康人群dna窗口深度统计子模块获得的分布统计模型,对每个固定窗口的健康人群中各单个样本dna的深度进行评估,舍去明显偏离的位点,并根据没有舍去的位点的窗口深度分布情况,得到校正的分布统计模型;
优选的,分布统计模型校正子模块,还包括用于统计没有舍去的位点的窗口深度的负二项分布情况,以此得到校正的分布统计模型。
需要说明的是,分布统计模型校正子模块的目的是去除与其它大部分的健康人群dna的窗口深度存在明显偏离的部分单个样本dna的位点,这样可以更好的保障分布统计模型的准确性,避免个别明显偏离的单个样本dna位点影响拷贝数变异检测结果的准确性。可以理解,虽然采用的健康人群dna测序数据中所有单个样本dna都是采用待测样本dna相同的测序方法获得的测序数据;但是,不排除在测序过程中可能存在错误或误差,影响个别单个样本dna的测序准确性。因此,通过分布统计模型校正子模块,可以过滤去除这部分与正常的健康人群dna的窗口深度明显偏离的位点,从而保障分布统计模型和拷贝数变异检测结果的准确性。
本申请的拷贝数变异检测装置中,分布统计模型构建模块可以仅仅在首次采用本申请的拷贝数变异检测装置时使用,在已经构建好健康人群dna窗口深度的分布统计模型的情况下,后续只需要根据本申请检测装置中的其它模块将待测样本dna测序数据与构建好的分布统计模型进行差异显著性分析即可。
因此,本申请的第四方面公开了一种健康人群dna窗口深度分布统计模型的构建装置,包括健康人群dna数据获取模块和健康人群dna窗口深度统计模块,健康人群dna数据获取模块,包括用于获取健康人群dna测序数据及健康人群中各单个样本dna在人类参考基因组上各个位置的比对数据;健康人群dna窗口深度统计模块,包括用于根据健康人群dna数据获取模块获取的健康人群dna测序数据,统计各单个样本dna在固定窗口区域内的深度,得到每个窗口的深度在健康人群的分布情况,统计完成所有窗口,即获得本申请的分布统计模型。
优选的,本申请的构建装置中,健康人群dna窗口深度统计模块,还包括用于统计健康人群中各单个样本dna在固定窗口区域内深度的负二项分布情况,以此得到健康人群的各窗口的深度分布统计模型。
优选的,本申请的健康人群dna窗口深度分布统计模型的构建装置还包括分布统计模型校正模块,分布统计模型校正模块包括用于根据健康人群dna窗口深度统计模块获得的分布统计模型,对每个固定窗口的健康人群中各单个样本dna的深度进行评估,舍去明显偏离的位点,并根据没有舍去的位点的窗口深度分布情况,得到校正的分布统计模型。
优选的,本申请的构建装置中,分布统计模型校正模块,还包括用于统计没有舍去的位点的窗口深度的负二项分布情况,以此得到校正的分布统计模型。
本申请的第五方面公开了一种用于拷贝数变异检测的装置,包括存储器和处理器:其中,存储器,用于存储程序;处理器,用于通过执行存储器存储的程序以实现本申请的检测拷贝数变异的方法或者本申请的健康人群dna窗口深度分布统计模型的构建方法。
本申请的第六方面公开了一种计算机可读存储介质,该存储介质中包括健康人群dna窗口深度分布统计模型和/或程序,该程序能够被处理器执行以实现实现本申请的检测拷贝数变异的方法或者本申请的健康人群dna窗口深度分布统计模型的构建方法。
其中,健康人群dna窗口深度分布统计模型即本申请的健康人群dna窗口深度分布统计模型的构建方法构建的分布统计模型。
本申请的有益效果在于:
本申请的检测拷贝数变异的方法,通过对健康人群dna测序数据进行统计分析,能够准确的区分真假拷贝数变异,结合另一维度结果支撑,从不同的角度确保拷贝数变异检测结果的准确性,降低了拷贝数变异检测的假阳性或假阴性。并且,本申请的检测方法,无需亲缘对照样本,能够对单个样本进行拷贝数变异检测,能够满足拷贝数变异研究和临床检测使用的需求。
附图说明
图1是本申请实施例中检测拷贝数变异的方法的流程框图;
图2是本申请实施例中拷贝数变异检测方法的数据获取步骤的流程框图;
图3是本申请实施例中健康人群dna窗口深度分布统计模型的构建方法的流程框图;
图4是本申请实施例中拷贝数变异检测的装置的结构框图;
图5是本申请实施例中健康人群dna窗口深度分布统计模型的构建装置的结构框图;
图6是本申请实施例中拷贝数变异检测结果图。
具体实施方式
现有的拷贝数变异检测,其难点在于需要亲缘的正常对照,并且会在实验过程引入的错误的深度变化,使其难以与真实的拷贝数变异区分开,从而造成假阳性或假阴性。
本申请发明人经过深入研究发现,健康人群dna在每一个区域,即固定窗口内,其深度变化在一定的范围内;而发生拷贝数变异的样本dna,其在相同的固定窗口区域内的深度,与健康人群的深度存在差异。
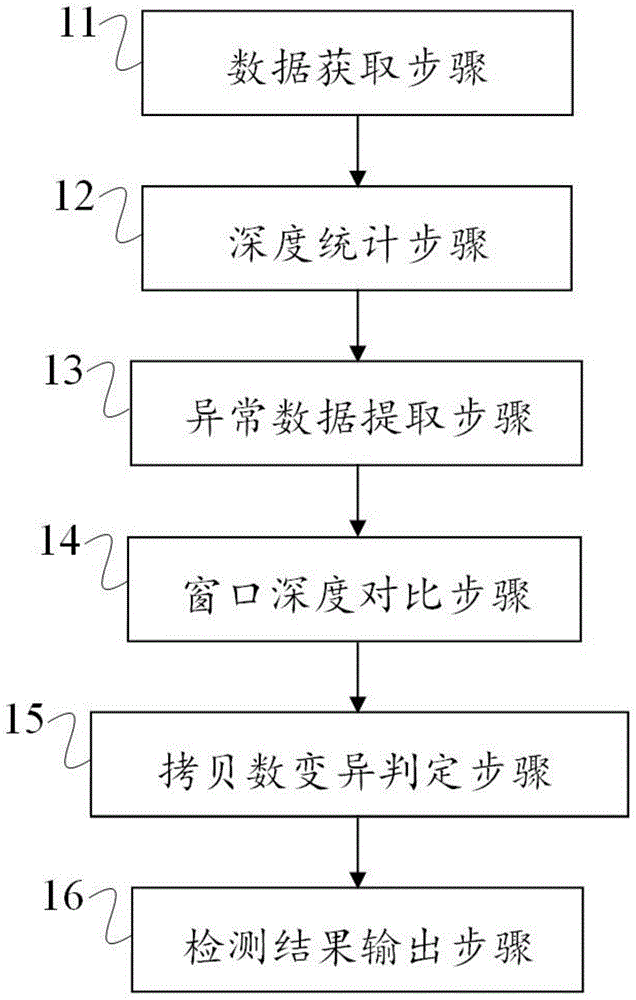
基因以上研究发现,本申请提出了一种检测拷贝数变异的方法,如图1所示,包括数据获取步骤11、深度统计步骤12、异常数据提取步骤13、窗口深度对比步骤14、拷贝数变异判定步骤15和检测结果输出步骤16。
其中,数据获取步骤11,包括获取待测样本dna测序数据及其在人类参考基因组上各个位置的比对数据。在本申请的一种实现方式中,由于首次采用本申请的检测方法需要构建健康人群dna窗口深度的分布统计模型;因此,数据获取步骤11还包括,获取健康人群dna测序数据及健康人群中各单个样本dna在人类参考基因组上各个位置的比对数据。
在本申请的一种实现方式中,本申请的数据获取步骤11,如图2所示,由获取子步骤111、过滤子步骤112、比对子步骤113、去重子步骤114和校正子步骤115组成。其中,获取子步骤111,包括获取待测样本dna测序数据,如果需要构建分布统计模型,则同时获取健康人群dna测序数据;过滤子步骤112,包括对获取子步骤获取的测序数据进行质检,过滤去除低质量的测序数据;比对子步骤113,包括将经过过滤子步骤的测序数据与人类参考基因组比对,获取待测样本dna的测序片段在人类参考基因组中的位置;去重子步骤114,包括根据比对子步骤的结果,去除比对到人类参考基因组相同位置的重复的测序片段;校正子步骤115,包括根据去重子步骤的结果,重新校正测序数据与人类参考基因组的比对结果。
深度统计步骤12,包括根据数据获取步骤获取的待测样本dna测序数据,统计待测样本dna在固定窗口区域内的深度。在本申请的一种实现方式中,由于需要构建健康人群dna窗口深度的分布统计模型;因此,深度统计步骤12还包括,统计健康人群dna中各单个样本dna在相应的固定窗口区域内的深度,得到每个窗口的深度在健康人群的分布统计模型。本申请的一种实现方式中,深度统计步骤12还包括统计待测样本dna在固定窗口区域内深度的负二项分布情况;对于分布统计模型构建来说,则统计健康人群中各单个样本dna在固定窗口区域内深度的负二项分布情况,以此得到健康人群的各窗口的深度分布统计模型。
异常数据提取步骤13,包括根据数据获取步骤中获取的待测样本dna在人类参考基因组上各个位置的比对数据,提取比对异常的数据序列,作为支持拷贝数变异的第一维度结果。本申请的一种实现方式中,具体提取了插入片段大小异常的序列数据、比对方向异常的序列数据,以及序列数据比对到不同位置的序列数据,作为拷贝数变异的第一维度结果。
窗口深度对比步骤14,包括根据深度统计步骤的结果,将待测样本dna的窗口深度与统计的健康人群dna窗口深度的分布统计模型进行比较,获得与分布统计模型显示出差异的窗口结果。
拷贝数变异判定步骤15,包括根据窗口深度对比步骤的结果,分析与分布统计模型显示出差异的窗口,判断其差异是否显著;如果差异显著,且能够得到异常数据提取步骤中第一维度结果的支持,则判断为阳性拷贝数变异。本申请的一种实现方式中,判断差异是否显著采用的是贝叶斯因子为判别条件,即先提出零假设和备择假设,以贝叶斯因子为判别条件,当贝叶斯因子小于设定值时,认为备择假设成立而零假设不成立,即判定为差异显著。
检测结果输出步骤16,包括根据拷贝数变异判定步骤的判断结果,输出待测样本dna的所有阳性拷贝数变异。本申请的一种实现方式中,具体的输出阳性拷贝数变异及其断点信息,根据需求还可以选择性的输出第一维度结果以及与分布统计模型显示出差异的窗口数据。
在本申请拷贝数变异检测方法的基础上,本申请进一步的提出了一种健康人群dna窗口深度分布统计模型的构建方法,如图3所示,包括健康人群dna数据获取步骤31和健康人群dna窗口深度统计步骤32,在进一步的改进方案中,还包括分布统计模型校正步骤33。
其中,健康人群dna数据获取步骤31,包括获取健康人群dna测序数据及健康人群中各单个样本dna在人类参考基因组上各个位置的比对数据。其中,健康人群的统计数量原则上越多越好,但是,处于成本和后续步骤的处理数据量,具体的统计数量可以根据检测条件和检测目的而定。
健康人群dna窗口深度统计步骤32,包括根据健康人群dna数据获取步骤获取的健康人群dna测序数据,统计各单个样本dna在固定窗口区域内的深度,得到每个窗口的深度在健康人群的分布情况,统计完成所有窗口,即获得分布统计模型。本申请的一种实现方式中,健康人群dna窗口深度统计步骤32,还包括统计健康人群中各单个样本dna在固定窗口区域内深度的负二项分布情况,以此得到健康人群的各窗口的深度分布统计模型。
分布统计模型校正步骤33,包括根据健康人群dna窗口深度统计步骤获得的分布统计模型,对每个固定窗口的健康人群中各单个样本dna的深度进行评估,舍去明显偏离的位点,并根据没有舍去的位点的窗口深度分布情况,得到校正的分布统计模型。本申请的一种实现方式中,分布统计模型校正步骤33,还包括统计没有舍去的位点的窗口深度的负二项分布情况,以此得到校正的分布统计模型。
基于本申请的拷贝数变异检测方法,本申请进一步的提出了一种拷贝数变异检测的装置,如图4所示,包括数据获取模块41、深度统计模块42、异常数据提取模块43、窗口深度对比模块44、拷贝数变异判定模块45和检测结果输出模块46。其中,数据获取模块41,包括用于获取待测样本dna测序数据及其在人类参考基因组上各个位置的比对数据;深度统计模块42,包括用于根据数据获取模块获取的待测样本dna测序数据,统计待测样本dna在固定窗口区域内的深度;异常数据提取模块43,包括用于根据数据获取模块中获取的待测样本dna在人类参考基因组上各个位置的比对数据,提取比对异常的数据序列,作为支持拷贝数变异的第一维度结果;窗口深度对比模块44,包括用于根据深度统计模块的结果,将待测样本dna的窗口深度与统计的健康人群dna窗口深度的分布统计模型进行比较,获得与分布统计模型显示出差异的窗口结果;拷贝数变异判定模块45,包括用于根据窗口深度对比模块的结果,分析与所述分布统计模型显示出差异的窗口,判断其差异是否显著;如果差异显著,且能够得到第一维度结果的支持,则判断为阳性拷贝数变异;检测结果输出模块46,包括用于根据拷贝数变异判定模块的判断结果,输出待测样本dna的所有阳性拷贝数变异。
在本申请的一种改进方案中,本申请的拷贝数变异检测的装置还可以进一步包括分布统计模型构建模块,分布统计模型构建模块的各个子模块可以整合到本申请拷贝数变异检测装置的各模块中,也可以单独组合成一个独立的分布统计模型构建模块,甚至在本申请的一种实现方式中,分布统计模型构建模块可以是一个独立的装置。
因此,在本申请的拷贝数变异检测的装置的基础上,本申请提供了一种健康人群dna窗口深度分布统计模型的构建装置,如图5所示,包括健康人群dna数据获取模块51和健康人群dna窗口深度统计模块52,改进方案中还包括分布统计模型校正模块53。其中,健康人群dna数据获取模块51,包括用于获取健康人群dna测序数据及健康人群中各单个样本dna在人类参考基因组上各个位置的比对数据;健康人群dna窗口深度统计模块52,包括用于根据健康人群dna数据获取模块获取的健康人群dna测序数据,统计各单个样本dna在固定窗口区域内的深度,得到每个窗口的深度在健康人群的分布情况,统计完成所有窗口,即获得分布统计模型;分布统计模型校正模块53,包括用于根据健康人群dna窗口深度统计模块获得的分布统计模型,对每个固定窗口的健康人群中各单个样本dna的深度进行评估,舍去明显偏离的位点,并根据没有舍去的位点的窗口深度分布情况,得到校正的分布统计模型。
本申请的拷贝数变异检测方法,其全部或部分功能可以通过计算机程序的方式实现,因此,本申请还提供了一种用于拷贝数变异检测的装置,包括存储器和处理器:其中,存储器,用于存储程序;处理器,用于通过执行存储器存储的程序以实现本申请的检测拷贝数变异的方法或本申请的健康人群dna窗口深度分布统计模型的构建方法。
在本申请的拷贝数变异检测方法全部或部分功能通过计算机程序的方式实现的情况下,并且进一步的提供了一种计算机可读存储介质,该存储介质中存储有健康人群dna窗口深度分布统计模型和/或程序,程序能够被处理器执行以实现实现本申请的检测拷贝数变异的方法或本申请的健康人群dna窗口深度分布统计模型的构建方法。
本申请中提及的科技术语具有与本领域技术人员通常理解的含义相同的含义,如有冲突以本申请的定义为准。一般而言,本申请采用的科技术语具有如下含义:
负二项分布:负二项分布是统计学上一种描述在一系列独立同分布的伯努利试验中,失败次数到达指定次数(记为r)时成功次数的离散概率分布。例如,如果定义掷骰子随机变量x值为x=1时为失败,所有x≠1为成功,这时反复掷骰子直到1出现3次,即失败次数r=3,此时非1数字出现次数的概率分布即为负二项分布。
人类参考基因组:全基因组测序是对未知基因组序列的物种进行个体的基因组测序。1986年,renatodulbecco等最早提出人类基因组测序;美国能源部(doe)与美国国家卫生研究院(nih),分别在1986年与1987年加入人类基因组计划;日本在1981年也已经开始研究相关问题;直到1990年开始国际合作,1996年,多个国家召开百慕达会议,以2005年完成测序为目标,分配各国负责的工作,并且宣布研究结果将会及时公布,并完全免费。
拷贝数变异(cnv):拷贝数变异(copynumbervariation,cnv)是由基因组发生重排而导致的,一般指长度为1kb以上的基因组大片段的拷贝数增加或者减少,主要表现为亚显微水平的缺失和重复。cnv是基因组结构变异(structuralvariation,sv)的重要组成部分。cnv位点的突变率远高于snp(singlenucleotidepolymorphism),是人类疾病的重要致病因素之一。
聚合酶链式反应(pcr):是一种在生物体外进行的放大扩增特定dna片段的分子生物学技术,pcr的最大特点是能将微量的dna大幅增加。pcr由1983年美国mullis首先提出设想,1985年由其发明聚合酶链反应,即简易dna扩增法,意味着pcr技术的真正诞生。到2013年,pcr已发展到第三代技术。
正链:与rna序列相同的那一个dna单链;复制中,正链就是与新链序列相同的原单链,非模板链。
下面通过具体实施例对本申请作进一步详细说明。以下实施例仅对本申请进行进一步说明,不应理解为对本申请的限制。
实施例
本例的拷贝数变异检测方法,包括以下步骤:
数据获取步骤,获取待测样本dna测序数据及健康人群dna测序数据,以及待测样本dna测序数据在人类参考基因组上各个位置的比对情况;通常,待测样本dna可以来自对待测生物样本dna进行测序而获得的数据;健康人群dna测序数据可以来自已经建立的健康人群dna数据库,或者来自对健康人群生物样本dna进行测序而获得的数据;原则上,待测生物样本dna和健康人群生物样本dna的测序方法相同,即平行测序。
深度统计步骤,其承接数据获取步骤,用于统计待测样本dna在固定窗口区域内的深度及健康人群中各单个样本dna在相应的固定窗口区域内深度,得到每个窗口的深度在健康人群的分布统计模型;
异常数据提取步骤,其承接数据获取步骤,用于将待测样本dna比对到人类参考基因组的比对文件中异常比对数据序列提取出来,获得可以支持拷贝数变异的第一维度结果;
窗口深度对比步骤,其承接数据获取步骤和深度统计步骤,用于将待测样本dna的窗口深度与健康人群各固定窗口深度分布进行比较,获得与健康人群固定窗口深度显出差异的窗口结果。
拷贝数变异判定步骤,其承接深度统计步骤和异常数据提取步骤,用于判定待测样本dna各窗口的深度异常是否为真实的拷贝数变异,获得判定结果;具体的,即判断待测样本dna的窗口深度与健康人群固定窗口深度的差异是否显著,如果差异显著,且能够得到第一维度结果的支持,则判定结果为真实的拷贝数变异,即阳性拷贝数变异。
检测结果输出步骤,其承接拷贝数变异判定步骤,用于输出拷贝数变异判定步骤的判断结果。
本例的检测方法中,数据获取步骤包括以下子步骤:
获取子步骤,包括获取待测样本dna测序数据;
过滤子步骤,包括对获取子步骤获取的待测样本dna测序数据进行质检,过滤去除低质量的测序数据,即过滤去除小于q30的测序数据,得到cleanfastqdata;
比对子步骤,包括将经过过滤子步骤的测序数据与人类参考基因组比对,获取待测样本dna的测序片段在人类参考基因组中的位置;即获取测序片段(reads)在人类参考基因组中对应的位置;具体而言,用bwa软件对cleanfastqdata进行比对得到sam格式文件,用samtools将sam格式文件转为bam格式,其中包含reads在人类参考基因组中对应的位置的信息,节省内存空间;
去重子步骤,包括根据比对子步骤的结果,去除比对到人类参考基因组相同位置的重复的测序片段;具体而言,去重子步骤处理bam文件,去除重复的reads,得到uniquebam文件;
校正子步骤,包括根据去重子步骤的结果,重新校正测序数据与人类参考基因组的比对结果。
本例的深度统计步骤,具体而言,包括利用参数化固定区域reads覆盖数量分布,令xijk代表与参考基因组上固定区域上的read数目。其中i代表第i号正常dna样本,j代表参考基因组第j个固定窗口位置。并且x符合如下分布:
xijk~nb(r,p)
参数r=rijk和p=pijk分别代表样本特定基因组区域的相应read数目,存在如下关系:
使用一组正常样本集,对于集合中的一个样本i,其他样本记为j(i)={h≠i},则xijk=∑h∈i(i)xhjk如果该样本固定窗口深度大于/小于一定的阈值,则会进行上一步替换。假设对照组固定窗口深度分布符合负二项分布,且均值ν=νijk,覆盖深度n=nijk=∑h∈i(i)nhjk、n’=n’ijk=∑h∈i(i)n'hjk,以上定义与独立样本服从负二项分布相一致,并且分布因子ρ相对较小。
x~nb(r,p)
根据正常样本集对每个固定窗口进行模型评估及参数固定。以此完成深度统计模块功能。
本例的检测方法中,拷贝数变异判定步骤包括以下子步骤:
突变显著性判定子步骤,用于判定待测样本dna各位点的突变的显著性,即判断与分布统计模型显示出差异的窗口,其差异是否显著。
突变类型判定子步骤,判定待测样本dna各位点的具有显著性的突变的类型是否真实,即判断存在显然差异的窗口是否能够得到第一维度结果的支持。
其中,突变显著性判定子步骤,具体而言,固定窗口深度差异变异预测的方法即模型选择的过程,一个真实的变异将出现在基因组上,且是在一个平稳的区域有着较大的差异。并且能够找到断点的统计模型。当某区域仅存在系统误差没有发生拷贝数变化的情况,零假设为样本均值和方差应该与正常样本集合的均值和方差保持一致。基于此,提出零假设和备择假设如下:
m0:μ=ν∨μ’=ν’
m1:μ,μ’>ν,ν’
并根据模型进行推断。
其中,以贝叶斯因子为判别条件,当贝叶斯因子<0.05(5*e-2),也就是零假设发生概率小于备择假设概率的20倍时,认为备择假设成立而零假设不成立,即该位置拷贝数变化为真实突变而非系统错误所导致的假阳性变化。
以及,当贝叶斯因子小于设定值时,认为备择假设成立而零假设不成立,即判定对比结果为有显著差异。
检测结果输出步骤输出的结果可以包括:真实突变位置,例如12号染色体上1444444绝对位置,参考基因组为hg19,以及突变类型,例如扩增/缺失。
试验例
本例对na12878标准品,采用novaseq进行测序,将获得的测序数据作为待测样本dna测序数据,采用前述的拷贝数变异检测方法对na12878标准品进行体细胞突变检测;并将检测获得的结果与真实的突变结果进行比较,用以评估本例拷贝数变异检测方法的准确性。
检测的拷贝数变异结果与真实的突变结果对比分析结果如图6所示,图6中,横坐标表示人类基因组上的位置,其中方框标注的为发生拷贝数变异的位置。图6的结果显示,本例的拷贝数变异检测方法能够真实准确的检测出na12878标准品的拷贝数变异情况,与真实的突变结果相吻合。
以上内容是结合具体的实施方式对本申请所作的进一步详细说明,不能认定本申请的具体实施只局限于这些说明。对于本申请所属技术领域的普通技术人员来说,在不脱离本申请构思的前提下,还可以做出若干简单推演或替换。
- 还没有人留言评论。精彩留言会获得点赞!