一种二氧化硅/环氧树脂复合材料界面热阻的预测新方法与流程
本发明属于热界面复合材料界面热阻预测技术领域,具体是指一种二氧化硅/环氧树脂复合材料界面热阻的预测新方法。
背景技术:
热界面材料可以分为很多种,但其中的聚合物基热界面材料由于具有电绝缘性能优异,质轻,易加工成型和低成本等优点越来越受到重视。然而由于大多数聚合物材料的热导率仅为0.1~0.3w/(m·k),导热能力较差,仅仅依赖聚合物自身的导热性能,难以满足电子元器件的散热要求,因此为了提高聚合物材料的导热性能通常是在聚合物基体材料中添加导热填料。而导热填料与高分子聚合物的界面相容性较差,导热填料粒子的表面不易被聚合物基体润湿,加之两者表面张力的差异,使得导热填料颗粒在高分子聚合物中难以充分有效地分散。因此,造成导热填料颗粒与聚合物基体的界面间有一定的空气间隙(空气热导率只有0.023w/(m·k),从而使得复合材料的界面热阻较大。目前,主要是通过界面改性的方法减小热阻。热界面材料的界面热阻是微尺度传热领域的热点问题,由于它反映的是界面声子的传输性质,因此直接影响材料的热传导性能,从而对微/纳米器件的设计和热优化产生影响。
目前,通过微尺度传热理论分析和实验方法尚未对热界面材料界面热阻的内部机制和变化规律建立完善的理论体系和研究方法,从实验研究的角度而言,界面热阻难以直接测量。近几年发展起来的分子动力学模拟是理论和实验有效的补充手段,它通过原子间相互作用势,求出每一个原子所受的力,在选定的时间步长、边界条件、初始位置和初始速度下,对有限数目的分子(原子)建立其牛顿动力学方程组,用数值方法求解得到这些原子经典运动轨迹和速度,然后对足够长时间的结果求统计平均,从而得到所需要的宏观物理量和力学量。
据文献报道,计算界面热阻的非平衡态分子动力学方法主要有以下两种模型:一种是周期性的反向非平衡分子动力学(rnemd)模型,模型一端和模型中部的粒子通过交换速度实现外加热流,周期性边界条件应用于x,y,z三个方向,模型中产生的局部温度具有对称性;另一种是绝热壁形式的非平衡分子动力学(nemd)模型,对模拟体系外加热流或温度梯度,周期性边界条件只应用于模型的y,z方向上,分别在x方向两端设置绝热壁,模型中产生的局部温度不具有对称性。周期性的rnemd模型由于采用的是对称体系,所以该方法的计算精度高,但计算量也大,而绝热壁的nemd模型计算量小,通过固定边界原子位置来实现绝热。本发明中的模型如图2所示,不同之处在于:通过在z方向上添加真空层来实现z方向上的绝热条件,通过交换体系两端的粒子速度来实现外加热流,计算量可进一步减小,这样,既能研究填料与基体界面热阻的大小,又能研究填料与基体单侧的界面相互作用能,计算得到的界面热阻通过与界面能量的对比分析,可以保证仿真结果的可靠性。
技术实现要素:
为解决上述现有难题,本发明为了解决目前通过理论和实验方法不能准确分析热界面材料传热机理的问题,提供一种计算热界面材料界面热阻的新方法。
本发明采用的技术方案如下:一种二氧化硅/环氧树脂复合材料界面热阻的预测新方法,包括以下步骤:
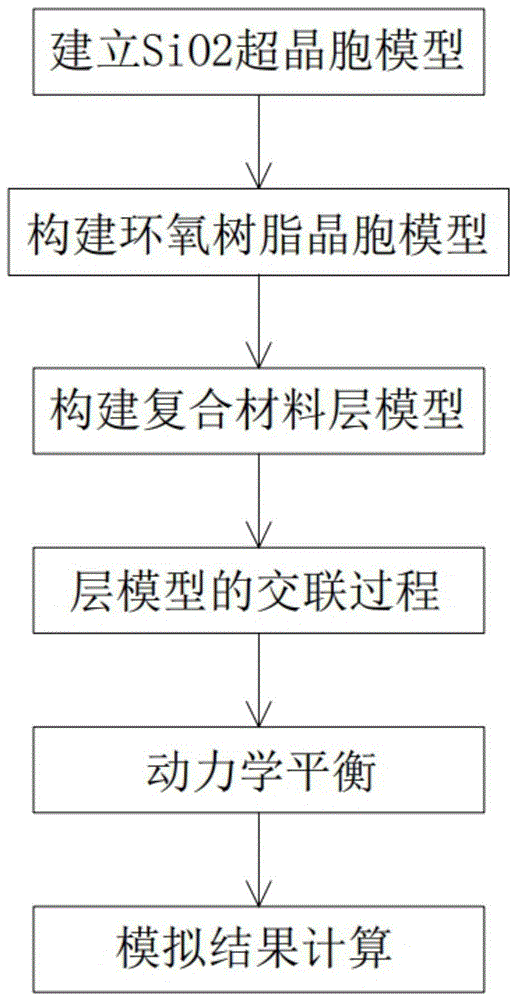
1)建立sio2超晶胞模型:根据α-sio2的空间群和晶格参数,建立sio2的晶体模型,并切割晶体构建超晶胞模型,然后对超胞模型进行几何优化;
2)构建环氧树脂晶胞模型:构建双酚a型环氧树脂和三乙烯四胺固化剂的单体分子模型,建立45个双酚a型环氧树脂分子和15个三乙烯四胺固化剂分子组成的初始密度为0.6g/cm3的3维周期性晶胞,对晶胞进行几何优化;
3)构建复合材料层模型:建立sio2和环氧树脂的层模型,选择优化算法对层模型进行几何优化;
4)层模型的交联过程:通过运行xink.pl交联脚本,对优化后层模型中的双酚a型环氧树脂和三乙烯四胺固化剂分子进行交联反应,得到环氧树脂分子链;
5)动力学平衡:对交联完成后的sio2/环氧树脂层状模型进行动力学模拟,使模型达到平衡;
6)模拟结果计算:通过运行tc.pl脚本,得到sio2/环氧树脂层状模型的热流密度和温度分布,计算得到界面热阻。
进一步地,步骤1)根据α-sio2的空间群p3221和晶格参数:
a=b=4.910,c=5.402;
在分数坐标下,硅原子和氧原子的位置参数分别为:
si:a=0.480781,b=0.480781,c=0;
o:a=0.150179,b=0.414589,c=0.116499;
得到sio2的晶体模型,剪切(00
进一步地,步骤2)构建双酚a型环氧树脂(dgeba)和三乙烯四胺(teta)固化剂分子模型,然后构建一个由45个dgeba分子和15个teta分子组成的初始密度为0.6g/cm3的3维周期性晶胞,选定compassii力场,模型精度设置为medium;选择smart优化算法对晶胞模型进行几何优化。
进一步地,步骤3)将上面得到的sio2超晶胞模型设置为layer1,环氧树脂晶胞模型设置为layer2,并在layer2下设置
进一步地,步骤4)在分子动力学软件中打开xink.pl交联脚本文件,定义反应位点,设置好交联参数,运行脚本文件进行交联模拟,得到环氧树脂分子链,然后将研究体系移到盒子中间。
进一步地,步骤5)对交联完成后的sio2/环氧树脂层状模型进行动力学模拟。
进一步地,步骤6)运行tc.pl脚本,对平衡后的模型进行界面热阻的计算;采用交换模型两端粒子速度实现外加热流的nemd方法,周期性边界条件只应用于模型的x,y方向上,在模型的z方向上引入30埃的真空层作为边界条件,得到层状模型的热流密度和温度分布,基于传统的傅里叶定律计算不同功能化和不同接枝率的二氧化硅与环氧树脂之间的界面热阻。
采用上述方案本发明取得有益效果如下:本发明一种二氧化硅/环氧树脂复合材料界面热阻的预测新方法,可研究不同功能化和不同接枝率的二氧化硅与环氧树脂之间的界面热阻,同时计算了界面能量和振动能谱,对界面问题的研究更加深入细致,且模型原子数目减少,计算量减少,但有界面能量和振动能谱印证界面热阻的模拟结果,以保证模拟结果的准确性,这有效节省了计算成本,并通过仿真计算和理论研究复合材料的界面热阻,不仅可以减少实验成本,对于设计具备优异性能的热界面材料具有重要的研究意义和应用价值。
附图说明
图1为本发明二氧化硅/环氧树脂复合材料界面热阻的预测新方法的流程图;
图2为二氧化硅/环氧树脂复合材料界面热阻的预测新方法的仿真模型示意图;
图3为二氧化硅/环氧树脂复合材料界面热阻的预测新方法的温度分布图。
具体实施方式
下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。
本发明采用的技术方案如下:一种二氧化硅/环氧树脂复合材料界面热阻的预测新方法,包括以下步骤:
1)建立sio2超晶胞模型:根据α-sio2的空间群和晶格参数,建立sio2的晶体模型,并切割晶体构建超晶胞模型,然后对超胞模型进行几何优化;
2)构建环氧树脂晶胞模型:构建双酚a型环氧树脂和三乙烯四胺固化剂的单体分子模型,建立45个双酚a型环氧树脂分子和15个三乙烯四胺固化剂分子组成的初始密度为0.6g/cm3的3维周期性晶胞,对晶胞进行几何优化;
3)构建复合材料层模型:建立sio2和环氧树脂的层模型,选择优化算法对层模型进行几何优化;
4)层模型的交联过程:通过运行xink.pl交联脚本,对优化后层模型中的双酚a型环氧树脂和三乙烯四胺固化剂分子进行交联反应,得到环氧树脂分子链;
5)动力学平衡:对交联完成后的sio2/环氧树脂层状模型进行动力学模拟,使模型达到平衡;
6)模拟结果计算:通过运行tc.pl脚本,得到sio2/环氧树脂层状模型的热流密度和温度分布,计算得到界面热阻。
进一步地,步骤1)根据α-sio2的空间群p3221和晶格参数:
a=b=4.910,c=5.402;
在分数坐标下,硅原子和氧原子的位置参数分别为:
si:a=0.480781,b=0.480781,c=0;
o:a=0.150179,b=0.414589,c=0.116499;
得到sio2的晶体模型,剪切(00
步骤2)构建双酚a型环氧树脂(dgeba)和三乙烯四胺(teta)固化剂分子模型,然后构建一个由45个dgeba分子和15个teta分子组成的初始密度为0.6g/cm3的3维周期性晶胞,选定compassii力场,模型精度设置为medium;选择smart优化算法对晶胞模型进行几何优化。
步骤3)将上面得到的sio2超晶胞模型设置为layer1,环氧树脂晶胞模型设置为layer2,并在layer2下设置
步骤4)在分子动力学软件中打开xink.pl交联脚本文件,定义反应位点,设置好交联参数,运行脚本文件进行交联模拟,得到环氧树脂分子链,然后将研究体系移到盒子中间。
步骤5)对交联完成后的sio2/环氧树脂层状模型进行动力学模拟。
步骤6)运行tc.pl脚本,对平衡后的模型进行界面热阻的计算;采用交换模型两端粒子速度实现外加热流的nemd方法,周期性边界条件只应用于模型的x,y方向上,在模型的z方向上引入30埃的真空层作为边界条件,得到层状模型的热流密度和温度分布,基于传统的傅里叶定律计算不同功能化和不同接枝率的二氧化硅与环氧树脂之间的界面热阻。
具体使用时,建立模型:采用分子动力学软件建立不同功能化和不同接枝率的二氧化硅与环氧树脂复合材料模型,运行xink.pl交联脚本对双酚a型环氧树脂(dgeba)和三乙烯四胺(teta)固化剂分子进行交联反应,得到环氧树脂分子链,然后进行动力学平衡,最后通过运行tc.pl脚本计算界面热阻,计算二氧化硅/环氧树脂复合材料界面热阻的仿真模型示意图如图2;仿真计算:对平衡后的模型进行界面热阻的计算;采用交换模型两端粒子速度实现外加热流的nemd方法,但边界条件不同于原来的边界条件,周期性边界条件只应用于模型的x、y方向,而在模型的z方向引入一个
动量守恒公式如下:
能量守恒公式如下:
从公式(1)和(2),可以得到:
值得注意的是,当mc=mh时,冷、热端粒子的新速度分别表示为:
其中下标c和h分别表示冷端和热端中的粒子,mc和mh分别表示在冷端和热端中被选择的粒子的质量,
通过计算单位时间单位面积内的能量交换,可以得到热流密度,公式如下:
其中〈jz(t)〉是z方向的热流密度,a是横截面积,t是粒子速度进行交换的时间间隔;
根据理想气体气动理论的温度关系公式:
得到整个仿真体系的局部温度计算公式:
其中,tk表示第k层的温度,nk是第k层的粒子数,kb是玻尔兹曼常数;界面热阻计算:根据上述公式(7)和(9)分别得到的热流密度和局部温度可计算界面热阻:
其中rint表示界面热阻,δt是不同位置的温度差。
实施列1
未经修饰的二氧化硅/环氧树脂复合材料界面热阻的计算过程,具体步骤如下:
建立sio2超晶胞模型:根据α-sio2的空间群p3221(第154号)和晶格参数:
a=b=4.910,c=5.402;
在分数坐标下,硅原子和氧原子的位置参数分别为:
si:a=0.480781,b=0.480781,c=0;
o:a=0.150179,b=0.414589,c=0.116499;
得到sio2的晶体模型,剪切(00
构建环氧树脂晶胞模型:构建双酚a型环氧树脂(dgeba)和三乙烯四胺(teta)固化剂分子模型,然后构建一个由45个dgeba分子和15个teta分子组成的初始密度为0.6g/cm3的3维周期性晶胞,其中compassii力场被选中,模型精度设置为medium;选择smart优化算法对晶胞模型进行几何优化,随着计算步长的增加,总的能量值趋于稳定时,得到收敛曲线,即模型达到平衡稳定状态;
构建复合材料层模型:将上面得到的sio2超晶胞模型设置为layer1,环氧树脂晶胞模型设置为layer2,并在layer2下设置
层模型的交联过程:在分子动力学软件中打开xink.pl交联脚本文件,在优化后层模型中的双酚a型环氧树脂(dgeba)和三乙烯四胺(teta)固化剂分子上定义反应位点,即对dgeba分子上的末端碳原子和teta分子上的氮原子进行重新命名,分别命名为r1和r2;设置的初始截断距离为
动力学平衡:对交联完成后的sio2/环氧树脂层状模型进行动力学模拟,在分子动力学软件中分别进行anneal和dynamics计算;nvt退火温度范围为300k~600k,时间步长为1fs,总模拟时长为200ps,力场为compassii,控温的方法为andersen;然后选择正则系综(nvt系综)对体系进行1次平衡,随后选择微正则系综(nve系综)再次进行平衡,2次模拟均设置温度为298k,步长分别为1fs和0.1fs,模拟时间长分别为2000ps和10ps,力场为compassii,控温的方法为andersen。
模拟结果计算:dynamics计算完成后选择interaction_energy.pl脚本计算界面能量;选择速度自相关函数收集原子轨迹,并对收集的轨迹数据进行分析,研究界面声子的匹配性;运行tc.pl脚本,对平衡后的模型进行界面热阻的计算;
未经修饰的二氧化硅/环氧树脂复合材料界面模型,计算得到的界面能量值为-284.81kcal/mol,计算振动能谱发现界面声子匹配性较差,界面热阻值约为0.625×10-8m2kw-1。
实施例2
kh550修饰的二氧化硅/环氧树脂复合材料界面热阻的计算过程,具体步骤如下:
建立sio2超晶胞模型:根据α-sio2的空间群p3221(第154号)和晶格参数:
a=b=4.910,c=5.402。
在分数坐标下,硅原子和氧原子的位置参数分别为:
si:a=0.480781,b=0.480781,c=0;
o:a=0.150179,b=0.414589,c=0.116499;
得到sio2的晶体模型,剪切(00
构建环氧树脂晶胞模型:构建双酚a型环氧树脂(dgeba)和三乙烯四胺(teta)固化剂分子模型,然后构建一个由45个dgeba分子和15个teta分子组成的初始密度为0.6g/cm3的3维周期性晶胞,其中compassii力场被选中,模型精度设置为medium;选择smart优化算法对晶胞模型进行几何优化,随着计算步长的增加,总的能量值趋于稳定时,得到收敛曲线,即模型达到平衡稳定状态;
构建复合材料层模型:将上面得到的sio2超晶胞模型设置为layer1,环氧树脂晶胞模型设置为layer2,并在layer2下设置
层模型的交联过程:在分子动力学软件中打开xink.pl交联脚本文件,在优化后层模型中的双酚a型环氧树脂(dgeba)和三乙烯四胺(teta)固化剂分子上定义反应位点,即对dgeba分子上的末端碳原子和teta分子上的氮原子进行重新命名,分别命名为r1和r2;设置的初始截断距离为
动力学平衡:对交联完成后的sio2/环氧树脂层状模型进行动力学模拟,在分子动力学软件中分别进行anneal和dynamics计算;nvt退火温度范围为300k~600k,时间步长为1fs,模拟时间长为200ps,力场为compassii,控温的方法为andersen;然后选择正则系综(nvt系综)对体系进行1次平衡,随后选择微正则系综(nve系综)再次进行平衡,2次模拟均设置温度为298k,步长分别为1fs和0.1fs,模拟时间长分别为2000ps和10ps,力场为compassii,控温的方法为andersen;
模拟结果计算:dynamics计算完成后选择interaction_energy.pl脚本计算界面能量;选择速度自相关函数收集原子轨迹,并对收集的轨迹数据进行分析,研究界面声子的匹配性;运行tc.pl脚本,对平衡后的模型进行界面热阻的计算;
kh550修饰的二氧化硅/环氧树脂复合材料界面模型,计算得到的界面能量值为-353.03kcal/mol,计算振动能谱发现界面声子匹配性较好,界面热阻值约为0.505×10-8m2kw-1。
以上所述仅为本发明的实施例,并非因此限制本发明的专利范围,凡是利用本发明说明书内容所作的等效结构或等效流程变换,或直接或间接运用在其它相关的技术领域,均同理包括在本发明的专利保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!