机器学习方法、系统及设备
1.本发明属于环境污染评估技术领域,特别是涉及一种机器学习方法、系统及设备。
背景技术:
2.重金属污染是一个极其严重的环境问题,含有重金属离子的废水、污泥等通过土壤、空气,尤其是食物链对人类的生存和身心健康造成了严重危害。近年来,吸附技术有了重大的发展,由于纳米材料吸附量高、分离简单、循环再生效率高等特定,在重金属废水处理中有着重要的应用前景。吸附材料存在各种活性成分如孔径、基团等,通过与吸附的金属离子形成离子键或共价键达到吸附重金属离子的目的。因此,对吸附材料进行有机修饰后,进一步增加它的活性位点,可以设计出更好的吸附剂。寻找或开发对重金属离子有较强吸附作用的理想吸附材料成为了当下的研究热点与难点。
3.现有的研究技术主要分为实验测量和理论计算。为了开发对重金属离子吸附能力强的材料,在实验上,研究人员在特定条件下合成新型的候选材料,配置重金属离子的溶液,再通过测量吸附前后的重金属离子浓度来判断候选材料的吸附能力。该类技术可以评估一个吸附材料的吸附能力,缺点是耗时长、成本高、难度大。为了降低试错成本,越来越多的科研工作者开始借助于理论计算,在前期辅助新型材料的设计。
4.具体地,现有技术包括以下缺陷:
5.第一,实验成本高;现有技术在利用实验制备吸附材料、测量吸附量等时需要昂贵的化学材料成本。
6.第二,效率低;有实验制备、合成、测量等繁琐的步骤,且一般都需要xps等精密仪器来操作,试验周期长。
7.第三,对吸附位点的认识不足;实验上很难测定重金属离子在吸附材料的具体吸附位点,而理论上仅仅研究了最佳的吸附位点,对吸附材料整体的吸附能力缺乏认识。
8.第四,理论研究任意位点的吸附能,需要大量的第一性原理计算,耗时长且成本高。
9.第五,泛化能力差;现有技术通常只能针对某种重金属离子进行研究,设计理想吸附材料,对其他重金属离子适用性不强。
10.因此,如何提供一种机器学习方法、系统及设备,以解决现有技术出现的成本高,效率低,能耗大、使用不便,泛化能力差等缺陷,实已成为本领域技术人员亟待解决的技术问题。
技术实现要素:
11.鉴于以上所述现有技术的缺点,本发明的目的在于提供一种机器学习方法、系统及设备,用于解决现有技术中成本高,效率低,能耗大、使用不便,泛化能力差的问题。
12.为实现上述目的及其他相关目的,本发明一方面提供一种机器学习方法,用于预测吸附材料上不同重金属离子的吸附能力;所述机器学习方法包括:进行第一性原理计算,
以获取吸附材料与多个重金属离子相结合的最优结构;在具有最优结构的吸附材料上随机放置重金属离子,获取带有重金属离子的复合结构,并计算不同重金属离子对应的复合结构的吸附能,以构建用于机器学习的数据集;构建任意带有重金属离子的复合结构的主吸附能预测模型;所述主吸附能预测模型用以预测吸附能;根据已构建的主吸附能预测模型,训练剩余带有重金属离子的复合结构的吸附能预测模型;统计所有已训练带有重金属离子的复合结构的吸附能预测模型的统计学数据,以评估所述吸附材料对重金属离子的吸附能力。
13.于本发明的一实施例中,在进行第一性原理计算的步骤之前,所述机器学习方法还包括:建立所述吸附材料的结构模型。
14.于本发明的一实施例中,所述进行第一性原理计算,以获取吸附材料与多个重金属离子相结合的最优结构的步骤包括:以能量为优化目标,按照共轭梯度下降方式不断迭代,以优化所述吸附材料和多种重金属离子的原子位置;当相邻两次能量之差小于预设能量差阈值时,将能量最小的结构设置为吸附材料与一重金属离子结合的最优结构。
15.于本发明的一实施例中,所述计算不同重金属离子对应的复合结构的吸附能,以构建用于机器学习的数据集的步骤包括:从不同重金属离子中选取任一重金属离子;按照第一预设计算个数,计算选取的重金属离子对应的最优结构的吸附能;按照第二预设计算个数,计算剩余重金属离子对应的最优结构的吸附能;其中,所述第一预设计算个数大于所述第二预设计算个数。
16.于本发明的一实施例中,选取的重金属离子对应的最优结构的吸附能=选取的重金属离子与吸附材料相结合的最优结构的整体能量-吸附材料的能量-选取的重金属离子的能量;剩余重金属离子对应的最优结构的吸附能=剩余重金属离子中一重金属离子与吸附材料相结合的最优结构的整体能量-吸附材料的能量-剩余重金属离子中一重金属离子的能量。
17.于本发明的一实施例中,所述构建任意带有重金属离子的复合结构的主吸附能预测模型为构建选取的重金属离子的复合结构的吸附能预测模型:根据构建的所述数据集,计算每一个原子的局部化学环境,将所有的原子的局部化学环境组成一矩阵,以构建主吸附能预测模型的输入;将主吸附能预测模型的输入与每一个原子对应的子网络进行矩阵运算,获取子网络的贡献值,将所有子网络的贡献值汇总为吸附能,以作为所述主吸附能预测模型的输出,并获取已训练好的模型参数。
18.于本发明的一实施例中,所述根据已构建的主吸附能预测模型,训练剩余带有重金属离子的复合结构的吸附能预测模型的步骤包括将迁移学习引入至已构建的主吸附能预测模型;该步骤包括:利用已训练好的模型参数初始化剩余带有重金属离子的复合结构的吸附能预测模型;调节剩余带有重金属离子的复合结构的吸附能预测模型的模型参数,直至两者的模型参数相同。
19.于本发明的一实施例中,所述机器学习方法还包括将已训练好的吸附能预测模型中任一吸附能预测模型作为评估通用框架,通过第一性原理计算、训练吸附能预测模型、迁移学习、统计能量平均值来预测吸附材料对任意重金属离子的吸附能力。
20.本发明另一方面提供一种机器学习系统,用于预测吸附材料上不同重金属离子的吸附能力;所述机器学习系统包括:计算模块,用于进行第一性原理计算,以获取吸附材料
与多个重金属离子相结合的最优结构;第一构建模块,用于在具有最优结构的吸附材料上随机放置重金属离子,获取带有重金属离子的复合结构,并计算不同重金属离子对应的复合结构的吸附能,以构建用于机器学习的数据集;第二构建模块,用于构建任意带有重金属离子的复合结构的主吸附能预测模型;所述主吸附能预测模型用以预测吸附能;训练模块,用于根据已构建的主吸附能预测模型,训练剩余带有重金属离子的复合结构的吸附能预测模型;统计模块,用于统计所有已训练带有重金属离子的复合结构的吸附能预测模型的统计学数据,以评估所述吸附材料对重金属离子的吸附能力。
21.本发明最后一方面提供一种机器学习设备,包括:处理器及存储器;所述存储器用于存储计算机程序,所述处理器用于执行所述存储器存储的计算机程序,以使所述机器学习设备执行所述机器学习方法。
22.如上所述,本发明所述的机器学习方法、系统及设备,具有以下有益效果:
23.第一,本发明避免了繁琐、昂贵的实验环节,完全基于理论计算,降低了成本,同时降低了对第一性原理计算的依赖,缩短了研究周期。
24.第二,通过机器学习方法建立高精度、高效率的吸附能预测模型,对某一重金属离子在吸附材料的任意吸附位点进行准确预测,为设计新型吸附材料提高理论指导。
25.第三,本发明结合迁移学习方法,使得预测模型适用于其他重金属离子,模型具有很强的泛化能力。
26.第四,本发明构建的模型简单,可进一步在工业界得到广泛应用。
附图说明
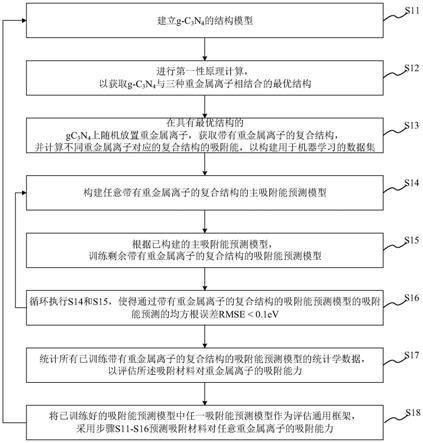
27.图1显示为本发明的机器学习方法于一实施例中的流程示意图。
28.图2a显示为本发明的g-c3n4分别与pb(ii)、hg(ii)、cd(ii)三种重金属离子相结合的最优结构的平视图。
29.图2b显示为本发明的g-c3n4分别与pb(ii)、hg(ii)、cd(ii)三种重金属离子相结合的最优结构的斜视图。
30.图3显示为本发明的基于局部化学环境构建的pb(ii)/g-c3n4的主吸附能预测模型示意图。
31.图4显示为本发明的利用迁移学习算法得到hg(ii)/g-c3n4和cd(ii)/g-c3n4的吸附能预测模型框架示意图。
32.图5显示为本发明的机器学习系统于一实施例中的原理结构示意图。
33.元件标号说明
34.5机器学习系统51第一构建模块52计算模块53第二构建模块54训练模块55统计模块56预测模块s11~s18步骤
具体实施方式
35.以下通过特定的具体实例说明本发明的实施方式,本领域技术人员可由本说明书所揭露的内容轻易地了解本发明的其他优点与功效。本发明还可以通过另外不同的具体实施方式加以实施或应用,本说明书中的各项细节也可以基于不同观点与应用,在没有背离本发明的精神下进行各种修饰或改变。需说明的是,在不冲突的情况下,以下实施例及实施例中的特征可以相互组合。
36.需要说明的是,以下实施例中所提供的图示仅以示意方式说明本发明的基本构想,遂图式中仅显示与本发明中有关的组件而非按照实际实施时的组件数目、形状及尺寸绘制,其实际实施时各组件的型态、数量及比例可为一种随意的改变,且其组件布局型态也可能更为复杂。
37.本发明所述机器学习方法、系统及设备的技术原理如下:
38.本发明以吸附材料(例如,g-c3n4吸附材料)为研究对象,在第一性原理计算的基础上,考察重金属离子不同位点的吸附强弱关系,结合深度神经网络建立吸附材料(例如,g-c3n4吸附材料)对某一重金属离子的高精度、高效率的吸附能预测模型,并将迁移学习引入吸附能预测中,得到吸附材料(例如,g-c3n4吸附材料)对其他重金属离子的吸附预测模型。融合大数据统计学知识,准确评估吸附材料(例如,g-c3n4吸附材料)对重金属离子的吸附能力。以此来大幅降低了吸附材料的设计成本,为设计新型吸附材料来吸附重金属离子提供有力指导。本发明不仅适用于吸附材料(例如,g-c3n4吸附材料),也适用于其他吸附材料对不同重金属离子的吸附能力预测。
39.实施例一
40.本实施例提供一种机器学习方法,用于预测吸附材料上不同重金属离子的吸附能力;所述机器学习方法包括:
41.进行第一性原理计算,以获取吸附材料与任一重金属离子相结合的最优结构;
42.在具有最优结构的吸附材料上随机放置重金属离子,获取带有重金属离子的复合结构,并计算不同重金属离子对应的复合结构的吸附能,以构建用于机器学习的数据集;
43.构建任意带有重金属离子的复合结构的主吸附能预测模型;所述主吸附能预测模型用以预测吸附能;
44.根据已构建的主吸附能预测模型,训练剩余带有重金属离子的复合结构的吸附能预测模型;
45.统计所有已训练带有重金属离子的复合结构的吸附能预测模型的统计学数据,以评估所述吸附材料对重金属离子的吸附能力。
46.以下将结合图示对本实施例所提供的机器学习方法进行详细描述。本实施例所述机器学习方法用于预测吸附材料上不同重金属离子的吸附能力。于实际应用中,所述吸附材料可以采用g-c
x
n
y
,例如,si3n4、ge3n4、c3p4、si3p4、mos2、c2n2、g-c3n4(c3n4具有5种相结构,其中g相是g-c3n4,是一种典型的聚合物半导体)等二维材料。
47.请参阅图1,显示为机器学习方法于一实施例中的流程示意图。如图1所示,所述机器学习方法具体包括以下步骤:
48.s11,建立所述g-c3n4的结构模型。
49.具体地,建立平行四边形的g-c3n4结构模型。
50.s12,进行第一性原理计算,以获取g-c3n4与三种重金属离子相结合的最优结构。于本实施例中,三种重金属离子分别为pb(ii)、hg(ii)、cd(ii)三种离子。请参阅图2a和2b,分别显示为g-c3n4分别与pb(ii)、hg(ii)、cd(ii)三种重金属离子相结合的最优结构的平视图和斜视图。
51.在本实施例中,所述第一性原理是指根据原子核和电子相互作用的原理及其基本运动规律,运用量子力学原理,从具体要求出发,经过近似处理后直接求解薛定谔方程的算法。经常用来计算材料体系的基态性质等。
52.具体地,步骤s11以能量为优化目标,按照共轭梯度下降方式不断迭代,以优化所述吸附材料和多种重金属离子的原子位置;当相邻两次能量之差小于预设能量差阈值(例如,小于1e-6ev)时,将能量最小的结构设置为合物半导体与一重金属离子结合的最优结构。
53.在本实施例中,按照共轭梯度下降方式是指在优化过程中,以吸附材料和重金属离子的结构,即他们的原子xyz坐标作为输入,就可以计算得到他们的能量。能量对xyz坐标的导数就可以得到每个原子在xyz方向上的受力,称为原子力。根据受力大小,原子就会运动,运动之后又会产生新的坐标,即新的结构。直到两次结构之间的能量差小于阈值,优化就停止。
54.在本实施例中,在利用共轭梯度下降算法优化结构过程中,采用现在主流的广义梯度近似general gradient approximation(gga)(属于密度泛函理论中的一类,密度泛函理论density functional theory(dft),是一种研究多电子体系电子结构的量子力学方法。密度泛函理论在物理和化学上都有广泛的应用,特别是用来研究分子和凝聚态的性质,是凝聚态物理计算材料学和计算化学领域最常用的方法之一),其在原有泛函的基础上加入了密度函数的梯度,而密度函数的梯度,可以理解为电子动能、或是电子密度随空间的改变,gga中的perdew-berke-ernzerhof(pbe)交换关联泛函,可以预测更准确的体系能量、结合能、活化能。在处理计算体系中原子的电子态时,即计算离子-电子相互作用能,在对能带结构进行数值计算时所引入的一个虚拟的势,即赝势,赝势选择投影扩充波投影扩充波projector augmented-wave(paw)。
55.s13,在具有最优结构的g-c3n4上随机设置重金属离子(于本实施例中,随机放置pb(ii)、hg(ii)、cd(ii)),获取带有重金属离子的复合结构,并计算不同重金属离子对应的复合结构的吸附能,以构建用于机器学习的数据集。在本实施例中,所述数据集包括最优结构的g-c3n4和对应的吸附能。
56.具体地,所述s13包括以下步骤:
57.从不同重金属离子中选取任一重金属离子。
58.于本实施例中,选取pb(ii)
59.按照第一预设计算个数,计算选取的重金属离子对应的最优结构的吸附能。
60.于本实施例中,选取的重金属离子对应的最优结构为pb(ii)/g-c3n4。
61.所述第一预设计算个数设置为7000~10000个,7000-10000个结构能够保证庞大的机器学习数据集,使得最终预测的精度更高。
62.选取的重金属离子对应的最优结构的吸附能=选取的重金属离子与吸附材料相结合的最优结构的整体能量-吸附材料的能量-选取的重金属离子的能量,
63.计算公式具体为:
64.δe=e
sub+met-e
sub-e
met
65.其中,δe为选取的重金属离子对应的最优结构的吸附能,e
sub+met
为选取的重金属离子与吸附材料相结合的最优结构的整体能量,e
sub
为吸附材料的能量,e
met
为选取的重金属离子的能量。
66.按照第二预设计算个数,计算剩余重金属离子对应的最优结构的吸附能;其中,所述第一预设计算个数大于所述第二预设计算个数。
67.于本实施例中,剩余重金属离子为hg(ii)和cd(ii)。
68.剩余重金属离子对应的最优结构的吸附能=剩余重金属离子中一重金属离子与吸附材料相结合的最优结构的整体能量-吸附材料的能量-剩余重金属离子中一重金属离子的能量。
69.s14,构建任意带有重金属离子的复合结构的主吸附能预测模型;所述主吸附能预测模型用以预测吸附能。
70.于本实施例中,构建pb(ii)/g-c3n4的主吸附能预测模型。
71.具体地,所述s14包括:
72.根据构建的所述数据集,计算每一个原子的局部化学环境,将所有的原子的局部化学环境组成一矩阵,以构建主吸附能预测模型的输入。在本实施例中,选取原子的局部化学环境作为结构特征,该结构特征能够很好地满足周期性材料中的平移不变性、旋转不变性、置换不变性。
73.其中,计算每一个原子的局部化学环境的计算过程如下:
74.对于第i个原子和其邻居原子j,第i个原子的局部化学环境g
ij
表示为:
[0075][0076][0077][0078]
e(x)=x/||x||
[0079]
上述公式中的下标表示两者的差,x
ij
表示第i个原子和第j个原子的x坐标之差,a
(i)
和b
(i)
表示距离第i个原子最近的两个原子,r
ia
表示第i个原子的坐标和a原子的坐标矢量之差,r
ib
表示第i个原子的坐标和b原子的坐标矢量之差。邻居原子之间计算的截止距离设为两个原子距离小于的才视为邻居原子;r
ij
是第i个原子和第j个原子的坐标向量之差,即(x
ij
,y
ij
,z
ij
),也可写成(x
i-x
j
,y
i-y
j
,z
i-z
j
)。其中j是i的邻居原子,即与i的距离小于阈值的原子;r
ia(i)
,这里a(i)表示离第i个原子最近的原子;r
ib(i)
,这里b(i)表示离第i个原子第二近的原子;e(x)表示向量x的方向向量。||x||是(x2+y2+z2)^0.5,是向量的长度。e(x)=(x/||x||,y/||x||,z/||x||)称为“旋转矩阵”。
[0080]
将主吸附能预测模型的输入与每一个原子对应的子网络进行矩阵运算,获取子网
络的贡献值,将所有子网络的贡献值汇总为吸附能,以作为所述主吸附能预测模型的输出,并获取已训练好的模型参数。请参阅图3,显示为基于局部化学环境构建的pb(ii)/g-c3n4的主吸附能预测模型示意图。如图3所示,对于pb(ii)/g-c3n4,有3个子网络,分别是pb、c、n。神经网络的结构为3~5层隐含层,每层10~50个神经元,训练过程中以adam优化器(一种优化算法,用于求解最优化问题)进行迭代,选取32~128为批量训练单元,训练时初始学习率为0.002~0.003,即每次更新参数的步长。在训练之前,神经网络中的神经元所代表的参数数值是随机初始化的,然后根据输出-输入的对应关系来调整神经元的参数,即训练过程。训练好神经网络后,得到一个模型参数,对于一个新的结构,在计算好局部化学环境的矩阵之后,利用矩阵和这个模型参数进行矩阵运算,就可以预测得到吸附能,这个过程非常快,比dft计算一个吸附能快近10000倍。
[0081]
s15,根据已构建的主吸附能预测模型,训练剩余带有重金属离子的复合结构的吸附能预测模型。
[0082]
于本实施例中,所述s15包括:
[0083]
利用已训练好的模型参数初始化剩余带有重金属离子的复合结构的吸附能预测模型。
[0084]
调节剩余带有重金属离子的复合结构的吸附能预测模型的模型参数,直至模型收敛,或模型的误差rmse小于0.1。
[0085]
由于hg(ii)/g-c3n4和cd(ii)/g-c3n4的数据量很小,采用迁移学习算法,利用已训练好的pb(ii)/g-c3n4的主吸附能预测模型的模型参数初始化hg(ii)/g-c3n4和cd(ii)/g-c3n4的模型参数。再对hg(ii)/g-c3n4和cd(ii)/g-c3n4的模型参数进行微调,为了保证参数震荡幅度很小,达到微调的目的,此时学习率调整为0.001~0.0015,hg(ii)/g-c3n4和cd(ii)/g-c3n4的模型参数与pb(ii)/g-c3n4相同,如神经元个数等,接着就是训练这两个模型。于本实施例中,将迁移学习用在吸附能预测模型中,可解决机器学习预测材料性质中数据量小带来的预测不准、过拟合等问题。
[0086]
请参阅图4,显示为利用迁移学习算法得到hg(ii)/g-c3n4和cd(ii)/g-c3n4的吸附能预测模型框架示意图。
[0087]
s16,循环执行s14和s15,使得通过带有重金属离子的复合结构的吸附能预测模型的吸附能预测的均方根误差rmse<0.1ev,即预测的吸附能和实际的吸附能的均方根误差:
[0088][0089]
其中,m是能量的个数,也等于结构的个数,相当于pb中的7000-10000;e
nn,i
是用神经网络预测的第i结构的能量,即第i个预测能量。e
dft,i
是用dft计算(即第一性原理计算)的第i个结构的能量,即第i个计算能量。e
rmse
计算就是这两者的均方根误差。
[0090]
s17,统计所有已训练带有重金属离子的复合结构的吸附能预测模型的统计学数据,以评估所述吸附材料对重金属离子的吸附能力。
[0091]
具体地,利用训练好的hg(ii)/g-c3n4和cd(ii)/g-c3n4吸附能预测模型进一步预测7000~10000个对应的结构,使得三种体系的吸附能量个数相同,此时再利用统计学知识,计算并分析三种体系能量的最小值、最大值、标准差、平均值。平均值稳定、可靠,可以综合考虑所有的能量数据,所包含的信息最多。因此,选取能量的平均值来衡量吸附能力的大
小。
[0092]
s18,将已训练好的吸附能预测模型中任一吸附能预测模型作为评估通用框架,采用步骤s11-s16预测吸附材料对任意重金属离子的吸附能力。
[0093]
例如,对于新的重金属离子如cu,也可以只进行少量的dft计算,然后用pb的吸附能预测模型参数来进行迁移学习,从而得到cu的吸附能预测模型,然后快速预测7000-10000个cu的吸附能,取平均值,便可知吸附材料对cu的整体吸附能力。
[0094]
本实施例所述机器学习方法具有以下有益效果:
[0095]
第一,本实施例所述机器学习方法避免了繁琐、昂贵的实验环节,完全基于理论计算,降低了成本,同时降低了对第一性原理计算的依赖,缩短了研究周期。
[0096]
第二,通过机器学习方法建立高精度、高效率的吸附能预测模型,对某一重金属离子在吸附材料的任意吸附位点进行准确预测,为设计新型吸附材料提高理论指导。
[0097]
第三,本实施例所述机器学习方法结合迁移学习方法,使得预测模型适用于其他重金属离子,模型具有很强的泛化能力。
[0098]
第四,本实施例所述机器学习方法构建的模型简单,可进一步在工业界得到广泛应用。
[0099]
实施例二
[0100]
本实施例提供一种机器学习系统,用于预测吸附材料上不同重金属离子的吸附能力;所述机器学习系统包括:
[0101]
计算模块,用于进行第一性原理计算,以获取吸附材料与多个重金属离子相结合的最优结构;
[0102]
第一构建模块,用于在具有最优结构的吸附材料上随机设置重金属离子,获取带有重金属离子的复合结构,并计算不同重金属离子对应的复合结构的吸附能,以构建用于机器学习的数据集;
[0103]
第二构建模块,用于构建任意带有重金属离子的复合结构的主吸附能预测模型;所述主吸附能预测模型用以预测吸附能;
[0104]
训练模块,用于根据已构建的主吸附能预测模型,训练剩余带有重金属离子的复合结构的吸附能预测模型;
[0105]
统计模块,用于统计所有已训练带有重金属离子的复合结构的吸附能预测模型的统计学数据,以评估所述吸附材料对重金属离子的吸附能力。
[0106]
以下将结合图示对本实施例所提供机器学习系统进行详细描述。请参阅图5,显示为机器学习系统于一实施例中的原理结构示意图。如图5所示,所述机器学习系统5包括第一构建模块51、计算模块52、第二构建模块53、训练模块54、统计模块55及预测模块56。
[0107]
所述第一构建模块51用于建立所述g-c3n4的结构模型。
[0108]
具体地,所述第一构建模块51建立平行四边形的g-c3n4结构模型。
[0109]
所述计算模块52用于进行第一性原理计算,以获取g-c3n4与三种重金属离子相结合的最优结构。于本实施例中,三种重金属离子分别为pb(ii)、hg(ii)、cd(ii)三种离子。请参阅图2a和2b,分别显示为g-c3n4分别与pb(ii)、hg(ii)、cd(ii)三种重金属离子相结合的最优结构的平视图和斜视图
[0110]
在本实施例中,所述第一性原理是指根据原子核和电子相互作用的原理及其基本
运动规律,运用量子力学原理,从具体要求出发,经过近似处理后直接求解薛定谔方程的算法。经常用来计算材料体系的基态性质等。
[0111]
具体地,所述计算模块52以能量为优化目标,按照共轭梯度下降方式不断迭代,以优化所述吸附材料和多种重金属离子的原子位置;当相邻两次能量之差小于预设能量差阈值(例如,小于1e-6ev)时,将能量最小的结构设置为合物半导体与一重金属离子结合的最优结构。
[0112]
所述第一构建模块51还用于在具有最优结构的g-c3n4上随机设置重金属离子(于本实施例中,随机放置pb(ii)、hg(ii)、cd(ii)),获取带有重金属离子的复合结构,并计算不同重金属离子对应的复合结构的吸附能,以构建用于机器学习的数据集。在本实施例中,所述数据集包括最优结构的g-c3n4和对应的吸附能。
[0113]
具体地,所述第一构建模块51从不同重金属离子中选取任一重金属离子,按照第一预设计算个数,计算选取的重金属离子对应的最优结构的吸附能;按照第二预设计算个数,计算剩余重金属离子对应的最优结构的吸附能;其中,所述第一预设计算个数大于所述第二预设计算个数。
[0114]
选取的重金属离子对应的最优结构的吸附能=选取的重金属离子与吸附材料相结合的最优结构的整体能量-吸附材料的能量-选取的重金属离子的能量,
[0115]
计算公式具体为:
[0116]
δe=e
sub+met-e
sub-e
met
[0117]
其中,δe为选取的重金属离子对应的最优结构的吸附能,e
sub+met
为选取的重金属离子与吸附材料相结合的最优结构的整体能量,e
sub
为吸附材料的能量,e
met
为选取的重金属离子的能量。
[0118]
剩余重金属离子对应的最优结构的吸附能=剩余重金属离子中一重金属离子与吸附材料相结合的最优结构的整体能量-吸附材料的能量-剩余重金属离子中一重金属离子的能量。
[0119]
所述第二构建模块53用于构建任意带有重金属离子的复合结构的主吸附能预测模型;所述主吸附能预测模型用以预测吸附能。
[0120]
具体地,所述第二构建模块53根据构建的所述数据集,计算每一个原子的局部化学环境,将所有的原子的局部化学环境组成一矩阵,以构建主吸附能预测模型的输入。在本实施例中,选取原子的局部化学环境作为结构特征,该结构特征能够很好地满足周期性材料中的平移不变性、旋转不变性、置换不变性。
[0121]
其中,计算每一个原子的局部化学环境的计算过程如下:
[0122]
对于第i个原子和其邻居原子j,第i个原子的局部化学环境g
ij
表示为:
[0123][0124]
[0125][0126]
e(x)=x/||x||
[0127]
上述公式中的下标表示两者的差,x
ij
表示第i个原子和第j个原子的x坐标之差,a
(i)
和b
(i)
表示距离第i个原子最近的两个原子,r
ia
表示第i个原子的坐标和a原子的坐标矢量之差,r
ib
表示第i个原子的坐标和b原子的坐标矢量之差。邻居原子之间计算的截止距离设为两个原子距离小于的才视为邻居原子;;r
ij
是第i个原子和第j个原子的坐标向量之差,即(x
ij
,y
ij
,z
ij
),也可写成(x
i-x
j
,y
i-y
j
,z
i-z
j
)。其中j是i的邻居原子,即与i的距离小于阈值的原子;r
ia(i)
,这里a(i)表示离第i个原子最近的原子;r
ib(i)
,这里b(i)表示离第i个原子第二近的原子;e(x)表示向量x的方向向量。||x||是(x2+y2+z2)^0.5,是向量的长度。e(x)=(x/||x||,y/||x||,z/||x||)称为“旋转矩阵”。
[0128]
将主吸附能预测模型的输入与每一个原子对应的子网络进行矩阵运算,获取子网络的贡献值,将所有子网络的贡献值汇总为吸附能,以作为所述主吸附能预测模型的输出,并获取已训练好的模型参数。
[0129]
与所述第一构建模块51和所述第二构建模块53耦合的训练模块54用于根据已构建的主吸附能预测模型,训练剩余带有重金属离子的复合结构的吸附能预测模型。
[0130]
于本实施例中,所述训练模块54用于利用已训练好的模型参数初始化剩余带有重金属离子的复合结构的吸附能预测模型。调节剩余带有重金属离子的复合结构的吸附能预测模型的模型参数,直至两者的模型参数相同。
[0131]
与所述训练模块54耦合的统计模块55用于循环调用所述第一构建模块51和所述第二工构建模块52,使得通过带有重金属离子的复合结构的吸附能预测模型的吸附能预测的均方根误差rmse<0.1ev,即预测的吸附能和实际的吸附能的均方根误差:
[0132][0133]
所述统计模块55还用于统计所有已训练带有重金属离子的复合结构的吸附能预测模型的统计学数据,以评估所述吸附材料对重金属离子的吸附能力。
[0134]
具体地,所述统计模块55利用训练好的hg(ii)/g-c3n4和cd(ii)/g-c3n4吸附能预测模型进一步预测7000~10000个对应的结构,使得三种体系的吸附能量个数相同,此时再利用统计学知识,计算并分析三种体系能量的最小值、最大值、标准差、平均值。平均值稳定、可靠,可以综合考虑所有的能量数据,所包含的信息最多。因此,选取能量的平均值来衡量吸附能力的大小。
[0135]
与所述第一构建模块51、所述计算模块52、所述第二构建模块53、所述训练模块54和所述统计模块55耦合的预测模块56用于将已训练好的吸附能预测模型中任一吸附能预测模型作为评估通用框架,调用第一构建模块51,计算模块52,第二构建模块53、训练模块54及统计模块55来预测吸附材料对任意重金属离子的吸附能力。
[0136]
需要说明的是,应理解以上系统的各个模块的划分仅仅是一种逻辑功能的划分,实际实现时可以全部或部分集成到一个物理实体上,也可以物理上分开。且这些模块可以
全部以软件通过处理元件调用的形式实现,也可以全部以硬件的形式实现,还可以部分模块通过处理元件调用软件的形式实现,部分模块通过硬件的形式实现。例如:x模块可以为单独设立的处理元件,也可以集成在上述系统的某一个芯片中实现。此外,x模块也可以以程序代码的形式存储于上述系统的存储器中,由上述系统的某一个处理元件调用并执行以上x模块的功能。其它模块的实现与之类似。这些模块全部或部分可以集成在一起,也可以独立实现。这里所述的处理元件可以是一种集成电路,具有信号的处理能力。在实现过程中,上述方法的各步骤或以上各个模块可以通过处理器元件中的硬件的集成逻辑电路或者软件形式的指令完成。以上这些模块可以是被配置成实施以上方法的一个或多个集成电路,例如:一个或多个特定集成电路(application specific integrated circuit,简称asic),一个或多个微处理器(digital singnal processor,简称dsp),一个或者多个现场可编程门阵列(field programmable gate array,简称fpga)等。当以上某个模块通过处理元件调度程序代码的形式实现时,该处理元件可以是通用处理器,如中央处理器(central processing unit,简称cpu)或其它可以调用程序代码的处理器。这些模块可以集成在一起,以片上系统(system-on-a-chip,简称soc)的形式实现。
[0137]
实施例三
[0138]
本实施例提供一种机器学习设备,包括:处理器、存储器、收发器、通信接口或/和系统总线;存储器和通信接口通过系统总线与处理器和收发器连接并完成相互间的通信,存储器用于存储计算机程序,通信接口用于和其他设备进行通信,处理器和收发器用于运行计算机程序,使机器学习设备执行如上所述机器学习方法的各个步骤。
[0139]
上述提到的系统总线可以是外设部件互连标准(peripheral component interconnect,简称pci)总线或扩展工业标准结构(extended industry standard architecture,简称eisa)总线等。该系统总线可以分为地址总线、数据总线、控制总线等。为便于表示,图中仅用一条粗线表示,但并不表示仅有一根总线或一种类型的总线。通信接口用于实现数据库访问装置与其他设备(如客户端、读写库和只读库)之间的通信。存储器可能包含随机存取存储器(random access memory,简称ram),也可能还包括非易失性存储器(non-volatile memory),例如至少一个磁盘存储器。
[0140]
上述的处理器可以是通用处理器,包括中央处理器(central processing unit,简称cpu)、网络处理器(network processor,简称np)等;还可以是数字信号处理器(digital signal processing,简称dsp)、专用集成电路(application specific integrated circuit,简称asic)、现场可编程门阵列(field programmable gate array,简称fpga)或者其他可编程逻辑器件、分立门或者晶体管逻辑器件、分立硬件组件。
[0141]
本发明所述的机器学习方法的保护范围不限于本实施例列举的步骤执行顺序,凡是根据本发明的原理所做的现有技术的步骤增减、步骤替换所实现的方案都包括在本发明的保护范围内。
[0142]
本发明还提供一种机器学习系统,所述机器学习系统可以实现本发明所述的机器学习方法,但本发明所述的机器学习方法的实现装置包括但不限于本实施例列举的机器学习系统的结构,凡是根据本发明的原理所做的现有技术的结构变形和替换,都包括在本发明的保护范围内。
[0143]
本发明所述机器学习方法、系统及设备具有以下有益效果:
[0144]
第一,本发明避免了繁琐、昂贵的实验环节,完全基于理论计算,降低了成本,同时降低了对第一性原理计算的依赖,缩短了研究周期。
[0145]
第二,通过机器学习方法建立高精度、高效率的吸附能预测模型,对某一重金属离子在吸附材料的任意吸附位点进行准确预测,为设计新型吸附材料提高理论指导。
[0146]
第三,本发明结合迁移学习方法,使得预测模型适用于其他重金属离子,模型具有很强的泛化能力。
[0147]
第四,本发明构建的模型简单,可进一步在工业界得到广泛应用。综上所述,本发明有效克服了现有技术中的种种缺点而具高度产业利用价值。
[0148]
上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属技术领域中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1