一种构建异交物种高密度基因型图谱的方法与流程
本发明涉及生物领域,尤其涉及分子育种和基因工程领域。
背景技术:
随着测序技术的发展,现代基因工具的应用极大地促进了各种作物的遗传改良,全基因组测序和重测序被广泛应用于遗传育种以及农艺性状的挖掘。目前,许多遗传研究方法都集中在具有自花传粉习性的植物上,通过连续自花传粉收集或产生自交系进行遗传研究和育种。比较典型的方法有适合对ems诱变的隐性突变基因进行分析的mutmap(1)和对自交群体进行分析的seg-map(2)等。这些方法主要包括三个步骤:1)测序2)基因分型3)基因定位。但是mutmap仅适用于诱变材料,而seg-map只适合应用于二倍体自交作物的基因分型和定位,即要求遗传群体最初的亲本是纯合的。在自然界及农业生产中,还有许多重要作物具有异花授粉的特性。由于自交不亲和、无性系繁殖方式或世代时间长等原因,这些植物没有可用于遗传研究和育种的自交系,使得农艺上重要基因的鉴定相对困难,特别是在多倍体基因组结构复杂的作物中(比如红薯、土豆)、药用植物(如苦艾、罂粟)和几乎所有的经济林木(用于生产茶叶、水果和坚果)。另外本方法同样适用于部分多子代的动物群体例如果蝇、家蚕、各种鱼类等。针对这些物种的重要基因的鉴定仍未有有效的方法。因而开发一种能够基于全基因组低成本测序来定位异交作物的重要农艺位点具有重要价值。
技术实现要素:
本发明的目的是提供一种能够大规模的获得基因型图谱的方法。经过大量研究,本发明提出来一种根据低覆盖高通量测序的结果进行基因分型的方法,得到了精确的基因型图谱,并且还可以进一步结合其他软件方法进行基因的连锁定位和后续基因功能的挖掘及基因组学的研究。
在本发明方法提出之前,异交物种群体基因型图谱的确定必须依赖于群体里每个子代个体的深度测序。
以典型的六倍体作物甘薯异交种群体为例:本发明方法之前,由于甘薯的高倍性,对其农艺性状的遗传研究较少。在同源多倍体作物中,遗传系统并不简单,因为其减数分裂过程相当复杂。在甘薯中,有六组同源染色体(但只有一套简并的参考基因组序列)与许多重组组合的可能性。与只有4个单倍型组合的二倍体异交种群体不同,六倍体甘薯在每个局部区域共能产生400个单倍型组合的可能性,因此要进行有效的单倍型定相是极其复杂的。
本发明方法使用tn5酶建立了全基因组测序文库(参见,hennigbp,veltenl,rackei,tucs,thomsm,rybinv,besirh,remansk,steinmetzlm:large-scalelow-costngslibrarypreparationusingarobusttn5purificationandtagmentationprotocol.g3-genesgenomgenet.2018;8:79-89)。利用二代测序方法得到低覆盖的目标作物子代群体测序结果,分别使用默认参数的bwa包(版本0.7.1,参见lih,durbinr:fastandaccurateshortreadalignmentwithburrows-wheelertransform.bioinformatics.2009;25:1754-1760)比对到其基因组。使用picard工具的“markdupl”模块(版本1.119)删除由于pcr扩增引入的错误扩增序列。利用genomeanalysistk软件(版本3.4.0)的“indelrealigner”对测序数据与参考基因组进行重新比对,并在每个群体的genomeanalysistk中使用“unifiedgenotyper”进行基因型调用。使用基因型调用数据(vcf格式文件)作为输入文件构建基因型图谱。在一定小的染色体区域内不会发生重组,根据这一原理,将目标植物的染色体分为多个区段(几百kb到1m左右),根据每个区域段内snp(单核苷酸的多态性)来源或根据snp计算的相关系数或者亲缘系数为依据,对子代的基因型进行判断,从而得到群体的单倍型高分辨图谱,用于后续的分析。
本方法适用于来自异交植物的遗传群体。通过群体规模的低覆盖测序,可以实现植物异交群体的基因型和单倍型综合分析,成本低,准确率高。
具体而言,本发明提供一种构建异交物种高密度基因型图谱的方法,其特征在于,所述方法包括下述步骤:
步骤1、对目标植物的亲本材料进行高覆盖测序,对目标植物的子代群体材料进行低覆盖测序,获得相应测序结果;
步骤2:将亲子代群体测序获得的序列比对到目标植物的现有基因组上,并得到变异文件,所述变异文件中包含每个被测序材料测得的变异位点信息;
步骤3:基于亲本材料的测序结果,对所有亲子代测序比对结果中的snp位点进行筛选,将筛选出的snp位点进行区段划分,并基于划分后各区段内基因片段的亲缘关系进行基因型分类;
步骤4:对各个区段中每个材料的基因型进行单倍体类型统一和缺失基因型填充。
在一种优选实现方式中,所述步骤3包括:
步骤3.1:基于亲本材料的测序结果,从目标植物测得的基因序列中,挑选至少一个亲本是杂合的snp位点;
步骤3.2:从所挑选出的位点中,过滤掉子代缺失严重的位点和低质量的变异位点;
步骤3.3:以预定序列长度作为窗口对所有材料的测序结果进行分区,所述窗口设置使得每个窗口内的标记数目不小于预定值且不小于预定的局部基因组区域长度,并计算每个窗口内两两材料之间的亲缘关系,用作相关系数;
步骤3.4:每个区段内根据材料之间的相关系数对各区段内的材料进行聚类。
在另一种优选实现方式中,所述步骤4包括:将每个窗口内不同测序材料的聚类结果进行比对,将相同材料的分类结果进行统一,并根据缺失位点的上下游类型填充缺失位点的基因型。
在另一种优选实现方式中,对于双交种群体植物,将其位点分为两类,第一类为在四种亲本单倍型中基因型分离比为3:1的变异位点,第二类为2:2的变异位点。
在另一种优选实现方式中,对于同源多倍体植物,所述步骤3还包括:
步骤3.1、筛选两个亲本中至少一个亲本的六个单倍型是1:5的snp位点,即单一型单核苷酸多态性simplexsnp或双单一单核苷酸多态性double-simplexsnp位点;
步骤3.2、根据每种材料基因序列中标记的数目对其基因序列进行分区段。
在另一种优选实现方式中,所述方法还包括基于异交植物的类型分别为每种植物选择不同的分区段窗口。
在另一种优选实现方式中,基因填充的步骤包括:
如果缺失的位点在整条基因序列的头部则将缺失基因型与其最近的已知基因型设置为一致,如果缺失的位点在整条基因序列尾部则将缺失基因型与其最近的已知基因型设置为一致,如果缺失部位上端已知基因型与缺失部位下端已知基因型一致,则将缺失部分填充为该基因型;
如果缺失部位上端已知基因型与缺失部位下端已知基因型不一致,则缺失部分上半部分填充上端已知基因型,缺失部分下半部分填充下端已知基因型。
在另一种优选实现方式中,所述筛选包括基于亲本是否为杂合位点进行筛选。
技术效果
本发明方法具有定位准确、成本低、可以大规模实施的优势。本申请的发明人对基因型图谱构建的准确性进行了对比,证实本发明方法的准确性明显高于现有方法。
具体而言,发明人将广泛用于各种遗传群体单个snp计算的beagle软件包应用于甘薯基因型数据集。并且以群体中两个深度序列的基因型(分别为75×和84×覆盖)为标准,对其准确性进行了评估,发现本发明方法准确性远远高于beagle(本发明方法>90%、beagle<45%)。发明人进一步比较了使用本发明方法输入的基因型数据和使用beagle数据的遗传作图,发现本发明方法的作图能力大大优于beagle的作图能力。
关于成本,本发明方法将子代个体进行低覆盖测序,并不需要将所有基因型都测出,只需挑出特异的位点,根据已有的低覆盖位置信息进行填充即可得到精确的位点基因型,大大降低了成本。
利用本发明方法鉴定异交作物中重要的农艺基因。能对已有的数量遗传学研究方法进行补充,为研究异交物种功能基因组学知识打下基础,用以识别控制农艺性状的基因和等位基因,并了解其内在机制;同时有助于异交植物的分子设计育种。
本发明方法可以应用于各种已测序异交植物,拥有染色体级别的参考基因组序列。
优选地,本发明方法可以应用于所有异交植物群体和多倍体作物(四倍体、六倍体)群体,双交种群体等包括结合其他技术进行后续功能基因组学研究
更优选地,本发明方法可以应用于亲本有明确的单倍体定相信息,或亲本来自两个已知自交系的杂交(即双交种,如本项目示例所使用的两个杂交水稻或两个杂交玉米进一步杂交生成的子代群体)。
附图说明
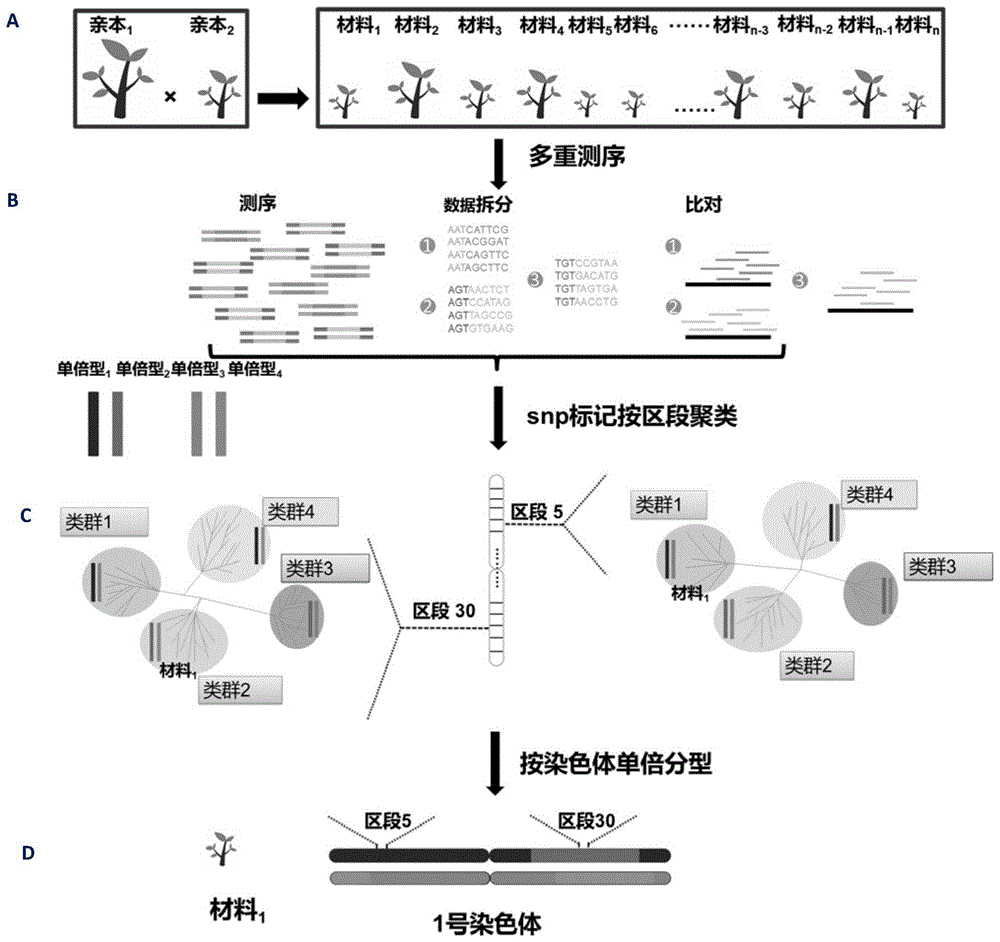
图1为普通异交植物的基因型图谱构建过程;
图2为双交种群体的基因型图谱构建过程;
图3为同源多倍体植物的基因型图谱构建过程;
具体实施方式
下面结合实施例及附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。
本发明中用到的主要软件包outcrossseq为市场可获得并且免费提供的,可在以下网址免费获取:
http://www.xhhuanglab.cn/tool/outcrossseq.html
本发明方法适用于
a)双交种群体(二倍体)如杂交水稻、杂交玉米等;
b)种植年限较长或其他原因无法获取初始四个亲本,但是可获得杂交的两个亲本的群体(二倍体)如核桃、茶树、杨树等;
c)同源多倍体群体如甘薯、土豆、甘蔗等;
d)部分动物家系群体如f1代的果蝇、家蚕、鱼类等。
利用本发明方法能够获得精确的基因型图谱并且结合相应表型进行农艺性状的定位,采用本发明方法发明人已经在不同的异交作物中定位到了多组基因,比如:
甘薯定位到已知基因:ibmyb1(3)、ibfbw2(4)、ibexp4a(5)等,此外还获得28个候选基因位点。以甘薯基因的表达量作为表型时,可以检测到表达数量性状基因座一共获得了267个eqtl.
核桃定位到候选基因:一共获得7个qtl位点,其中jrzfp1(6)、jrtcp4(7,8)根据已知报道选为候选基因
水稻定位到已知基因:oscol4(9)、hd1(10)、ghd7.1(11)、ghd8(12)等。
玉米定位到候选基因:一共定位到93个候选基因区域,其中47个与之前文章报道过的一致(13)。
实施例1
本实施例中,将对异交二倍体中的基因定位过程进行详细描述。
概括来讲,异交二倍体中的基因定位过程包括如下步骤:
步骤1:对目标植物的亲本进行高覆盖测序(>20×),对目标植物的子代群体进行低覆盖测序(<5×),获得相应测序结果。
步骤2:将子代群体(或亲子代群体)测序获得的短读长序列比对到目标植物的现有基因组上,并得到包含变异位点的变异文件。
步骤3:基于亲本材料的测序结果,对所有亲子代测序比对结果中的snp位点进行筛选,并且根据被测序材料中所有子代群体的亲缘关系进行聚类,然后对基因进行分区。对于异交二倍体每个区段分为四种基因型。比如,筛选出双亲都是杂合的snp位点,按片段长度对各材料的序列进行分区(比如300k为一个区段),根据区段内的snp位点差异计算每个区段内材料与材料之间的亲缘关系,利用亲缘关系对材料进行聚类。聚类可以采用r包hclust进行。
步骤4:对划分后的基因型进行类型统一和缺失填充。按染色体进行单倍体分型,得到准确的单倍型图谱。将基因型结果和表型结果输入gacd软件进行定位,定位得到候选基因。
下面结合图1,以核桃为例对本实施例中步骤3和4中聚类以及基因型定位的具体过程进行详细说明。核桃是典型的异交二倍体,两个亲本的单倍型为(h1h2×h3h4),在任何位点的基因型都有四种重组方式:h1-h3,h1-h4,h2-h3和h2-h4。
步骤1:首先,按照上述方式对核桃进行亲子代测序;
步骤2:基于测序结果与现有基因组比对获得变异信息。
步骤3.1:基于亲本材料的测序结果,由于双亲中的纯合snp导致群体中的固定基因型(例如,在父本中为aa基因型,在母本中为tt基因型,或在父本中为tt基因型,在母本中为tt基因型)。所以从核桃的基因序列中,挑选至少一个亲本是杂合的位点(例如,tt和at分别用于父本和母本;或at用于双亲)留下用于后续分析。
步骤3.2:
从所挑选出的位点中,过滤掉子代缺失严重的位点(缺失率可根据实验目的和要求进行调节)和低质量的位点
步骤3.3:将300kb(窗口大小可调节)作为窗口大小对基因组进行分区,同时保证每个窗口内的标记数目不小于3000个(标记数目可调节),并计算每个窗口内两两材料之间的亲缘关系(两个个体之间具有相同基因型的单核苷酸多态性数量/总单核苷酸多态性数量,删除任何一个个体中缺失数据的位点),产生了一个n×n的亲属关系矩阵(n代表个体数,该矩阵中包含了每个个体与任意一个其他个体的亲缘关系)用作相关系数。
步骤3.4:每个区段内根据材料之间相关系数用类平均法进行聚类,通过不断降低阈值,将材料分成四类,聚类中的四个组可以对应于四个单倍型组合(h1h3、h1h4、h2h3和h2h4)。
步骤4:将每个窗口内的聚类的结果进行统一,并根据缺失位点的上下游类型填充缺失位点的基因型。即,对于不同窗口中将同一材料分配在不同类别的情况,对该材料在各个窗口内的材料类别进行统一。
填充方式:如果某个材料缺失位点的上一个基因型和缺失位点下一个基因型一致则缺失位点也填充一样的基因型。否则根据材料间的亲缘关系填充与其亲缘关系较近的基因型。得到完整的基因型矩阵。
实施例2
本实施例中,将对双交种群体植物中的基因定位过程进行详细描述。
概括来讲,双交种群体中的基因定位过程包括如下步骤:
步骤1:对目标植物的亲本进行高覆盖测序(>15×),对目标植物的子代群体进行低覆盖测序(<1×),。
步骤2:采用实施例1类似方式将子代群体测序获得的序列与现有基因组进行基因比对。
步骤3:基于亲本材料的测序结果,对所有亲子代测序比对结果中的snp位点进行筛选,将选出的snp位点进行区段划分,并基于划分后各区段内基因片段的亲缘关系进行基因型分类。对于双交种群体,进一步地,根据亲本基因型将snp标记分为两类,再根据选出的snp标记分区段进行基因型判定。
步骤4:根据已知基因型填充未知区段。
下面结合图2,以水稻为例对本实施例中基因定位的具体过程进行详细说明。
水稻四个亲本的单倍型分别为(h1h1,h2h2,h3h3,h4h4)。其中h1与h2杂交得到f1(h1h2),其中h3与h4杂交得到f1(h3h4),h1h2×h3h4得到双交f1。双交f1任何位点的基因型都有四种重组方式:h1-h3,h1-h4,h2-h3和h2-h4。
步骤1,首先,按照上述方式对水稻四个亲本进行高覆盖测序,子代群体进行低覆盖测序。将测序结果用bwa比对到水稻基因组,利用picard删除由于pcr扩增引入的错误扩增序列。利用genomeanalysistk进行重新比对并获取变异信息,然后按照下述步骤进行位点选择和基因图谱构建。
步骤2:进行基因型比对。
步骤3:进一步地,对于双交种群体采用下述方式进行位点筛选,将其分为两类。
步骤3.1:挑选四种单倍型中基因型分离比为3:1的位点:
例如,亲本1-亲本2-亲本3-亲本4:a-t-t-t、g-g-g-c、t-t-c-t等;
挑选四种单倍型中基因型分离比为2:2的位点:(a-a-t-t的位点不合要求)
亲本1-亲本2-亲本3-亲本4:a-t-a-t、a-t-t-a等;
将300kb(窗口大小可调节)作为窗口大小对基因组进行分区,同时保证每个窗口内的标记数目不小于150个,选择每个窗口内3:1的标记,并且鉴定出双交子代测到的碱基是3:1中的1所对应亲本的支持数(比如,对于snp位:假设是at的情况,a对应3,t对应1,其代表的是包含t的位点来自与哪个亲本),作为亲本1的标记数。例如:材料1某位点的碱基是a,该位点四个亲本的基因型:a-t-t-t,该材料在该位点的来源则为亲本1。
通过上述判断来源方式,进行来源判断,比如某个材料在该区段内
来源于亲本1的标记:145个
来源于亲本2的标记:2个
来源于亲本3的标记:0个
来源于亲本4的标记:5个
则该区段的一个亲本为亲本1。
步骤3.2:
接下来确定第二个亲本的来源。选择每个窗口内2:2的标记,例如经过上一步的判断,其中一个亲本是亲本1,该位点四个亲本的基因型是c-g-c-g,子代在该位点测到的是g,则另一个亲本的来源只能是亲本4。
通过上述判断来源方式,比如某个材料在该区域(由于已经确定了一个来源是亲本1则另一个来源只能是亲本3或亲本4)
来源于亲本3的标记:67个
来源于亲本4的标记:3个
则该区域的另一个亲本为亲本4
在该区域内该材料的单倍型组合类型则为:h1-h4
步骤4:
根据已知基因型填充未知基因型,如果缺失的在整条基因序列的头部则判定缺失基因型与其最近的已知基因型一致,如果缺失的在整条基因序列尾部则缺失基因型与其最近的已知基因型一致,如果缺失部位上端已知基因型与缺失部位下端已知基因型一致,则缺失部分填充为该基因。
如果缺失部位上端已知基因型与缺失部位下端已知基因型不一致,则缺失部分上半部分填充上端已知基因型,缺失部分下半部分填充下端已知基因型。从而得到完整的基因型矩阵。
实施例3
本实施例中,将对同源多倍体植物中的基因定位过程进行详细描述。
概括来讲,该基因定位过程包括如下步骤:
1:亲本进行高覆盖测序(>60×),子代群体进行低覆盖测序(<5×)(比对基因组方式与核桃模式相同)。
2:将亲子代群体测序获得的序列比对到目标植物的现有基因组上,并得到变异文件,所述变异文件中包含每个被测序材料测得的基因序列和变异位点信息;
3:基于亲本材料的测序结果,对所有亲子代测序比对结果中的snp位点进行筛选,将选出的snp位点进行区段划分,并基于划分后各区段内基因片段的亲缘关系进行基因型分类;
4:根据已知基因型填充未知基因型;
下面以红薯为例进行过程的详细说明。
步骤1、对红薯双亲高覆盖测序,子代群体低覆盖测序。。
步骤2、将测序结果用bwa比对到现有甘薯基因组,picard删除由于pcr扩增引入的错误扩增序列。利用genomeanalysistk进行重新比对并获取变异信息
步骤3、基于亲本材料的测序结果,对所有亲子代测序比对结果中的snp位点进行筛选并进行区段划分和分类(gatk多倍体模式进行调用,查找变异位点,子代用正常模式);
具体而言,首先,3.1、筛选两个亲本中至少一个亲本的六个单倍型是1:5的snp位点,即单一型单核苷酸多态性simplexsnp或双单一单核苷酸多态性double-simplexsnp位点。
甘薯有六组同源染色体(但只有一组参考基因组序列)。六倍体的甘薯自交非常复杂,局部区域可以产生400种
步骤3.2、筛选单一型单核苷酸多态性和双单一单核苷酸多态性的位点(simplexordouble-simplexsinglenucleotidepolymorphisms)。
simplexsnp:两个亲本基因型为(atttttxtttttt)、(tttattxtttttt)、(ttttttxttattt)、(ttttttxtttta)等
double-simplexsnp:两个亲本基因型为(atttttxattttt)、(tttattxttttat)、(tttattxattttt)、(tattttxttttta)等
根据gatk找出的变异位点文件(vcf文件),亲本用六倍体的形式(0/0/0/0/0/1),子代用二倍体形式(0/1)。
步骤3.2:根据亲本的变异信息过滤出simplexsnp和double-simplexsnp位点,该种方式杂交产生的子代群体杂合的概率范围是1/12~2/12,由于子代测序覆盖过低,测到的杂合概率偏低,所以挑选子代群体杂合概率在0.04~0.28之间的snp位点。
步骤3.3:每固定数目的snp位点(例如300、500个)作为一个区段,计算每个窗口内snp位点间的相关系数。本实施例中,每300个标记分成一个区段。计算这300个标记两两之间的相关系数(该步骤用perl模块:statistics::basic)。
步骤3.4:每个区段内根据标记之间相关系数用类平均法进行聚类,挑选聚类后标记数大于10个(可更改)的簇
步骤4:在每个簇内每个材料根据相邻的已知基因型的标记对缺失位点进行填充,从而得到低缺失的基因型图谱。
根据每个簇内的snp总数、缺失率及杂合基因型比例等变量因素,分成40种情形进行填充。
例如,如果一个簇内总共有大于80个标记,缺失基因型<80%,并且杂合基因型比例=0%,那么填充的基因型也都为纯和。
根据基因结果和表现结果利用fastgwa进行定位,定位得到候选基因:ibmyb1、fbw2、ibexp4a。
虽然上面结合本发明的优选实施例对本发明的原理进行了详细的描述,本领域技术人员应该理解,上述实施例仅仅是对本发明的示意性实现方式的解释,并非对本发明包含范围的限定。实施例中的细节并不构成对本发明范围的限制,在不背离本发明的精神和范围的情况下,任何基于本发明技术方案的等效变换、简单替换等显而易见的改变,均落在本发明保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!