氧化还原双敏感三硫键桥连二聚体前药及其自组装纳米粒的制作方法
本发明属于医药技术领域,涉及氧化还原双敏感三硫键桥连前药及其自组装纳米粒的构建,具体涉及三硫键桥连的氧化还原双敏感二聚体前药和二聚体前药自组装纳米粒的构建,以及其在药物传递中的应用。
背景技术:
化疗是癌症治疗中最常用的策略之一,尤其是对于那些不能通过手术切除和转移扩散的肿瘤。但大部分化疗药是细胞毒性药物,且存在溶解度低、稳定性差、治疗窗窄和药动学性质不佳等缺点。而现有的制剂策略递送效率低,肿瘤靶向性较差,导致化疗临床效果不佳且毒副作用严重。例如,紫杉醇(paclitaxel,ptx)作为一线化疗药在临床上被广泛用于治疗非小细胞肺癌和乳腺癌等。但是,由于紫杉醇的水溶性极低,市售的溶液剂泰素(taxol)使用聚氧乙烯蓖麻油和乙醇作为增溶剂和助溶剂,会引起很严重的辅料相关的毒副作用,极大地限制了其在临床上的应用。因此,如何改善化疗药物的不良性质并提高递送效率是临床上亟待解决的难题。
近年来,前体药物和纳米技术在药物传递领域的广泛应用极大地丰富了抗肿瘤药物的递送策略,且已经有多个制剂成功上市,如伊立替康(sn-38前体药物),紫杉醇白蛋白纳米粒,阿霉素脂质体等。前体药物本身没有生物活性或活性很低,经过体内代谢后变为有活性的物质。前药策略可以通过巧妙的结构修饰来改善化疗药物的不良性质,包括溶解度低,稳定性差,毒副作用大等。此外,基于纳米技术构建新型的纳米给药系统可以显著改善药物的药动学性质,延长化疗药物的体内循环时间,并能够通过主动靶向或被动靶向提高药物在肿瘤部位的蓄积,增加药物的细胞摄取,控制药物的释放速度,进而提高抗肿瘤效果,降低毒副作用。在此基础上,基于小分子前体药物的自组装纳米药物递送系统将前体药物和纳米技术的优点结合到一起,以其载药量高、稳定性好、毒副作用低等优势,已成为近几年化疗药物递送研究的热点。
疏水性的刚性药物倾向于长程有序的堆积,在水溶液中具有较大的聚集沉淀趋势。在前药设计中,将结构缺陷引入到药物结构当中,调节分子结构灵活性可以促进前药自组装成纳米粒。将药物与脂肪酸或者角鲨烯等碳链共轭连接是构建前药自组装纳米粒常用的手段,可以通过引入的碳链来调节药物分子结构的灵活性。此外,将两个活性药物共价连接形成二聚体前药可以进一步提高前药自组装纳米粒递送系统的载药量。但是,二聚体前药之间较强的分子间作用力常常限制了其自组装能力,如何设计具有良好自组装性质的二聚体前药自组装纳米粒仍然是个巨大的挑战。此外,不论是前药还是纳米递药系统,智能触发药物在靶部位的选择性释放对于制剂的有效性和安全性都非常重要。与正常细胞相比,肿瘤细胞内存在更高浓度的活性氧(ros)和谷胱甘肽(gsh),这种特殊的肿瘤细胞氧化还原微环境已被广泛用于设计刺激-响应型药物递送系统。目前,针对肿瘤氧化还原微环境敏感的化学桥连种类较少。因此,开发新型智能响应型化学桥连具有重要的科学意义和实用价值。
技术实现要素:
本发明所解决的技术问题是将三硫键引入前药和自组装纳米粒中,设计三硫键桥连的氧化还原双敏感二聚体前药,并将该二聚体前药用于自组装纳米粒,从而实现载药量高、稳定性好、毒副作用低和肿瘤部位特异性快速释药的效果,进而提高抗肿瘤活性。同时,以含有单硫键,二硫键的二聚体前药作为对照,考察不同化学桥连在键角/二面角、氧化还原敏感响应能力以及抗肿瘤活性等方面的差异,以及对前药自组装纳米粒的稳定性、药物释放、细胞毒性、药动学、组织分布以及药效学产生的影响。
本发明的目的是设计和合成含有三硫键桥连的氧化还原双敏感二聚体前药,制备前药自组装纳米药物传递系统,探讨不同化学桥连对前药自组装纳米粒的稳定性、药物释放、细胞毒性、药动学、组织分布以及药效学产生的影响,综合筛选出效果最佳的化学桥连,为开发肿瘤微环境智能响应型药物递送系统提供新的策略和更多的选择,满足临床中对高效化疗制剂的迫切需求。
为了实现上述目的,本发明提供了如通式(i)所示的氧化还原双敏感三硫键桥连的二聚体前药。
其中,x为nh、o,drug为含羟基或者氨基的难溶性药物。
本发明所述通式所示结构的化合物中:含有氨基、或羟基的药物选自活性药物,包括抗肿瘤药物如紫衫烷类、蒽醌类、核苷类、喜树碱类、铂类、长春碱类、泊苷类、青蒿素类化合物等;抗代谢类药物如嘧啶,嘌呤,他滨等;抗炎类药及其他难溶性药物如卤泛群、灰黄霉素、环抱霉素a等及其它们的衍生物。
本发明选择紫杉醇和阿霉素作为模型药物,以所选三硫键为例,将药物通过(a)3,3'-硫代二丙酸,(b)3,3'-二硫代二丙酸,(c)3,3'-三硫代二丙酸相连制备含有单硫键,二硫键,三硫键桥连的氧化还原双敏感二聚体前药。
以紫杉醇(羟基)为例,通式(i)所示的化合物结构为:
以阿霉素(氨基)为例,通式(i)所示的化合物结构为:
以单硫键和二硫键为对照,合成紫杉醇和阿霉素二聚体对照结构如下:
本发明提供含有单硫键,二硫键,三硫键桥连的氧化还原双敏感二聚体前药合成方法,包括如下步骤:
(a)合成3,3'-三硫代二丙酸:将硫代硫酸钠水溶液缓缓滴加到溴丙酸水溶液中,在50-60℃下搅拌反应4-5h,再将硫化钠溶液缓缓滴加至上述反应液中,25-30℃下反应12-24h。
(b)合成紫杉醇二聚体前药:将3,3'-硫代二丙酸、3,3'-二硫代二丙酸和3,3'-三硫代二丙酸分别溶于二氯甲烷中,再将4-二甲氨基吡啶(dmap)及1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edci)溶于无水二氯甲烷中滴加至上述溶液中,活化1-2小时,然后加入紫杉醇,室温条件下搅拌24-48小时,所得产物经制备液相分离纯化,上述反应全程都在n2保护下进行。
(c)合成阿霉素二聚体前药:将3,3'-硫代二丙酸、3,3'-二硫代二丙酸和3,3'-三硫代二丙酸分别溶于n,n-二甲基甲酰胺(dmf)中,再将o-苯并三氮唑-四甲基脲六氟磷酸酯(hbtu)及n,n-二异丙基乙胺(dipea)溶于dmf中滴加至上述溶液中,冰浴1-2小时,然后加入阿霉素,室温条件下搅拌24-48小时,所得产物经制备液相分离纯化,上述反应全程都在n2保护下进行。
本发明中所述的紫杉醇或阿霉素可以用其他含有活性羟基或氨基的抗癌药物,如其他紫杉烷类化合物、核苷类化合物、蒽环类化合物或喜树碱类化合物所代替。
本发明还提供了所述的系列紫杉醇和阿霉素二聚体前药自组装纳米粒,所述的前药自组装纳米粒可以是非peg化的二聚体前药纳米粒、peg修饰的二聚体前药纳米粒等。制备方法为纳米沉淀法,包括高速搅拌法,超声法等。
本发明提供的紫杉醇和阿霉素二聚体前药自组装纳米粒的制备方法如下:
将一定量的紫杉醇二聚体前药或阿霉素二聚体前药与peg修饰剂混合物溶解到适量的乙醇(或者dmso)中,搅拌下,将该乙醇溶液缓缓滴加到水中,前药自发形成均匀的纳米粒。最后,采用透析法除去制剂中的乙醇,得到不含任何有机溶剂的纳米胶体溶液。所述的peg修饰剂为tpgs、dspe-peg、plga-peg和pe-peg等,优选的peg修饰剂为dspe-peg。所述peg的分子量为1000-5000,优选为1000、2000和5000,更优选peg分子量为2000。
二聚体前药与peg修饰剂的重量比为:90:10~70:30,在此范围条件下,前药纳米粒可以发挥较好的抗肿瘤效果。
具体地,所述的紫杉醇和阿霉素二聚体前药自组装纳米粒的制备方法如下:(1)非peg化的小分子前药自组装纳米粒的制备方法:将一定量的前药溶解到适量的乙醇中,搅拌下,将该乙醇溶液缓缓滴加到水中,前药自发形成均匀的纳米粒。采用透析法除去制剂中的乙醇,得到不含任何有机溶剂的纳米胶体溶液。(2)peg修饰的小分子前药自组装纳米粒的制备方法:将一定量的peg修饰剂(tpgs、dspe-peg、plga-peg或pe-peg)和前药溶解到适量的乙醇中,搅拌下,将该乙醇溶液缓缓滴加到水中,前药自发形成均匀的纳米粒。采用透析法除去制剂中的乙醇,得到不含任何有机溶剂的纳米胶体溶液。
本发明的优势在于:(1)设计合成了含有三硫键桥连的氧化还原双敏感二聚体前药和含有单硫键,二硫键桥连的对照前药,合成方法简单易行;(2)制备了均匀的二聚体前药自组装纳米粒,制备方法简单易行,实现药物的高效包载,具有超高载药量和稳定性;(3)考察了不同化学桥连在键角/二面角、氧化还原敏感响应能力以及抗肿瘤活性等方面的差异,以及对前药自组装纳米粒的稳定性、药物释放、细胞毒性、药动学、组织分布以及药效学产生的影响。综合筛选出效果最佳的化学桥连,为开发肿瘤微环境智能响应型药物递送系统提供新的策略和更多的选择,满足临床中对高效化疗制剂的迫切需求。
附图说明
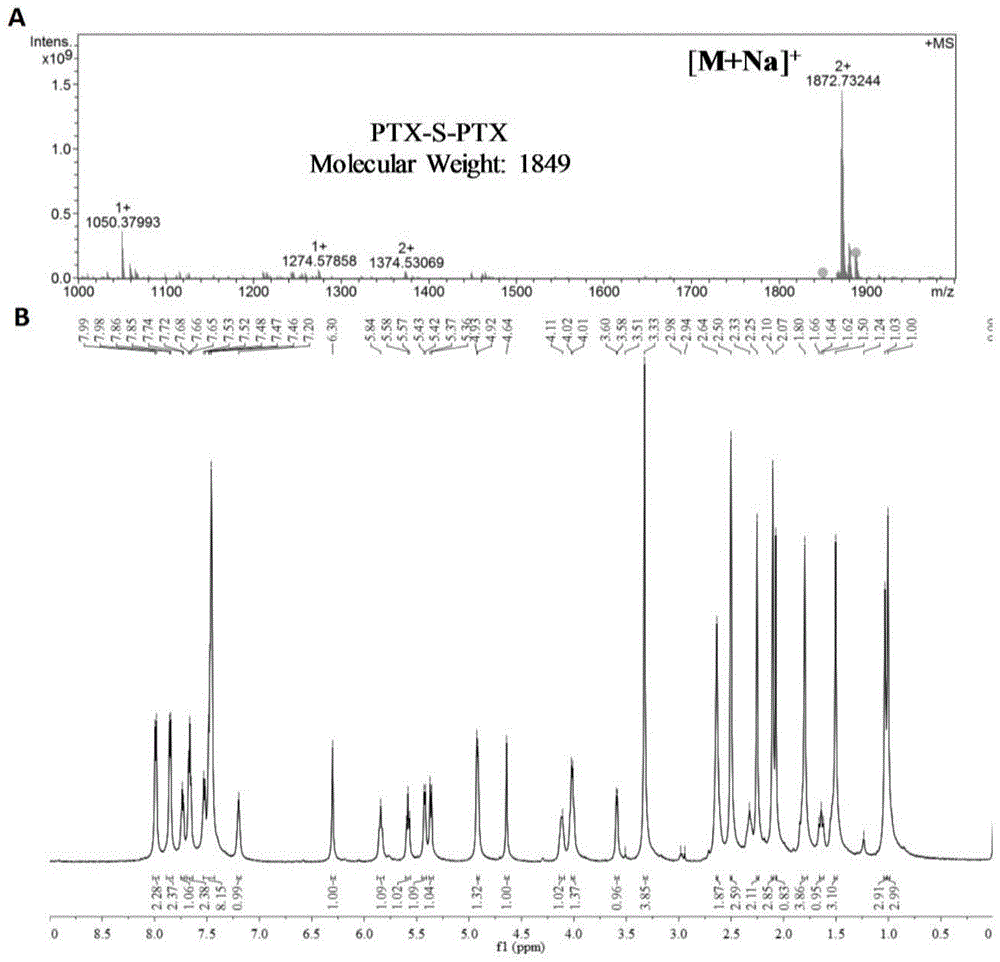
图1为本发明实施例1的单硫键桥连紫杉醇二聚体前药(ptx-s-ptx)的1hnmr谱图和质谱图。
图2为本发明实施例2的二硫键桥连紫杉醇二聚体前药(ptx-ss-ptx)的1hnmr谱图和质谱图。
图3为本发明实施例3的三硫键桥连紫杉醇二聚体前药(ptx-sss-ptx)的1hnmr谱图和质谱图。
图4为本发明实施例4的单硫键桥连阿霉素二聚体前药(dox-s-dox)的1hnmr谱图和质谱图。
图5为本发明实施例5的二硫键键桥连阿霉素二聚体前药(dox-ss-dox)的1hnmr谱图和质谱图。
图6为本发明实施例6的三硫键桥连的阿霉素二聚体前药(dox-sss-dox)的1hnmr谱图和质谱图。
图7为本发明实施例7的二聚体前药自组装纳米粒的外观与peg修饰的纳米粒的透射电子显微镜图。
图8为本发明实施例8的peg修饰的紫杉醇二聚体前药自组装纳米粒的粒径-胶体稳定性图。
图9为本发明实施例9的peg修饰的二聚体前药自组装纳米粒的体外释放试验图
图10为本发明实施例10的peg修饰的二聚体前药自组装纳米粒的细胞毒性图。
图11为本发明实施例10的peg修饰的紫杉醇二聚体前药自组装纳米粒的肿瘤细胞内药物释放图。
图12为本发明实施例11的peg修饰的二聚体前药自组装纳米粒的血药浓度-时间曲线图。
图13为本发明实施例12的peg修饰的紫杉醇二聚体前药自组装纳米粒的在体抗肿瘤实验图。
图14为本发明实施例13的peg修饰的阿霉素二聚体前药自组装纳米粒的在体抗肿瘤实验图。
具体实施方式
下面通过实施例的方式进一步说明本发明,但并不因此将发明限制在所述的实施例范围之中。
实施例1:单硫键桥连紫杉醇二聚体前药(ptx-s-ptx)的合成
将适量硫代二丙酸加入到50ml圆底烧瓶中,并用5ml二氯甲烷溶解,并加入适量的edci和dmap,室温条件下搅拌1小时,再加入适量的edci和dmap溶于10ml无水二氯甲烷中,然后加入适量紫杉醇,在室温下再搅拌24小时,通过薄层色谱和高效液相色谱监测反应过程,目标产物通过制备液相色谱分离纯化。上述反应全程都在n2保护下进行。
采用质谱法以及核磁共振氢谱法来确定实施例1中前药的结构,结果如图1所示。核磁共振波谱解析结果如下:ptx-s-ptx(c100h108n2o30s)
1hnmr(400mhz,dmso-d6,ppm):δ1.00(s,3h,(c-16)-ch3),1.03(s,3h,(c-17)-ch3),1.50(s,3h,(c-19)-ch3),1.62-1.80(m,5h,(c-6)-ch,1-ohand(c-18)-ch3),2.07(m,1h,(c-14)-ch),2.10(s,3h,10-oac),2.25-2.64(m,6h,4-oac,(c-14)-chandsch2ch2coo(ptx)),3.33(m,4h,(c-6)-ch,7-oh,andsch2ch2coo(ptx)),3.60(d,1h,j=7hz,(c-3)-ch),4.02(d,1h,j=8hz,(c-20)-ch),4.11(d,1h,j=8hz,(c-20)-ch),4.64(m,1h,(c-7)-ch),4.93(dd,1h,j=9hz,j=2hz,(c-5)-ch),5.36(d,1h,j=3hz,(c-2’)-ch),5.42(d,1h,j=7hz,(c-2)-ch),5.58(dd,1h,j=9hz,j=3hz,(c-3’)-ch),5.84(m,1h,(c-10)-ch),6.30(m,1h,(c-13)-ch),7.20(d,1h,j=9hz,3’-nh),7.46-7.68(m,11h,ph-h),7.86(d,2h,j=8hz,phh),7.98(d,2h,j=8hz,phh)。ms(esi)m/zforc100h108n2o30sna[m+na]+:1872.73244。
实施例2:二硫键桥连紫杉醇二聚体前药(ptx-ss-ptx)的合成
将适量二硫代二丙酸加入到50ml圆底烧瓶中,并用5ml二氯甲烷溶解,并加入适量的edci和的dmap,室温条件下搅拌1小时,再加入适量的edci和dmap溶于10ml无水二氯甲烷中,然后加入适量紫杉醇,在室温下再搅拌24小时,通过薄层色谱和高效液相色谱监测反应过程,目标产物通过制备液相色谱分离纯化。上述反应全程都在n2保护下进行。
采用质谱法以及核磁共振氢谱法来确定实施例2中前药的结构,结果如图2所示。核磁共振波谱解析结果如下:ptx-ss-ptx(c100h108n2o30s2)
1hnmr(400mhz,dmso-d6,ppm):δ1.00(s,3h,(c-16)-ch3),1.03(s,3h,(c-17)-ch3),1.50(s,3h,(c-19)-ch3),1.62-1.80(m,5h,(c-6)-ch,1-ohand(c-18)-ch3),2.07(m,1h,(c-14)-ch),2.10(s,3h,10-oac),2.25-2.85(m,6h,4-oac,(c-14)-chandsch2ch2coo(ptx)),3.32(m,4h,(c-6)-ch,7-oh,andsch2ch2coo(ptx)),3.60(d,1h,j=7hz,(c-3)-ch),4.00(d,1h,j=8hz,(c-20)-ch),4.04(d,1h,j=8hz,(c-20)-ch),4.63(m,1h,(c-7)-ch),4.92(dd,1h,j=9hz,j=2hz,(c-5)-ch),5.37(d,1h,j=3hz,(c-2’)-ch),5.42(d,1h,j=7hz,(c-2)-ch),5.58(dd,1h,j=9hz,j=3hz,(c-3’)-ch),5.84(m,1h,(c-10)-ch),6.30(m,1h,(c-13)-ch),7.20(d,1h,j=9hz,3’-nh),7.45-7.74(m,11h,ph-h),7.84(d,2h,j=8hz,phh),7.98(d,2h,j=8hz,phh)。ms(esi)m/zforc100h108n2o30s2na[m+na]+:1904.73200。
实施例3:三硫键桥连紫杉醇二聚体前药(ptx-sss-ptx)的合成
将适量三硫代二丙酸加入到50ml圆底烧瓶中,并用5ml二氯甲烷溶解,并加入适量的edci和的dmap,室温条件下搅拌1小时,再加入适量的edci和dmap溶于10ml无水二氯甲烷中,然后加入适量紫杉醇,在室温下再搅拌24小时,通过薄层色谱和高效液相色谱监测反应过程,目标产物通过制备液相色谱分离纯化。上述反应全程都在n2保护下进行。
采用质谱法以及核磁共振氢谱法来确定实施例3中前药的结构,结果如图3所示。核磁共振波谱解析结果如下:ptx-sss-ptx(c100h108n2o30s3)
1hnmr(400mhz,dmso-d6,ppm):δ1.00(s,3h,(c-16)-ch3),1.03(s,3h,(c-17)-ch3),1.50(s,3h,(c-19)-ch3),1.62-1.79(m,5h,(c-6)-ch,1-ohand(c-18)-ch3),2.07(m,1h,(c-14)-ch),2.10(s,3h,10-oac),2.25-3.05(m,6h,4-oac,(c-14)-chandsch2ch2coo(ptx)),3.33(m,4h,(c-6)-ch,7-oh,andsch2ch2coo(ptx)),3.59(d,1h,j=7hz,(c-3)-ch),4.02(d,1h,j=8hz,(c-20)-ch),4.10(d,1h,j=8hz,(c-20)-ch),4.64(m,1h,(c-7)-ch),4.93(dd,1h,j=9hz,j=2hz,(c-5)-ch),5.38(d,1h,j=3hz,(c-2’)-ch),5.41(d,1h,j=7hz,(c-2)-ch),5.58(dd,1h,j=9hz,j=3hz,(c-3’)-ch),5.84(m,1h,(c-10)-ch),6.30(m,1h,(c-13)-ch),7.20(d,1h,j=9hz,3’-nh),7.45-7.75(m,11h,ph-h),7.86(d,2h,j=8hz,phh),7.98(d,2h,j=8hz,phh)。ms(esi)m/zforc100h108n2o30s3na[m+na]+:1935.60788。
实施例4:单硫键桥连阿霉素二聚体前药(dox-s-dox)的合成
将适量硫代二丙酸加入到50ml圆底烧瓶中,并用10ml的n,n-二甲基甲酰胺溶解,并加入适量的hbtu和的dipea,冰浴条件下搅拌1小时,再加入适量的阿霉素,在室温下再搅拌24小时,通过薄层色谱和高效液相色谱监测反应过程,目标产物通过制备液相色谱分离纯化。上述反应全程都在n2保护下进行。
采用质谱法以及核磁共振氢谱法来确定实施例4中前药的结构,结果如图4所示。核磁共振波谱解析结果如下:dox-s-dox(c60h64n2o24s)
1h-nmr(400mhz,dmso-d6,ppm)ofdsd:13.92(s,1h),13.17(s,1h),7.84(d,j=7.4hz,1h),7.81(d,j=8.2hz,1h),7.56(d,j=7.2hz,1h),7.49(t,j=7.4hz,1h),5.32(s,1h),5.21(s,1h),4.81(d,2h),4.67(s,1h),4.51(s,2h),4.09(s,1h),3.85-3.93(m,4h),3.40(s,3h),2.87(d,2h),2.76(s,2h),2.19(s,1h),2.15(s,1h),1.81(t,1h),1.43(s,1h),1.09(s,3h)。ms(esi)m/zforc60h64n2o24sna[m+na]+:1251.34388。
实施例5:二硫键桥连阿霉素二聚体前药(dox-ss-dox)的合成
将适量二硫代二丙酸加入到50ml圆底烧瓶中,并用10ml的n,n-二甲基甲酰胺溶解,并加入适量的hbtu和的dipea,冰浴条件下搅拌1小时,再加入适量的阿霉素,在室温下再搅拌24小时,通过薄层色谱和高效液相色谱监测反应过程,目标产物通过制备液相色谱分离纯化。上述反应全程都在n2保护下进行。
采用质谱法以及核磁共振氢谱法来确定实施例5中前药的结构,结果如图5所示。核磁共振波谱解析结果如下:dox-ss-dox(c60h64n2o24s2)
1h-nmr(400mhz,dmso-d6,ppm)ofdssd:13.87(s,1h),13.14(s,1h),7.83(d,j=7.4hz,1h),7.73(d,j=8.2hz,1h),7.65(d,j=7.2hz,1h),7.56(t,j=7.4hz,1h),5.21(s,1h),4.81(s,1h),4.67(d,2h),4.70(s,1h),4.53(s,2h),4.13(s,1h),3.89-3.95(m,4h),3.39(s,3h),2.89(d,2h),2.58(s,2h),2.23(s,1h),2.16(s,1h),1.84(t,1h),1.43(s,1h),1.12(s,3h)。ms(esi)m/zforc60h64n2o24s2na[m+na]+:1283.31267。
实施例6:三硫键桥连阿霉素二聚体前药(dox-sss-dox)的合成
将适量三硫代二丙酸加入到50ml圆底烧瓶中,并用10ml的n,n-二甲基甲酰胺溶解,并加入适量的hbtu和的dipea,冰浴条件下搅拌1小时,再加入适量的阿霉素,在室温下再搅拌24小时,通过薄层色谱和高效液相色谱监测反应过程,目标产物通过制备液相色谱分离纯化。上述反应全程都在n2保护下进行。
采用质谱法以及核磁共振氢谱法来确定实施例6中前药的结构,结果如图6所示。核磁共振波谱解析结果如下:dox-sss-dox(c60h64n2o24s3)
1h-nmr(400mhz,dmso-d6,ppm)ofdsssd:13.89(s,1h),13.16(s,1h),7.81(d,j=7.4hz,1h),7.76(d,j=8.2hz,1h),7.68(d,j=7.2hz,1h),7.54(t,j=7.4hz,1h),5.38(s,1h),5.21(s,1h),4.85(d,2h),4.70(s,1h),4.58(s,2h),4.14(s,1h),3.87-3.96(m,4h),3.40(s,3h),2.93(d,2h),2.83(s,2h),2.20(s,1h),2.09(s,1h),1.84(t,1h),1.44(s,1h),1.10(s,3h)。ms(esi)m/zforc60h64n2o24s3na[m+na]+:1315.28935。
实施例7:非peg化或peg修饰的不同化学键连接的二聚体前药自组装纳米粒的制备
(1)紫杉醇二聚体前药
非peg化紫杉醇前药纳米粒:将2mg不同化学键连接的紫杉醇二聚体前药溶于1ml的乙醇中,搅拌下,将该乙醇溶液缓缓滴加到4ml的去离子水中,考察前药自组装能力。如图7a所示,在没有表面活性剂帮助下,在ptx-s-ptxnps和ptx-ss-ptxnps中均有沉淀析出,不能独立组装成稳定的纳米粒,只有ptx-sss-ptxnps可以形成稳定的纳米粒,表明三硫键桥连的紫杉醇前药具有较好的自组装能力和稳定性。
peg化紫杉醇前药纳米粒:精密称取dspe-peg2k2mg和不同化学键连接的紫杉醇二聚体前药8mg,用1ml乙醇将其溶解,搅拌下,将该乙醇溶液缓缓滴加到4ml去离子水中,自发形成均匀的纳米粒ptx-s-ptxnps,ptx-ss-ptxnps和ptx-sss-ptxnps。在25℃的条件下用去离子水透析除去纳米制剂中的有机溶剂。如表1所示,纳米粒的粒径都在80nm左右,粒径分布小于0.2,表面电荷在-20mv左右,载药量都在70%以上。通过透射电子显微镜测定实施例7中制备的二聚体前药自组装纳米粒的粒径和形态,结果如图7b,透射电镜图表明载药纳米粒为均一的球形,粒径在80nm左右。
(2)阿霉素二聚体前药
非peg化阿霉素前药纳米粒:将2mg的阿霉素前药溶于1ml的dmf中,搅拌下,将该dmf溶液缓缓滴加到4ml的去离子水中,考察前药自组装能力。如图7a所示,在没有表面活性剂帮助下,在dox-s-doxnps和dox-ss-doxnps中均有沉淀析出,不能独立组装成稳定的纳米粒,只有dox-sss-dox可以形成稳定的纳米粒,表明三硫键桥连的阿霉素前药具有较好的自组装能力和稳定性。
peg化阿霉素前药纳米粒:精密称取dspe-peg2k2mg和阿霉素二聚体前药8mg,用1ml的dmso将其溶解,搅拌下,将该dmso溶液缓缓滴加到4ml去离子水中,自发形成均匀的纳米粒dox-ss-doxnps和dox-sss-doxnps。然而,dox-s-dox在表面活性剂dspe-peg2k的辅助下,仍然不能组装成稳定的纳米粒,最终析出大量沉淀(如图7a)。在25℃的条件下用去离子水透析除去纳米制剂中的有机溶剂。如表1所示,纳米粒的粒径都在80nm左右,粒径分布接近0.2,表面电荷在-20mv左右,载药量都在67%以上。通过透射电子显微镜测定实施例7中制备的二聚体前药自组装纳米粒的粒径和形态,结果如图7b,透射电镜图表明载药纳米粒为均一的球形,粒径在75nm左右。综上,dox-s-dox前药自组装能力和胶体稳定性最差,其次是dox-ss-dox,而dox-sss-dox组装能力与稳定性最好。
表1.peg修饰的二聚体前药自组装纳米粒的粒径、粒径分布、表面电荷和载药量
实施例8:peg修饰的不同化学键连接的紫杉醇二聚体前药自组装纳米粒的胶体稳定性试验
将实施例7中制备的peg修饰的不同化学键连接的紫杉醇二聚体前药自组装纳米粒取出1ml,加入到20ml含有10%fbs的磷酸盐缓冲液(pbs,ph为7.4)中,在37℃的条件下孵育24小时,并且在预定的时间点(0,2,4,6,8,12和24小时)通过动态光散射法测定其粒径变化。结果如图8所示,ptx-sss-ptxnps胶体稳定性最好,在24小时内粒径没有发生明显的变化。相比之下,ptx-s-ptxnps和ptx-ss-ptxnps纳米粒的胶体稳定性较差,随着孵化时间的延长,纳米粒的粒径增大。
实施例9:peg修饰的不同化学键连接的二聚体前药自组装纳米粒的体外释放试验。
(1)紫杉醇二聚体前药
以含30%乙醇的ph7.4的磷酸盐缓冲液(pbs)为释放介质,考察不同化学键连接的紫杉醇二聚体前药自组装纳米粒的体外释放情况。将1ml实施例7中制备的peg修饰的紫杉醇二聚体前药自组装纳米粒(紫杉醇含量为200μg/ml)加入到30ml释放介质中,在37℃条件下,于设定的时间点取样,通过高效液相色谱测定释放出的紫杉醇浓度。向释放介质中加入一定浓度的双氧水(h2o2,1mm,10mm)或二硫苏糖醇(dtt,0.1mm,1mm,10mm),以分别考察纳米粒在氧化条件和还原条件下的释放情况。结果如图9a-b所示,硫键桥连的前药纳米粒都具有氧化还原双重响应性,能够在h2o2或dtt的作用下快速释放出紫杉醇。其中,氧化敏感性大小顺序为单硫键>二硫键>三硫键。还原敏感性则相反,三硫键>二硫键>单硫键。单硫键(-s-)的硫化学价为-2价,二硫键的硫为-1价,三硫键中间的硫为0价,两边的硫为-1价。化学价越低越容易被氧化,化学价越高越容易被还原。此外,三硫键具有三个硫原子,均可以作为响应触发点。因此三硫键具有还原超敏特性,在较低浓度还原物质下依旧能快速释放出紫杉醇。此外,ptx-sss-ptxnps具有良好的稳定性,在缺乏还原物质的释放介质中,释放出来的母药最少,比较稳定。
(2)阿霉素二聚体前药
以含30%乙醇的ph7.4的磷酸盐缓冲液(pbs)为释放介质,考察阿霉素不同化学键连接的二聚体前药自组装纳米粒的体外释放情况。将1ml实施例7中制备的peg修饰的阿霉素二聚体前药自组装纳米粒(阿霉素含量为200μg/ml)加入到30ml释放介质中,在37℃条件下,于设定的时间点取样,通过高效液相色谱测定释放度。阿霉素二聚体前药通过共轭连接阿霉素的氨基,形成的酰胺键在介质中难以水解,因此释放的最终产物是阿霉素硫醇(dox-sh),研究表明该中间体具有与阿霉素相当的细胞毒。向释放介质中加入一定浓度的gsh,分别为0.1mm,0.5mm,1mm,以考察纳米粒在还原条件下的释放情况。结果如图9c所示,二硫键和三硫键桥连的阿霉素前药纳米粒都具有还原响应性,能够在gsh的作用下快速释放出阿霉素硫醇。其中,三硫键表现出对gsh的超敏响应特性,比二硫键更敏感。二硫键的硫为-1价,三硫键中间的硫为0价,两边的硫为-1价。三硫键的化学价越高越容易被还原,具有更高的电子储存能力。此外,三硫键具有三个硫原子,均可以作为响应触发点。因此三硫键具有还原超敏特性,在较低浓度还原物质下能快速释药物。此外,阿霉素二聚体前药纳米粒具有良好的稳定性,酰胺键连接的前药比较稳定,在缺乏还原物质的释放介质中,几乎没有药物释放释放,前药非常稳定。
实施例10:peg修饰的不同化学键连接的二聚体前药自组装纳米粒的细胞毒性
(1)紫杉醇二聚体前药
采用mtt法考察peg修饰的不同化学键连接的二聚体前药自组装纳米粒对三种肿瘤细胞,人鳞状上皮癌(kb)细胞,小鼠乳腺癌(4t1)细胞,小鼠黑色瘤(b16)细胞和人正常肝(l02)细胞的毒性。首先将形态良好的细胞消化,用培养液稀释至5000cells/ml细胞吹匀后于96孔板中每孔加入细胞悬液100μl,置培养箱中孵育24h使其贴壁。待细胞贴壁后加泰素或实施例7中制备的紫杉醇前药纳米粒。本实验中药物溶液与纳米粒制剂的配制和稀释均用相应的培养液,并用0.22μm滤膜无菌过滤。受试溶液每孔加入100μl,每个浓度3个平行孔。对照组,即不加待测药液,单一补加100μl培养液,置培养箱中和细胞共同孵育。于加药后48h,将96孔板取出,每孔加入5mg/mlmtt溶液20μl,置培养箱中孵育4h后甩板,将96孔板倒扣于滤纸上充分吸干残留液体后,每孔加入200μldmso于振荡器上振荡10min以溶解蓝紫色结晶物。设定a1孔(只含有200μldmso)为调零孔。使用酶标仪在570nm处测定各孔调零后的吸光度值。
细胞毒性结果如图10a所示。与泰素组相比,前药纳米粒的细胞毒性降低。这是因为紫杉醇需要一定时间从前药纳米粒中释放出来,限制了紫杉醇药效的发挥。与ptx-ss-ptxnps和ptx-s-ptxnps相比,ptx-sss-ptxnps对肿瘤细胞具有更高的细胞毒性。前药纳米粒细胞毒性与紫杉醇从纳米粒中的释放速度相关,药物释放速度越快,细胞毒性越强。因此,考察了前药纳米粒在4t1细胞中紫杉醇的释放速度。从图11可知,细胞毒性与紫杉醇的释放速率正相关,ptx-sss-ptxnps释放紫杉醇的速度较快,ptx-ss-ptxnps和ptx-s-ptxnps释放相对较慢。因此ptx-sss-ptxnps具有比ptx-ss-ptxnps和ptx-s-ptxnps更强的肿瘤细胞毒性。此外,与泰素相比,前药纳米粒对正常细胞的毒性比较低,尤其是ptx-sss-ptxnps对正常细胞的毒性最低。ptx-sss-ptxnps具有良好的胶体稳定性,在正常细胞内保持较好的完整性,延迟了其在正常细胞内的释放。因此,ptx-sss-ptxnps具有最高的选择性毒性,对于肿瘤细胞毒性较强,但是对于正常细胞毒性较低。
(2)阿霉素二聚体前药
阿霉素二聚体前药细胞毒实验操作同上。如图10b所示,与阿霉素溶液剂组相比,前药纳米粒的细胞毒性降低。这是因为阿霉素需要一定时间从前药纳米粒中释放出来,限制了阿霉素药效的发挥。在肿瘤细胞中,dox-sss-doxnps表现出比dox-ss-doxnps更高的细胞毒性;在正常细胞中,dox-sss-doxnps表现出比dox-ss-doxnps更低的细胞毒性。因此,dox-sss-doxnps具有最高的选择性毒性,对于肿瘤细胞毒性较强,但是对于正常细胞毒性较低。这是由于三硫键在肿瘤细胞内高表达的gsh下,快速释放药物,具有超敏响应特性。同时,在正常细胞中良好的胶体稳定性使得其利于保持完整纳米结构形态,缓解了其释放。
实施例11:peg修饰的不同化学键连接的二聚体前药自组装纳米粒的药代动力学研究
(1)紫杉醇二聚体前药
取体重在200-250g之间的sd大鼠,随机分组,给药前禁食12h,自由饮水。分别静脉注射taxol以及实施例7中制备的peg化的紫杉醇二聚体前药自组装纳米粒。紫杉醇的剂量为5mg/kg。于规定的时间点眼眶取血,分离获得血浆。通过液相色谱-质谱联用仪测定血浆中的药物浓度。
实验结果如图12a所示,由于半衰期短,泰素中的紫杉醇迅速从血液中清除。相比之下,紫杉醇前药自组装纳米粒的循环时间明显延长。同时,化学连接键对前药纳米粒的药动学行为有显著影响。ptx-sss-ptxnps具有更高的auc和更长的循环时间。这是因为三硫键前药自组装纳米粒具有较强的胶体稳定性,延长了其体内保留。单硫键和二硫键前药纳米粒具有弱的胶体稳定性,在血液循环中纳米结构会快速解体,随后被清除,因此ptx-s-ptxnps和ptx-ss-ptxnps的auc更低。
(2)阿霉素二聚体前药
取体重在200-250g之间的sd大鼠,随机分组,给药前禁食12h,自由饮水。分别静脉注射阿霉素溶液剂以及实施例7中制备的peg化的阿霉素二聚体前药自组装纳米粒。阿霉素的剂量为2mg/kg。于规定的时间点眼眶取血,分离获得血浆。通过液相色谱-质谱联用仪测定血浆中的药物浓度。
实验结果如图12b所示,由于半衰期短,阿霉素迅速从血液中清除。相比之下,阿霉素二聚体前药自组装纳米粒的循环时间明显延长。与紫杉醇二聚体前药纳米粒药动学行为相似,dox-sss-doxnps具有更高的auc和更长的循环时间。因为dox-sss-doxnps具有较强的胶体稳定性,延长了其体内保留时间,提高了auc。然而dox-ss-doxnps具有弱的胶体稳定性,在血液循环中纳米结构会快速解体,随后被清除,因而auc更低。
实施例12:peg修饰的不同化学键连接的紫杉醇二聚体前药自组装纳米粒的在体抗肿瘤实验
以4t1荷瘤balb/c小鼠为模型,尾静脉给药紫杉醇二聚体自组装纳米粒,设置taxol和生理盐水静注组为对照组。结果如图13a-b所示,taxol组与生理盐水组相比,具有一定的抑瘤作用。ptx-s-ptxnps和ptx-ss-ptxnps均表现出了较强的抑瘤作用,但是在肿瘤负荷上与taxol组无显著性差别。值得注意的是,ptx-sss-ptxnps表现出最强的抗肿瘤效果,明显强于ptx-s-ptxnps和ptx-ss-ptxnps组。这是由于ptx-sss-ptxnps具有良好的胶体稳定性,提高了其药动学行为,具有较高的auc。此外,ptx-sss-ptxnps在肿瘤细胞中具有较快的释药速率,提高了其细胞毒性。综上,纳米粒的稳定性、细胞毒性、药动学分布和肿瘤部位响应释药能力都会影响最终的抗肿瘤效果。如图13c所示,各制剂组无体重降低现象,表明其安全性良好。此外,在黑色素荷瘤c57bl/6小鼠模型中(如图13d-f),观察到与4t1瘤模型相似的药效结果,再次证明了三硫键桥连紫杉醇二聚体前药自组装纳米粒的优势。
实施例13:peg修饰的不同化学键连接的阿霉素二聚体前药自组装纳米粒的在体抗肿瘤实验
以4t1荷瘤balb/c小鼠为模型,尾静脉给药阿霉素二聚体自组装纳米粒,设置阿霉素溶液剂和生理盐水静注组为对照组。结果如图14a-b所示,阿霉素组与生理盐水组相比,具有显著的抑瘤作用。但是阿霉素溶液剂具有严重的副作用,给药后小鼠体重下降近15%(如图14c)。dox-ss-doxnps表现出了较强的抑瘤作用,与阿霉素组无显著性差别,但是降低了其毒性,小鼠体重与生理盐水组相比没有明显变化。值得注意的是,dox-sss-doxnps表现出最强的抗肿瘤效果,强于阿霉素组和dox-ss-doxnps组。这是由于dox-sss-doxnps具有良好的胶体稳定性,提高了其药动学行为,具有较高的auc。此外,dox-sss-doxnps在具有gsh超敏的释药特性,提高了其细胞毒性。提示我们,药动学行为与肿瘤部位的特异性释放对于良好的抗肿瘤效果很重要。此外,dox-sss-doxnps组小鼠体重没有明显变化,表明其良好的安全性。
基于上述阿霉素二聚体前药纳米粒抗肿瘤的安全性和有效性,在黑色素荷瘤c57bl/6小鼠模型中,前药纳米粒设置了高低两个剂量。如图14d-e所示,低剂量下,阿霉素溶液剂组和dox-ss-doxnps均表现出很强的抗肿瘤效果,但是两者之间没有显著差异。如图14f所示,与生理盐水对照组相比,阿霉素溶液剂给药后的小鼠体重明显下降,超过15%,显示出严重的副作用。相比之下,各纳米粒给药组小鼠体重正常,具有良好的安全性。在黑色素模型中,dox-sss-doxnps仍然表现出最强的抗肿瘤效果,这与上述4t1瘤模型的药效结果相似。此外,高剂量组dox-ss-doxnps和dox-sss-doxnps均表现出比阿霉素更强的抗肿瘤作用。这些结果表明,dox-sss-doxnps具有增强的抗肿瘤作用的同时,降低了阿霉素的毒副作用,提高了安全性。再次启发我们,良好的药效学结果需要药物递送各个过程的协同,良好的药动学行为与肿瘤部位的特异性快速释放对抗肿瘤药效具有相当重要性。
- 还没有人留言评论。精彩留言会获得点赞!