人PSMD7基因的用途及相关产品的制作方法
本发明属于生物医药研究领域,具体涉及人psmd7基因的用途及相关产品。
背景技术:
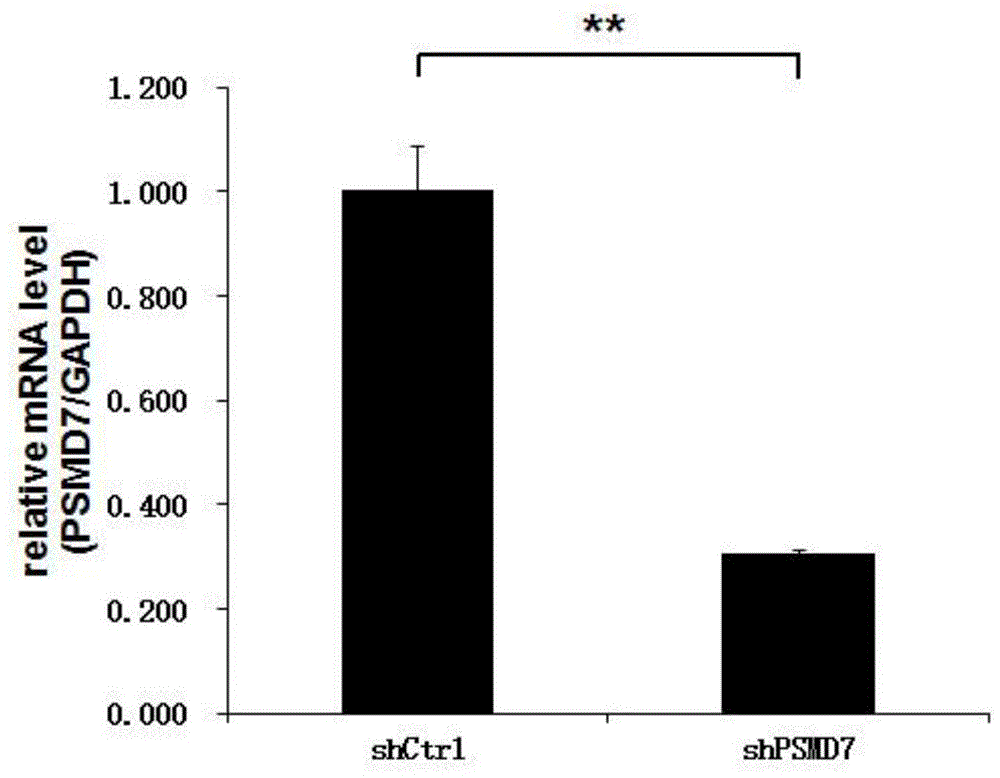
:psmd7(proteasome26ssubunit,non-atpase7)是一种蛋白编码基因,是一种26s蛋白酶体的组成部分——19s蛋白酶体的亚基。而26s蛋白酶体参与atp依赖泛素化降解有关,通过去除可能损害细胞功能的错误折叠或受损的蛋白质,以及去除功能上非必须蛋白,在维持蛋白质内环境稳定方面起着关键作用。psmd7编码蛋白参与许多细胞过程,包括细胞周期进展、纤毛/鞭毛解聚等过程。已知与psmd7相关的疾病包括气管炎和亨廷顿病等。据文献报道,psmd7与乳腺癌、食管癌等癌症的发生发展有关。在一项研究中,通过tcga乳腺侵袭腺癌人群的数据分析发现,psmd7是乳腺癌当中的一个独立的预后靶标,与患者总生存期、无进展生存期相关。在另一项研究中,通过细胞功能实验验证发现食管癌中下调psmd7可通过mtor/p70s6k信号通路诱导食管癌细胞的凋亡,抑制食管癌肿瘤生成。目前还未有psmd7基因用于胰腺癌治疗的相关报道。技术实现要素:为了克服现有技术中所存在的问题,本发明的目的在于提供人psmd7基因的用途及相关产品。为了实现上述目的以及其他相关目的,本发明采用如下技术方案:本发明的第一方面,提供人psmd7基因作为靶标在制备胰腺癌治疗药物中的用途。所述人psmd7基因作为靶标在制备胰腺癌治疗药物具体是指:将psmd7基因作为作用对象,对药物或制剂进行筛选,以找到可以抑制人psmd7基因表达的药物作为胰腺癌治疗备选药物。如本发明所述的psmd7基因小分子干扰rna(sirna)即是以人psmd7基因为作用对象筛选获得的,可用作具有抑制胰腺癌细胞增殖作用的药物。除此之外,诸如抗体药物,小分子药物等也可将psmd7基因作为作用对象。所述胰腺癌治疗药物为能够特异性抑制psmd7基因的转录或翻译,或能够特异性抑制psmd7蛋白的表达或活性的分子,从而降低胰腺癌细胞中psmd7基因的表达水平,达到抑制胰腺癌细胞的增殖、生长、分化和/或存活的目的。所述通过psmd7基因制备获得的胰腺癌治疗药物包括但不限于:核酸分子、碳水化合物、脂类、小分子化学药、抗体药、多肽、蛋白或干扰慢病毒。所述核酸包括但不限于:反义寡核苷酸、双链rna(dsrna)、核酶、核糖核酸内切酶iii制备的小干扰rna或者短发夹rna(shrna)。所述胰腺癌治疗药物的施用量为足够降低人psmd7基因的转录或翻译,或者足够降低人psmd7蛋白的表达或活性的剂量。以使人psmd7基因的表达至少被降低50%、80%、90%、95%或99%。采用前述胰腺癌治疗药物治疗胰腺癌的方法,主要是通过降低人psmd7基因的表达水平抑制胰腺癌细胞的增殖来达到治疗的目的。具体的,治疗时,将能有效降低人psmd7基因表达水平的物质给药于患者。在一种实施方式中,所述psmd7基因的靶标序列如seqidno:1所示。具体为:5’-tgaggaagttggagttgaa-3’。本发明的第二方面,提供psmd7抑制剂在制备至少具备以下功效之一的产品中的用途:治疗胰腺癌;抑制胰腺癌细胞的增殖能力;抑制胰腺癌细胞克隆;促进胰腺癌细胞凋亡;抑制胰腺癌细胞转移能力;抑制胰腺癌生长。所述产品必然包括psmd7抑制剂,并以psmd7抑制剂作为前述功效的有效成分。所述产品中,发挥前述功用的有效成分可仅为psmd7抑制剂,亦可包含其他可起到前述功用的分子。亦即,psmd7抑制剂为所述产品的唯一有效成分或有效成分之一。所述产品可以为单成分物质,亦可为多成分物质。所述产品的形式无特殊限制,可以为固体、液体、凝胶、半流质、气雾等各种物质形式。所述产品主要针对的对象为哺乳动物。所述哺乳动物优选为啮齿目动物、偶蹄目动物、奇蹄目动物、兔形目动物、灵长目动物等。所述灵长目动物优选为猴、猿或人。所述产品包括但不限于药物、保健品、食品等。所述psmd7抑制剂可以为核酸分子、抗体、小分子化合物。如本发明实施例列举的,所述psmd7抑制剂可以为降低胰腺癌细胞中psmd7基因表达的核酸分子。具体的,可以是双链rna或shrna。本发明的第三方面,提供了一种治疗胰腺癌的方法,为向对象施用psmd7抑制剂。所述的对象可以为哺乳动物或哺乳动物的胰腺癌细胞。所述哺乳动物优选为啮齿目动物、偶蹄目动物、奇蹄目动物、兔形目动物、灵长目动物等。所述灵长目动物优选为猴、猿或人。所述胰腺癌细胞可以为离体胰腺癌细胞。所述对象可以是罹患胰腺癌的患者或者期待治疗的胰腺癌的个体。或者所述对象为胰腺癌患者或者期待治疗胰腺癌的个体的离体胰腺癌细胞。所述psmd7抑制剂可以在接受胰腺癌治疗前、中、后向对象施用。本发明第四方面公开了一种降低胰腺癌细胞中psmd7基因表达的核酸分子,所述核酸分子包含双链rna或shrna。其中,所述双链rna中含有能够与psmd7基因杂交的核苷酸序列;所述shrna中含有能够与psmd7基因杂交的核苷酸序列。进一步的,所述双链rna包含第一链和第二链,所述第一链和所述第二链互补共同形成rna二聚体,并且所述第一链的序列与psmd7基因中的靶序列基本相同。所述psmd7基因中的靶序列即为核酸分子用于特异性沉默psmd7基因表达时,被所述核酸分子识别并沉默的mrna片段所对应的psmd7基因中的片段。进一步的,所述双链rna的靶序列如seqidno:1所示。具体为:5’-tgaggaagttggagttgaa-3’。更进一步的,所述双链rna第一链的序列如seqidno:2所示。具体为5’-ugaggaaguuggaguugaa-3’。进一步的,所述双链rna为小干扰rna(sirna)。seqidno:2为以seqidno:1所示的序列为rna干扰靶序列设计的、针对人psmd7基因的小干扰rna的一条链,另一条链即第二链的序列与第一链序列互补,该sirna可以起到特异性沉默胰腺癌细胞中内源psmd7基因表达的作用。所述shrna包括正义链片段和反义链片段,以及连接所述正义链片段和反义链片段的茎环结构,所述正义链片段和所述反义链片段的序列互补,并且所述正义链片段的序列与psmd7基因中的靶序列基本相同。进一步的,所述shrna的靶序列如seqidno:1所示。所述shrna经酶切加工后可成为小干扰rna(sirna)进而起到特异性沉默胰腺癌细胞中内源psmd7基因表达的作用。进一步的,所述shrna的茎环结构的序列可选自以下任一:uucaagaga、aug、ccc、uucg、ccacc、ctcgag、aagcuu和ccacacc。更进一步的,所述shrna的序列如seqidno:3所示。具体为5’-gcugaggaaguuggaguugaacucgaguucaacuccaacuuccucagc-3’。进一步的,所述psmd7基因来源于人。本发明第五方面,公开了一种psmd7基因干扰核酸构建体,含有编码前述核酸分子中的shrna的基因片段,能表达所述shrna。所述的psmd7基因干扰核酸构建体可以是将编码前述人psmd7基因shrna的基因片段克隆入已知载体获得。进一步的,所述psmd7基因干扰核酸构建体为psmd7基因干扰慢病毒载体。本发明公开的psmd7基因干扰慢病毒载体是将编码前述psmd7基因shrna的dna片段克隆入已知载体获得,所述已知载体多为慢病毒载体,所述psmd7基因干扰慢病毒载体经过病毒包装成为有感染力的病毒颗粒后,感染胰腺癌细胞,进而转录出本发明所述shrna,通过酶切加工等步骤,最终获得所述sirna,用于特异性沉默psmd7基因的表达。进一步的,所述psmd7基因干扰慢病毒载体还含有启动子序列和/或编码胰腺癌细胞中可被检测的标记物的核苷酸序列;较优的,所述可被检测的标记物如绿色荧光蛋白(gfp)。进一步的,所述慢病毒载体可以选自:plko.1-puro、plko.1-cmv-tgfp、plko.1-puro-cmv-tgfp、plko.1-cmv-neo、plko.1-neo、plko.1-neo-cmv-tgfp、plko.1-puro-cmv-tagcfp、plko.1-puro-cmv-tagyfp、plko.1-puro-cmv-tagrfp、plko.1-puro-cmv-tagfp635、plko.1-puro-ubc-turbogfp、plko.1-puro-ubc-tagfp635、plko-puro-iptg-1xlaco、plko-puro-iptg-3xlaco、plp1、plp2、plp/vsv-g、pentr/u6、plenti6/block-it-dest、plenti6-gw/u6-laminshrna、pcdna1.2/v5-gw/lacz、plenti6.2/n-lumio/v5-dest、pgcsil-gfp或plenti6.2/n-lumio/v5-gw/lacz中的任一。本发明实施例具体列举了以pgcsil-gfp为载体构建的人psmd7基因干扰慢病毒载体,命名为pgcsil-gfp-psmd7-sirna。本发明的psmd7基因sirna可用于抑制胰腺癌细胞的增殖,进一步地可以用作治疗胰腺癌的药物或制剂。psmd7基因干扰慢病毒载体则可用于制备所述psmd7基因sirna。当用作治疗胰腺癌的药物或制剂时,是将安全有效量的所述核酸分子施用于哺乳动物。具体剂量还应考虑给药途径、病人健康状况等因素,这些都是熟练医师技能范围之内的。本发明第六方面,公开了一种psmd7基因干扰慢病毒,由前述psmd7基因干扰核酸构建体在慢病毒包装质粒、细胞系的辅助下,经过病毒包装而成。该慢病毒可感染胰腺癌细胞并产生针对psmd7基因的小分子干扰rna,从而抑制胰腺癌细胞的增殖。该psmd7基因干扰慢病毒可用于制备预防或治疗胰腺癌的药物。本发明的第七方面,提供前述核酸分子,或前述psmd7基因干扰核酸构建体,或前述psmd7基因干扰慢病毒的用途,为:用于制备预防或治疗胰腺癌的药物,或用于制备降低胰腺癌细胞中psmd7基因表达的试剂盒。所述预防或治疗胰腺癌的药物的应用为胰腺癌的治疗提供了一种方法,具体为一种预防或治疗对象体内胰腺癌的方法,包括将有效剂量的所述的药物施用于对象中。进一步的,所述药物用于预防或治疗对象体内胰腺癌时,需要将有效剂量的所述的药物施用于对象中。采用该方法,所述胰腺癌的生长、增殖、复发和/或转移被抑制。进一步的,所述胰腺癌的生长、增殖、复发和/或转移的至少10%、20%、30%、40%、50%、60%、70%、80%、90%、95%或99%的部分被抑制。所述方法的对象可以为人。本发明的第八方面,提供一种用于预防或治疗胰腺癌的组合物,其有效物质含有:前述的核酸分子;和/或,前述psmd7基因干扰核酸构建体;和/或,前述psmd7基因干扰慢病毒,以及药学上可接受的载体、稀释剂或赋形剂。所述组合物可以为药物组合物。当所述组合物用于预防或治疗对象体内胰腺癌时,需要将有效剂量的所述的组合物施用于对象中。采用该方法,所述胰腺癌的生长、增殖、复发和/或转移被抑制。进一步的,所述胰腺癌的生长、增殖、复发和/或转移的至少10%、20%、30%、40%、50%、60%、70%、80%、90%、95%或99%的部分被抑制。所述组合物的形式无特殊限制,可以为固体、液体、凝胶、半流质、气雾等各种物质形式。所述组合物主要针对的对象为哺乳动物。所述哺乳动物优选为啮齿目动物、偶蹄目动物、奇蹄目动物、兔形目动物、灵长目动物等。所述灵长目动物优选为猴、猿或人。综上所述,本发明设计了针对人psmd7基因的rnai靶点序列,构建相应的psmd7rnai载体,其中rnai载体pgcsil-gfp-psmd7-sirna能够显著下调psmd7基因在mrna水平和蛋白水平的表达。使用慢病毒(lentivirus,简写为lv)作为基因操作工具携带rnai载体pgcsil-gfp-psmd7-sirna能够靶向地将针对psmd7基因的rnai序列高效导入胰腺癌aspc-1细胞、panc-1细胞,降低psmd7基因的表达水平,显著抑制上述肿瘤细胞的增殖能力。因此慢病毒介导的psmd7基因沉默是恶性肿瘤潜在的临床非手术治疗方式。与现有技术相比,本发明具有如下有益效果:本发明经过广泛而深入的研究发现,采用rnai方法下调人psmd7基因的表达后可有效地抑制胰腺癌细胞的增殖、促进细胞凋亡,可以有效地控制胰腺癌的生长进程。本发明提供的sirna或者包含该sirna序列的核酸构建体、慢病毒能够特异性抑制胰腺癌细胞的增殖能力、抑制胰腺癌细胞克隆、促进胰腺癌细胞凋亡、抑制胰腺癌细胞转移能力、抑制胰腺癌生长;从而治疗胰腺癌,为胰腺癌治疗开辟新的方向。附图说明图1-1:rt-pcr检测aspc-1细胞mrna水平靶基因消减效率。图1-2:rt-pcr检测panc-1细胞mrna水平靶基因消减效率。图2-1:westernblot检测aspc-1细胞靶点降低psmd7基因蛋白水平表达情况。图2-2:westernblot检测panc-1细胞靶点降低psmd7基因蛋白水平表达情况。图3-1:celigo细胞计数法验证psmd7基因对aspc-1细胞增殖影响。(celigo连续5天记录细胞图片)图3-2:celigo细胞计数法验证psmd7基因对aspc-1细胞增殖影响。(shpsmd7组与shctrl对照组细胞数目随时间变化的曲线)图3-3:celigo细胞计数法验证psmd7基因对panc-1细胞增殖影响。(celigo连续5天记录细胞图片)图3-4:celigo细胞计数法验证psmd7基因对panc-1细胞增殖影响。(shpsmd7组与shctrl对照组细胞数目随时间变化的曲线)图4-1:mtt实验检测psmd7基因对aspc-1细胞的增殖能力的影响。图4-2:mtt实验检测psmd7基因对panc-1细胞的增殖能力的影响。图5-1:细胞克隆形成法检测psmd7基因对aspc-1细胞增殖能力的影响,shrna慢病毒感染aspc-1细胞,培养14天后,观察克隆数,为数码相机记录图。图5-2:细胞克隆形成法检测psmd7基因对aspc-1细胞增殖能力的影响,shrna慢病毒感染aspc-1细胞,培养14天后,观察克隆数,柱状结果以细胞克隆数量平均值±标准差显示。图5-3:细胞克隆形成法检测psmd7基因对panc-1细胞增殖能力的影响,shrna慢病毒感染panc-1细胞,培养14天后,观察克隆数,为数码相机记录图。图5-4:细胞克隆形成法检测psmd7基因对panc-1细胞增殖能力的影响,shrna慢病毒感染panc-1细胞,培养14天后,观察克隆数,柱状结果以细胞克隆数量平均值±标准差显示。图6-1:annexinv-apc流式细胞凋亡检测shpsmd7对aspc-1细胞凋亡的影响,为流式细胞凋亡示意图。图6-2:annexinv-apc流式细胞凋亡检测shpsmd7对aspc-1细胞凋亡的影响,柱状结果以细胞百分比平均值±标准差显示。图6-3:annexinv-apc流式细胞凋亡检测shpsmd7对panc-1细胞凋亡的影响,为流式细胞凋亡示意图。图6-4:annexinv-apc流式细胞凋亡检测shpsmd7对panc-1细胞凋亡的影响,柱状结果以细胞百分比平均值±标准差显示。图7-1:transwell实验显示psmd7消减影响aspc-1细胞的转移能力。(显微镜拍照观察图)图7-2:transwell实验显示psmd7消减影响aspc-1细胞的转移能力。(柱状图表示psmd7处理组转移细胞数与对照组(shctrl)转移细胞数比值分析结果)图7-3:transwell实验显示psmd7消减影响panc-1细胞的转移能力。(显微镜拍照观察图)图7-4:transwell实验显示psmd7消减影响panc-1细胞的转移能力。(柱状图表示psmd7处理组转移细胞数与对照组(shctrl)转移细胞数比值分析结果)图8-1:划痕愈合实验检测shpsmd7基因对aspc-1细胞转移水平的影响。图为通过celigo扫板获得迁移0h和48h后的图片。图8-2:划痕愈合实验检测shpsmd7基因对aspc-1细胞迁移率的影响。图8-3:划痕愈合实验检测shpsmd7基因对panc-1细胞转移水平的影响。图为通过celigo扫板获得迁移0h和48h后的图片。图8-4:划痕愈合实验检测shpsmd7基因对panc-1细胞迁移率的影响。附图中,柱形图代表三次实验的平均值,误差线表示标准偏差(sd)。**,shctrl与目的基因shrna慢病毒处理组相比,p<0.01。*,shctrl与目的基因shrna慢病毒处理组相比,0.01≤p<0.05。具体实施方式本发明人发现,采用rnai方法下调人psmd7基因的表达后可有效地抑制肿瘤细胞的增殖、克隆形成,促进细胞凋亡,降低肿瘤细胞转移和划痕愈合能力等,可以有效地控制肿瘤的生长进程,这一研究成果表明psmd7基因是原癌基因,可作为肿瘤治疗的靶点。发明人进一步合成和测试了多种针对psmd7基因的sirna,筛选出了可有效抑制psmd7的表达进而抑制人胰腺癌aspc-1细胞、胰腺癌panc-1细胞增殖和生长的sirna,在此基础上完成了本发明。psmd7抑制剂指对于psmd7具有抑制效果的分子。对于psmd7具有抑制效果包括但不限于:抑制psmd7的表达或活性。抑制psmd7活性是指使psmd7活力下降。优选地,相比抑制前,psmd7活力下降至少10%,较佳的降低至少30%,再佳的降低至少50%,更佳的降低至少70%,最佳的降低至少90%。抑制psmd7表达具体的可以是抑制psmd7基因的转录或翻译,具体的,可以是指:使psmd7的基因不转录,或降低psmd7的基因的转录活性,或者使psmd7的基因不翻译,或降低psmd7的基因的翻译水平。本领域技术人员可以使用常规方法对psmd7的基因表达进行调节,如基因敲除、同源重组,干扰rna等。psmd7的基因表达的抑制可以通过pcr及westernblot检测表达量验证。优选地,与野生型相比,psmd7基因表达降低至少10%,较佳的降低至少30%,再佳的降低至少50%,更佳的降低至少70%,又佳的降低至少90%,最佳地psmd7基因完全没有表达。小分子化合物本发明中指由几个或几十个原子组成,分子质量在1000以下的化合物。制备预防或治疗胰腺癌的药物可以利用降低胰腺癌细胞中psmd7基因表达的核酸分子;和/或,psmd7基因干扰核酸构建体;和/或,psmd7基因干扰慢病毒,作为有效成分,制备预防或治疗胰腺癌的药物。通常,所述药物中除了有效成分外,根据不同剂型的需要,还会包括一种或多种药学上可接受的载体或辅料。“药学上可接受的”是指当分子本体和组合物适当地给予动物或人时,它们不会产生不利的、过敏的或其它不良反应。“药学上可接受的载体或辅料”应当与所述有效成分相容,即能与其共混而不会在通常情况下大幅度降低药物的效果。可作为药学上可接受的载体或辅料的一些物质的具体例子是糖类,如乳糖、葡萄糖和蔗糖;淀粉,如玉米淀粉和土豆淀粉;纤维素及其衍生物,如甲基纤维素钠、乙基纤维素和甲基纤维素;西黄蓍胶粉末;麦芽;明胶;滑石;固体润滑剂,如硬脂酸和硬脂酸镁;硫酸钙;植物油,如花生油、棉籽油、芝麻油、橄榄油、玉米油和可可油;多元醇,如丙二醇、甘油、山梨糖醇、甘露糖醇和聚乙二醇;海藻酸;乳化剂,如tween;润湿剂,如月桂基硫酸钠;着色剂;调味剂;压片剂、稳定剂;抗氧化剂;防腐剂;无热原水;等渗盐溶液;和磷酸盐缓冲液等。这些物质根据需要用于帮助配方的稳定性或有助于提高活性或它的生物有效性或在口服的情况下产生可接受的口感或气味。本发明中,除非特别说明,药物剂型并无特别限定,可以被制成针剂、口服液、片剂、胶囊、滴丸、喷剂等剂型,可通过常规方法进行制备。药物剂型的选择应与给药方式相匹配。在进一步描述本发明具体实施方式之前,应理解,本发明的保护范围不局限于下述特定的具体实施方案;还应当理解,本发明实施例中使用的术语是为了描述特定的具体实施方案,而不是为了限制本发明的保护范围。下列实施例中未注明具体条件的试验方法,通常按照常规条件,或者按照各制造商所建议的条件。当实施例给出数值范围时,应理解,除非本发明另有说明,每个数值范围的两个端点以及两个端点之间任何一个数值均可选用。除非另外定义,本发明中使用的所有技术和科学术语与本
技术领域:
技术人员通常理解的意义相同。除实施例中使用的具体方法、设备、材料外,根据本
技术领域:
的技术人员对现有技术的掌握及本发明的记载,还可以使用与本发明实施例中所述的方法、设备、材料相似或等同的现有技术的任何方法、设备和材料来实现本发明。除非另外说明,本发明中所公开的实验方法、检测方法、制备方法均采用本
技术领域:
常规的分子生物学、生物化学、染色质结构和分析、分析化学、细胞培养、重组dna技术及相关领域的常规技术。实施例1针对人psmd7基因rnai慢病毒的制备1.筛选针对人psmd7基因的有效的sirna靶点从genbank调取psmd7(nm_002811)基因信息;设计针对psmd7基因的有效的sirna靶点。表1-1列出了筛选出的针对psmd7基因的有效sirna靶点序列。表1-1靶向于人psmd7基因的sirna靶点序列seqidnotargetseq(5’-3’)1tgaggaagttggagttgaa2.慢病毒载体的制备针对sirna靶点(以seqidno:1为例)合成两端含agei和ecori酶切位点粘端的双链dnaoligo序列(表1-2);以agei和ecori限制性内切酶作用于pgcsil-gfp载体(上海吉凯基因医学科技股份有限公司提供),使其线性化,琼脂糖凝胶电泳鉴定酶切片段。表1-2两端含agei和ecori酶切位点粘端的双链dnaoligo通过t4dna连接酶将双酶切线性化(酶切体系如表1-4所示,37℃,反应1h)的载体dna和纯化好的双链dnaoligo连接,在适当的缓冲体系(连接体系如表1-5所示)中于16℃连接过夜,回收连接产物。将连接产物转化氯化钙制备的新鲜的大肠杆菌感受态细胞(转化操作参考:分子克隆实验指南第二版55-56页)。在连接转化产物长出菌克隆表面沾一下,溶于10μllb培养基,混匀取1μl作为模板;在以慢病毒载体中rnai序列的上下游,设计通用pcr引物,上游引物序列:5’-cctatttcccatgattccttcata-3’(seqidno:6);下游引物序列:5’-gtaatacggttatccacgcg-3’(seqidno:7),进行pcr鉴定实验(pcr反应体系如表1-6,反应条件如表1-7)。对pcr鉴定阳性的克隆进行测序和比对分析,比对正确的克隆即为构建成功的针对seqidno:1的表达rnai的载体,命名为pgcsil-gfp-psmd7-sirna。构建pgcsil-gfp-scr-sirna阴性对照质粒,阴性对照sirna靶序列为5’-ttctccgaacgtgtcacgt-3’(seqidno:8)。构建pgcsil-gfp-scr-sirna阴性对照质粒时,针对scrsirna靶点合成两端含agei和ecori酶切位点粘端的双链dnaoligo序列(表1-3),其余构建方法、鉴定方法及条件均同pgcsil-gfp-psmd7-sirna。表1-3两端含agei和ecori酶切位点粘端的双链dnaoligo表1-4pgcsil-gfp质粒酶切反应体系试剂体积(μl)pgcsil-gfp质粒(1μg/μl)2.010×buffer5.0100×bsa0.5agei(10u/μl)1.0ecori(10u/μl)1.0ddh2o40.5total50.0表1-5载体dna和双链dnaoligo连接反应体系试剂阳性对照(μl)自连对照(μl)连接组(μl)线性化的载体dna(100ng/μl)1.01.01.0退火的双链dnaoligo(100ng/μl)1.0-1.010×t4噬菌体dna连接酶缓冲液1.01.01.0t4噬菌体dna连接酶1.01.01.0ddh2o16.017.016.0total20.020.020.0表1-6-1pcr反应体系表1-7pcr反应体系程序设定3.包装psmd7-sirna慢病毒以qiagen公司的质粒抽提试剂盒提取rnai质粒pgcsil-gfp-psmd7-sirna的dna,配制成100ng/μl储存液。转染前24h,用胰蛋白酶消化对数生长期的人胚肾细胞293t细胞,以含10%胎牛血清的dmem完全培养基调整细胞密度为1.5×105细胞/ml,接种于6孔板,37℃,5%co2培养箱内培养。待细胞密度达70%-80%时即可用于转染。转染前2h,吸出原有培养基,加入1.5ml新鲜的完全培养基。按照sigma-aldrich公司的missionlentiviralpackagingmix试剂盒的说明,向一灭菌离心管中加入packingmix(pvm)20μl,pei12μl,无血清dmem培养基400μl,取20μl上述抽提的质粒dna,加至上述pvm/pei/dmem混合液。将上述转染混和物在室温下孵育15min,转移至人胚肾细胞293t细胞的培养基中,37℃,5%co2培养箱内培养16h。弃去含有转染混和物的培养介质,pbs溶液洗涤,加入完全培养基2ml,继续培养48h。收集细胞上清液,centriconplus-20离心超滤装置(millipore)纯化和浓缩慢病毒,步骤如下:(1)4℃,4000g离心10min,除去细胞碎片;(2)0.45μm滤器过滤上清液于40ml超速离心管中;(3)4000g离心,10-15min,至需要的病毒浓缩体积;(4)离心结束后,将过滤杯和下面的滤过液收集杯分开,将过滤杯倒扣在样品收集杯上,离心2min离心力不超过1000g;(5)把离心杯从样品收集杯上移开,样品收集杯中的即为病毒浓缩液。将病毒浓缩液分装后于-80摄氏度保存。病毒浓缩液中含有的sirna的第一链的序列如seqidno:2所示。对照慢病毒的包装过程同psmd7-sirna慢病毒,仅以pgcsil-gfp-scr-sirna载体代替pgcsil-gfp-psmd7-sirna载体。实施例2实时荧光定量rt-pcr法检测基因的沉默效率处于对数生长期的人胰腺癌aspc-1细胞、panc-1细胞进行胰酶消化,制成细胞悬液(细胞数约为5×104/ml)接种于6孔板中,培养至细胞融合度达到约30%。根据侵染复数值(moi,aspc-1:20,panc-1:20),加入适宜量的实施例1制备的慢病毒,培养24h后更换培养基,待侵染时间达到5天后,收集细胞。根据invitrogen公司的trizol操作说明书,抽提总rna。根据promega公司的m-mlv操作说明书,将rna逆转录获得cdna(逆转录反应体系见表2-1,42℃反应1h,然后在70℃水浴锅中水浴10min使逆转录酶失活)。采用tp800型realtimepcr仪(takara)进行实时定量检测。psmd7基因的引物如下:上游引物5’-acctggaaaaagtcgccaca-3’(seqidno:11)和下游引物5’-atcagcgaggccaagtacac-3’(seqidno:12)。以管家基因gapdh为内参,引物序列如下:上游引物5’-tgacttcaacagcgacaccca-3’(seqidno:13)和下游引物5’-caccctgttgctgtagccaaa-3’(seqidno:14)。按表2-2中的比例配置反应体系。表2-1逆转录反应体系试剂体积(μl)5×rtbuffer4.010mmdntps2.0rnasin0.4m-mlv-rtase1.0rnase-free2.6total10.0表2-2real-timepcr反应体系试剂体积(μl)sybrpremixextaq:6.0引物mix(5μm):0.3cdna0.6ddh2o5.1total12.0sybrpremixextaq:6.0设定程序为两步法real-timepcr:预变性95℃,15s;之后每一步变性95℃,5s;退火延伸60℃,30s;共进行40个循环。每次在延伸阶段读取吸光值。pcr结束后,95℃变性15s,然后冷却至60℃,使dna双链充分结合。从60℃开始到95℃,每一步增加0.5℃,保持4s,同时读取吸光值,制作熔解曲线。采用2-δδct分析法计算侵染了psmd7mrna的表达丰度。侵染对照病毒的细胞作为对照。实验结果如图1-1和图1-2所示,表明人胰腺癌aspc-1细胞中psmd7mrna的表达水平下调了69.5%;人胰腺癌panc-1细胞中psmd7mrna的表达水平下调了80.7%。实施例3westernblot检测靶点降低psmd7基因蛋白水平表达1.细胞总蛋白抽提(1)将对照病毒和针对psmd7干扰靶点的rnai病毒,分别感染目的细胞(人胰腺癌aspc-1细胞、胰腺癌panc-1细胞,侵染复数moi,aspc-1:20,panc-1:20)。接收细胞样本,用pbs洗涤两次。取适当量的ripa裂解液,使用前数分钟内加入pmsf,使pmsf的最终浓度为1mm。(使用ripa裂解液,说明书链接:http://www.beyotime.com/ripa-lysis-bufferm.htm)(2)加入适当量的ripa裂解液,冰上裂解10-15min。细胞刮刮下细胞转移入新的ep管中,然后超声破碎细胞(40w共20次,每次1s,间隔2s)。(3)4℃、12000g,离心15min,取上清bca法测定蛋白浓度(bcaproteinassaykit说明书链接:http://www.beyotime.com/p0010s.htm)加入新的裂解液将每个样品蛋白浓度调为一致,一般为2μg/μl。然后加入1/5体积的6xloadingbuffer混匀,100度金属浴煮10min,短暂离心后-80℃保存备用。2.sds-page(1)制胶:根据目的蛋白分子量大小配制不同浓度的胶,具体体系如表3-1、表3-2、表3-3所示:表3-1sds-page分离胶(8ml体系)分离胶(8ml体系)8%9%10%12%13%15%h2o3.73.43.12.62.31.830%page2.12.42.73.23.541.5mol/ltris(ph8.8)22222210%sds0.080.080.080.080.080.0810%aps0.080.080.080.080.080.08temed0.0050.0040.0040.0040.0040.004表3-2sds-page分离胶(10ml体系)分离胶(10ml体系)8%9%10%12%13%15%h2o4.64.343.32.92.330%page2.733.344.451.5mol/ltris(ph8.8)2.52.52.52.52.52.510%sds0.10.10.10.10.10.110%aps0.10.10.10.10.10.1temed0.0060.0040.0040.0040.0040.004表3-3sds-page浓缩胶浓缩胶(5%)3ml4ml5mlh2o2.12.73.430%page0.50.670.831.0mol/ltris(ph6.8)0.380.50.6310%sds0.030.040.0510%aps0.030.040.05temed0.0030.0040.005(2)上样:胶凝固后,拔去梳子,电泳缓冲液清洗上样孔,将准备好的样品上样。(3)电泳:浓缩胶80ma,20min;分离胶120ma,1h。3.免疫印迹(湿转)电泳结束后,使用转移电泳装置,在4℃、300ma恒流条件下电转150min,将蛋白转移到pvdf膜上。4.抗体杂交:(1)封闭:用封闭液(含5%脱脂牛奶的tbst溶液)室温封闭pvdf膜1h或4℃过夜。(2)一抗孵育:封闭液稀释抗体,然后与封闭好的pvdf膜室温孵育2h或4℃过夜,并用tbst洗膜4次,每次8min。(3)二抗孵育:用封闭液稀释相应的二抗,室温下孵育pvdf膜1.5h,并用tbst洗膜4次,每次8min。5.x光显影(采用cst公司20xreagentand20xperoxide#7003试剂盒,说明书链接:https://www.cst-c.com.cn/products/wb-ip-reagents/20x-lumiglo-reagent-and-20x-peroxide/7003?site-search-type=products):(1)将试剂盒中a液和b液按1:1比例混合颠倒混匀,放置数分钟后可使用。(2)将膜取出,吸水纸擦干,平铺入暗盒,滴加适量混匀的ecl发光液,铺上保鲜膜(避免产生气泡),放上x光片(避免x光片的移动),关上暗盒,曝光1s-数min(曝光时间需要多尝试几次,根据肉眼能否看见荧光以及荧光的强弱无适当调整曝光时间)。(3)取出x光片,放入显影液中,出现条带后取出,在清水中漂洗几秒钟,后放入定影液中至少2min。(4)取出x光片,晾干,分析。如图2-1和图2-2所示,westernblot实验表明靶点对psmd7基因的内源表达有敲减作用,因而是有效靶点。实施例4检测侵染了psmd7-sirna慢病毒的肿瘤细胞的增殖能力(celigo实验)处于对数生长期的人胰腺癌aspc-1细胞、panc-1细胞进行胰酶消化,制成细胞悬液(细胞数约为5×104/ml)接种于6孔板中,培养至细胞融合度达到约30%。根据侵染复数(moi,aspc-1:20,panc-1:20),加入适宜量的病毒,培养24h后更换培养基,待侵染时间达到3天后,收集处于对数生长期的各实验组细胞。完全培养基重悬成细胞悬液(2×104/ml),以细胞密度约为1500个/孔,接种96孔板。每组5个复孔,每孔100μl。铺好板后,置37℃、5%co2培养箱培养。从铺板后第二天开始,每天用celigo仪器(nexcelom)检测读板一次,连续检测读板5天。通过调整analysissettings的输入参数,准确地计算出每次扫描孔板中的带绿色荧光的细胞的数量,对数据进行统计绘图,绘出细胞增殖曲线(结果如图3-1、3-2、3-3和3-4所示)。结果表明,慢病毒侵染组人胰腺癌aspc-1细胞、panc-1细胞在细胞体外培养5天后,增殖速度显著减缓,远低于对照组胰腺癌细胞的增殖速度,其中,人胰腺癌aspc-1细胞的活力细胞数目下降了49.21%,人胰腺癌panc-1细胞的活力细胞数目下降了81.31%,表明psmd7基因沉默导致人胰腺癌细胞增殖能力被抑制。实施例5检测侵染了psmd7-sirna慢病毒的肿瘤细胞的增殖能力(mtt实验)处于对数生长期的人胰腺癌aspc-1细胞、panc-1细胞进行胰酶消化,制成细胞悬液(细胞数约为5×104/ml)接种于6孔板中,培养至细胞融合度达到约30%。根据侵染复数(moi,aspc-1:20,panc-1:20),加入适宜量的病毒,培养24h后更换培养基,收集处于对数生长期的各实验组细胞胰酶消化后,完全培养基重悬成细胞悬液,并计数。根据细胞生长快慢决定铺板细胞密度(2000cell/well),每组3重复,统一铺好后,待细胞完全沉淀下来后,在显微镜下观察各实验组的细胞密度,如果密度不均匀,则固定一组,微调其他组细胞的量再次铺板(如:发现con组细胞较多,降低细胞量再次铺板),放入细胞培养箱中培养。从铺板后第二天开始,培养终止前4h加入20μl5mg/ml的mtt于孔中,无需换液。4h后完全吸去培养液,注意不要吸掉孔板底部的甲瓒颗粒,加100μldmso溶解甲瓒颗粒。振荡器振荡2-5min,酶标仪490/570nm检测od值。数据统计分析。结果如图4-1和4-2所示,检测侵染了psmd7-sirna慢病毒的人胰腺癌aspc-1细胞的细胞活力下降比例为46.76%;人胰腺癌panc-1细胞的细胞活力下降比例为76.58%。表明psmd7基因沉默导致人胰腺癌细胞增殖能力被抑制。实施例6侵染psmd7-sirna慢病毒的肿瘤细胞克隆形成能力的检测(克隆形成实验)将人胰腺癌aspc-1细胞、胰腺癌panc-1细胞胰酶消化后接种于12孔板中,细胞密度为10-15%。第二天换为新鲜的培养基,内含5ug/mlpolybrene。将psmd7-sirna慢病毒按照侵染复数(moi,aspc-1:20,panc-1:20)加入到培养板中,感染12-24h后换新鲜的培养基。感染72h后,荧光显微镜下观察荧光,感染效率达到80%。将处于对数生长期的感染病毒后的细胞胰酶消化后,完全培养基重悬成细胞悬液;细胞计数后接种于6孔板中(1000个细胞/孔),将接种好的细胞于培养箱中继续培养到8天,中途隔3天进行换液并观察细胞状态;实验终止前荧光显微镜下对细胞克隆进行拍照;实验终止时用多聚甲醛固定细胞,pbs洗涤细胞后,giemsa染色,拍照。结果如图5-1、5-2、5-3和5-4所示,与对照干扰(nc组)相比,rna干扰降低psmd7基因的表达(kd组)后,人胰腺癌细胞aspc-1细胞、panc-1细胞形成的克隆斑数目显著减少、克隆斑的体积明显减小;表明psmd7基因沉默导致人胰腺癌细胞形成克隆的能力下降。平板克隆形成实验检测降低psmd7基因的表达后,人胰腺癌细胞的克隆形成能力下降。实施例7侵染psmd7-sirna慢病毒的肿瘤细胞凋亡水平检测(facs细胞凋亡检测)将人胰腺癌aspc-1细胞、胰腺癌panc-1细胞胰酶消化后接种于12孔板中细胞密度为10-15%。第二天换为新鲜的培养基,内含5ug/mlpolybrene。将psmd7-sirna慢病毒按照侵染复数(moi,aspc-1:20,panc-1:20)加入到培养板中,感染12-24h后换新鲜的培养基。感染72h后,荧光显微镜下观察荧光,感染效率达到90%。染复数加入到培养板中,感染12-24h后换新鲜的培养基。感染72h后,荧光显微镜下观察荧光,感染效率达到90%。将处于对数生长期的细胞胰酶消化后,完全培养基重悬成细胞悬液;与上清细胞收集于同一5ml离心管中,每组设三个复孔(为保证上机细胞数足够,细胞数目≥5×105/处理)。1300rmp离心5min,弃上清,4℃预冷的pbs洗涤细胞沉淀。1×bindingbuffer洗涤细胞沉淀一次,1300rmp、3min离心,收集细胞。200μl1×bindingbuffer(ebioscience,88-8007)重悬细胞沉淀。加入10μlannexinv-apc(ebioscience,88-8007)染色,室温避光10-15min。根据细胞量,补加400-800μl1×bindingbuffer,上流式细胞仪进行检测。对结果进行分析。如图6-1、6-2、6-3和图6-4所示,annexinv单染法检测降低psmd7基因的表达后,人胰腺癌aspc-1细胞、panc-1细胞的细胞凋亡比例的变化。发现下调psmd7基因表达后人胰腺癌细胞的凋亡比例增加。与对照干扰(nc组),rna干扰降低基因的表达(kd组)后,凋亡人胰腺癌细胞数显著增多;表明psmd7基因沉默导致人胰腺癌细胞凋亡。实施例8侵染psmd7-sirna慢病毒的肿瘤细胞转移水平检测(transwell转移)取出试剂盒,将所需数目的小室置于新的24孔板中,上室加100μl无血清培养基,37℃培养箱中放置1h。制备无血清人胰腺癌aspc-1细胞、胰腺癌panc-1细胞(侵染psmd7-sirna慢病毒后的细胞,侵染复数moi,aspc-1:20,panc-1:20)悬浮液,并计数,细胞数根据预实验调整,一般为105/孔(24孔板)。小心除去上室中培养基并加入100μl细胞悬液,下室内加入600μl30%fbs培养基。同时,使用该细胞悬液铺一块mts96孔板,每孔约接种5000个细胞,接种后即测定od570,作为转移参照。37℃培养箱培养一段时间(具体时间根据预实验调整,本实验中均为48小时)。倒扣小室于吸水纸上以去除培养基,用棉拭子轻轻移去小室内非转移细胞,滴2-3滴giemsa染色液到膜的下表面染色转移细胞3-5min后,将小室浸泡冲洗数次,空气晾干。显微镜拍照:每个transwell小室,随机选取视野,拍100x照片4张,200x照片9张。以200x的照片来计数,进行数据分析,比较实验组与对照组细胞转移能力的差异:计算各组转移细胞数(migratorycellsperfield),标准差,t-test分析得到p值,判断是否有显著性差异(p<0.05,有显著性差异,否则无显著性差异)结果如图7-1、7-2、7-3和7-4所示,与对照干扰(nc组)相比,rna干扰降低psmd7基因的表达(kd组)后,发现下调psmd7基因表达后人胰腺癌aspc-1细胞、panc-1细胞的转移能力降低。实施例9侵染慢病毒的肿瘤细胞转移水平检测(划痕愈合实验)按照实验设计的组别,在孔中加入约4×104个感染后的人胰腺癌aspc-1细胞、panc-1细胞(侵染复数moi,aspc-1:20,panc-1:20,),以次日细胞达到90%以上汇合度为准。第二天换低浓度血清培养基,使用划痕仪对准96孔板的下端中央部位,向上轻推形成划痕。使用无血清培养基轻轻漂洗2-3遍,加入低浓度血清培养基(如0.5%fbs),拍照。37℃、5%co2培养箱培养,根据预实验选择合适的时间点拍照(0h、8h、24h)。荧光显微镜拍照(以96孔中心阴影区域为参照,划痕在图片的正中)。根据划痕后的图片,计算各组细胞迁移率。如图8-1、8-2、8-3和8-4所示,与对照干扰(nc组)相比,rna干扰降低psmd7基因的表达(kd组)后,发现下调psmd7基因表达后人胰腺癌aspc-1细胞、panc-1细胞的转移能力降低。以上所述,仅为本发明的较佳实施例,并非对本发明任何形式上和实质上的限制,应当指出,对于本
技术领域:
的普通技术人员,在不脱离本发明方法的前提下,还将可以做出若干改进和补充,这些改进和补充也应视为本发明的保护范围。凡熟悉本专业的技术人员,在不脱离本发明的精神和范围的情况下,当可利用以上所揭示的技术内容而做出的些许更动、修饰与演变的等同变化,均为本发明的等效实施例;同时,凡依据本发明的实质技术对上述实施例所作的任何等同变化的更动、修饰与演变,均仍属于本发明的技术方案的范围内。序列表<110>江南大学附属医院<120>人psmd7基因的用途及相关产品<160>14<170>siposequencelisting1.0<210>1<211>19<212>dna<213>人工序列(artificialsequence)<400>1tgaggaagttggagttgaa19<210>2<211>19<212>rna<213>人工序列(artificialsequence)<400>2ugaggaaguuggaguugaa19<210>3<211>48<212>rna<213>人工序列(artificialsequence)<400>3gcugaggaaguuggaguugaacucgaguucaacuccaacuuccucagc48<210>4<211>58<212>dna<213>人工序列(artificialsequence)<400>4ccgggctgaggaagttggagttgaactcgagttcaactccaacttcctcagctttttg58<210>5<211>58<212>dna<213>人工序列(artificialsequence)<400>5aattcaaaaagctgaggaagttggagttgaactcgagttcaactccaacttcctcagc58<210>6<211>24<212>dna<213>人工序列(artificialsequence)<400>6cctatttcccatgattccttcata24<210>7<211>20<212>dna<213>人工序列(artificialsequence)<400>7gtaatacggttatccacgcg20<210>8<211>19<212>dna<213>人工序列(artificialsequence)<400>8ttctccgaacgtgtcacgt19<210>9<211>54<212>dna<213>人工序列(artificialsequence)<400>9ccggttctccgaacgtgtcacgtctcgagacgtgacacgttcggagaatttttg54<210>10<211>54<212>dna<213>人工序列(artificialsequence)<400>10aattcaaaaattctccgaacgtgtcacgtctcgagacgtgacacgttcggagaa54<210>11<211>20<212>dna<213>人工序列(artificialsequence)<400>11acctggaaaaagtcgccaca20<210>12<211>20<212>dna<213>人工序列(artificialsequence)<400>12atcagcgaggccaagtacac20<210>13<211>21<212>dna<213>人工序列(artificialsequence)<400>13tgacttcaacagcgacaccca21<210>14<211>21<212>dna<213>人工序列(artificialsequence)<400>14caccctgttgctgtagccaaa21当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1